

Molecular Dynamics Simulation Research on Fe Atom Precipitation Behaviour of Cu-Fe Alloys during the Rapid Solidification Processes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Simulation Method
3. Results and Discussion
3.1. The Influence of the Fe Content on the Cu95Fe5 Alloy Structure
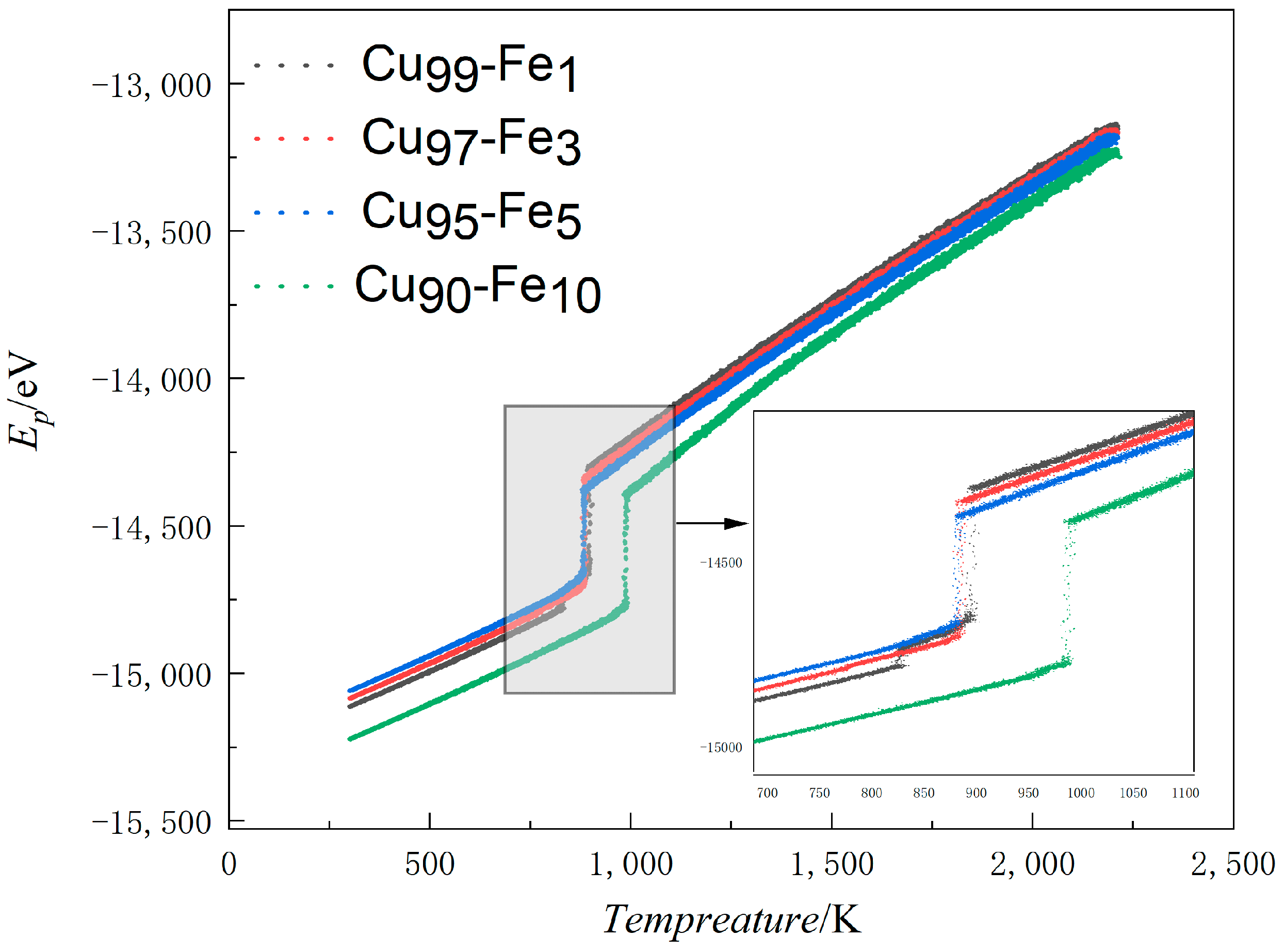
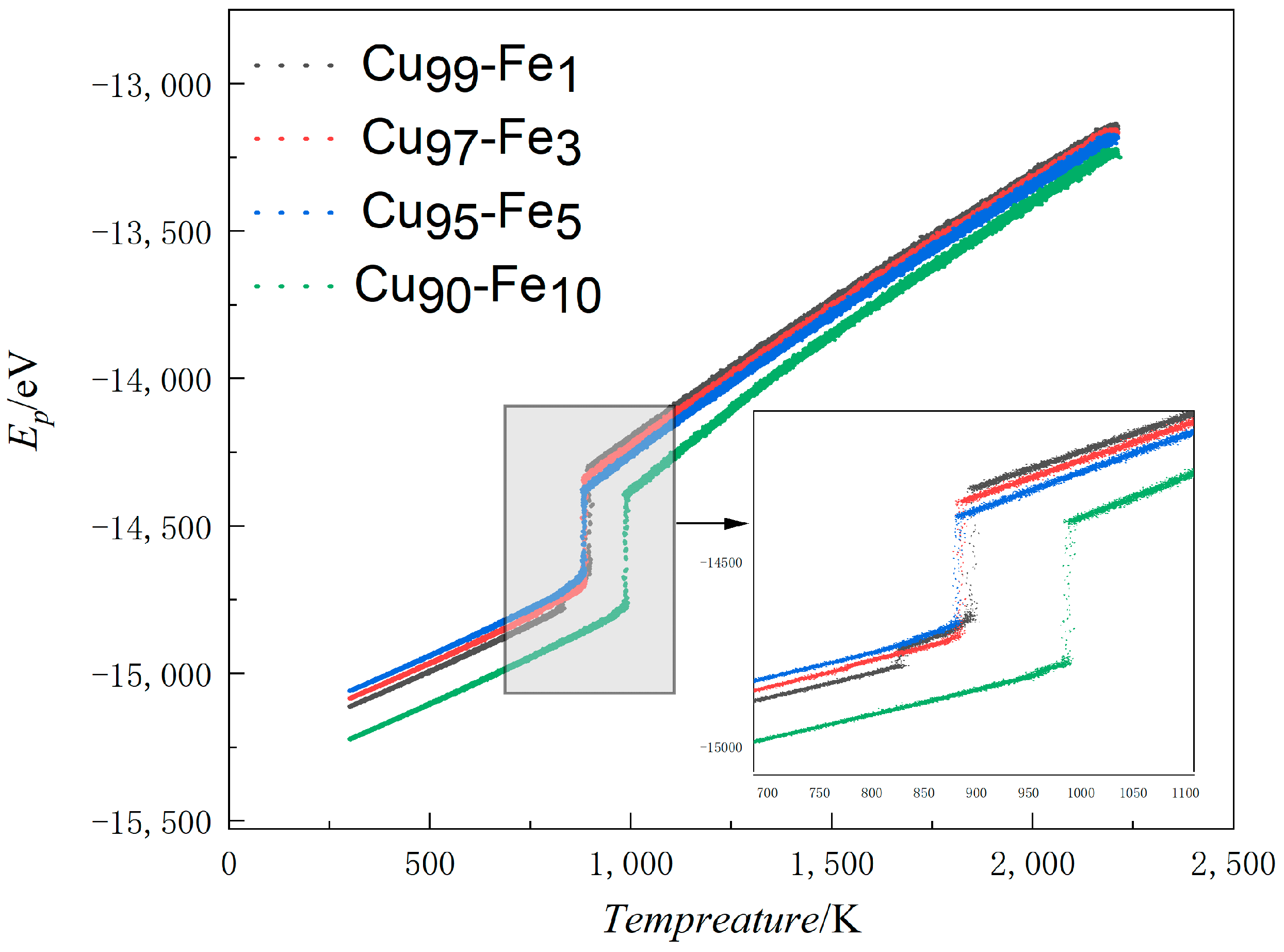
3.1.1. Average Atomic Potential Energy
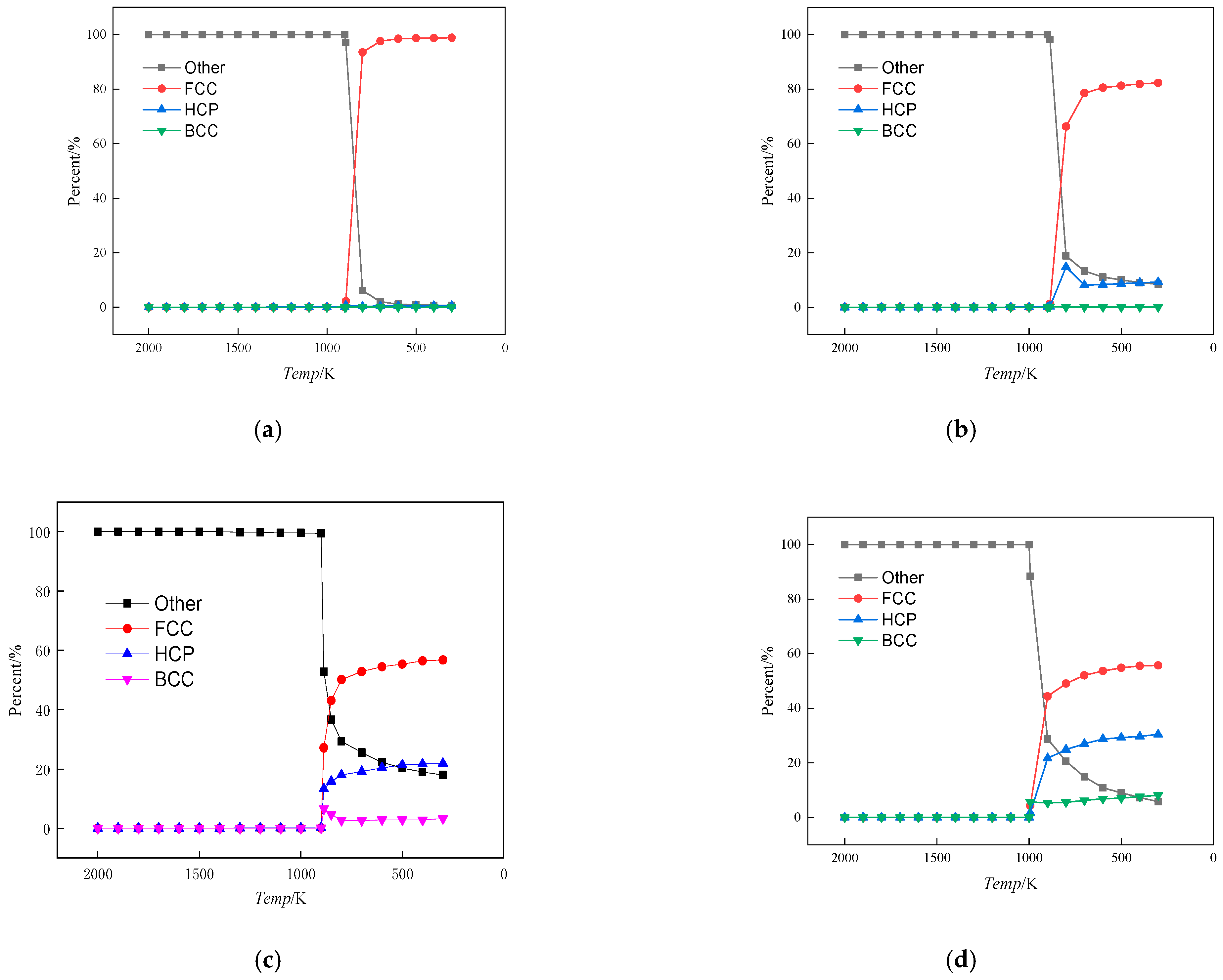
3.1.2. Analysis of Crystal Structure
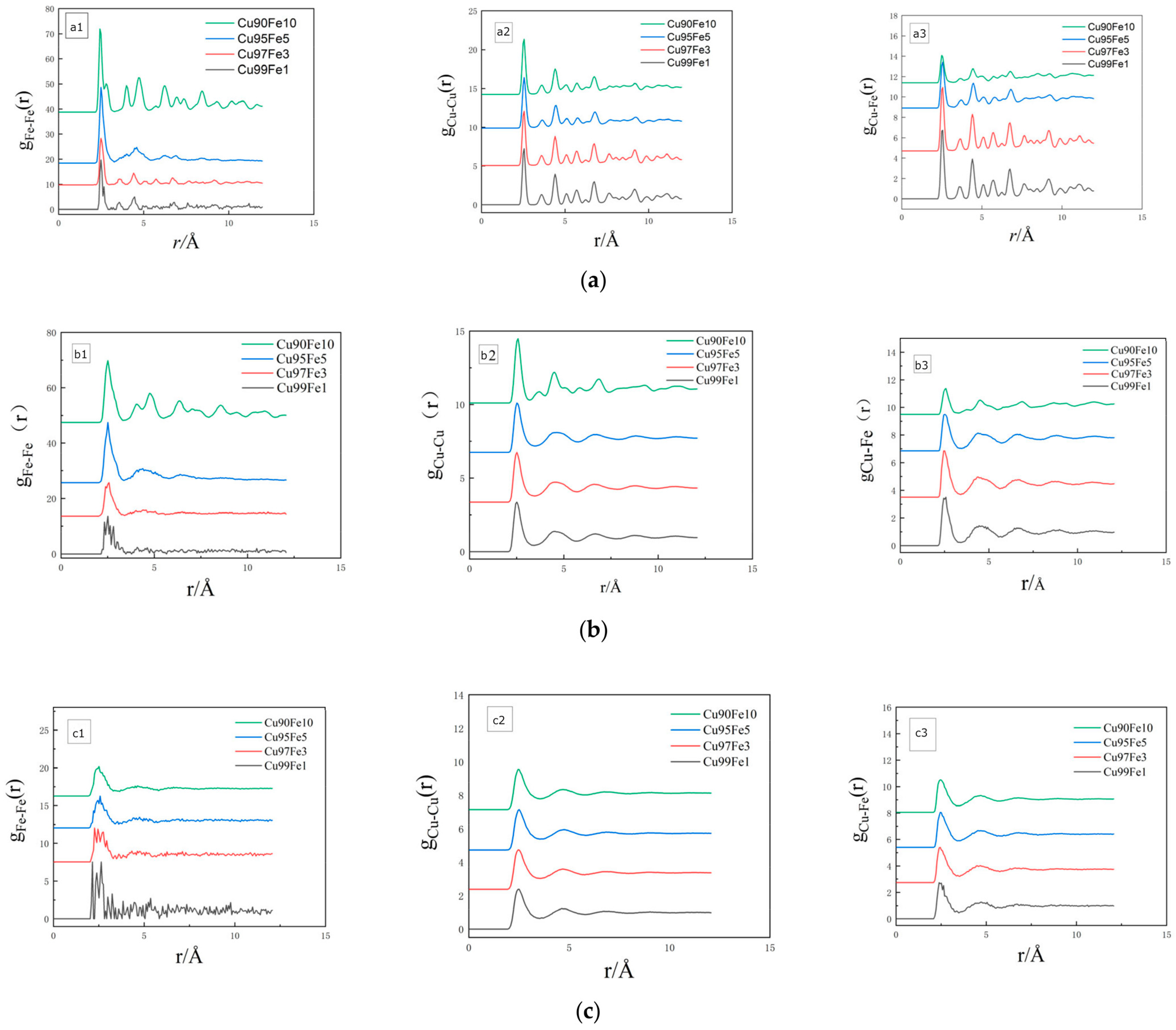
3.1.3. Radial Distribution Function Analysis
3.1.4. Allotropic Analysis
3.1.5. Mean-Square Displacement Analysis
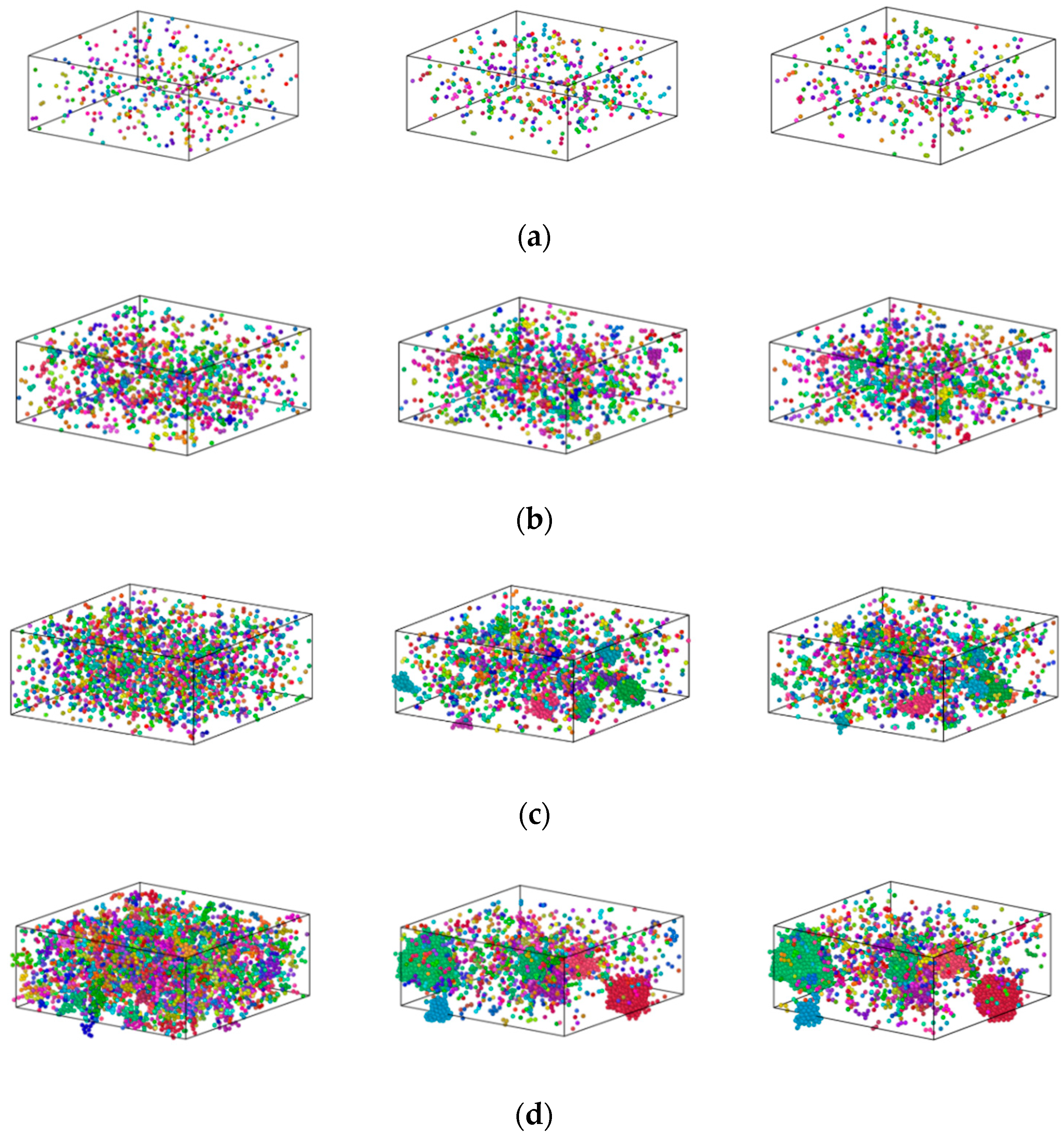
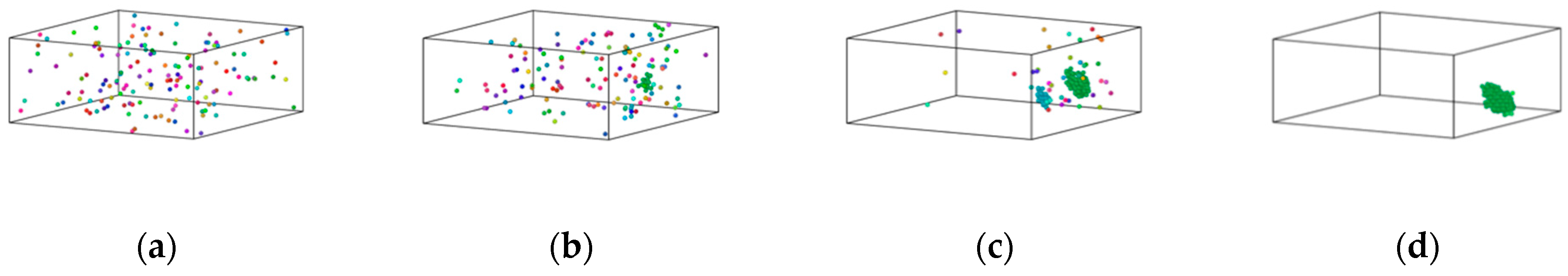
3.2. Formation Mechanism of Fe Clusters in Cu-Fe Alloys during Rapid Cooling
4. Conclusions
- (1)
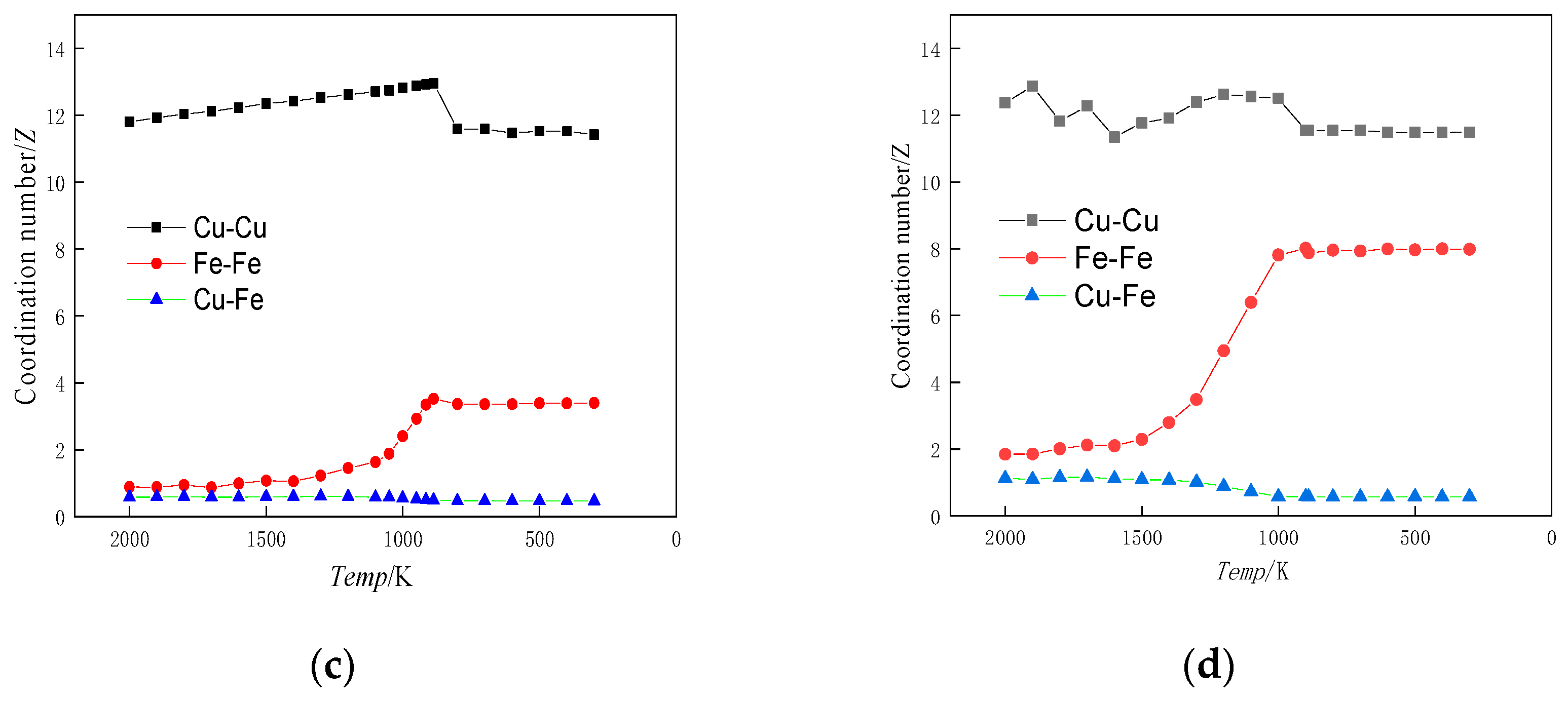
- The Fe-Fe interatomic interaction force was found to be crucial for driving Fe cluster formation. The radial distribution function analysis and coordination number of the alloy during solidification demonstrate that the interaction force between Fe-Fe atoms was considerably stronger than that between other atom pairs. Further, the tendency for Fe atom aggregation increased with a higher content of Fe atoms in the alloy. The mutual attraction and aggregation of Fe-Fe is a continuous process that commences when the alloy is in a liquid state.
- (2)
- When the Fe content reached 1%, lowering the alloy temperature did not lead to the aggregation of Fe atoms. Instead, Fe atoms maintained a uniform distribution throughout the matrix, forming a solid solution. The crystal structure remained dominated by FCC-based Cu crystals. At 3% Fe content, a decrease in alloy temperature resulted in Fe atom aggregation. However, the crystal structure, primarily based on BCC, did not undergo significant changes and continued to be dominated by FCC-based Cu crystals. When the Fe content was between 5% and 10%, the Fe atoms formed clusters as the temperature of the alloy decreased.
- (3)
- The higher the iron content, the more apparent the clusters became, and precipitation of Fe atoms occurred in three stages. In the first stage, the increase in the number of iron clusters occurred as a result of the interplay between iron-iron atomic attraction and the thermal motion of atoms. During this phase, the size of the clusters stabilised. In the second stage, non-diffusible iron atoms underwent rearrangement influenced by the local atomic structure. This stage encompassed non-diffusive rearrangement of atoms, particularly those within condensed and growing small clusters. The third stage could be characterised by the basic stability of cluster size and number following the crystallisation of the alloy.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moon, J.; Choi, Y.; Sasaki, T.; Joo, M.; Shin, H.; Lee, J.S.; Ohkubo, T.; Hono, K.; Baek, S.M.; Kim, H.S. Corrosion-resistant Cu-Fe-based immiscible medium-entropy alloy with tri-layer passivation. Corros. Sci. 2021, 193, 109888. [Google Scholar] [CrossRef]
- Yang, H.Y.; Ma, Z.C.; Lei, C.H.; Meng, L.; Fang, Y.T.; Liu, J.B.; Wang, H.T. High strength and high conductivity Cu alloys: A review. Sci. China Technol. Sci. 2020, 63, 41–53. [Google Scholar] [CrossRef]
- Hauptmann, A. Metals and Alloys. In Archaeometallurgy—Materials Science Aspects; Hauptmann, A., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 381–431. [Google Scholar]
- Han, Y.; Pan, L.; Zhang, H.; Zeng, Y.; Yin, Z. Effect of lubricant additives of Cu, Fe and bimetallic CuFe nanoparticles on tribological properties. Wear 2022, 508–509, 204485. [Google Scholar] [CrossRef]
- Adam, O.; Jan, V.; Spotz, Z.; Cupera, J.; Pouchly, V. Ultrafine-grained Cu50(FeCo)50 immiscible alloy with excellent thermal stability. Mater. Charact. 2021, 182, 111532. [Google Scholar] [CrossRef]
- Nizinkovskyi, R.; Halle, T.; Krüger, M. Influence of elasticity on the morphology of fcc-Cu precipitates in Fe-Cu alloys. A phase-field study. J. Nucl. Mater. 2022, 566, 153764. [Google Scholar] [CrossRef]
- Huang, S.; Qin, L.; Zhao, J.; Xiang, K.; Luo, S.; Michalik, S.; Mi, J. Revealing atomic structure evolution of an Al-1.5Fe alloy in the liquid state using X-ray total scattering and empirical potential structure refinement. IOP Conf. Ser. Mater. Sci. Eng. 2023, 1274, 012007. [Google Scholar] [CrossRef]
- Tian, Y.Z.; Yang, Y.; Peng, S.Y.; Pang, X.Y.; Li, S.; Jiang, M.; Li, H.X.; Wang, J.W.; Qin, G.W. Managing mechanical and electrical properties of nanostructured Cu-Fe composite by aging treatment. Mater. Charact. 2023, 196, 112600. [Google Scholar] [CrossRef]
- Lu, D.; Osman, M.S.; Khater, M.M.A.; Attia, R.A.M.; Baleanu, D. Analytical and numerical simulations for the kinetics of phase separation in iron (Fe–Cr–X (X=Mo,Cu)) based on ternary alloys. Phys. A Stat. Mech. Appl. 2020, 537, 122634. [Google Scholar] [CrossRef]
- Chatterjee, A.; Popov, D.; Velisavljevic, N.; Misra, A. Phase Transitions of Cu and Fe at Multiscales in an Additively Manufactured Cu-Fe Alloy under High-Pressure. Nanomaterials 2022, 12, 1514. [Google Scholar] [CrossRef]
- Huang, S.; Luo, S.; Qin, L.; Shu, D.; Sun, B.; Lunt, A.J.G.; Korsunsky, A.M.; Mi, J. 3D local atomic structure evolution in a solidifying Al-0.4 Sc dilute alloy melt revealed in operando by synchrotron X-ray total scattering and modelling. Scr. Mater. 2022, 211, 114484. [Google Scholar] [CrossRef]
- Chen, K.X.; Li, Z.X.; Wang, Z.D. Morphological Evolution of Fe-Rich Precipitates in a Cu-2.0Fe Alloy During Isothermal Treat ment. Acta Met. Sin. 2023, 59, 1665–1674. [Google Scholar]
- Zhang, H.C.; Wang, X.F.; Li, H.R.; Li, C.Q.; Li, Y.G. Molecular Dynamics Study on the Impact of Cu Clusters at the BCC-Fe Grain Boundary on the Tensile Properties of Crystal. Metals 2020, 10, 1533. [Google Scholar] [CrossRef]
- Nguyen-Trong, D.; Nguyen-Tri, P. Factors affecting the structure, phase transition and crystallization process of AlNi nanoparticles. J. Alloys Compd. 2020, 812, 152133. [Google Scholar] [CrossRef]
- Xu, H.D.; Bao, H.W.; Li, Y.; Bai, H.Z.; Ma, F. Atomic scale insights into the rapid crystallization and precipitation behaviors in FeCu binary alloys. J. Alloys Compd. 2021, 882, 160725. [Google Scholar] [CrossRef]
- Kumar, S. Structural Evolution of Iron–Copper (Fe–Cu) Bimetallic Janus Nanoparticles during Solidification: An Atomistic Investigation. J. Phys. Chem. C 2020, 124, 1053–1063. [Google Scholar] [CrossRef]
- Hirel, P. Atomsk: A tool for manipulating and converting atomic data files. Comput. Phys. Commun. 2015, 197, 212–219. [Google Scholar] [CrossRef]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in ‘t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Bonny, G.; Pasianot, R.C.; Castin, N.; Malerba, L. Ternary Fe-Cu-Ni many-body potential to model reactor pressure vessel steels: First validation by simulated thermal annealing. Philos. Mag. 2009, 89, 3531–3546. [Google Scholar] [CrossRef]
- Polak, W.Z. Efficiency in identification of internal structure in simulated monoatomic clusters: Comparison between common neighbor analysis and coordination polyhedron method. Comput. Mater. Sci. 2022, 201, 110882. [Google Scholar] [CrossRef]
- Wang, M.J.; Huang, X.X.; Wu, S.P.; Dai, G.X. Molecular dynamics simulations of tensile mechanical properties and microstructures of Al-4.5Cu alloy: The role of temperature and strain rate. Model. Simul. Mater. Sci. Eng. 2022, 30, 045004. [Google Scholar] [CrossRef]
- Mitsui, Y.; Onoue, M.; Kobayashi, R.; Sato, K.; Kuzuhara, S.; Ito, W.; Takahashi, K.; Koyama, K. High Magnetic Field Effects on Cu-precipitation Behavior of Fe-1mass%Cu at 773 K. ISIJ Int. 2022, 62, 413–417. [Google Scholar] [CrossRef]
- Makarenko, K.I.; Konev, S.D.; Dubinin, O.N.; Shishkovsky, I.V. Mechanical characteristics of laser-deposited sandwich structures and quasi-homogeneous alloys of Fe-Cu system. Mater. Des. 2022, 224, 111313. [Google Scholar] [CrossRef]
- Shao, Q.; Guo, J.; Chen, J.; Zhang, Z. Atomic-scale investigation on the structural evolution and deformation behaviors of Cu–Cr nanocrystalline alloys processed by high-pressure torsion. J. Alloys Compd. 2020, 832, 154994. [Google Scholar] [CrossRef]
- Kurz, W.; Rappaz, M.; Trivedi, R. Progress in modelling solidification microstructures in metals and alloys. Part II: Dendrites from 2001 to 2018. Int. Mater. Rev. 2021, 66, 30–76. [Google Scholar] [CrossRef]
- Tang, J.B.; Ahmadi, A.; Alizadeh, A.; Abedinzadeh, R.; Abed, A.M.; Smaisim, G.F.; Hadrawi, S.K.; Nasajpour-Esfahani, N.; Toghraie, D. Investigation of the mechanical properties of different amorphous composites using the molecular dynamics simulation. J. Mater. Res. Technol. 2023, 24, 1390–1400. [Google Scholar] [CrossRef]
- Rawat, S.; Chaturvedi, S. Effect of temperature on the evolution dynamics of voids in dynamic fracture of single crystal iron: A molecular dynamics study. Philos. Mag. 2021, 101, 657–672. [Google Scholar] [CrossRef]
- Rajput, A.; Paul, S.K. Effect of void in deformation and damage mechanism of single crystal copper: A molecular dynamics study. Model. Simul. Mater. Sci. Eng. 2021, 29, 085013. [Google Scholar] [CrossRef]
- Vargas Rubio, K.I.; Medrano Roldán, H.; Reyes Jáquez, D. Simulación mediante dinámica molecular de un nanocluster obtenido de la industria minera. Acta Univ. 2022, 31, e3010. [Google Scholar]
- Celtek, M.; Sengul, S.; Domekeli, U.; Guder, V. Molecular dynamics simulations of glass formation, structural evolution and diffusivity of the Pd-Si alloys during the rapid solidification process. J. Mol. Liq. 2023, 372, 121163. [Google Scholar] [CrossRef]
- Syarif, J.; Gillette, V.; Hussien, H.A.; Badawy, K.; Jisrawi, N. Molecular dynamics simulation of the amorphization and alloying of a mechanically milled Fe-Cu system. J. Non Cryst. Solids 2022, 580, 121410. [Google Scholar] [CrossRef]
- Trong, D.N.; Lu, T.; Tran, T. Study on the Influence of Factors on the Structure and Mechanical Properties of Amorphous Aluminium by Molecular Dynamics Method. Adv. Mater. Sci. Eng. 2019, 2021, 5564644. [Google Scholar]
- Bochkarev, A.; Lysogorskiy, Y.; Menon, S.; Qamar, M.; Mrovec, M.; Drautz, R. Efficient parametrization of the atomic cluster expansion. Phys. Rev. Mater. 2022, 6, 013804. [Google Scholar] [CrossRef]
- Duan, L.J.; Liu, Y.C.; Duan, J.S. Calculation of radii and atom numbers of different coordination shells in cubic crystals. Mater. Today Commun. 2020, 22, 100768. [Google Scholar] [CrossRef]
- Hansson, P.; Ahadi, A.; Melin, S. Molecular dynamic modelling of the combined influence from strain rate and temperature at tensile loading of nanosized single crystal Cu beams. Mater. Today Commun. 2022, 31, 103277. [Google Scholar] [CrossRef]
- Ranaweera, S.A.; Donnadieu, B.; Henry, W.P.; White, M.G. Effects of electron-donating ability of binding sites on coordination number: The interactions of a cyclic Schiff base with copper ions. Acta Crystallogr. Sect. C Struct. Chem. 2023, 79, 142–148. [Google Scholar] [CrossRef]
- Rogachev, A.S.; Fourmont, A.; Kovalev, D.Y.; Vadchenko, S.G.; Kochetov, N.A.; Shkodich, N.F.; Baras, F.; Politano, O. Mechanical alloying in the Co-Fe-Ni powder mixture: Experimental study and molecular dynamics simulation. Powder Technol. 2022, 399, 117187. [Google Scholar] [CrossRef]
- Dias, M.; Carvalho, P.A.; Gonçalves, A.P.; Alves, E.; Correia, J.B. Hybrid molecular dynamic Monte Carlo simulation and experimental production of a multi-component Cu–Fe–Ni–Mo–W alloy. Intermetallics 2023, 161, 107960. [Google Scholar] [CrossRef]
- Geslin, P.A.; Rodney, D. Microelasticity model of random alloys. Part I: Mean square displacements and stresses. J. Mech. Phys. Solids 2021, 153, 104479. [Google Scholar] [CrossRef]
- Seoane, A.; Farkas, D.; Bai, X.M. Influence of compositional complexity on species diffusion behavior in high-entropy solid-solution alloys. J. Mater. Res. 2022, 37, 1403–1415. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Gao, X.; Jin, Y.; Zhang, Z.; Lai, Z.; Zhang, H.; Li, Y. Molecular Dynamics Simulation Research on Fe Atom Precipitation Behaviour of Cu-Fe Alloys during the Rapid Solidification Processes. Materials 2024, 17, 719. https://doi.org/10.3390/ma17030719
Wang X, Gao X, Jin Y, Zhang Z, Lai Z, Zhang H, Li Y. Molecular Dynamics Simulation Research on Fe Atom Precipitation Behaviour of Cu-Fe Alloys during the Rapid Solidification Processes. Materials. 2024; 17(3):719. https://doi.org/10.3390/ma17030719
Chicago/Turabian StyleWang, Xufeng, Xufeng Gao, Yaxuan Jin, Zhenhao Zhang, Zhibo Lai, Hanyu Zhang, and Yungang Li. 2024. "Molecular Dynamics Simulation Research on Fe Atom Precipitation Behaviour of Cu-Fe Alloys during the Rapid Solidification Processes" Materials 17, no. 3: 719. https://doi.org/10.3390/ma17030719
APA StyleWang, X., Gao, X., Jin, Y., Zhang, Z., Lai, Z., Zhang, H., & Li, Y. (2024). Molecular Dynamics Simulation Research on Fe Atom Precipitation Behaviour of Cu-Fe Alloys during the Rapid Solidification Processes. Materials, 17(3), 719. https://doi.org/10.3390/ma17030719