
Effect of TiO2 and CaO Addition on the Crystallization and Flexural Strength of Novel Leucite Glass-Ceramics

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Glass Synthesis
2.2. Differential Scanning Calorimetry
2.3. Crystallisation Studies
2.4. Glass-Ceramic Powder Production for Flexural Strength Specimen Fabrication
2.5. X-ray Diffraction
2.6. Flexural Strength Specimen Fabrication
2.7. Biaxial Flexural Strength Testing
2.8. Dilatometry
2.9. Secondary Electron Imaging
3. Results
3.1. DSC Results
3.2. Dilatometry Results
3.3. XRD Results
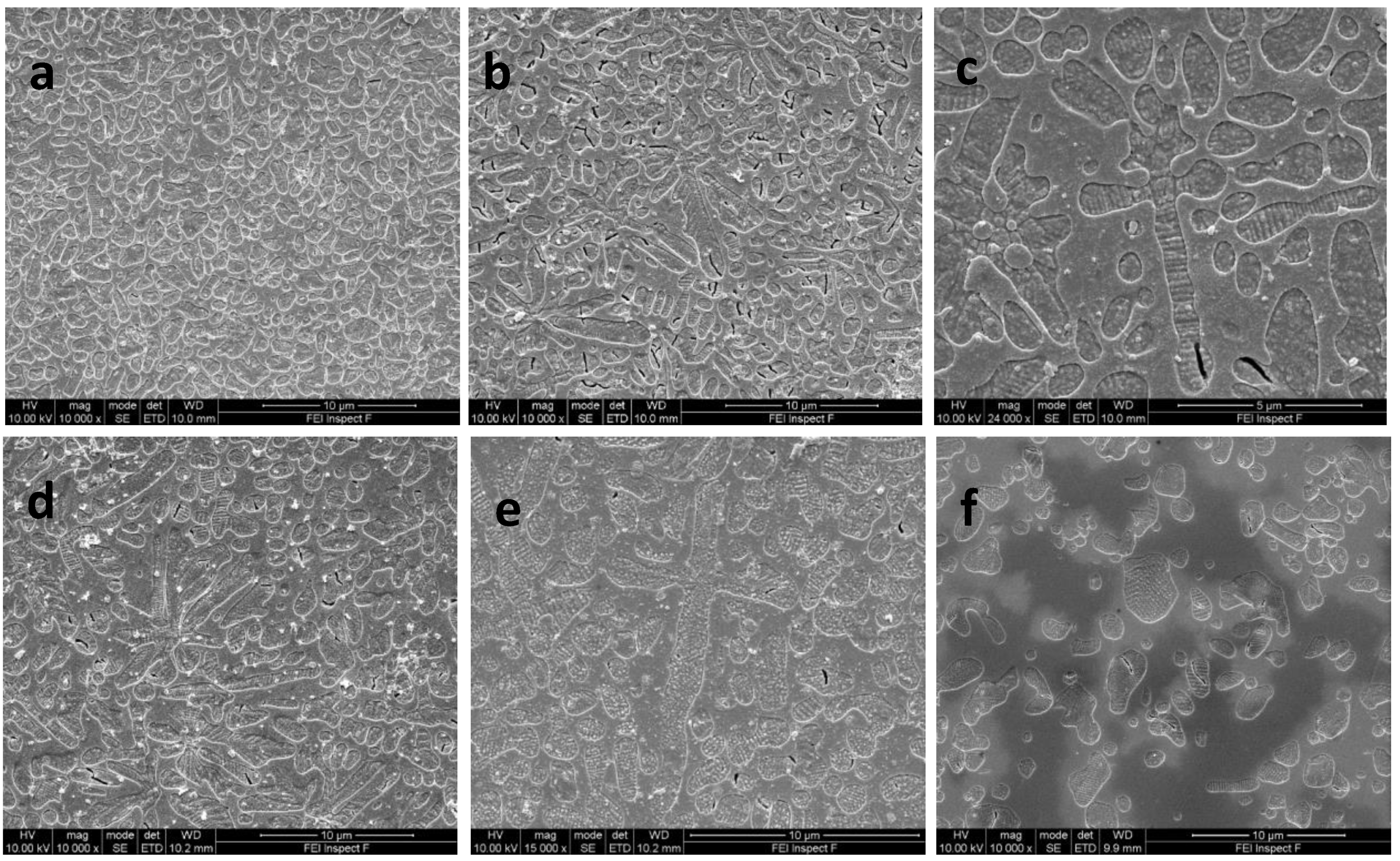
3.4. Scanning Electron Microscopy Results
3.5. BFS Results
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weinstein, L.K.; Weinstein, A.B. Fused Porcelain-to-Metal Teeth. Google Patents US3052982A, 11 September 1962. [Google Scholar]
- Mackert, J.R.; Russell, C.M. Leucite crystallization during processing of a heat-pressed dental ceramic. Int. J. Prosthod. 1996, 9, 261-–265. [Google Scholar]
- Ong, J.; Farley, D.; Norling, B. Quantification of leucite concentration using X-ray diffraction. Dent. Mater. 2000, 16, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wu, J.; Rao, P.; Fei, C.; Chen, D. The Influence of Nanosized Leucite on Dental Porcelain Properties. Key Eng. Mater. 2007, 280–283, 1605–1608. [Google Scholar]
- Novotna, M.; Klouzkova, A.; Maixner, J.; Satava, V. Preparation of leucite powders with controlled particle size distribution. Ceramics-Silikaty 2005, 49, 252–258. [Google Scholar]
- Yao, L.; Peng, C.; Wu, J. Fabrication of Superfine Leucite-Reinforced Dental Material by Hydrothermal Precursor and Low-Temperature Frit. J. Am. Ceram. Soc. 2011, 94, 3694–3697. [Google Scholar] [CrossRef]
- Burk, B.; Burnett, A.P. Leucite-Containing Porcelains and Method of Making Same. US Patent 4101330, 18 July 1978. [Google Scholar]
- Hasselman, D.; Fulrath, R. Proposed fracture theory of a dispersion-strengthened glass matrix. J. Am. Ceram. Soc. 1966, 49, 68–72. [Google Scholar] [CrossRef]
- Morena, R.; Lockwood, P.; Evans, L.; Fairhurst, C. Toughening of dental porcelain by tetragonal ZrO2 additions. J. Am. Ceram. Soc. 1986, 69, C-75–C-77. [Google Scholar] [CrossRef]
- Faber, K.T.; Evans, A.G. Crack deflection processes—I. Theory. Acta Metall. 1983, 31, 565–576. [Google Scholar] [CrossRef]
- Beham, G. IPS Empress: A new ceramic technology. Ivoclar-Vivadent-Rep. 1990, 6, 3–15. [Google Scholar]
- Cattell, M.J.; Chadwick, T.C.; Knowles, J.C.; Clarke, R.L. The crystallization of an aluminosilicate glass in the K2O–Al2O3–SiO2 system. Dent. Mater. 2005, 21, 811–822. [Google Scholar] [CrossRef]
- Höland, W.; Frank, M.; Rheinberger, V. Surface crystallization of leucite in glasses. J. Non-Cryst. Solids 1995, 180, 292–307. [Google Scholar] [CrossRef]
- Kohoutkova, M.; Kloužková, A.; Kostka, P.; Mrazova, M. Synthesis and characterization of an amorphous precursor for leucite dental ceramics. J. Non-Cryst. Solids 2008, 354, 741–748. [Google Scholar] [CrossRef]
- Cattell, M.J.; Chadwick, T.C.; Knowles, J.C.; Clarke, R.L.; Samarawickrama, D.Y. The nucleation and crystallization of fine grained leucite glass-ceramics for dental applications. Dent. Mater. 2006, 22, 925–933. [Google Scholar] [CrossRef]
- Chen, X.; Chadwick, T.; Wilson, R.; Hill, R.; Cattell, M. Crystallization of high-strength fine-sized leucite glass-ceramics. J. Dent. Res. 2010, 89, 1510–1516. [Google Scholar] [CrossRef]
- Theocharopoulos, A.; Chen, X.; Hill, R.; Cattell, M.J. Reduced wear of enamel with novel fine and nano-scale leucite glass-ceramics. J. Dent. 2013, 41, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.R. Ceramics in restorative and prosthetic dentistry. Annu. Rev. Mater. Sci. 1997, 27, 443–468. [Google Scholar] [CrossRef]
- Rouf, M.; Hermansson, L.; Carlsson, R. Crystallisation of glasses in the primary phase field of leucite in the K2O-Al2O3-SiO2 system. Trans. J. Br. Ceram Soc. 1978, 77, 36–39. [Google Scholar]
- Beall, G.H.; Pinckney, L.R. Nanophase glass-ceramics. J. Am. Ceram. Soc. 1999, 82, 5–16. [Google Scholar] [CrossRef]
- Stookey, S. Catalyzed crystallization of glass in theory and practice. Ind. Eng. Chem. 1959, 51, 805–808. [Google Scholar] [CrossRef]
- Kleebusch, E.; Rüssel, C.; Patzig, C.; Höche, T. Evidence of epitaxial growth of high-quartz solid solution on ZrTiO4 nuclei in a Li2O-Al2O3-SiO2 glass. J. Alloys Compd. 2018, 748, 73–79. [Google Scholar] [CrossRef]
- Stookey, S.D. Method of Making Ceramics and Product Thereof. Google Patent US 2920971A, 12 January 1960. [Google Scholar]
- Beall, G.H.; Duke, D.A. Glass Science and Technology; Academic Press Inc.: New York, NY, USA, 1983; pp. 403–445. [Google Scholar]
- Cattell, M.J.; Patzig, C.; Bissasu, S.; Tsoutsos, A.; Karpukhina, N. Nucleation efficacy and flexural strength of novel leucite glass-ceramics. Dent. Mater. 2020, 36, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Salama, S.N.; Salman, S.M.; Darwish, H. Effect of nucleation catalysts on crystallisation characteristics of aluminosilicate glasses. Ceramics-Silikáty 2002, 46, 15–23. [Google Scholar]
- Cattell, M.J.; Chadwick, T.C.; Knowles, J.C.; Clarke, R.L. The development and testing of glaze materials for application to the fit surface of dental ceramic restorations. Dent. Mater. 2009, 25, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Appen, A. Toward the method for calculating the properties of glass. Glass Ceram. 1961, 18, 235–237. [Google Scholar] [CrossRef]
- Winkelmann, A.; Schott, O. Über thermische Widerstandscoefficienten verschiedener Gläser in ihrer Abhängigkeit von der chemischen Zusammensetzung. Ann. Phys. 1894, 287, 730–746. [Google Scholar] [CrossRef]
- Theocharopoulos, A.; Chen, X.; Karpukhina, N.; Hill, R.; Cattell, M.J. Leucite Glass Ceramics. U.S. Patent US20160137547A1, 2 January 2018. [Google Scholar]
- Kissinger, H.E. Variation of peak temperature with heating rate in differential thermal analysis. J. Res. Natl. Bur. Stand. 1956, 57, 217–221. [Google Scholar] [CrossRef]
- Cullity, B.D.; Stock, S.R. Elements of X-ray Diffraction, 3rd ed.; Prentice-Hall: New York, NY, USA, 2001; pp. 439–441. [Google Scholar]
- Timoshenko, S.; Woinowsky-Krieger, S. Theory of Plates and Shells; McGraw-Hill: New York, NY, USA, 1959; pp. 70–71. [Google Scholar]
- Rodrigues, L.R.; Acosta, M.H.R.; Zanotto, E.D. Recent crucial discoveries and perspectives on crystal nucleation in supercooled liquids and oxide glasses. Prog. Mater. Sci. 2023, 139, 101185. [Google Scholar] [CrossRef]
- Zanotto, E.D. Effect of liquid phase separation on crystal nucleation in glass-formers. Case closed. Ceram. Int. 2020, 46 Pt A, 24779–24791. [Google Scholar] [CrossRef]
- Ojovan, M.I. Viscosity and Glass Transition in Amorphous Oxides. Adv. Condens. Matter Phys. 2008, 2008, 1–23. [Google Scholar] [CrossRef]
- Theocharopoulos, A. Synthesis of Low-Wear and High Strength Nano-Scale Leucite Glass-Ceramics for Dentistry. Ph.D. Thesis, University of London, London, UK, 2011. [Google Scholar]
- Vyazovkin, S. Kissinger Method in Kinetics of Materials: Things to Beware and Be Aware of. Molecules 2020, 25, 2813. [Google Scholar] [CrossRef]
- DeCeanne, A.V.; Rodrigues, L.R.; Wilkinson, C.J.; Mauro, J.C.; Zanotto, E.D. Examining the role of nucleating agents within glass-ceramic systems. J. Non-Cryst. Solids 2022, 591, 121714. [Google Scholar] [CrossRef]
- Zhang, Y.; Lv, M.; Chen, D.; Wu, J. Leucite crystallization kinetics with kalsilite as a transition phase. Mater. Lett. 2007, 61, 2978–2981. [Google Scholar] [CrossRef]
- Löschmann, J.; Fielitz, P.; Helsch, G.; Bornhöft, H.; Cassar, D.R.; Borchardt, G.; Deubener, J. Accelerated crystal growth in a lithia aluminosilicate glass. Acta Mater. 2022, 230, 117837. [Google Scholar] [CrossRef]
- Marghussian, V. 1—Glass Crystallization. In Nano-Glass Ceramics; Marghussian, V., Ed.; William Andrew Publishing: Oxford, UK, 2015; pp. 1–62. [Google Scholar]
- Ostwald, W. Studien über die Bildung und Umwandlung fester Körper. 1 Abhandlung: Übersättigung und Überkaltung. Z. Für Phys. Chem. 1897, 22, 289–330. [Google Scholar] [CrossRef]
- Zhang, Q.; Peng, X.; Nie, Y.; Zheng, Q.; Shangguan, J.; Zhu, C.; Bustillo, K.C.; Ercius, P.; Wang, L.; Limmer, D.T.; et al. Defect-mediated ripening of core-shell nanostructures. Nat. Commun. 2022, 13, 2211. [Google Scholar] [CrossRef] [PubMed]
- Ueberricke, L.; Murata, T.; Ikeda, H.; Nakane, S.; Deubener, J. Crystal growth in oxide melts—From CALPHAD thermodynamic modeling to statistical prediction. Acta Mater. 2024, 273, 119960. [Google Scholar] [CrossRef]
- Wisniewski, W.; Rüssel, C. Oriented surface nucleation in inorganic glasses—A review. Prog. Mater. Sci. 2021, 118, 100758. [Google Scholar] [CrossRef]
- McMillan, P. Glass-Ceramics; Academic Press: London, UK, 1979; pp. 100–106. [Google Scholar]
- Mackert, J., Jr.; Twiggs, S.W.; Russell, C.M.; Williams, A.L. Evidence of a critical leucite particle size for microcracking in dental porcelains. J. Dent. Res. 2001, 80, 1574–1579. [Google Scholar] [CrossRef] [PubMed]
- Shareef, M.; Van Noort, R.; Messer, P.; Piddock, V. The effect of microstructural features on the biaxial flexural strength of leucite reinforced glass-ceramics. J. Mater. Sci. Mater. Med. 1994, 5, 113–118. [Google Scholar] [CrossRef]
- Cattell, M.J.; Knowles, J.; Clarke, R.; Lynch, E. The biaxial flexural strength of two pressable ceramic systems. J. Dent. 1999, 27, 183–196. [Google Scholar] [CrossRef]
- Theocharopoulos, A.; Chen, X.; Wilson, R.M.; Hill, R.; Cattell, M.J. Crystallization of high-strength nano-scale leucite glass-ceramics. Dent. Mater. 2013, 29, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Green, D.J. Brittle Fracture. An Introduction to the Mechanical Properties of Ceramics. Cambridge Solid State Science Series; Cambridge University Press: Cambridge, UK, 1998; pp. 210–284. [Google Scholar]
- Denry, I.L.; JRMackert, J.; Holloway, J.A.; Rosenstiel, S.F. Effect of Cubic Leucite Stabilization on the Flexural Strength of Feldspathic Dental Porcelain. J. Dent. Res. 1996, 75, 1928–1935. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glass | SiO2 | Al2O3 | K2O | CaO | TiO2 | Na2O | Li2O | MgO |
|---|---|---|---|---|---|---|---|---|
| B1 | 69.6 | 10.2 | 12.5 | 1.0 | 1.0 | 4.0 | 1.1 | 0.5 |
| B2 | 68.2 | 10.0 | 12.3 | 2.0 | 2.0 | 3.9 | 1.1 | 0.5 |
| B3 | 66.8 | 9.7 | 12.0 | 3.0 | 3.0 | 3.8 | 1.1 | 0.5 |
| Glass | Nucleation Temperature (°C) | Crystal Growth Temperature (°C) |
|---|---|---|
| B1 | 606 | 868 |
| B2 | 613 | 903 |
| B3 | 618 | 916 |
| Material | Predicted TEC (°C) | TEC (×10−6/K, 100–400 °C) | Glass Transition Temperature (°C) | Softening Point (°C) |
|---|---|---|---|---|
| B1 glass | 10.32 | 9.93 | 576.0 | 664.4 |
| B2 glass | 10.35 | 10.01 | 582.7 | 675.3 |
| B3 glass | 10.37 | 10.06 | 587.8 | 675.4 |
| B1 glass-ceramic | 20.23 | 471.7 | 611.8 | |
| B2 glass-ceramic | 19.61 | 486.8 | 657.1 | |
| B3 glass-ceramic | 24.87 | 508.7 | 658.5 |
| Glass- Ceramics | Mean a–Axis Unit Cell Dimension Å (SD) | Mean c–Axis Unit Cell Dimension Å (SD) | Mean Unit Cell Volume Å (SD) | ɛa | ɛc |
|---|---|---|---|---|---|
| B1 | 13.1082 (0.0005) | 13.7205 (0.0009) | 2357.50 (0.21) | 0.33% | −0.25% |
| B2 | 13.1016 (0.0005) | 13.7248 (0.0008) | 2355.89 (0.20) | 0.28% | −0.22% |
| B3 | 13.1009 (0.0006) | 13.7319 (0.0009) | 2356.88 (0.22) | 0.27% | −0.17% |
| Glass-Ceramic B1 | Nucleation Hold (h) | Leucite Area Fraction (%) | Median Crystal Size (μm2) | Interquartile Range (Q1, Q3) (μm2) | Crystal Number |
| 0.5 | 61.4 | 0.784 a/A | 0.352, 1.655 | 1367 | |
| 1 | 57.4 | 0.617 b/A | 0.304, 1.223 | 1787 | |
| 2 | 60.3 | 0.603 b/A | 0.294, 1.283 | 1687 | |
| 3 | 60.3 | 0.596 b/A | 0.262, 1.301 | 1789 | |
| Glass-Ceramic B2 | Nucleation Hold (h) | Leucite Area Fraction (%) | Median Crystal Size (μm2) | Interquartile Range (Q1, Q3) (μm2) | Crystal Number |
| 0.5 | 56.9 | 0.950 a/B | 0.426, 2.024 | 910 | |
| 1 | 56.7 | 0.806 b/B | 0.376, 1.584 | 1004 | |
| 2 | 60.0 | 1.083 a/B | 0.525, 2.354 | 745 | |
| 3 | 60.6 | 1.012 a/B | 0.494, 1.920 | 983 | |
| Glass-Ceramic B3 | Nucleation Hold (h) | Leucite Area Fraction (%) | Median Crystal Size (μm2) | Interquartile Range (Q1, Q3) (μm2) | Crystal Number |
| 0.5 | 59.6 | 1.080 a/B | 0.542, 2.347 | 735 | |
| 1 | 55.1 | 1.145 a/C | 0.506, 2.267 | 841 | |
| 2 | 55.8 | 1.080 a/B | 0.467, 2.207 | 822 | |
| 3 | 60.8 | 1.308 b/C | 0.674, 2.369 | 732 |
| Glass-Ceramics | Mean Flexural Strength (MPa) | SD (MPa) | Weibull Modulus (m) | C.I. for m (95%) | σ0 (MPa) | C.I. For σ0 (95%) | r2 |
|---|---|---|---|---|---|---|---|
| B1 | 225.3 ab | 24.7 | 10.9 a | 8.8–13.6 | 235.6 ab | 228.9–242.6 | 0.976 |
| B2 | 211.6 a | 26.6 | 9.4 ac | 7.6–11.9 | 222.6 a | 215.2–230.1 | 0.962 |
| B3 | 235.1 b | 14.7 | 18.9 b | 14.9–24.1 | 241.6 b | 237.6–245.6 | 0.974 |
| IPS Empress Esthetic | 165.5 c | 30.6 | 6.3 c | 5.0–7.9 | 177.5 c | 168.9–186.6 | 0.977 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almuhamadi, J.; Almusali, M.H.; Chen, X.; Theocharopoulos, A.L.; Alostath, H.F.; Karpukhina, N.; Cattell, M.J. Effect of TiO2 and CaO Addition on the Crystallization and Flexural Strength of Novel Leucite Glass-Ceramics. Materials 2024, 17, 3422. https://doi.org/10.3390/ma17143422
Almuhamadi J, Almusali MH, Chen X, Theocharopoulos AL, Alostath HF, Karpukhina N, Cattell MJ. Effect of TiO2 and CaO Addition on the Crystallization and Flexural Strength of Novel Leucite Glass-Ceramics. Materials. 2024; 17(14):3422. https://doi.org/10.3390/ma17143422
Chicago/Turabian StyleAlmuhamadi, Jamila, Mustafa H. Almusali, Xiaohui Chen, Antonios L. Theocharopoulos, Hawraa F. Alostath, Natalia Karpukhina, and Michael J. Cattell. 2024. "Effect of TiO2 and CaO Addition on the Crystallization and Flexural Strength of Novel Leucite Glass-Ceramics" Materials 17, no. 14: 3422. https://doi.org/10.3390/ma17143422
APA StyleAlmuhamadi, J., Almusali, M. H., Chen, X., Theocharopoulos, A. L., Alostath, H. F., Karpukhina, N., & Cattell, M. J. (2024). Effect of TiO2 and CaO Addition on the Crystallization and Flexural Strength of Novel Leucite Glass-Ceramics. Materials, 17(14), 3422. https://doi.org/10.3390/ma17143422