
Very Large Pore Mesoporous Bioactive Silicate Glasses: Comparison of Behavior toward Classical Mesoporous Bioactive Glasses in Terms of Drug Loading/Release and Bioactivity




Abstract
1. Introduction
2. Materials and Methods
2.1. General Procedures
2.2. Synthesis of LPMSs
2.3. Load Tests and Release Tests
2.4. Gallium, Calcium, Silica, and Phosphorous Release in SBF
2.5. Bioactivity Evaluation after Soaking with SBF
2.6. Physical–Chemical Characterization of Powders
2.6.1. X-ray Fluorescence Spectroscopy (XRF)
2.6.2. Scanning Electron Microscopy (SEM)
2.6.3. Textural Properties
2.6.4. Elemental Analysis (EA)
2.6.5. Thermogravimetric Analysis (TG-DTA)
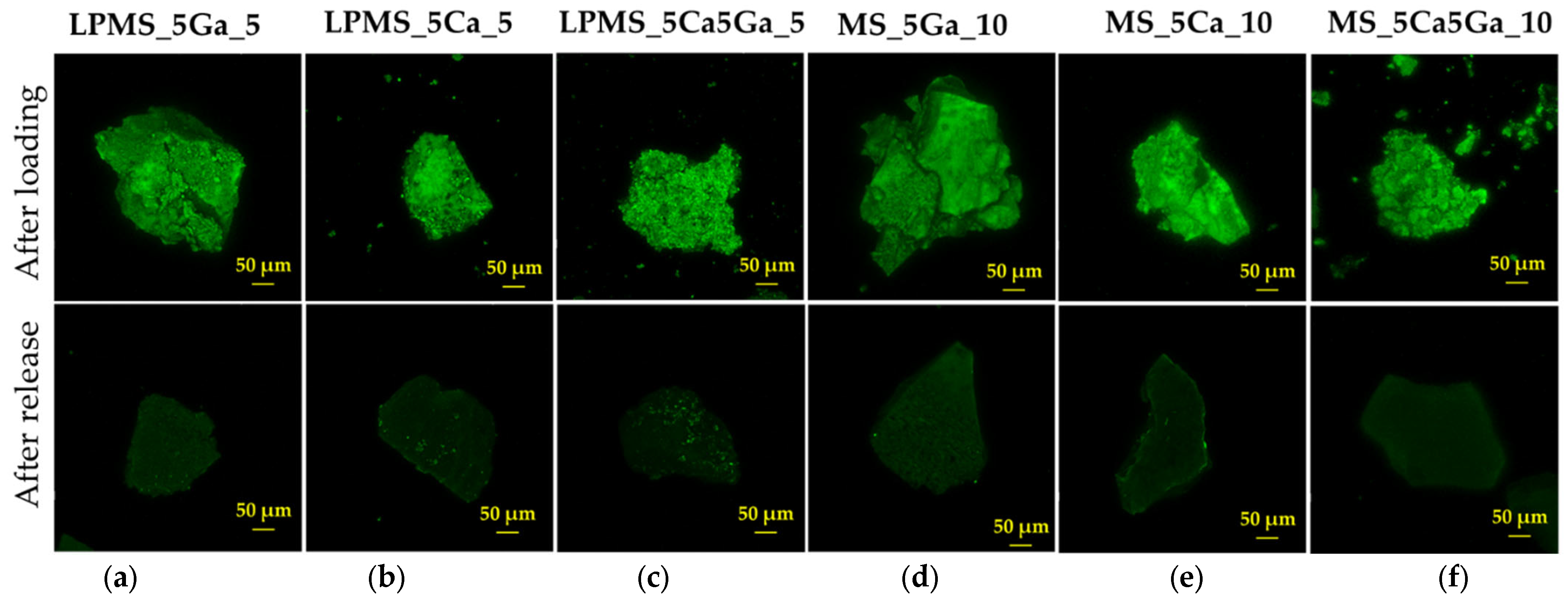
2.6.6. Confocal Laser Scanning Microscopy (CLSM)
3. Results and Discussion
3.1. LPMSs’ Morphology Characterization with SEM
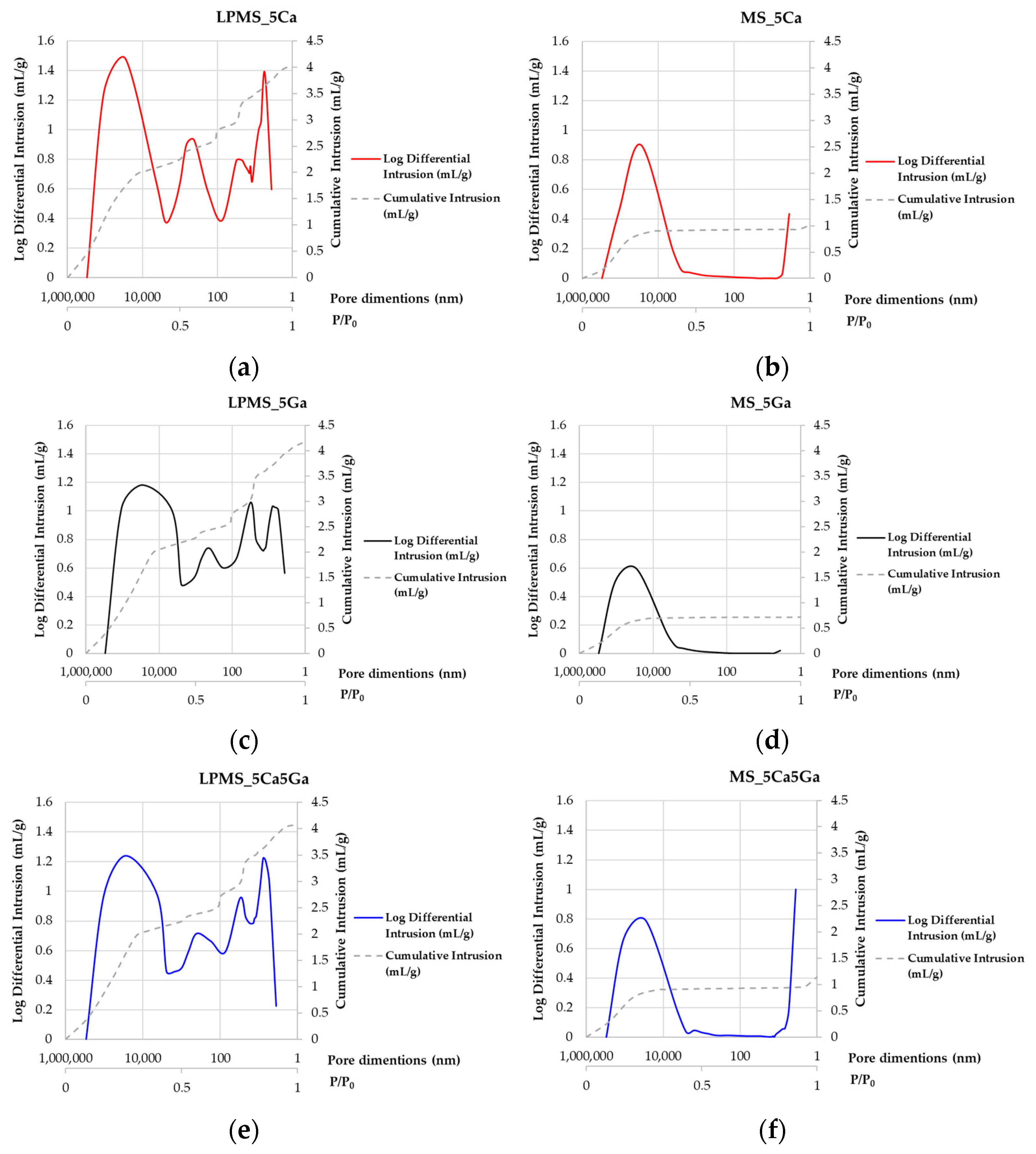
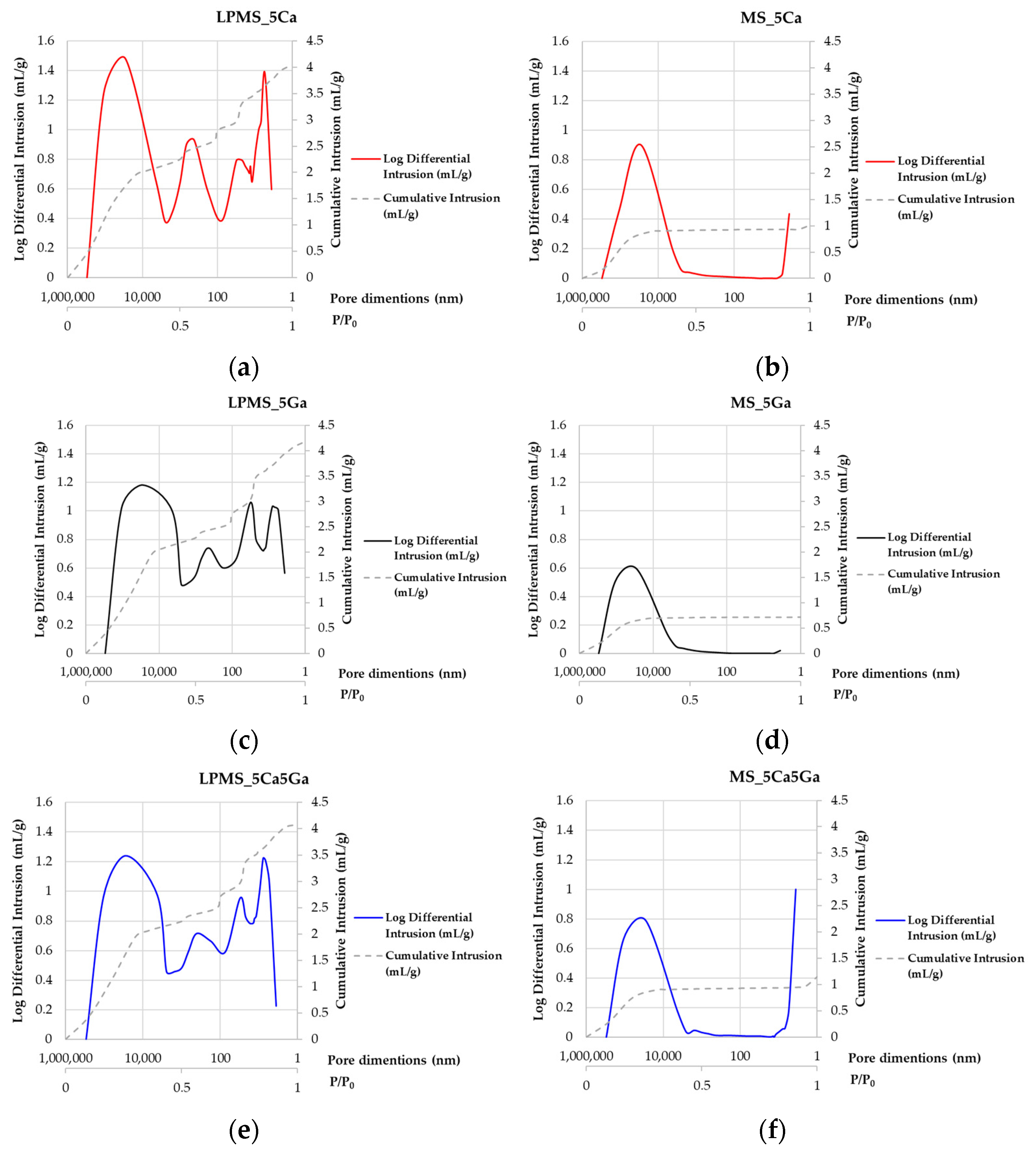
3.2. SSABET and Hg Porosimeter Characterization
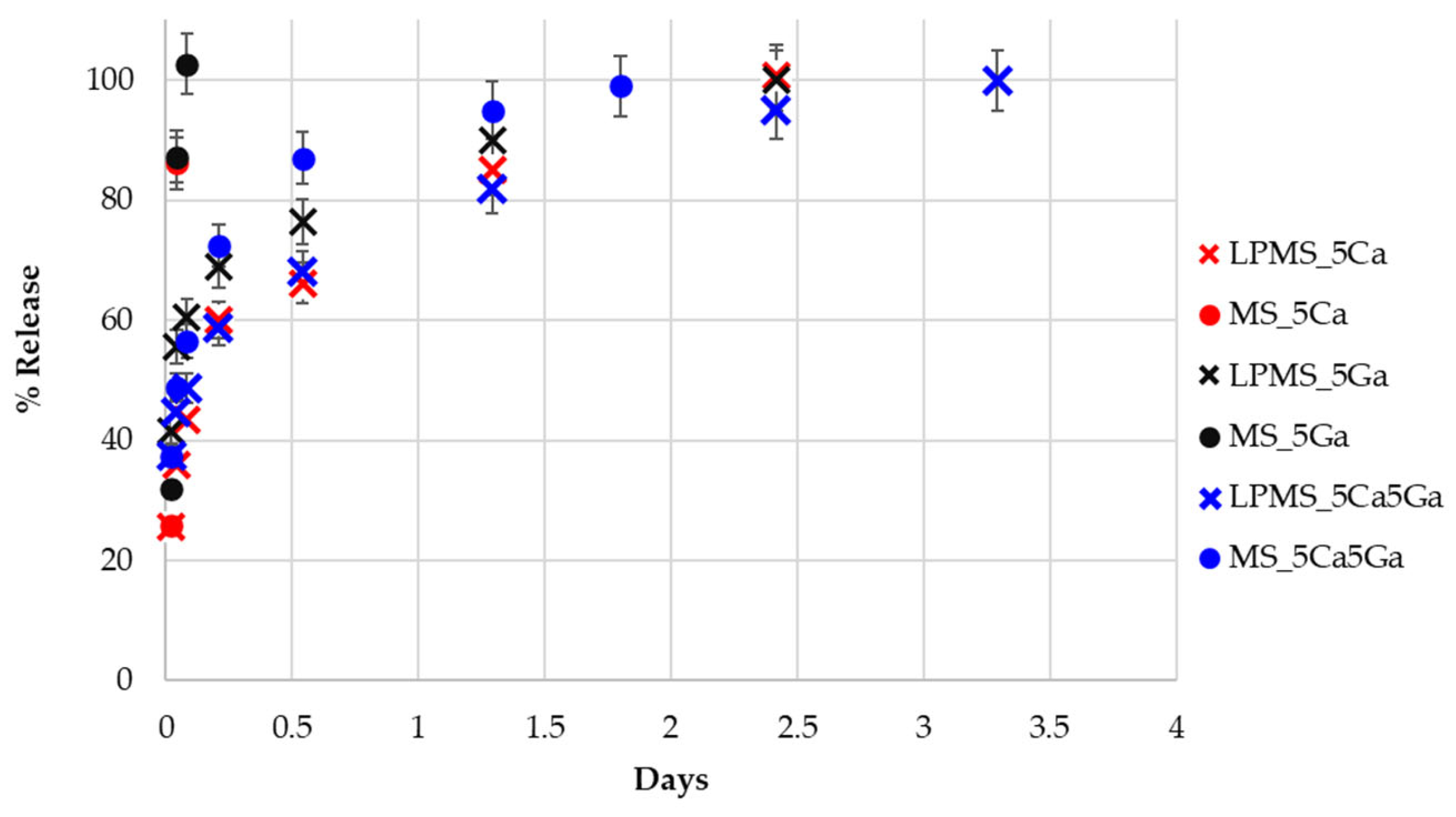
3.3. Nisin Load and Release Tests
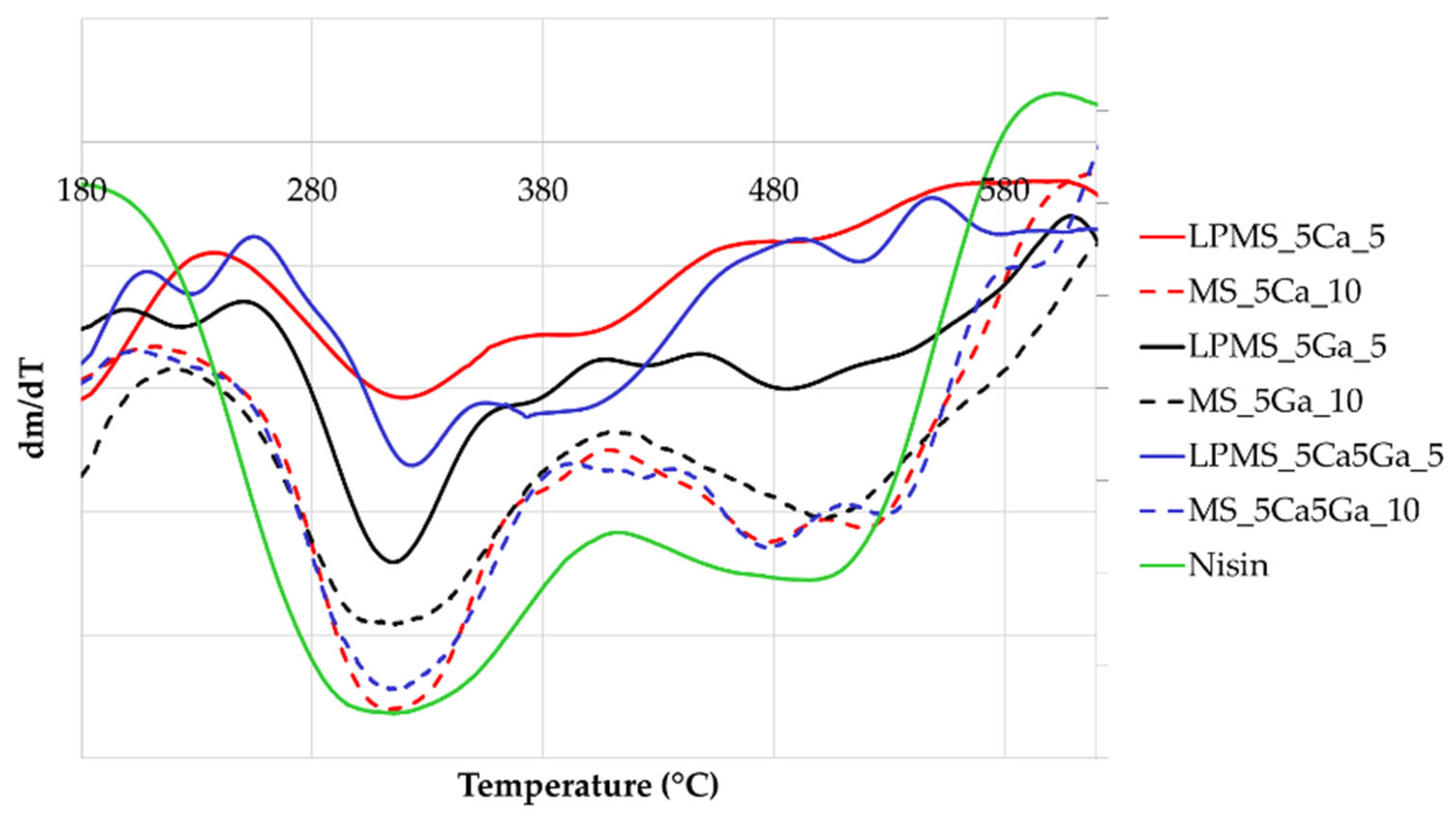
3.4. Thermogravimetric Analysis (TG-DTA)
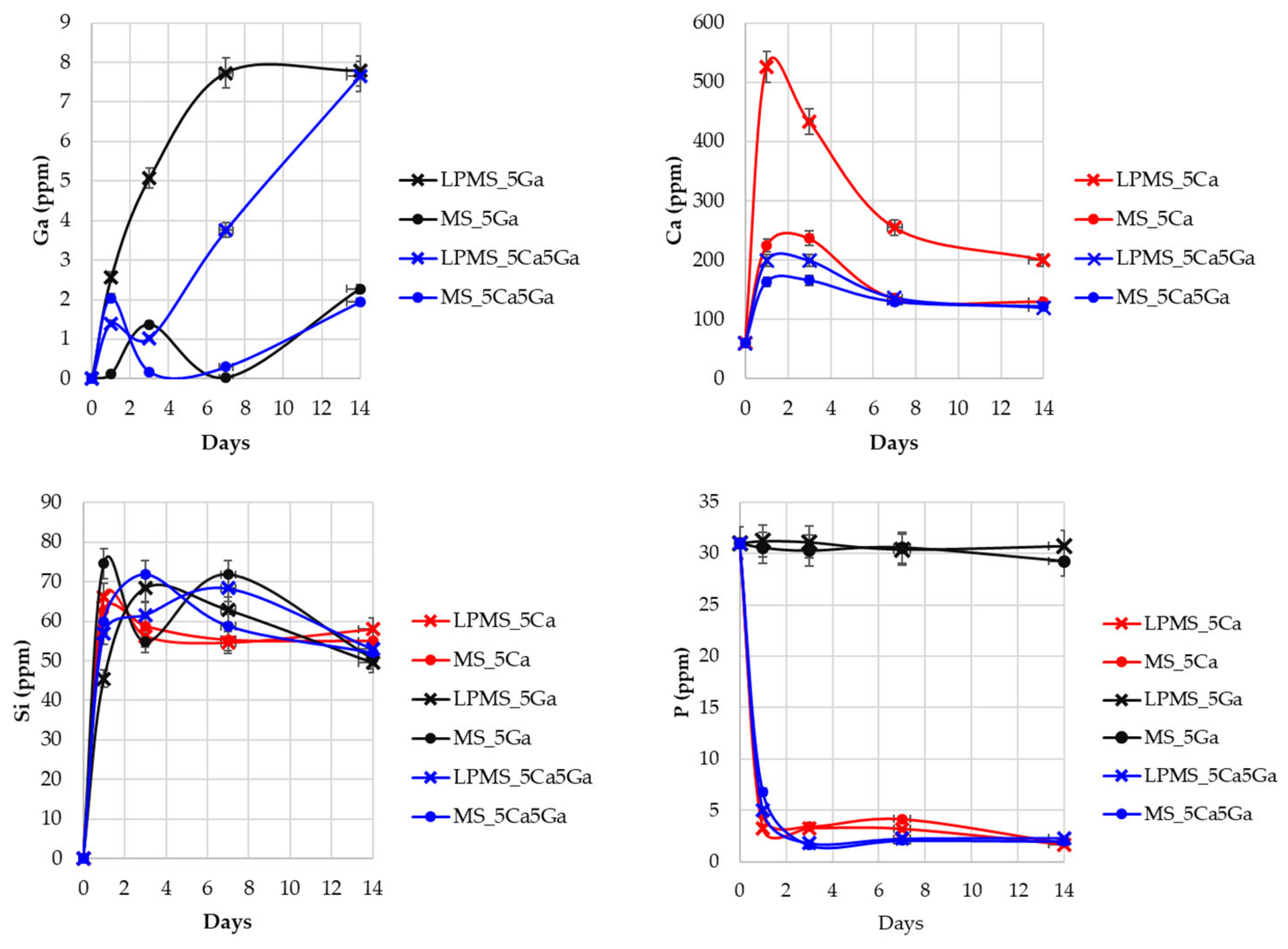
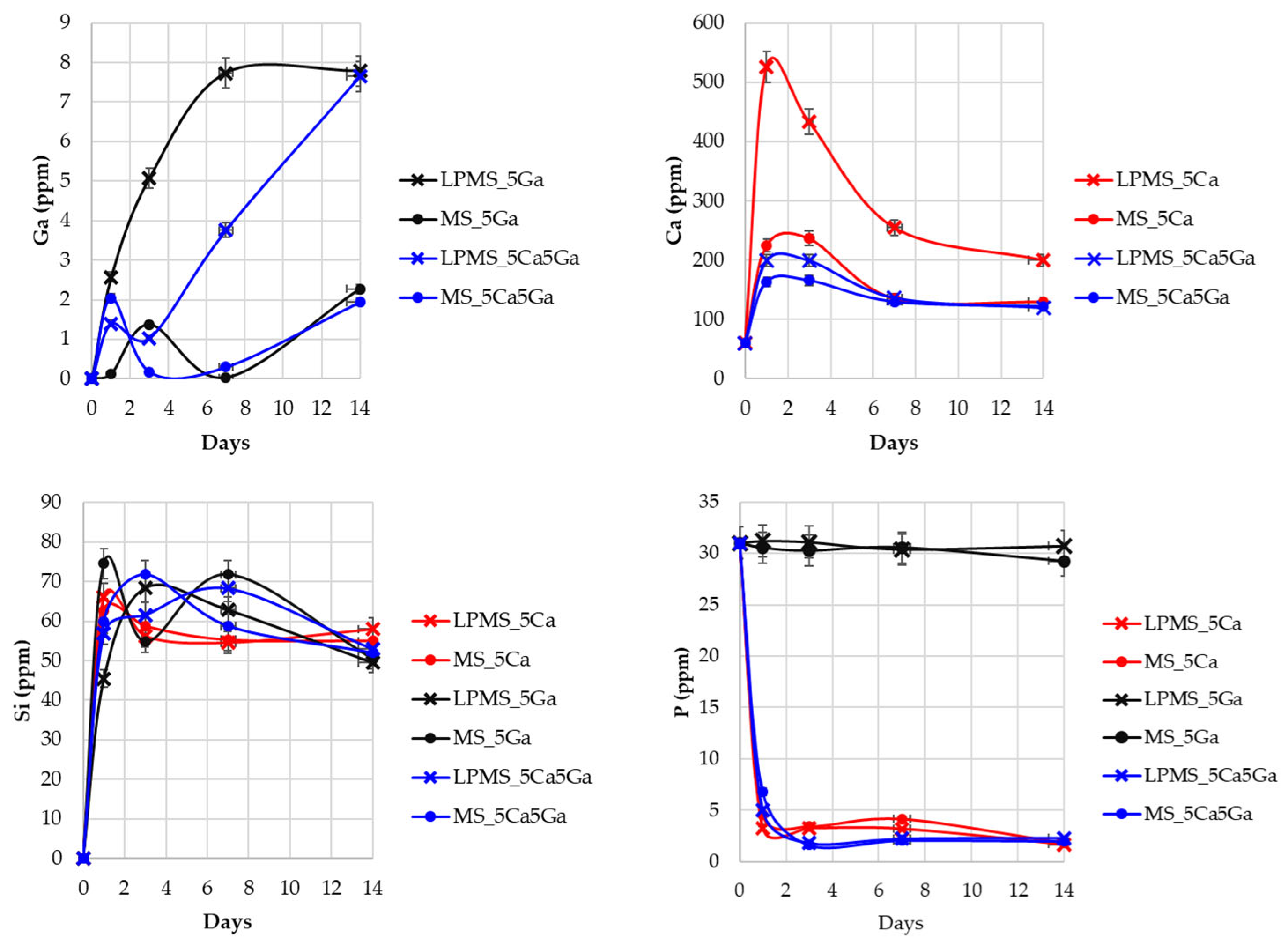
3.5. Gallium, Calcium, Silica, and Phosphorous Release in SBF
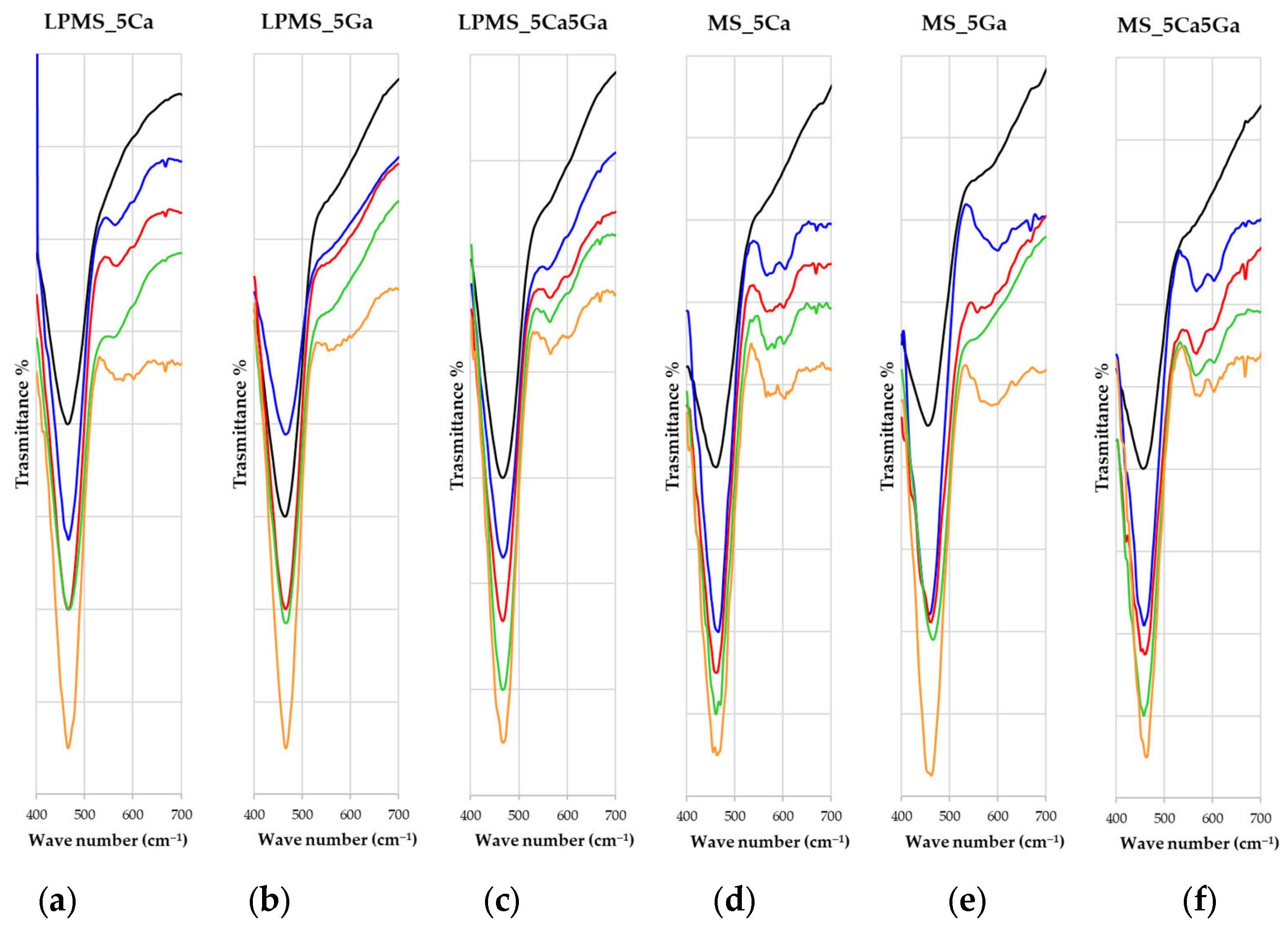
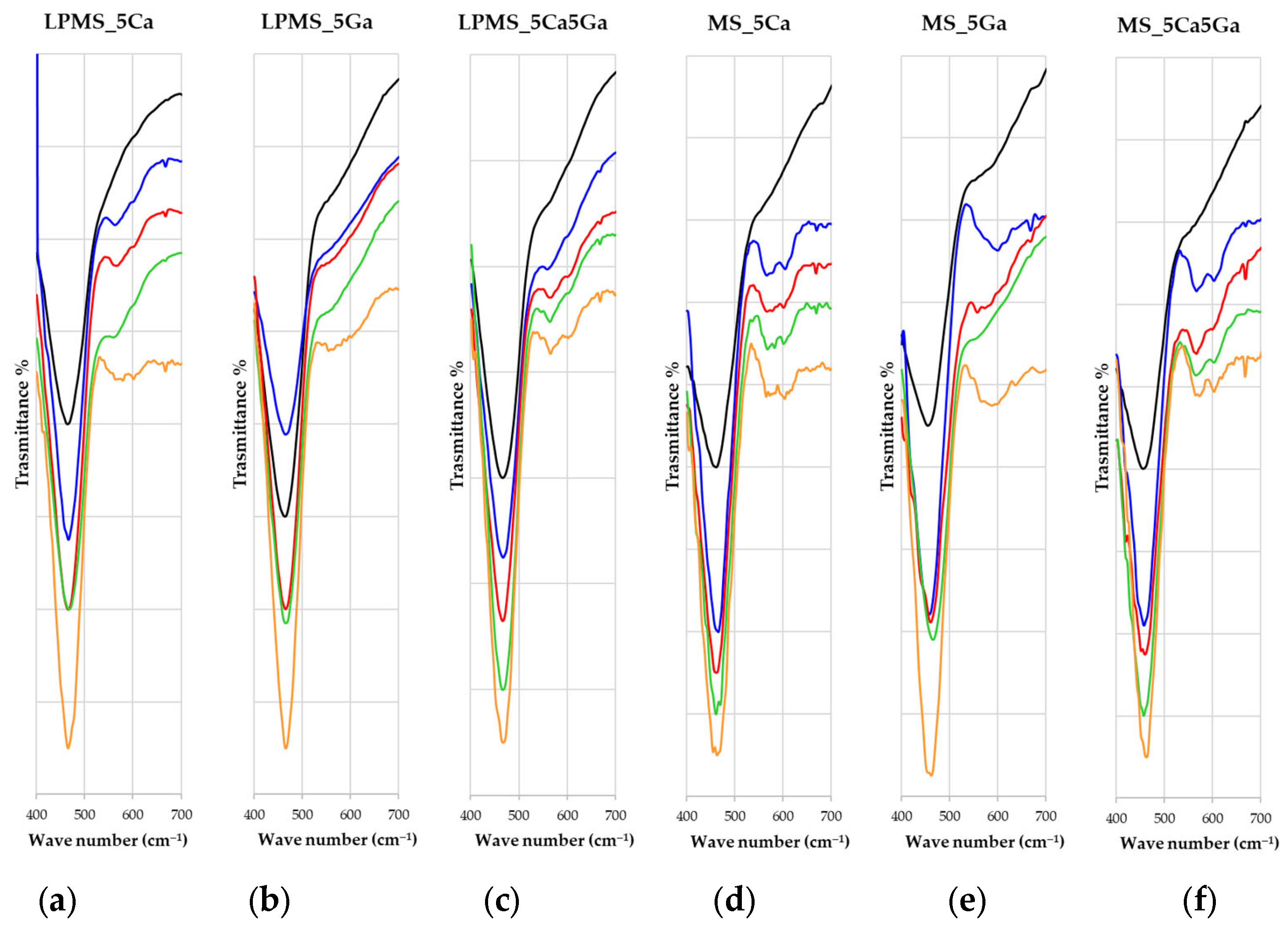
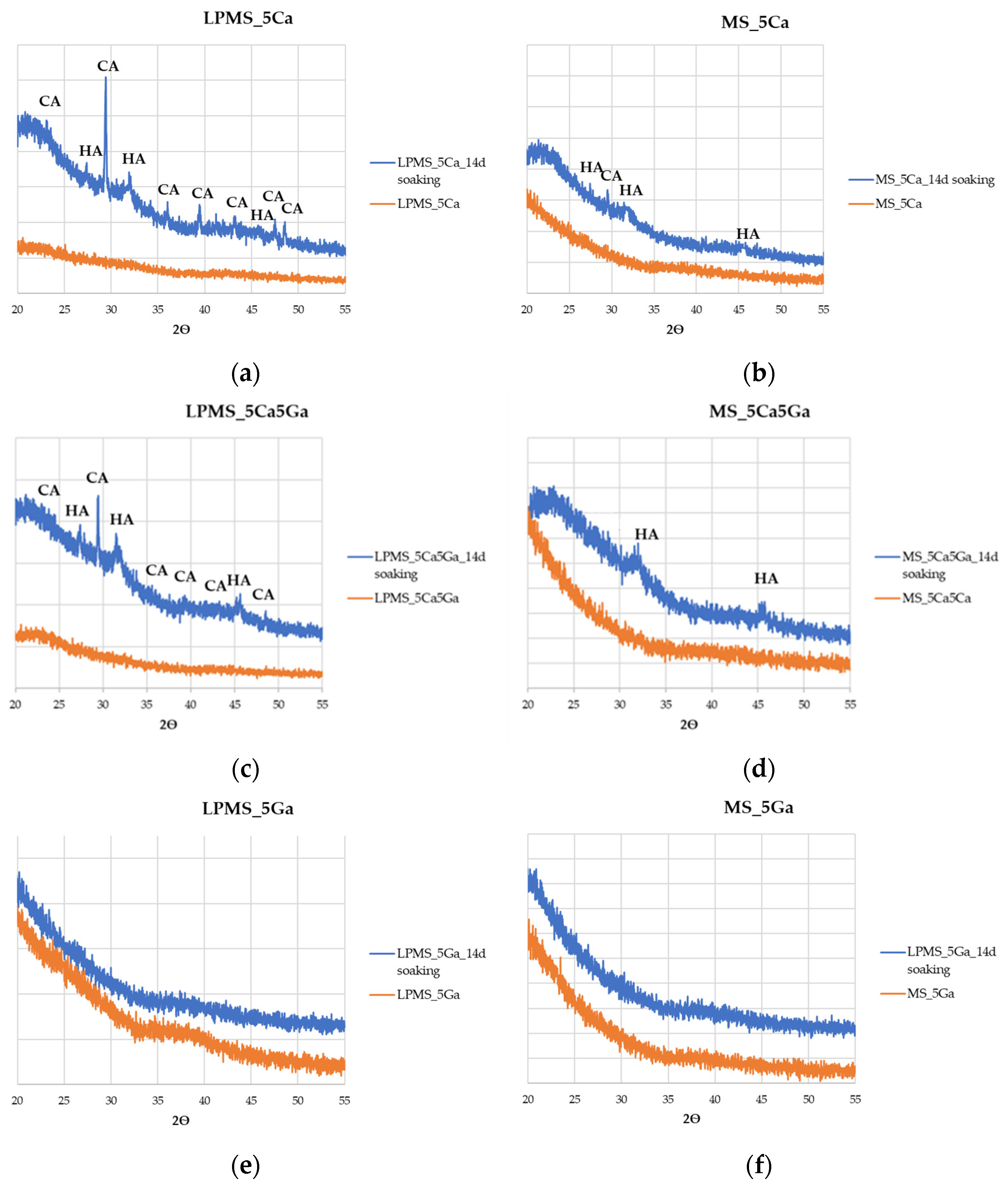
3.6. Bioactivity Evaluation after Soaking with SBF
4. Conclusions, Limitations and Prospects
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Johnell, O.; Kanis, J.A. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos. Int. 2006, 17, 1726–1733. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.-T.; Chiu, Y.-C.; Lee, A.K.-X.; Lin, Y.-H.; Huang, T.-H.; Liu, Y.-C.; Shie, M.-Y. The synergistic effects of Xu Duan combined Sr-contained calcium silicate/poly-ε-caprolactone scaffolds for the promotion of osteogenesis marker expression and the induction of bone regeneration in osteoporosis. Mater. Sci. Eng. C 2021, 119, 111629. [Google Scholar] [CrossRef] [PubMed]
- Vallet-Regí, M.; Colilla, M.; Izquierdo-Barba, I.; Vitale-Brovarone, C.; Fiorilli, S. Achievements in Mesoporous Bioactive Glasses for Biomedical Applications. Pharmaceutics 2022, 14, 2636. [Google Scholar] [CrossRef]
- Turnbull, G.; Clarke, J.; Picard, F.; Riches, P.; Jia, L.; Han, F.; Li, B.; Shu, W. 3D bioactive composite scaffolds for bone tissue engineering. Bioact. Mater. 2018, 3, 278–314. [Google Scholar] [CrossRef]
- Hollister, S.J. Scaffold Design and Manufacturing: From Concept to Clinic. Adv. Mater. 2009, 21, 3330–3342. [Google Scholar] [CrossRef]
- Gao, C.; Peng, S.; Feng, P.; Shuai, C. Bone biomaterials and interactions with stem cells. Bone Res. 2017, 5, 17059. [Google Scholar] [CrossRef]
- Vallet-Regi, M.; Salinas, A.J. Mesoporous bioactive glasses for regenerative medicine. Mater. Today Bio 2021, 11, 100121. [Google Scholar] [CrossRef]
- Salinas, A.J.; Vallet-Regi, M.; Heikkilä, J. Use of bioactive glasses as bone substitutes in orthopedics and traumatology. In Bioactive Glasses, 2nd ed.; Ylänen, H., Ed.; Woodhead Publishing Series in Biomaterials; Woodhead Publishing: Sawston, UK, 2018; pp. 337–364. [Google Scholar] [CrossRef]
- Montazerian, M.; Zanotto, E.D. A guided walk through Larry Hench’s monumental discoveries. J. Mater. Sci. 2017, 52, 8695–8732. [Google Scholar] [CrossRef]
- Lewiecki, E.M. Safety of Long-Term Bisphosphonate Therapy for the Management of Osteoporosis. Drugs 2011, 71, 791–814. [Google Scholar] [CrossRef]
- Geng, Z.; Ji, L.; Li, Z.; Wang, J.; He, H.; Cui, Z.; Yang, X.; Liu, C. Nano-needle strontium-substituted apatite coating enhances osteoporotic osseointegration through promoting osteogenesis and inhibiting osteoclastogenesis. Bioact. Mater. 2021, 6, 905–915. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Qiu, C.; Lu, T.; Shi, X.; Ye, J. Conjunction of gallium doping and calcium silicate mediates osteoblastic and osteoclastic performances of tricalcium phosphate bioceramics. Biomed. Mater. 2021, 17, 015012. [Google Scholar] [CrossRef]
- Zhu, H.; Zheng, K.; Boccaccini, A.R. Multi-functional silica-based mesoporous materials for simultaneous delivery of biologically active ions and therapeutic biomolecules. Acta Biomater. 2021, 129, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Lin, K.; Wang, Z.; Chang, J.; Wang, L.; Lu, J.; Ning, C. Reconstruction of calvarial defect of rabbits using porous calcium silicate bioactive ceramics. Biomaterials 2008, 29, 2588–2596. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Xia, D.; Ni, Z.; Ou, T.; Wang, Y.; Zhang, H.; Mao, L.; Lin, K.; Xu, S.; Liu, J. Calcium silicate bioactive ceramics induce osteogenesis through oncostatin M. Bioact. Mater. 2021, 6, 810–822. [Google Scholar] [CrossRef] [PubMed]
- Hench, L.L. Bioceramics. J. Am. Ceram. Soc. 1998, 81, 1705–1728. [Google Scholar] [CrossRef]
- Ni, S.; Chang, J.; Chou, L.; Zhai, W. Comparison of osteoblast-like cell responses to calcium silicate and tricalcium phosphate ceramics in vitro. J. Biomed. Mater. Res. Part B Appl. Biomater. 2007, 80B, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Chitambar, C.R. Gallium-containing anticancer compounds. Future Med. Chem. 2012, 4, 1257–1272. [Google Scholar] [CrossRef]
- Kurtuldu, F.; Mutlu, N.; Boccaccini, A.; Galusek, D. Gallium containing bioactive materials: A review of anticancer, antibacterial, and osteogenic properties. Bioact. Mater. 2022, 17, 125–146. [Google Scholar] [CrossRef]
- Bernstein, L.R. Mechanisms of Therapeutic Activity for Gallium. Pharmacol. Rev. 1998, 50, 665–682. [Google Scholar]
- Collery, P. Metal Ions in Biology and Medicine, 1st ed.; John Libbey: Paris, France, 1990; Volume 6. [Google Scholar]
- Petrisor, G.; Ficai, D.; Motelica, L.; Trusca, R.D.; Bîrcă, A.C.; Vasile, B.S.; Voicu, G.; Voicu, G.; Oprea, O.C.; Semenescu, A.; et al. Mesoporous Silica Materials Loaded with Gallic Acid with Antimicrobial Potential. Nanomaterials 2022, 12, 1648. [Google Scholar] [CrossRef] [PubMed]
- Grnberg, B.; Emmler, T.; Gedat, E.; Shenderovich, I.; Findenegg, G.H.; Limbach, H.-H.; Buntkowsky, G. Hydrogen Bonding of Water Confined in Mesoporous Silica MCM-41 and SBA-15 Studied by 1H Solid-State NMR. Chem. Eur. J. 2004, 10, 5689–5696. [Google Scholar] [CrossRef] [PubMed]
- Carrozza, D.; Malavasi, G.; Ferrari, E. Very Large Pores Mesoporous Silica as New Candidate for Delivery of Big Therapeutics Molecules, Such as Pharmaceutical Peptides. Materials 2023, 16, 4151. [Google Scholar] [CrossRef] [PubMed]
- Patil, B.S.; Kulkarni, U.; Bhavik, P. Formulation and Evaluation of Diclofenac Potassium Matrix Tablets. Int. J. Pharm. Sci. Res. 2010, 1, 88–92. [Google Scholar] [CrossRef]
- Tarn, D.; Yu, C.-J.; Lu, J.; Hartz, A.; Tamanoicd, F.; Zink, J.I. In vitro delivery of calcium ions by nanogated mesoporous silica nanoparticles to induce cancer cellular apoptosis. Mol. Syst. Des. Eng. 2017, 2, 384–392. [Google Scholar] [CrossRef]
- Phama, A.L.-T.; Sedlaka, D.L.; Doyle, F.M. Dissolution of mesoporous silica supports in aqueous solutions: Implications for mesoporous silica-based water treatment processes. Appl. Catal. B Environ. 2012, 126, 258–264. [Google Scholar] [CrossRef]
- Zhai, W.; He, C.; Wu, L.; Zhou, Y.; Chen, H.; Chang, J.; Zhang, H. Degradation of hollow mesoporous silica nanoparticles in human umbilical vein endothelial cells. J. Biomed. Mater. Res. B Appl. Biomater. 2012, 100B, 1397–1403. [Google Scholar] [CrossRef]
- Braun, K.; Pochert, A.; Beck, M.; Fiedler, R.; Gruber, J.; Linden, M. Dissolution kinetics of mesoporous silica nanoparticles in different simulated body fluids. J. Sol-Gel Sci. Technol. 2016, 79, 319–327. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Arcos, D.; Vallet-Regí, M. Sol–gel silica-based biomaterials and bone tissue regeneration. Acta Biomater. 2010, 6, 2874–2888. [Google Scholar] [CrossRef]
- Brown, S.M.; Lard, E.W. A comparison of nitrogen and mercury pore size distributions of silicas of varying pore volume. Powder Technol. 1974, 9, 187–190. [Google Scholar] [CrossRef]
- Sing, K.S.W. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Mays, T.J. A new classification of pore sizes. In Studies in Surface Science and Catalysis; Llewellyn, P.L., Rodriquez-Reinoso, F., Rouqerol, J., Seaton, N., Eds.; Characterization of Porous Solids; Elsevier: Amsterdam, The Netherlands, 2007; Volume 160, pp. 57–62. [Google Scholar] [CrossRef]
- ALOthman, Z.A. A Review: Fundamental Aspects of Silicate Mesoporous Materials. Materials 2012, 5, 2874–2902. [Google Scholar] [CrossRef]
- Kumar, K.V.; Gadipelli, S.; Wood, B.; Ramisetty, K.A.; Stewart, A.A.; Howard, C.A.; Brett, D.J.L.; Rodriguez-Reinoso, F. Characterization of the adsorption site energies and heterogeneous surfaces of porous materials. J. Mater. Chem. A 2019, 7, 10104–10137. [Google Scholar] [CrossRef]
- Mazzella, A.; Tilocca, C.; Ciccu, R. Analisi Comparativa delle Potenzialità di Confinamento della CO2 per via Geologica e Mineralogica All’Interno del Bacino Minerario del Sulcis; Report RdS/2011/284; ENEA: Poznań, Poland, 2011. [Google Scholar] [CrossRef]
- El-Say, K.M. Maximizing the encapsulation efficiency and the bioavailability of controlled-release cetirizine microspheres using Draper-Lin small composite design. Drug. Des. Dev. Ther. 2016, 24, 825–839. [Google Scholar] [CrossRef]
- Ibrahim, A.H.; Smått, J.-H.; Govardhanam, N.P.; Ibrahim, H.M.; Ismael, H.R.; Afouna, M.I.; Samy, A.M.; Rosenholm, J.M. Formulation and optimization of drug-loaded mesoporous silica nanoparticle-based tablets to improve the dissolution rate of the poorly water-soluble drug silymarin. Eur. J. Pharm. Sci. 2020, 142, 105103. [Google Scholar] [CrossRef]
- Shen, S.; Wu, Y.; Liu, Y.; Wu, D. High drug-loading nanomedicines: Progress, current status, and prospects. Int. J. Nanomed. 2017, 12, 4085–4109. [Google Scholar] [CrossRef]
- Hosseinpour, S.; Gomez-Cerezo, M.N.; Cao, Y.; Lei, C.; Dai, H.; Walsh, L.J.; Ivanovski, S.; Xu, C. A Comparative Study of Mesoporous Silica and Mesoporous Bioactive Glass Nanoparticles as Non-Viral MicroRNA Vectors for Osteogenesis. Pharmaceutics 2022, 14, 2302. [Google Scholar] [CrossRef]
- Li, X.; Zhang, L.; Dong, X.; Liang, J.; Shi, J. Preparation of mesoporous calcium doped silica spheres with narrow size dispersion and their drug loading and degradation behavior. Microporous Mesoporous Mater. 2007, 102, 151–158. [Google Scholar] [CrossRef]
- Vialpando, M.; Aerts, A.; Persoons, J.; Martens, J.; Van Den Mooter, G. Evaluation of ordered mesoporous silica as a carrier for poorly soluble drugs: Influence of pressure on the structure and drug release. J. Pharm. Sci. 2011, 100, 3411–3420. [Google Scholar] [CrossRef]
- Badea, E.; Carşote, C.; Hadîmbu, E.; Șendrea, C.; Lupaș, M.-C. The effect of halloysite nanotubes dispersions on vegetable-tanned leather thermal stability. Herit. Sci. 2019, 7, 68. [Google Scholar] [CrossRef]
- Minandri, F.; Bonchi, C.; Frangipani, E.; Imperi, F.; Visca, P. Promises and failures of gallium as an antibacterial agent. Future Microbiol. 2014, 9, 379–397. [Google Scholar] [CrossRef]
- Campoccia, D.; Montanaro, L.; Arciola, C.R. The significance of infection related to orthopedic devices and issues of antibiotic resistance. Biomaterials 2006, 27, 2331–2339. [Google Scholar] [CrossRef]
- Pourshahrestani, S.; Zeimaran, E.; Kadri, N.A.; Gargiulo, N.; Samuel, S.; Naveen, S.V.; Kamarulc, T.; Towler, M.R. Gallium-containing mesoporous bioactive glass with potent hemostatic activity and antibacterial efficacy. J. Mater. Chem. B 2016, 4, 71–86. [Google Scholar] [CrossRef]
- Antunes, L.C.S.; Imperi, F.; Minandri, F.; Visca, P. In Vitro and In Vivo Antimicrobial Activities of Gallium Nitrate against Multidrug-Resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 2012, 56, 5961–5970. [Google Scholar] [CrossRef]
- Peng, X.-X.; Gao, S.; Zhang, J.-L. Gallium (III) Complexes in Cancer Chemotherapy. Eur. J. Inorg. Chem. 2022, 6, e202100953. [Google Scholar] [CrossRef]
- Hart, M.M.; Smith, C.F.; Yancey, S.T.; Adamson, R.H. Toxicity and Antitumor Activity of Gallium Nitrate and Periodically Related Metal Salts. J. Natl. Cancer Inst. 1971, 47, 1121–1128. [Google Scholar] [CrossRef]
- Lessa, J.A.; Parrilha, G.L.; Beraldo, H. Gallium complexes as new promising metallodrug candidates. Inorg. Chim. Acta 2012, 393, 53–63. [Google Scholar] [CrossRef]
- Santos, E.M.; Radin, S. Ducheyne Sol–gel derived carrier for the controlled release of proteins. Biomaterials 1999, 20, 1695–1700. [Google Scholar] [CrossRef]
- Prokopowicz, M.; Żegliński, J.; Gandhi, A.; Sawicki, W.; Tofail, S.A.M. Bioactive silica-based drug delivery systems containing doxorubicin hydrochloride: In vitro studies. Colloids Surf. B Biointerfaces 2012, 93, 249–259. [Google Scholar] [CrossRef]
- Shenderovich, I.G. For Whom a Puddle Is the Sea? Adsorption of Organic Guests on Hydrated MCM-41 Silica. Langmuir 2020, 36, 11383–11392. [Google Scholar] [CrossRef]
- Saravanapavan, P.; Hench, L.L. Low-temperature synthesis, structure, and bioactivity of gel-derived glasses in the binary CaO-SiO2 system. J. Biomed. Mater. Res. 2001, 54, 608–618. [Google Scholar] [CrossRef]
- Skipper, L.J.; Sowrey, F.E.; Pickup, D.M.; Fitzgerald, V. Structural studies of bioactivity in sol–gel-derived glasses by X-ray spectroscopy. J. Biomed. Mater. Res. Part A. 2004, 70A, 354–360. [Google Scholar] [CrossRef]
- Klepetsanis, P.G.; Drakia, P.; Koutsoukos, P.G. The Calcite—Hydroxyapatite System Crystal Growth Studies in Aqueous Solutions. In Mineral Scale Formation and Inhibition; Amjad, Z., Ed.; Springer: Boston, MA, USA, 1995; pp. 251–259. [Google Scholar] [CrossRef]
- Yanyan, S.; Guangxin, W.; Wuhui, L.; Yaming, W.; Hayakawa, S.; Osaka, A. Conversion of sub-µm calcium carbonate (calcite) particles to hollow hydroxyapatite agglomerates in K2HPO4 solutions. Nanotechnol. Rev. 2020, 9, 945–960. [Google Scholar] [CrossRef]
- Dickinson, S.R.; Henderson, G.E.; McGrath, K.M. Controlling the kinetic versus thermodynamic crystallization of calcium carbonate. J. Cryst. Growth 2002, 244, 369–378. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | TEOS (g) | Pluronic® F127 (g) | TMB (mL) | HCl (1.7%) w/w (mL) | Ca(NO3)2·4H2O (g) | Ga(NO3)3·9H2O (g) |
|---|---|---|---|---|---|---|
| LPMS_5Ca | 2.10 | 4.20 | 0.55 | 40 | 0.90 | \ |
| LPMS_5Ga | 2.10 | 4.20 | 0.55 | 40 | \ | 0.93 |
| LPMS_5Ca5Ga | 2.10 | 4.20 | 0.55 | 40 | 0.90 | 1.05 |
| Sample | LE% | LC% | Loading% | Loading% with EA |
|---|---|---|---|---|
| LPMS_5Ca_5 | 26.4 | 8.0 | 7.4 | 8.9 |
| LPMS_5Ga_5 | 27.6 | 8.4 | 7.7 | 9.4 |
| LPMS_5Ca5Ga_5 | 23.4 | 7.1 | 6.6 | 7.3 |
| MS_5Ca_5 | 8.1 | 2.8 | 2.7 | 3.6 |
| MS_5Ga_5 | 12.4 | 4.1 | 3.9 | 3.8 |
| MS_5Ca5Ga_5 | 13.1 | 4.3 | 5.1 | 4.8 |
| MS_5Ca_10 | 29.8 | 9.6 | 8.7 | 9.4 |
| MS_5Ga_10 | 20.5 | 6.4 | 6.0 | 7.5 |
| MS_5Ca5Ga_10 | 17.3 | 10.3 | 9.3 | 9.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrozza, D.; Ferrari, E.; Malavasi, G. Very Large Pore Mesoporous Bioactive Silicate Glasses: Comparison of Behavior toward Classical Mesoporous Bioactive Glasses in Terms of Drug Loading/Release and Bioactivity. Materials 2024, 17, 373. https://doi.org/10.3390/ma17020373
Carrozza D, Ferrari E, Malavasi G. Very Large Pore Mesoporous Bioactive Silicate Glasses: Comparison of Behavior toward Classical Mesoporous Bioactive Glasses in Terms of Drug Loading/Release and Bioactivity. Materials. 2024; 17(2):373. https://doi.org/10.3390/ma17020373
Chicago/Turabian StyleCarrozza, Debora, Erika Ferrari, and Gianluca Malavasi. 2024. "Very Large Pore Mesoporous Bioactive Silicate Glasses: Comparison of Behavior toward Classical Mesoporous Bioactive Glasses in Terms of Drug Loading/Release and Bioactivity" Materials 17, no. 2: 373. https://doi.org/10.3390/ma17020373
APA StyleCarrozza, D., Ferrari, E., & Malavasi, G. (2024). Very Large Pore Mesoporous Bioactive Silicate Glasses: Comparison of Behavior toward Classical Mesoporous Bioactive Glasses in Terms of Drug Loading/Release and Bioactivity. Materials, 17(2), 373. https://doi.org/10.3390/ma17020373