
First-Principles Investigation into the Interaction of H2O with α-CsPbI3 and the Intrinsic Defects within It

Abstract
1. Introduction
2. Calculation
3. Results and Discussion
3.1. Comparison of Adsorption of H2O on the Surface and in the Bulk of CsPbI3
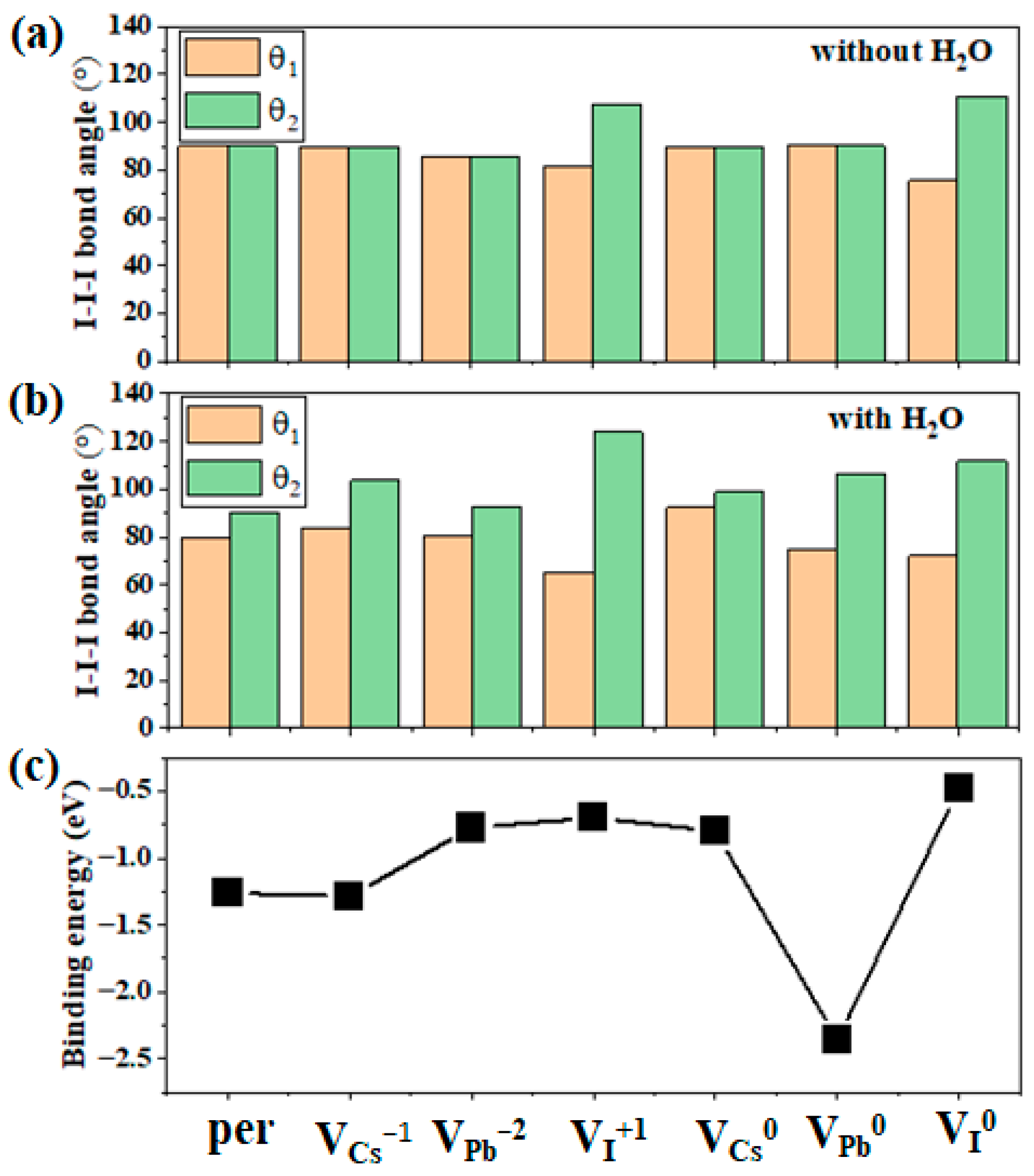
3.2. Effect of Intrinsic Vacancies within the Bulk Phase on H2O Insertion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jeon, N.J.; Na, H.; Jung, E.H.; Yang, T.-Y.; Lee, Y.G.; Kim, G.; Shin, H.-W.; Seok, S.I.; Lee, J.; Seo, J. A fluorene-terminated hole-transporting material for highly efficient and stable perovskite solar cells. Nat. Energy 2018, 3, 682–689. [Google Scholar] [CrossRef]
- Yang, W.S.; Park, B.-W.; Jung, E.H.; Jeon, N.J.; Kim, Y.C.; Lee, D.U.; Shin, S.S.; Seo, J.; Kim, E.K.; Noh, J.H.; et al. Iodide management in formamidinium-lead-halide–based perovskite layers for efficient solar cells. Science 2017, 356, 1376–1379. [Google Scholar] [CrossRef]
- Saliba, M.; Matsui, T.; Seo, J.-Y.; Domanski, K.; Correa-Baena, J.-P.; Nazeeruddin, M.K.; Zakeeruddin, S.M.; Tress, W.; Abate, A.; Hagfeldt, A.; et al. Cesium-containing triple cation perovskite solar cells: Improved stability, reproducibility and high efficiency. Energy Environ. Sci. 2016, 9, 1989–1997. [Google Scholar] [CrossRef]
- Lee, J.; Kim, D.; Kim, H.; Seo, S.; Cho, S.M.; Park, N. Formamidinium and Cesium Hybridization for Photo- and Moisture-Stable Perovskite Solar Cell. Adv. Energy Mater. 2015, 5, 1501310. [Google Scholar] [CrossRef]
- Bi, D.; Tress, W.; Dar, M.I.; Gao, P.; Luo, J.; Renevier, C.; Schenk, K.; Abate, A.; Giordano, F.; Baena, J.-P.C.; et al. Efficient luminescent solar cells based on tailored mixed-cation perovskites. Sci. Adv. 2016, 2, e1501170. [Google Scholar] [CrossRef] [PubMed]
- Kojima, A.; Teshima, K.; Shirai, Y.; Miyasaka, T. Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells. J. Am. Chem. Soc. 2009, 131, 6050–6051. [Google Scholar] [CrossRef] [PubMed]
- Conings, B.; Drijkoningen, J.; Gauquelin, N.; Babayigit, A.; D’Haen, J.; D’Olieslaeger, L.; Ethirajan, A.; Verbeeck, J.; Manca, J.; Mosconi, E.; et al. Intrinsic Thermal Instability of Methylammonium Lead Trihalide Perovskite. Adv. Energy Mater. 2015, 5, 1500477. [Google Scholar] [CrossRef]
- Nagabhushana, G.P.; Shivaramaiah, R.; Navrotsky, A. Direct calorimetric verification of thermodynamic instability of lead halide hybrid perovskites. Proc. Natl. Acad. Sci. USA 2016, 113, 7717–7721. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Luo, J.; Meloni, S.; Boziki, A.; Ashari-Astani, N.; Grätzel, C.; Zakeeruddin, S.M.; Röthlisberger, U.; Grätzel, M. Entropic stabilization of mixed A-cation ABX3 metal halide perovskites for high performance perovskite solar cells. Energy Environ. Sci. 2015, 9, 656–662. [Google Scholar] [CrossRef]
- Colella, S.; Mosconi, E.; Fedeli, P.; Listorti, A.; Gazza, F.; Orlandi, F.; Ferro, P.; Besagni, T.; Rizzo, A.; Calestani, G.; et al. MAPbI3-xClx Mixed Halide Perovskite for Hybrid Solar Cells: The Role of Chloride as Dopant on the Transport and Structural Properties. Chem. Mater. 2013, 25, 4613–4618. [Google Scholar] [CrossRef]
- Saliba, M.; Matsui, T.; Domanski, K.; Seo, J.-Y.; Ummadisingu, A.; Zakeeruddin, S.M.; Correa-Baena, J.-P.; Tress, W.R.; Abate, A.; Hagfeldt, A.; et al. Incorporation of rubidium cations into perovskite solar cells improves photovoltaic performance. Science 2016, 354, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, W.; Khan, J.; Niu, G.; Tang, J. Inorganic CsPbI3 Perovskite-Based Solar Cells: A Choice for a Tandem Device. Sol. RRL 2017, 1, 1700048. [Google Scholar] [CrossRef]
- Eperon, G.E.; Paterno, G.M.; Sutton, R.J.; Zampetti, A.; Haghighirad, A.A.; Cacialli, F.; Snaith, H.J. Inorganic caesium lead iodide perovskite solar cells. J. Mater. Chem. A 2015, 3, 19688–19695. [Google Scholar] [CrossRef]
- Zhou, W.; Zhao, Y.; Zhou, X.; Fu, R.; Li, Q.; Zhao, Y.; Liu, K.; Yu, D.; Zhao, Q. Light-Independent Ionic Transport in Inorganic Perovskite and Ultrastable Cs-Based Perovskite Solar Cells. J. Phys. Chem. Lett. 2017, 8, 4122–4128. [Google Scholar] [CrossRef]
- Chen, C.; Lin, H.; Chiang, K.; Tsai, W.; Huang, Y.; Tsao, C.; Lin, H. All-Vacuum-Deposited Stoichiometrically Balanced Inorganic Cesium Lead Halide Perovskite Solar Cells with Stabilized Efficiency Exceeding 11%. Adv. Mater. 2017, 29, 1605290. [Google Scholar] [CrossRef]
- Dastidar, S.; Hawley, C.J.; Dillon, A.D.; Gutierrez-Perez, A.D.; Spanier, J.E.; Fafarman, A.T. Quantitative Phase-Change Thermodynamics and Metastability of Perovskite-Phase Cesium Lead Iodide. J. Phys. Chem. Lett. 2017, 8, 1278–1282. [Google Scholar] [CrossRef]
- Frolova, L.A.; Anokhin, D.V.; Piryazev, A.A.; Luchkin, S.Y.; Dremova, N.N.; Stevenson, K.J.; Troshin, P.A. Highly Efficient All-Inorganic Planar Heterojunction Perovskite Solar Cells Produced by Thermal Coevaporation of CsI and PbI2. J. Phys. Chem. Lett. 2016, 8, 67–72. [Google Scholar] [CrossRef]
- Kim, Y.G.; Kim, T.-Y.; Oh, J.H.; Choi, K.S.; Kim, Y.-J.; Kim, S.Y. Cesium lead iodide solar cells controlled by annealing temperature. Phys. Chem. Chem. Phys. 2017, 19, 6257–6263. [Google Scholar] [CrossRef]
- Brgoch, J.; Lehner, A.J.; Chabinyc, M.; Seshadri, R. Ab Initio Calculations of Band Gaps and Absolute Band Positions of Polymorphs of RbPbI3 and CsPbI3: Implications for Main-Group Halide Perovskite Photovoltaics. J. Phys. Chem. C 2014, 118, 27721–27727. [Google Scholar] [CrossRef]
- Eaton, S.W.; Lai, M.; Gibson, N.A.; Wong, A.B.; Dou, L.; Ma, J.; Wang, L.-W.; Leone, S.R.; Yang, P. Lasing in robust cesium lead halide perovskite nanowires. Proc. Natl. Acad. Sci. USA 2016, 113, 1993–1998. [Google Scholar] [CrossRef]
- Trots, D.; Myagkota, S. High-temperature structural evolution of caesium and rubidium triiodoplumbates. J. Phys. Chem. Solids 2008, 69, 2520–2526. [Google Scholar] [CrossRef]
- Stoumpos, C.C.; Mao, L.; Malliakas, C.D.; Kanatzidis, M.G. Structure–Band Gap Relationships in Hexagonal Polytypes and Low-Dimensional Structures of Hybrid Tin Iodide Perovskites. Inorg. Chem. 2016, 56, 56–73. [Google Scholar] [CrossRef]
- Lin, J.; Lai, M.; Dou, L.; Kley, C.S.; Chen, H.; Peng, F.; Sun, J.; Lu, D.; Hawks, S.A.; Xie, C.; et al. Thermochromic halide perovskite solar cells. Nat. Mater. 2018, 17, 261–267. [Google Scholar] [CrossRef]
- Zhao, X.-G.; Dalpian, G.M.; Wang, Z.; Zunger, A. Polymorphous nature of cubic halide perovskites. Phys. Rev. B 2020, 101, 155137. [Google Scholar] [CrossRef]
- Yang, R.X.; Tan, L.Z. First-Principles Characterization of Surface Phonons of Halide Perovskite CsPbI3 and Their Role in Stabilization. J. Phys. Chem. Lett. 2021, 12, 9253–9261. [Google Scholar] [CrossRef]
- Kye, Y.-H.; Yu, C.-J.; Jong, U.-G.; Ri, K.-C.; Kim, J.-S.; Choe, S.-H.; Hong, S.-N.; Li, S.; Wilson, J.N.; Walsh, A. Vacancy-Driven Stabilization of the Cubic Perovskite Polymorph of CsPbI3. J. Phys. Chem. C 2019, 123, 9735–9744. [Google Scholar] [CrossRef]
- Jiang, C.; Wang, Y.; Zhou, R.; Wang, H.; Chen, Q. Air molecules in XPbI3 (X = MA, FA, Cs) perovskite: A degradation mechanism based on first-principles calculations. J. Appl. Phys. 2018, 124, 085105. [Google Scholar] [CrossRef]
- Li, W.; Liu, P.; Wang, F.; Pan, L.; Guo, H.; Chen, Y.; Yang, S.-E. Phase stability and impact of water on CsSnI3 perovskite. Appl. Phys. Express 2020, 13, 071003. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Huang, Y.; Yin, W.-J.; He, Y. Intrinsic Point Defects in Inorganic Cesium Lead Iodide Perovskite CsPbI3. J. Phys. Chem. C 2018, 122, 1345–1350. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, C.; Zhang, X.; Huang, D.; Shen, Q.; Cheng, Y.; Huang, W. Intrinsic point defects in inorganic perovskite CsPbI3 from first-principles prediction. Appl. Phys. Lett. 2017, 111, 162106. [Google Scholar] [CrossRef]
- Yang, K.; Zhang, Y.; Lu, H. First-principles study on the geometric, electronic and thermodynamic properties of the CsPbI3 (001) surfaces. IOP Conf. Ser. Earth Environ. Sci. 2021, 692, 022020. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Steiner, S.; Khmelevskyi, S.; Marsmann, M.; Kresse, G. Calculation of the magnetic anisotropy with projected-augmented-wave methodology and the case study of disordered Fe1-xCox alloys. Phys. Rev. B 2016, 93, 224425. [Google Scholar] [CrossRef]
- Shishkin, M.; Kresse, G. Self-consistent $GW$ calculations for semiconductors and insulators. Phys. Rev. B 2007, 75, 235102. [Google Scholar] [CrossRef]
- Chen, G.-Y.; Guo, Z.-D.; Gong, X.-G.; Yin, W.-J. Kinetic pathway of γ-to-δ phase transition in CsPbI3. Chem 2022, 8, 3120–3129. [Google Scholar] [CrossRef]
- Wang, N.; West, D.; Liu, J.; Li, J.; Yan, Q.; Gu, B.-L.; Zhang, S.B.; Duan, W. Microscopic origin of the p-type conductivity of the topological crystalline insulator SnTe and the effect of Pb alloying. Phys. Rev. B 2014, 89, 045142. [Google Scholar] [CrossRef]
- Wang, N.; Wu, Y. Intrinsic defects on α, γ and δ-CsPbI3 (001) surfaces and implications for the α/γ to δ phase transition. Phys. Chem. Chem. Phys. 2023, 25, 16077–16085. [Google Scholar] [CrossRef]
- Mosquera-Lois, I.; Kavanagh, S.R.; Klarbring, J.; Tolborg, K.; Walsh, A. Imperfections are not 0 K: Free energy of point defects in crystals. Chem. Soc. Rev. 2023, 52, 5812–5826. [Google Scholar] [CrossRef]
- Zunger, A.; Malyi, O.I. Understanding Doping of Quantum Materials. Chem. Rev. 2021, 121, 3031–3060. [Google Scholar] [CrossRef]
- Wei, S.-H.; Zhang, S.B. Chemical trends of defect formation and doping limit in II-VI semiconductors: The case of CdTe. Phys. Rev. B 2002, 66, 155211. [Google Scholar] [CrossRef]
- Van de Walle, C.G.; Neugebauer, J. First-principles calculations for defects and impurities: Applications to III-nitrides. J. Appl. Phys. 2004, 95, 3851–3879. [Google Scholar] [CrossRef]
- Freysoldt, C.; Grabowski, B.; Hickel, T.; Neugebauer, J.; Kresse, G.; Janotti, A.; Van de Walle, C.G. First-principles calculations for point defects in solids. Rev. Mod. Phys. 2014, 86, 253–305. [Google Scholar] [CrossRef]
- Makov, G.; Payne, M.C. Periodic boundary conditions in ab initio calculations. Phys. Rev. B 1995, 51, 4014–4022. [Google Scholar] [CrossRef] [PubMed]
- Afsari, M.; Boochani, A.; Hantezadeh, M. Electronic, optical and elastic properties of cubic perovskite CsPbI3: Using first principles study. Optik 2016, 127, 11433–11443. [Google Scholar] [CrossRef]
- Hellström, M.; Spångberg, D.; Hermansson, K.; Broqvist, P. Band-Filling Correction Method for Accurate Adsorption Energy Calculations: A Cu/ZnO Case Study. J. Chem. Theory Comput. 2013, 9, 4673–4678. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-G.; Wang, Z.; Malyi, O.I.; Zunger, A. Effect of static local distortions vs. dynamic motions on the stability and band gaps of cubic oxide and halide perovskites. Mater. Today 2021, 49, 107–122. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Initial Position of H2O Insertion | Binding Energy Ebind/eV |
|---|---|
| near Cs | −1.21 |
| near Pb | −1.25 |
| near I | −1.26 |
| Position of H2O Insertion | Binding Energy Ebind/eV |
|---|---|
| Near VCs−1 | −1.29 |
| Near VPb−2 | −0.78 |
| Near VI+1 | −0.70 |
| Near VCs0 | −0.80 |
| Near VPb0 | −2.36 |
| Near VI0 | −0.48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, N.; Wu, Y. First-Principles Investigation into the Interaction of H2O with α-CsPbI3 and the Intrinsic Defects within It. Materials 2024, 17, 1091. https://doi.org/10.3390/ma17051091
Wang N, Wu Y. First-Principles Investigation into the Interaction of H2O with α-CsPbI3 and the Intrinsic Defects within It. Materials. 2024; 17(5):1091. https://doi.org/10.3390/ma17051091
Chicago/Turabian StyleWang, Na, and Yaqiong Wu. 2024. "First-Principles Investigation into the Interaction of H2O with α-CsPbI3 and the Intrinsic Defects within It" Materials 17, no. 5: 1091. https://doi.org/10.3390/ma17051091
APA StyleWang, N., & Wu, Y. (2024). First-Principles Investigation into the Interaction of H2O with α-CsPbI3 and the Intrinsic Defects within It. Materials, 17(5), 1091. https://doi.org/10.3390/ma17051091