

Visualising Geopolymerisation Processes Using Scanning X-Ray Diffraction and Fluorescence Microscopy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Samples
2.1. Sample Presentation
2.2. Sample Preparation
3. In Situ X-Ray Imaging
4. Results
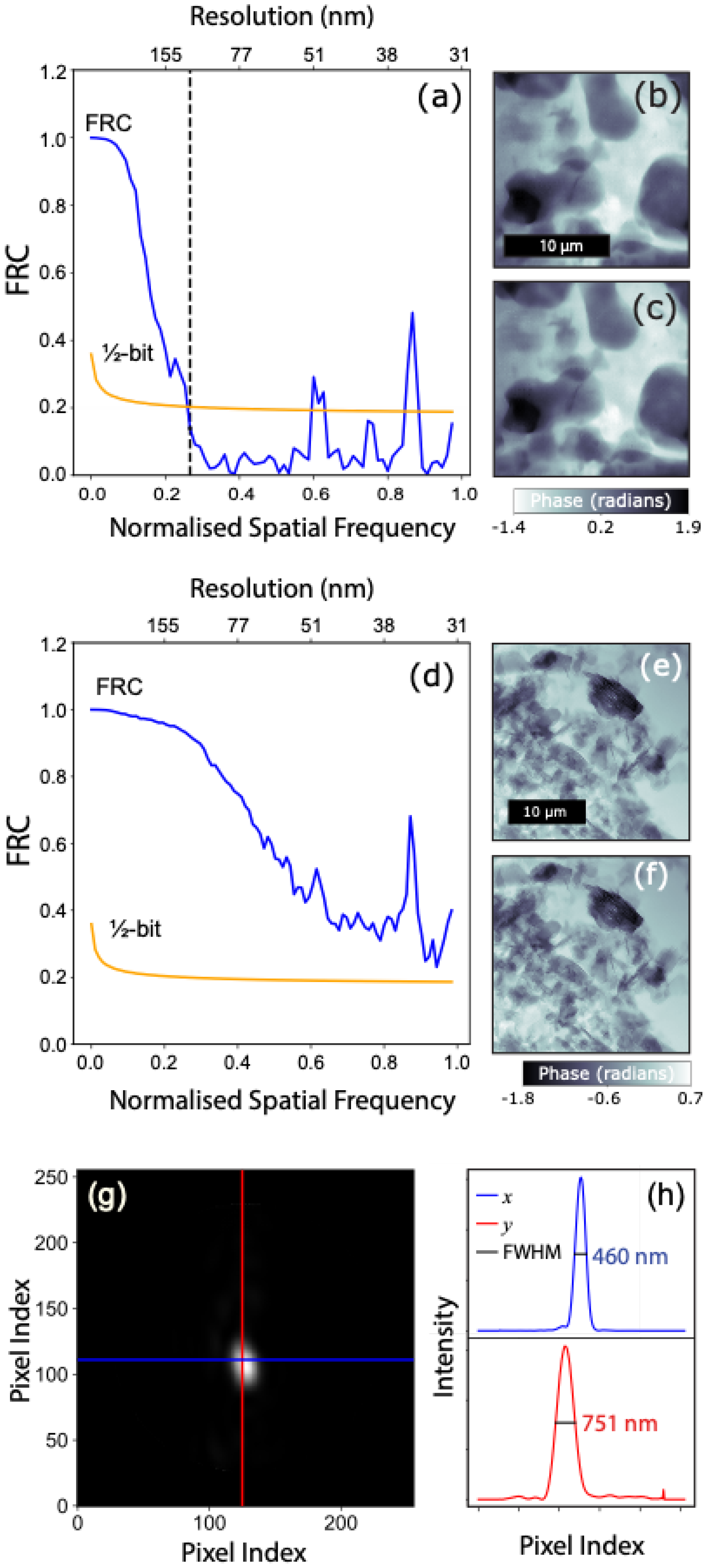
4.1. SXDM
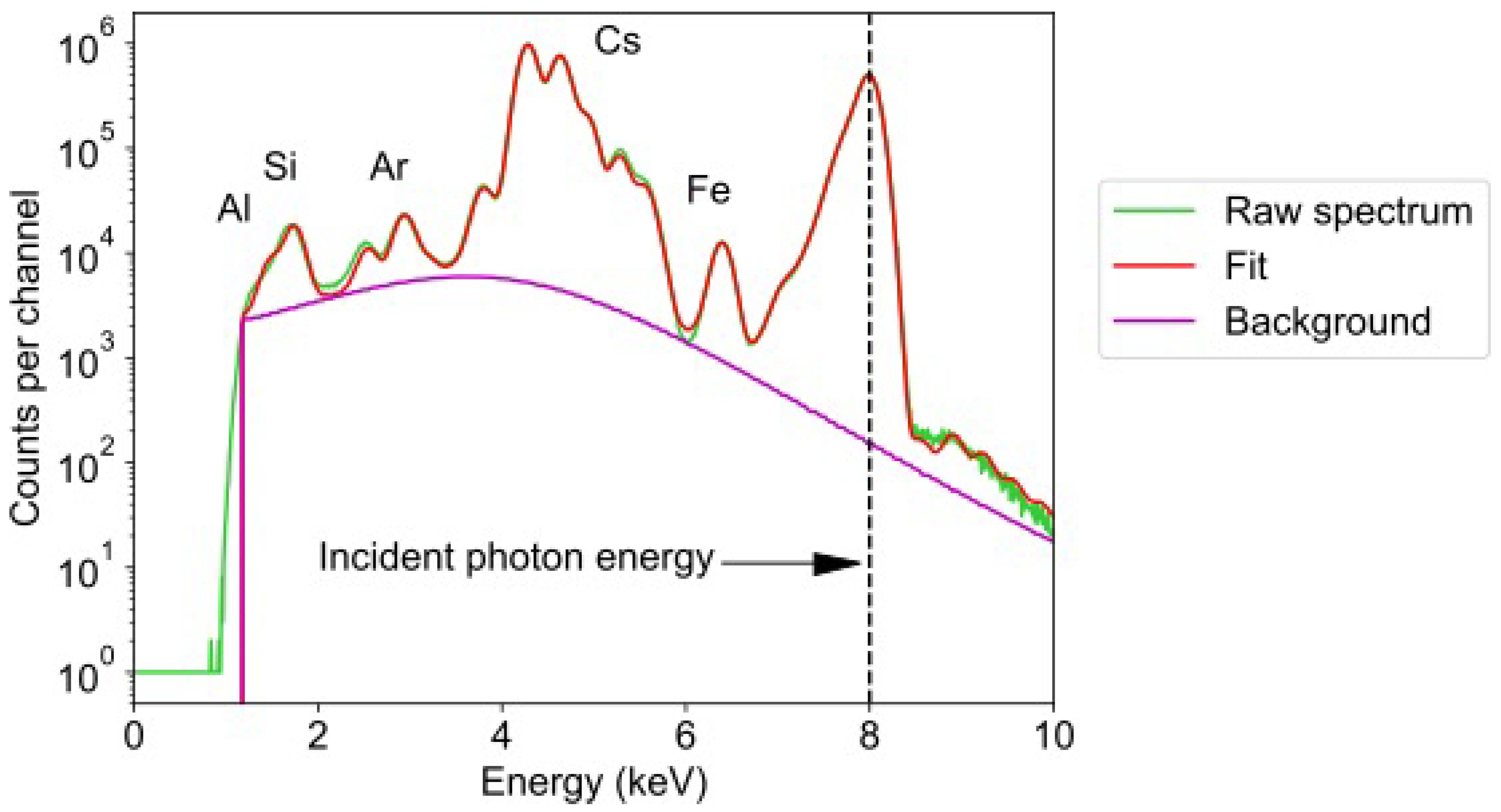
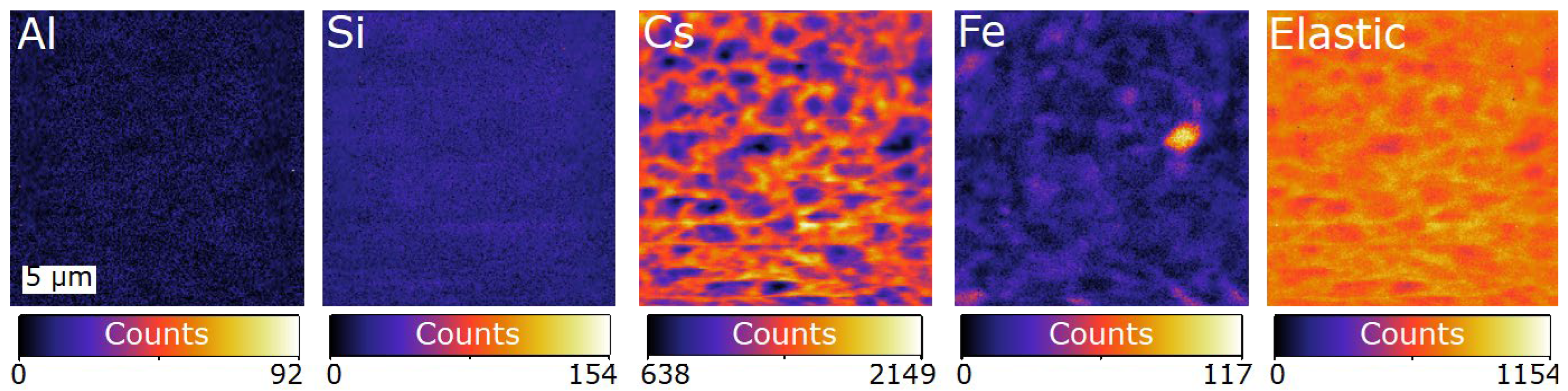
4.2. SXFM
4.3. Combinatorial Time-Series Images

4.4. SEM
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rangan, B. Studies on fly ash-based geopolymer concrete. Malays. Constr. Res. J. 2008, 3, 1–20. [Google Scholar]
- Blackford, M.G.; Hanna, J.V.; Pike, K.J.; Vance, E.R.; Perera, D.S. Transmission electron microscopy and nuclear magnetic resonance studies of geopolymers for radioactive waste immobilization. J. Am. Ceram. Soc. 2007, 90, 1193–1199. [Google Scholar] [CrossRef]
- Rickard, W.D.A.; van Riessen, A.; Walls, P. Thermal Character of Geopolymers Synthesized from Class F Fly Ash Containing High Concentrations of Iron and α-Quartz. Int. J. Appl. Ceram. Technol. 2010, 7, 81–88. [Google Scholar] [CrossRef]
- Kriven, W.M.; Bell, J.L. Effect of Alkali Choice on Geopolymer Properties. In Proceedings of the 28th International Conference on Advanced Ceramics and Composites B: Ceramic Engineering and Science Proceedings, Cocoa Beach, FL, USA, 25–30 January 2004; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2004; pp. 99–104. [Google Scholar]
- Provis, J.; Duxson, P.; Van Deventer, J.; Lukey, G. The role of mathematical modelling and gel chemistry in advancing geopolymer technology. Chem. Eng. Res. Des. 2005, 83, 853–860. [Google Scholar] [CrossRef]
- Duxson, P.; Provis, J.L.; Lukey, G.C.; Mallicoat, S.W.; Kriven, W.M.; van Deventer, J.S.J. Understanding the relationship between geopolymer composition, microstructure and mechanical properties. Colloids Surf. A Physicochem. Eng. Asp. 2005, 269, 47–58. [Google Scholar] [CrossRef]
- Rowles, M.; O’Connor, B. Chemical optimisation of the compressive strength of aluminosilicate geopolymers synthesised by sodium silicate activation of metakaolinite. J. Mater. Chem. 2003, 13, 1161–1165. [Google Scholar] [CrossRef]
- Williams, R.P.; van Riessen, A. Determination of the reactive component of fly ashes for geopolymer production using XRF and XRD. Fuel 2010, 89, 3683–3692. [Google Scholar] [CrossRef]
- Xu, H.; Van Deventer, J.S. Microstructural characterisation of geopolymers synthesised from kaolinite/stilbite mixtures using XRD, MAS-NMR, SEM/EDX, TEM/EDX, and HREM. Cem. Concr. Res. 2002, 32, 1705–1716. [Google Scholar] [CrossRef]
- Provis, J.L.; Myers, R.J.; White, C.E.; Rose, V.; van Deventer, J.S. X-ray microtomography shows pore structure and tortuosity in alkali-activated binders. Cem. Concr. Res. 2012, 42, 855–864. [Google Scholar] [CrossRef]
- Provis, J.L.; Hajimohammadi, A.; White, C.E.; Bernal, S.A.; Myers, R.J.; Winarski, R.P.; Rose, V.; Proffen, T.E.; Llobet, A.; van Deventer, J.S. Nanostructural characterization of geopolymers by advanced beamline techniques. Cem. Concr. Compos. 2013, 36, 56–64, special issue: Nanotechnology in Construction. [Google Scholar] [CrossRef]
- Das, S.; Yang, P.; Singh, S.S.; Mertens, J.C.; Xiao, X.; Chawla, N.; Neithalath, N. Effective properties of a fly ash geopolymer: Synergistic application of x-ray synchrotron tomography, nanoindentation, and homogenization models. Cem. Concr. Res. 2015, 78, 252–262. [Google Scholar] [CrossRef]
- van Riessen, A.; Rickard, W.; Williams, R.; van Riessen, G. Methods for geopolymer formulation development and microstructural analysis. J. Ceram. Sci. Technol. 2017, 8, 421–432. [Google Scholar]
- Hajimohammadi, A.; Masoumi, S.; Kim, T.; McCaslin, E.; Alnahhal, M.F.; Almer, J.D.; White, C.E. Chemo-mechanical properties of carbon fiber reinforced geopolymer interphase. J. Am. Ceram. Soc. 2022, 105, 1519–1532. [Google Scholar] [CrossRef]
- Provis, J.; van Deventer, J. Geopolymerisation kinetics. 1. in situ energy-dispersive x-ray diffractometry. Chem. Eng. Sci. 2007, 62, 2309–2317. [Google Scholar] [CrossRef]
- Provis, J.; van Deventer, J. Geopolymerisation kinetics. 2. reaction kinetic modelling. Chem. Eng. Sci. 2007, 62, 2318–2329. [Google Scholar] [CrossRef]
- Bell, J.L.; Sarin, P.; Provis, J.L.; Haggerty, R.P.; Driemeyer, P.E.; Chupas, P.J.; van Deventer, J.S.J.; Kriven, W.M. Atomic structure of a cesium aluminosilicate geopolymer: A pair distribution function study. Chem. Mater. 2008, 20, 4768–4776. [Google Scholar] [CrossRef]
- Williams, R.P.; van Riessen, A. The first 20 hours of geopolymerization: An in situ WAXS study of flyash-based geopolymers. Materials 2016, 9, 552. [Google Scholar] [CrossRef]
- Jones, M.W.M.; Phillips, N.W.; van Riessen, G.A.; Abbey, B.; Vine, D.J.; Nashed, Y.S.G.; Mudie, S.T.; Afshar, N.; Kirkham, R.; Chen, B.; et al. Simultaneous X-ray fluorescence and scanning X-ray diffraction microscopy at the Australian Synchrotron XFM beamline. J. Synchrotron Radiat. 2016, 23, 1151–1157. [Google Scholar]
- Jones, M.W.M.; Phillips, N.W.; Abbey, B.; Hare, D.J.; van Riessen, G.A.; Vine, D.J.; de Jonge, M.D.; McColl, G. Simultaneous nanostructure and chemical imaging of intact whole nematodes. Chem. Commun. 2019, 55, 1052–1055. [Google Scholar] [CrossRef]
- Jones, M.W.M.; van Riessen, G.A.; Phillips, N.W.; Schrank, C.E.; Hinsley, G.N.; Afshar, N.; Reinhardt, J.; de Jonge, M.D.; Kewish, C.M. High-speed free-run ptychography at the Australian Synchrotron. J. Synchrotron Radiat. 2022, 29, 480–487. [Google Scholar] [CrossRef]
- Bras, W.; Stanley, H. Unexpected effects in non crystalline materials exposed to x-ray radiation. J. Non Cryst. Solids 2016, 451, 153–160. [Google Scholar] [CrossRef]
- Benavent, V.; Frizon, F.; Poulesquen, A. Effect of composition and aging on the porous structure of metakaolin-based geopolymers. J. Appl. Crystallogr. 2016, 49, 2116–2128. [Google Scholar] [CrossRef]
- Barbosa, V.F.; MacKenzie, K.J.; Thaumaturgo, C. Synthesis and characterisation of materials based on inorganic polymers of alumina and silica: Sodium polysialate polymers. Int. J. Inorg. Mater. 2000, 2, 309–317. [Google Scholar] [CrossRef]
- Bell, J.L.; Driemeyer, P.E.; Kriven, W.M. Formation of Ceramics from Metakaolin-Based Geopolymers: Part I—Cs-Based Geopolymer. J. Am. Ceram. Soc. 2009, 92, 1–8. [Google Scholar] [CrossRef]
- Howard, D.L.; de Jonge, M.D.; Afshar, N.; Ryan, C.G.; Kirkham, R.; Reinhardt, J.; Kewish, C.M.; McKinlay, J.; Walsh, A.; Divitcos, J.; et al. The XFM beamline at the Australian Synchrotron. J. Synchrotron Radiat. 2020, 27, 1447–1458. [Google Scholar] [CrossRef]
- Ryan, C.G.; Kirkham, R.; Jonge, M.D.D.; Siddons, D.P.; Ent, A.V.D.; Pagés, A.; Boesenberg, U.; Kuczewski, A.J.; Dunn, P.; Jensen, M.; et al. The Maia Detector and Event Mode. Synchrotron Radiat. News 2018, 31, 21–27. [Google Scholar] [CrossRef]
- Enders, B.; Thibault, P. A computational framework for ptychographic reconstructions. Proc. R. Soc. A 2016, 472, 20160640. [Google Scholar] [CrossRef]
- Thibault, P.; Menzel, A. Reconstructing state mixtures from diffraction measurements. Nature 2013, 494, 68–71. [Google Scholar] [CrossRef]
- Elser, V. Phase retrieval by iterated projections. J. Opt. Soc. Am. A 2003, 20, 40–55. [Google Scholar] [CrossRef]
- Thibault, P.; Guizar-Sicairos, M. Maximum-likelihood refinement for coherent diffractive imaging. New J. Phys. 2012, 14, 063004. [Google Scholar] [CrossRef]
- Odstrčil, M.; Menzel, A.; Guizar-Sicairos, M. Iterative least-squares solver for generalized maximum-likelihood ptychography. Opt. Express 2018, 26, 3108–3123. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.; Cousens, D.; Sie, S.; Griffin, W.; Suter, G.; Clayton, E. Quantitative PIXE microanalysis of geological material using the CSIRO proton microprobe. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 1990, 47, 55–71. [Google Scholar] [CrossRef]
- Schropp, A.; Hoppe, R.; Patommel, J.; Samberg, D.; Seiboth, F.; Stephan, S.; Wellenreuther, G.; Falkenberg, G.; Schroer, C.G. Hard x-ray scanning microscopy with coherent radiation: Beyond the resolution of conventional x-ray microscopes. Appl. Phys. Lett. 2012, 100, 253112. [Google Scholar] [CrossRef]
- Banterle, N.; Bui, K.H.; Lemke, E.A.; Beck, M. Fourier ring correlation as a resolution criterion for super-resolution microscopy. J. Struct. Biol. 2013, 183, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.; Laird, J.; Fisher, L.; Kirkham, R.; Moorhead, G. Improved dynamic analysis method for quantitative PIXE and SXRF element imaging of complex materials. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2015, 363, 42–47. [Google Scholar] [CrossRef]
- Steins, P.; Poulesquen, A.; Diat, O.; Frizon, F. Structural Evolution during Geopolymerization from an Early Age to Consolidated Material. Langmuir 2012, 28, 8502–8510. [Google Scholar] [CrossRef]
- Donath, T.; Jung, D.Š.; Burian, M.; Radicci, V.; Zambon, P.; Fitch, A.N.; Dejoie, C.; Zhang, B.; Ruat, M.; Hanfland, M.; et al. EIGER2 hybrid-photon-counting X-ray detectors for advanced synchrotron diffraction experiments. J. Synchrotron Radiat. 2023, 30, 723–738. [Google Scholar] [CrossRef]
- Hinsley, G.N.; Kewish, C.M.; van Riessen, G.A. Dynamic coherent diffractive imaging using unsupervised identification of spatiotemporal constraints. Opt. Express 2020, 28, 36862–36872. [Google Scholar] [CrossRef]
- Hinsley, G.N.; Kewish, C.M.; van Riessen, G.A. Towards Kilohertz Synchrotron Coherent Diffractive Imaging. J. Appl. Crystallogr. 2022, 55, 479–483. [Google Scholar] [CrossRef]
- Williams, R.P.; Hart, R.D.; van Riessen, A. Quantification of the Extent of Reaction of Metakaolin-Based Geopolymers Using X-Ray Diffraction, Scanning Electron Microscopy, and Energy-Dispersive Spectroscopy. J. Am. Ceram. Soc. 2011, 94, 2663–2670. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Riessen, G.A.; Hinsley, G.N.; Kewish, C.M.; van Riessen, A. Visualising Geopolymerisation Processes Using Scanning X-Ray Diffraction and Fluorescence Microscopy. Materials 2024, 17, 5896. https://doi.org/10.3390/ma17235896
van Riessen GA, Hinsley GN, Kewish CM, van Riessen A. Visualising Geopolymerisation Processes Using Scanning X-Ray Diffraction and Fluorescence Microscopy. Materials. 2024; 17(23):5896. https://doi.org/10.3390/ma17235896
Chicago/Turabian Stylevan Riessen, Grant A., Gerard N. Hinsley, Cameron M. Kewish, and Arie van Riessen. 2024. "Visualising Geopolymerisation Processes Using Scanning X-Ray Diffraction and Fluorescence Microscopy" Materials 17, no. 23: 5896. https://doi.org/10.3390/ma17235896
APA Stylevan Riessen, G. A., Hinsley, G. N., Kewish, C. M., & van Riessen, A. (2024). Visualising Geopolymerisation Processes Using Scanning X-Ray Diffraction and Fluorescence Microscopy. Materials, 17(23), 5896. https://doi.org/10.3390/ma17235896