
A Modified Embedded-Atom Method Potential for a Quaternary Fe-Cr-Si-Mo Solid Solution Alloy



Abstract
1. Introduction
2. Materials and Methods
2.1. Potential Development and Fitting
2.2. Unary Potential
2.3. Binary Potential
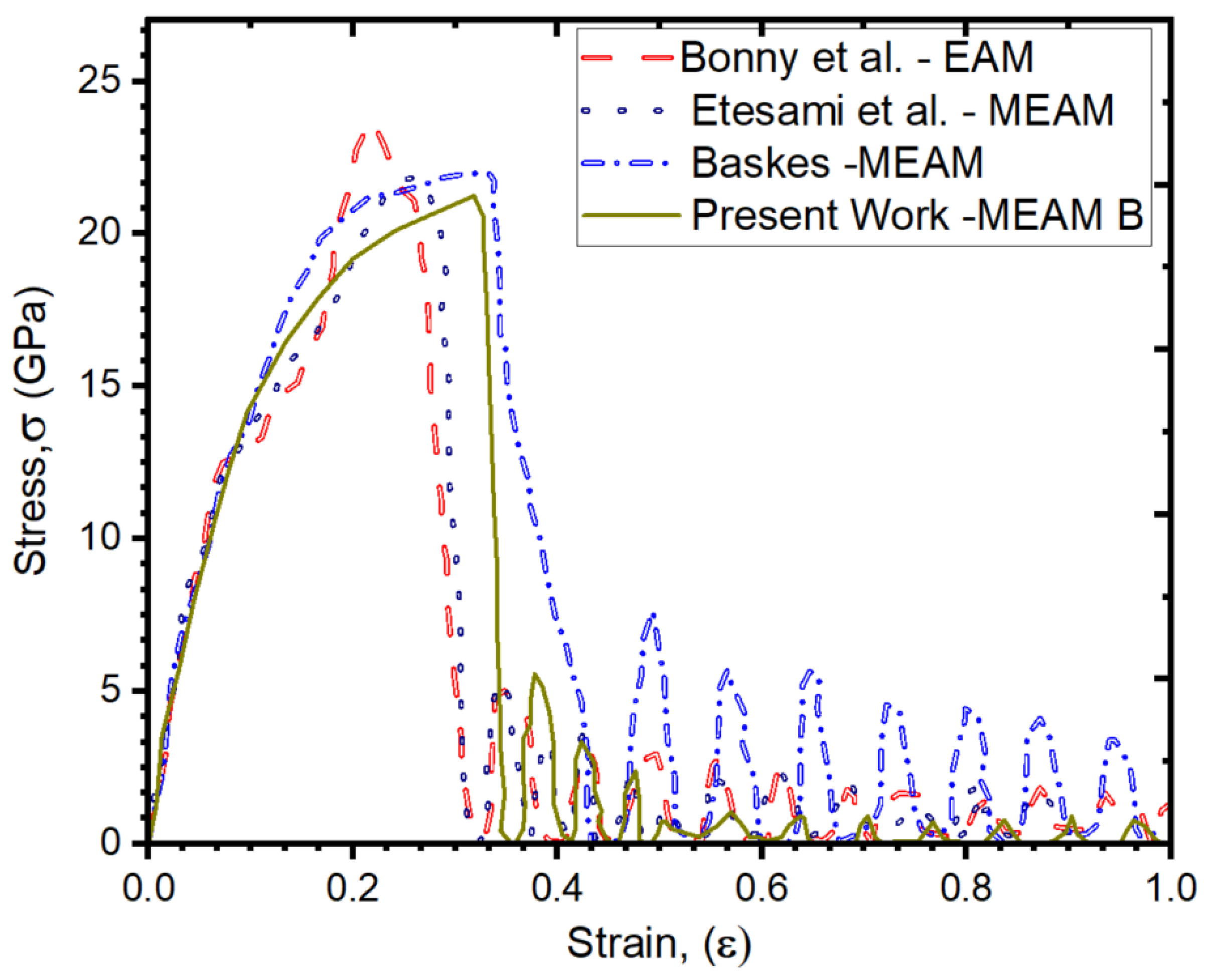
3. Results
3.1. Bulk Mechanical Properties
3.2. Our Results Indicate an Excellent Agreement with Previous Interatomic Potentials
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mansur, L.K.; Rowcliffe, A.F.; Nanstad, R.K.; Zinkle, S.J.; Corwin, W.R.; Stoller, R.E. Materials Needs for Fusion, Generation IV Fission Reactors and Spallation Neutron Sources-Similarities and Differences. J. Nucl. Mater. 2004, 329–333, 166–172. [Google Scholar] [CrossRef]
- Paul, S.; Schwen, D.; Short, M.P.; Momeni, K. Effect of Irradiation on Ni-Inconel/Incoloy Heterostructures in Multimetallic Layered Composites. J. Nucl. Mater. 2021, 547, 152778. [Google Scholar] [CrossRef]
- Zinkle, S.J.; Busby, J.T. Structural Materials for Fission & Fusion Energy. Mater. Today 2009, 12, 12–19. [Google Scholar]
- Odette, G.R.; Alinger, M.J.; Wirth, B.D. Recent Developments in Irradiation-Resistant Steels. Annu. Rev. Mater. Res. 2008, 38, 471–503. [Google Scholar] [CrossRef]
- Yvon, P.; Carré, F. Structural Materials Challenges for Advanced Reactor Systems. J. Nucl. Mater. 2009, 385, 217–222. [Google Scholar] [CrossRef]
- Terrani, K.A.; Zinkle, S.J.; Snead, L.L. Advanced Oxidation-Resistant Iron-Based Alloys for LWR Fuel Cladding. J. Nucl. Mater. 2014, 448, 420–435. [Google Scholar] [CrossRef]
- Zinkle, S.J.; Terrani, K.A.; Gehin, J.C.; Ott, L.J.; Snead, L.L. Accident Tolerant Fuels for LWRs: A Perspective. J. Nucl. Mater. 2014, 448, 374–379. [Google Scholar] [CrossRef]
- Yang, Z.; Li, J.P.; Zhang, J.X.; Lorimer, G.W.; Robson, J. Review on Research and Development of Magnesium Alloys. Acta Metall. Sin. (Engl. Lett.) 2008, 21, 313–328. [Google Scholar] [CrossRef]
- Ivanov, I.; Stefanov, Y.; Noncheva, Z.; Petrova, M.; Dobrev, T.; Mirkova, L.; Vermeersch, R.; Demaerel, J.P. Insoluble Anodes Used in Hydrometallurgy: Part I. Corrosion Resistance of Lead and Lead Alloy Anodes. Hydrometallurgy 2000, 57, 109–124. [Google Scholar] [CrossRef]
- Altuner, E.E.; Gur, T.; Şen, F. 10—Ternary/Quaternary Nanomaterials for Direct Alcohol Fuel Cells. In Nanomaterials for Direct Alcohol Fuel Cells: Characterization, Design, and Electrocatalysis, Micro and Nano Technologies; Elsevier: Amsterdam, The Netherlands, 2021; pp. 157–172. [Google Scholar] [CrossRef]
- Yan, G.; Tan, H.; Wang, Y.; Li, Y. Amorphous Quaternary Alloy Phosphide Hierarchical Nanoarrays with Pagoda-like Structure Grown on Ni Foam as PH-Universal Electrocatalyst for Hydrogen Evolution Reaction. Appl. Surf. Sci. 2019, 489, 519–527. [Google Scholar] [CrossRef]
- Rached, H. Prediction of a New Quaternary Heusler Alloy within a Good Electrical Response at High Temperature for Spintronics Applications: DFT Calculations. Int. J. Quantum. Chem. 2021, 121, e26647. [Google Scholar] [CrossRef]
- Murty, K.L.; Charit, I. Structural Materials for Gen-IV Nuclear Reactors: Challenges and Opportunities. J. Nucl. Mater. 2008, 383, 189–195. [Google Scholar] [CrossRef]
- Zinkle, S.J. Challenges in Developing Materials for Fusion Technology-Past, Present, and Future. In Proceedings of the Fusion Science and Technology, Nashville, TN, USA, 27–31 August 2013; Volume 64, pp. 65–75. [Google Scholar]
- Tavassoli, A.A.F. Materials Design Data for Fusion Reactors. J. Nucl. Mater. 1998, 258–263, 85–96. [Google Scholar] [CrossRef]
- Gigax, J.G.; Chen, T.; Kim, H.; Wang, J.; Price, L.M.; Aydogan, E.; Maloy, S.A.; Schreiber, D.K.; Toloczko, M.B.; Garner, F.A.; et al. Radiation Response of Alloy T91 at Damage Levels up to 1000 Peak Dpa. J. Nucl. Mater. 2016, 482, 257–265. [Google Scholar] [CrossRef]
- Pareige, C.; Kuksenko, V.; Pareige, P. Behaviour of P, Si, Ni Impurities and Cr in Self Ion Irradiated Fe-Cr Alloys-Comparison to Neutron Irradiation. J. Nucl. Mater. 2015, 456, 471–476. [Google Scholar] [CrossRef]
- Cui, S.; Jung, I.H. Thermodynamic Assessments of the Fe-Si-Cr and Fe-Si-Mg Systems. Metall. Mater. Trans. A Phys. Metall. Mater. Sci. 2017, 48, 4342–4355. [Google Scholar] [CrossRef]
- Hazza, M.I.; El-Dahshan, M.E. The Effect of Molybdenum on the Corrosion Behaviour of Some Steel Alloys. Desalination 1994, 95, 199–209. [Google Scholar] [CrossRef]
- Hojna, A.; Di Gabriele, F. On the Kinetics of LME for the Ferritic–Martensitic Steel T91 Immersed in Liquid PbBi Eutectic. J. Nucl. Mater. 2011, 413, 21–29. [Google Scholar] [CrossRef]
- Di Gabriele, F.; Hojna, A.; Chocholousek, M.; Klecka, J. Behavior of the Steel T91 under Multi Axial Loading in Contact with Liquid and Solid Pb. Metals 2017, 7, 342. [Google Scholar] [CrossRef]
- Devanathan, R.; Van Brutzel, L.; Chartier, A.; Guéneau, C.; Mattsson, A.E.; Tikare, V.; Bartel, T.; Besmann, T.; Stan, M.; Van Uffelen, P. Modeling and Simulation of Nuclear Fuel Materials. Energy Environ. Sci. 2010, 3, 1406–1426. [Google Scholar] [CrossRef]
- Samaras, M.; Hoffelner, W.; Victoria, M. Modelling of Advanced Structural Materials for GEN IV Reactors. J. Nucl. Mater. 2007, 371, 28–36. [Google Scholar] [CrossRef]
- Momeni, K. Enhanced Mechanical Properties of ZnO Nanowire-Reinforced Nanocomposites: A Size-Scale Effect. Acta Mech 2014, 225, 2549–2562. [Google Scholar] [CrossRef]
- Momeni, K. A Multiscale Approach to Nanocomposite Electrical Generators. Nano Energy 2014, 4, 132–139. [Google Scholar] [CrossRef]
- Momeni, K.; Mortazavi, S.M.Z. Optimal Aspect Ratio of Zinc Oxide Nanowires for a Nanocomposite Electrical Generator. J. Comput. Theor. Nanosci. 2012, 9, 1670–1674. [Google Scholar] [CrossRef]
- Momeni, K.; Yassar, R.S. Stress Distribution on a Single-Walled Carbon Nanohorn Embedded in an Epoxy Matrix Nanocomposite under Axial Force. J. Comput. Theor. Nanosci. 2010, 7, 1035–1041. [Google Scholar] [CrossRef]
- Momeni, K.; Alasty, A. Stress Distribution on Open-Ended Carbon Nanotubes. In Proceedings of the 2008 Proceedings of the ASME-2nd International Conference on Integration and Commercialization of Micro and Nanosystems, MicroNano 2008, Kowloon, Hong Kong, 3–5 June 2008. [Google Scholar]
- Momeni, K.; Yassar, R.S. Analytical Formulation of Stress Distribution in Cellulose Nanocomposites. J. Comput. Theor. Nanosci. 2009, 6, 1511–1518. [Google Scholar] [CrossRef]
- Momeni, K.; Mofidian, S.M.M.; Bardaweel, H. Systematic Design of High-Strength Multicomponent Metamaterials. Mater. Des. 2019, 183, 108124. [Google Scholar] [CrossRef]
- Momeni, K.; Odegard, G.M.; Yassar, R.S. Nanocomposite Electrical Generator Based on Piezoelectric Zinc Oxide Nanowires. J. Appl. Phys. 2010, 108, 114303. [Google Scholar] [CrossRef]
- Ding, H.; Zeng, C.; Raush, J.; Momeni, K.; Guo, S. Developing Fused Deposition Modeling Additive Manufacturing Processing Strategies for Aluminum Alloy 7075: Sample Preparation and Metallographic Characterization. Materials 2022, 15, 1340. [Google Scholar] [CrossRef]
- Mofidian, S.M.M.; Davani, S.; Momeni, K.; Bardaweel, H. 3D-Printed Strain Sensors: Electro-Mechanical Simulation and Design Analysis Using Nonlinear Material Model and Experimental Investigation. IEEE Sens. J. 2021, 21, 1675–1685. [Google Scholar] [CrossRef]
- Asthana, A.; Momeni, K.; Prasad, A.; Yap, Y.; Yassar, R. A Study on the Structure-Piezoresponse Property of a ZnO Nanobelt by In Situ Transmission Electron Microscopy. Microsc. Microanal. 2011, 17, 1724–1725. [Google Scholar] [CrossRef]
- Asthana, A.; Momeni, K.; Prasad, A.; Yap, Y.K.; Yassar, R.S. In Situ Observation of Size-Scale Effects on the Mechanical Properties of ZnO Nanowires. Nanotechnology 2011, 22, 265712. [Google Scholar] [CrossRef]
- Tajyar, A.; Brooks, N.; Vaseghi, M.; Hackel, L.; Momeni, K.; Davami, K. Multi-Cycling Nanoindentation in Additively Manufactured Inconel 625 before and after Laser Peening. Surf. Topogr. 2022, 10, 025031. [Google Scholar] [CrossRef]
- Davami, K.; Mohsenizadeh, M.; Munther, M.; Palma, T.; Beheshti, A.; Momeni, K. Dynamic Energy Absorption Characteristics of Additivelymanufactured Shape-Recovering Lattice Structures. Mater. Res. Express 2019, 6, 045302. [Google Scholar] [CrossRef]
- Momeni, K.; Asthana, A.; Prasad, A.; Yap, Y.K.; Shahbazian-Yassar, R. Structural Inhomogeneity and Piezoelectric Enhancement in ZnO Nanobelts. Appl. Phys. A Mater. Sci. Process 2012, 109, 95–100. [Google Scholar] [CrossRef]
- Munther, M.; Shaygan, M.; Centeno, A.; Neumaier, D.; Zurutuza, A.; Momeni, K.; Davami, K. Probing the Mechanical Properties of Vertically-Stacked Ultrathin Graphene/Al2O3 Heterostructures. Nanotechnology 2019, 30, 185703. [Google Scholar] [CrossRef]
- Rowe, R.A.; Tajyar, A.; Munther, M.; Johanns, K.E.; Allison, P.G.; Momeni, K.; Davami, K. Nanoscale Serration Characteristics of Additively Manufactured Superalloys. J. Alloys Compd. 2021, 854, 156723. [Google Scholar] [CrossRef]
- Sadeghirad, A.; Momeni, K.; Ji, Y.; Ren, X.; Chen, L.-Q.L.Q.; Lua, J. Multiscale Crystal-Plasticity Phase Field and Extended Finite Element Methods for Fatigue Crack Initiation and Propagation Modeling. Int. J. Fract. 2019, 216, 41–57. [Google Scholar] [CrossRef]
- Zhang, F.; Momeni, K.; AlSaud, M.A.; Azizi, A.; Hainey, M.F.; Redwing, J.M.; Chen, L.-Q.; Alem, N. Controlled Synthesis of 2D Transition Metal Dichalcogenides: From Vertical to Planar MoS 2. 2d Mater. 2017, 4, 025029. [Google Scholar] [CrossRef]
- Vilá, R.A.; Momeni, K.; Wang, Q.; Bersch, B.M.; Lu, N.; Kim, M.J.; Chen, L.Q.; Robinson, J.A. Bottom-up Synthesis of Vertically Oriented Two-Dimensional Materials. 2d Mater 2016, 3, 041003. [Google Scholar] [CrossRef]
- Hu, J.-M.; Yang, T.; Momeni, K.; Cheng, X.; Chen, L.; Lei, S.; Zhang, S.; Trolier-McKinstry, S.; Gopalan, V.; Carman, G.P.; et al. Fast Magnetic Domain-Wall Motion in a Ring-Shaped Nanowire Driven by a Voltage. Nano Lett. 2016, 16, 2341–2348. [Google Scholar] [CrossRef]
- Peng, R.-C.R.-C.; Hu, J.-M.J.-M.; Momeni, K.; Wang, J.-J.J.-J.; Chen, L.-Q.L.-Q.; Nan, C.-W. Fast 180° Magnetization Switching in a Strain-Mediated Multiferroic Heterostructure Driven by a Voltage. Sci. Rep. 2016, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Briggs, N.; Subramanian, S.; Lin, Z.; Li, X.; Zhang, X.; Zhang, K.; Xiao, K.; Geohegan, D.; Wallace, R.; Chen, L.-Q.; et al. A Roadmap for Electronic Grade 2D Materials. 2d Mater 2019, 6, 022001. [Google Scholar] [CrossRef]
- Asthana, A.; Momeni, K.; Prasad, A.; Yap, Y.K.; Yassar, R.S. On the Correlation of Crystal Defects and Band Gap Properties of ZnO Nanobelts. Appl. Phys. A Mater. Sci. Process 2011, 105, 909–914. [Google Scholar] [CrossRef]
- Ghosh, M.; Hendy, M.; Raush, J.; Momeni, K. A Phase-Field Model for In-Space Manufacturing of Binary Alloys. Materials 2022, 16, 383. [Google Scholar] [CrossRef] [PubMed]
- Momeni, K.; Neshani, S.; Uba, C.; Ding, H.; Raush, J.; Guo, S. Engineering the Surface Melt for In-Space Manufacturing of Aluminum Parts. J. Mater. Eng. Perform. 2022, 31, 6092–6100. [Google Scholar] [CrossRef]
- Momeni, K.; Levitas, V.I. A Phase-Field Approach to Nonequilibrium Phase Transformations in Elastic Solids: Via an Intermediate Phase (Melt) Allowing for Interface Stresses. Phys. Chem. Chem. Phys. 2016, 18, 12183–12203. [Google Scholar] [CrossRef]
- Momeni, K.; Levitas, V.I. A Phase-Field Approach to Solid–Solid Phase Transformations via Intermediate Interfacial Phases under Stress Tensor. Int. J. Solids Struct. 2015, 71, 39–56. [Google Scholar] [CrossRef]
- Momeni, K.; Levitas, V.I.; Warren, J.A. The Strong Influence of Internal Stresses on the Nucleation of a Nanosized, Deeply Undercooled Melt at a Solid–Solid Phase Interface. Nano Lett. 2015, 15, 2298–2303. [Google Scholar] [CrossRef]
- Momeni, K. A Diffuse Interface Approach to Phase Transformation via Virtual Melting. Ph.D. Thesis, Iowa State University, Ames, IA, USA, 2015. [Google Scholar]
- Momeni, K.; Levitas, V.I. Propagating Phase Interface with Intermediate Interfacial Phase: Phase Field Approach. Phys. Rev. B 2014, 89, 184102. [Google Scholar] [CrossRef]
- Levitas, V.I.; Momeni, K. Solid–Solid Transformations via Nanoscale Intermediate Interfacial Phase: Multiple Structures, Scale and Mechanics Effects. Acta Mater. 2014, 65, 125–132. [Google Scholar] [CrossRef]
- Paul, S.; Schwen, D.; Short, M.P.; Erickson, A.; Momeni, K. Effect of Differently Oriented Interlayer Phases on the Radiation Damage of Inconel-Ni Multimetallic Layered Composite. J. Alloys Compd. 2022, 915, 165432. [Google Scholar] [CrossRef]
- Paul, S.; Torsi, R.; Robinson, J.A.; Momeni, K. Effect of the Substrate on MoS2 Monolayer Morphology: An Integrated Computational and Experimental Study. ACS Appl. Mater. Interfaces 2022, 14, 18835–18844. [Google Scholar] [CrossRef]
- Sakib, N.; Paul, S.; Nayir, N.; van Duin, A.C.T.; Neshani, S.; Momeni, K. Role of Tilt Grain Boundaries on the Structural Integrity of WSe2 Monolayers. Phys. Chem. Chem. Phys. 2022, 24, 27241–27249. [Google Scholar] [CrossRef]
- Paul, S.; Momeni, K.; Levitas, V.I. Shear-Induced Diamondization of Multilayer Graphene Structures: A Computational Study. Carbon NY 2020, 167, 140–147. [Google Scholar] [CrossRef]
- Paul, S.; Momeni, K. Mechanochemistry of Stable Diamane and Atomically Thin Diamond Films Synthesis from Bi- and Multilayer Graphene: A Computational Study. J. Phys. Chem. C 2019, 123, 15751–15760. [Google Scholar] [CrossRef]
- Attariani, H.; Rezaei, S.E.E.; Momeni, K. Mechanical Property Enhancement of One-Dimensional Nanostructures through Defect-Mediated Strain Engineering. Extreme Mech. Lett. 2019, 27, 66–75. [Google Scholar] [CrossRef]
- Attariani, H.; Emad Rezaei, S.; Momeni, K. Defect Engineering, a Path to Make Ultra-High Strength Low-Dimensional Nanostructures. Comput. Mater. Sci. 2018, 151, 307–316. [Google Scholar] [CrossRef]
- Attariani, H.; Momeni, K.; Adkins, K. Defect Engineering: A Path toward Exceeding Perfection. ACS Omega 2017, 2, 663–669. [Google Scholar] [CrossRef]
- Ghosh, M.; Ghosh, S.; Attariani, H.; Momeni, K.; Seibt, M.; Mohan Rao, G. Atomic Defects Influenced Mechanics of II–VI Nanocrystals. Nano Lett. 2016, 16, 5969–5974. [Google Scholar] [CrossRef] [PubMed]
- Momeni, K.; Attariani, H.; Lesar, R.A.R.A. Structural Transformation in Monolayer Materials: A 2D to 1D Transformation. Phys. Chem. Chem. Phys. 2016, 18, 19873–19879. [Google Scholar] [CrossRef]
- Momeni, K.; Attariani, H. Electromechanical Properties of 1D ZnO Nanostructures: Nanopiezotronics Building Blocks, Surface and Size-Scale Effects. Phys. Chem. Chem. Phys. 2014, 16, 4522–4527. [Google Scholar] [CrossRef]
- Momeni, K.; Odegard, G.M.; Yassar, R.S. Finite Size Effect on the Piezoelectric Properties of ZnO Nanobelts: A Molecular Dynamics Approach. Acta Mater. 2012, 60, 5117–5124. [Google Scholar] [CrossRef]
- Momeni, K.; Ji, Y.; Nayir, N.; Sakib, N.; Zhu, H.; Paul, S.; Choudhury, T.H.; Neshani, S.; van Duin, A.C.T.; Redwing, J.M.; et al. A Computational Framework for Guiding the MOCVD-Growth of Wafer-Scale 2D Materials. NPJ Comput. Mater. 2022, 8, 240. [Google Scholar] [CrossRef]
- Momeni, K. Sensitivity of Additively Manufactured AA7075 to Variation in Feedstock Composition and Print Parameters. J. Manuf. Process 2022, 73, 555–562. [Google Scholar] [CrossRef]
- Momeni, K.; Ji, Y.; Chen, L.-Q. Computational Synthesis of 2D Materials Grown by Chemical Vapor Deposition. J. Mater. Res. 2022, 37, 114–123. [Google Scholar] [CrossRef]
- Ji, Y.; Momeni, K.; Chen, L.-Q. A Multiscale Insight into the Growth of H-BN: Effect of the Enclosure. 2d Mater 2021, 8, 035033. [Google Scholar] [CrossRef]
- Momeni, K.; Ji, Y.; Wang, Y.; Paul, S.; Neshani, S.; Yilmaz, D.E.; Shin, Y.K.; Zhang, D.; Jiang, J.-W.; Park, H.S.; et al. Multiscale Computational Understanding and Growth of 2D Materials: A Review. NPJ Comput. Mater. 2020, 6, 22. [Google Scholar] [CrossRef]
- Momeni, K.; Alasty, A. Introducing Structural Approximation Method for Modeling Nanostructures. J. Comput. Theor. Nanosci. 2010, 7, 423–428. [Google Scholar] [CrossRef][Green Version]
- Kwon, J.; Lee, G.; Shin, C. Multiscale Modeling of Rdiation Effects on Materials: Pressure Vessel Embrittlement. Nucl. Eng. Technol. 2009, 41, 11–20. [Google Scholar] [CrossRef]
- Daw, M.S.; Baskes, M.I. Semiempirical, Quantum Mechanical Calculation of Hydrogen Embrittlement in Metals. Phys. Rev. Lett. 1983, 50, 1285–1288. [Google Scholar] [CrossRef]
- Baskes, M.I. Modified Embedded-Atom Potentials for Cubic Materials and Impurities. Phys. Rev. B 1992, 46, 2727–2742. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.L.; Su, C.H.; Ju, S.P.; Liu, S.H.; Chen, H.T. Local Structural Evolution of Fe54C18Cr16Mo12 Bulk Metallic Glass during Tensile Deformation and a Temperature Elevation Process: A Molecular Dynamics Study. RSC Adv. 2015, 5, 103925–103935. [Google Scholar] [CrossRef]
- Samaras, M.; Victoria, M. Modelling in Nuclear Energy Environments. Mater. Today 2008, 11, 54–62. [Google Scholar] [CrossRef]
- Baskes, M.I. Atomistic Potentials for the Molybdenum-Silicon System. Mater. Sci. Eng. A 1999, 261, 165–168. [Google Scholar] [CrossRef]
- Mendelev, M.I.; Han, S.; Srolovitz, D.J.; Ackland, G.J.; Sun, D.Y.; Asta, M. Development of New Interatomic Potentials Appropriate for Crystalline and Liquid Iron. Philos. Mag. 2003, 83, 3977–3994. [Google Scholar] [CrossRef]
- Choi, W.M.; Jo, Y.H.; Sohn, S.S.; Lee, S.; Lee, B.J. Understanding the Physical Metallurgy of the CoCrFeMnNi High-Entropy Alloy: An Atomistic Simulation Study. NPJ Comput. Mater. 2018, 4, 1–9. [Google Scholar] [CrossRef]
- Jelinek, B.; Groh, S.; Horstemeyer, M.F.; Houze, J.; Kim, S.-G.G.; Wagner, G.J.; Moitra, A.; Baskes, M.I. Modified Embedded Atom Method Potential for Al, Si, Mg, Cu, and Fe Alloys. Phys. Rev. B 2012, 85, 245102. [Google Scholar] [CrossRef]
- Paul, S.; Muralles, M.; Schwen, D.; Short, M.; Momeni, K. A Modified Embedded-Atom Potential for Fe-Cr-Si Alloys. J. Phys. Chem. C 2021, 125, 22863–22871. [Google Scholar] [CrossRef]
- Qiang, R.; Leong, A.; Zhang, J.; Short, M.P. Corrosion Behavior of Fe–Cr–Si Alloys in Simulated PWR Primary Water Environment. J. Nucl. Mater. 2019, 526, 151735. [Google Scholar] [CrossRef]
- Paul, S.; Momeni, K.; Schwen, D.; Short, M.P. Radiation Damage Study of T91/Fe-Cr-Si Multimetallic Layered Composite for Generation IV Reactor Deployment. In Proceedings of the Energy Proceedings, Cambridge, CA, USA, 11–13 August 2021. [Google Scholar]
- Maresca, F.; Curtin, W.A. Theory of Screw Dislocation Strengthening in Random BCC Alloys from Dilute to “High-Entropy” Alloys. Acta Mater. 2020, 182, 144–162. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short—Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Daw, M.S.; Baskes, M.I. Embedded-Atom Method: Derivation and Application to Impurities, Surfaces, and Other Defects in Metals. Phys. Rev. B 1984, 29, 6443–6453. [Google Scholar] [CrossRef]
- Tersoff, J. New Empirical Approach for the Structure and Energy of Covalent Systems. Phys. Rev. B 1988, 37, 6991–7000. [Google Scholar] [CrossRef]
- Eich, S.M.; Beinke, D.; Schmitz, G. Embedded-Atom Potential for an Accurate Thermodynamic Description of the Iron-Chromium System. Comput. Mater. Sci. 2015, 104, 185–192. [Google Scholar] [CrossRef]
- Pun, G.P.P.; Mishin, Y. Optimized Interatomic Potential for Silicon and Its Application to Thermal Stability of Silicene. Phys. Rev. B 2017, 95, 1–21. [Google Scholar] [CrossRef]
- Ziegler, J.F.; Biersack, J.P. The Stopping and Range of Ions in Matter. In Treatise on Heavy-Ion Science; Springer: Berlin, Germany, 1985; pp. 93–129. [Google Scholar]
- Lee, B.-J.J.; Baskes, M.I.; Kim, H.; Cho, Y.K.; Lee, B.-J.J.; Kim, H.; Koo Cho, Y. Second Nearest-Neighbor Modified Embedded Atom Method Potentials for Bcc Transition Metals. Phys. Rev. B 2001, 64, 184102. [Google Scholar] [CrossRef]
- Wang, H.; Guo, X.; Zhang, L.; Wang, H.; Xue, J. Deep Learning Inter-Atomic Potential Model for Accurate Irradiation Damage Simulations. Appl. Phys. Lett. 2019, 114, 24101. [Google Scholar] [CrossRef]
- Simmons, G. Single Crystal Elastic Constants and Calculated Aggregate Properties. J. Grad. Res. Cent. 1965, 34, 1. [Google Scholar]
- Ryu, S.; Weinberger, C.R.; Baskes, M.I.; Cai, W. Improved Modified Embedded-Atom Method Potentials for Gold and Silicon. Model Simul. Mat. Sci. Eng. 2009, 17, 75008. [Google Scholar] [CrossRef]
- Schall, J.D.; Gao, G.; Harrison, J.A. Elastic Constants of Silicon Materials Calculated as a Function of Temperature Using a Parametrization of the Second-Generation Reactive Empirical Bond-Order Potential. Phys. Rev. B 2008, 77, 115209. [Google Scholar] [CrossRef]
- Lee, B.; Rudd, R.E. First-Principles Calculation of Mechanical Properties of Si001 Nanowires and Comparison to Nanomechanical Theory. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 75, 1–13. [Google Scholar] [CrossRef]
- Bolef, D.I.; De Klerk, J. Elastic Constants of Single-Crystal Mo. J. Appl. Phys. 1962, 33, 2311. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, G.; Punkkinen, M.P.J.; Hertzman, S.; Johansson, B.; Vitos, L. Elastic Anomalies in Fe-Cr Alloys. J. Phys. Condens. Matter 2013, 25, 195501. [Google Scholar] [CrossRef]
- Caracas, R.; Wentzcovitch, R. Equation of State and Elasticity of FeSi. Geophys. Res. Lett. 2004, 31, 20601–20604. [Google Scholar] [CrossRef]
- Lv, Z.Q.; Zhang, Z.F.; Zhang, Q.; Wang, Z.H.; Sun, S.H.; Fu, W.T. Structural, Electronic and Elastic Properties of the Laves Phases WFe2, MoFe2, WCr2 and MoCr2 from First-Principles. Solid State Sci. 2016, 56, 16–22. [Google Scholar] [CrossRef]
- Ren, B.; Lu, D.-H.H.; Zhou, R.; Ji, D.-P.P.; Hu, M.-Y.Y.; Feng, J. First Principles Study of Stability, Mechanical, and Electronic Properties of Chromium Silicides. Chin. Phys. B 2018, 27, 107102. [Google Scholar] [CrossRef]
- Zinoveva, G.P.; Andreeva, L.P.; Geld, P. V Elastic Constants and Dynamics of Crystal Lattice in Monosilicides with B20 Structure. Phys. Status Solidi (A) 1974, 23, 711–718. [Google Scholar] [CrossRef]
- Li, X.P.; Sun, S.P.; Wang, H.J.; Lei, W.N.; Jiang, Y.; Yi, D.Q. Electronic Structure and Point Defect Concentrations of C11b MoSi2 by First-Principles Calculations. J. Alloys Compd. 2014, 605, 45–50. [Google Scholar] [CrossRef]
- Zhu, Z.L.; Fu, H.Z.; Sun, J.F.; Liu, Y.F.; Shi, D.H.; Xu, G.L. First-Principles Calculations of Elastic and Thermal Properties of Molybdenum Disilicide. Chin. Phys. Lett. 2009, 26, 1–4. [Google Scholar] [CrossRef]
- Tanaka, K.; Onome, H.; Inui, H.; Yamaguchi, M.; Koiwa, M. Single-Crystal Elastic Constants of MoSi2 with the C11b Structure. Mater. Sci. Eng. A 1997, 239–240, 188–194. [Google Scholar] [CrossRef]
- Alouani, M.; Albers, R.C.; Methfessel, M. Calculated Elastic Constants and Structural Properties of Mo and MoSi2. Phys. Rev. B 1991, 43, 6500–6509. [Google Scholar] [CrossRef]
- Chung, D.H.; Buessem, W.R. The Voigt-Reuss-Hill Approximation and Elastic Moduli of Polycrystalline Mgo, CaF2, β-ZnS, ZnSe, and CdTe. J. Appl. Phys. 1967, 38, 2535–2540. [Google Scholar] [CrossRef]
- Stein, F.; Leineweber, A. Laves Phases: A Review of Their Functional and Structural Applications and an Improved Fundamental Understanding of Stability and Properties. J. Mater. Sci. 2021, 56, 5321–5427. [Google Scholar] [CrossRef]
- Huang, L.F.; Gong, P.L.; Zeng, Z. Correlation between structure, phonon spectra, thermal expansion, and thermomechanics of single-layer MoS2. Phys. Rev. B 2014, 90, 045409. [Google Scholar] [CrossRef]
- Zhou, X.W.; Nowak, C.; Skelton, R.S.; Foster, M.E.; Ronevich, J.A.; San Marchi, C.; Sills, R.B. An Fe–Ni–Cr–H Interatomic Potential and Predictions of Hydrogen-Affected Stacking Fault Energies in Austenitic Stainless Steels. Int. J. Hydrog. Energy 2022, 47, 651–665. [Google Scholar] [CrossRef]
- Petrova, A.E.; Krasnorussky, V.N.; Stishov, S.M. Elastic Properties of FeSi. J. Exp. Theor. Phys. 2010, 111, 427–430. [Google Scholar] [CrossRef][Green Version]
- Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Society. Sect. A 1932, 129, 484. [Google Scholar] [CrossRef]
- Lee, J.; Kim, T.; Hwang, I.S.; Ballinger, R.G.; Kim, J.H. Development of Pilgering Process of Hybrid-Layer Cladding for Advanced Small Modular Fast Reactor Application. In Proceedings of the International Atomic Energy Agency, Division of Nuclear Power, Nuclear Power Technology Section, Vienna, Austria, 26–29 June 2017; pp. 1–10. [Google Scholar]
- Bonny, G.; Castin, N.; Terentyev, D. Interatomic Potential for Studying Ageing under Irradiation in Stainless Steels: The FeNiCr Model Alloy. Model Simul. Mater. Sci. Eng. 2013, 21, 85004. [Google Scholar] [CrossRef]
- Alireza, E.S.; Asadi, E. Molecular dynamics for near melting temperatures simulations of metals using modified embedded-atom method. J. Phys. Chem. Solids 2018, 112, 61–72. [Google Scholar]
- Dholakia, M.; Chandra, S.; Valsakumar, M.C.; Mathi Jaya, S. Atomistic Simulations of Displacement Cascades in Y2O3 Single Crystal. J. Nucl. Mater. 2014, 454, 96–104. [Google Scholar] [CrossRef]
- Zhang, X.; Li, B.; Liu, H.X.; Zhao, G.H.; Yang, Q.L.; Cheng, X.M.; Wong, C.H.; Zhang, Y.M.; Lim, C.W.J. Atomic Simulation of Melting and Surface Segregation of Ternary Fe-Ni-Cr Nanoparticles. Appl. Surf. Sci. 2019, 465, 871–879. [Google Scholar] [CrossRef]
- Posner, E.S.; Rubie, D.C.; Frost, D.J.; Vlček, V.; Steinle-Neumann, G. High P–T Experiments and First Principles Calculations of the Diffusion of Si and Cr in Liquid Iron. Geochim. Cosmochim. Acta 2017, 203, 323–342. [Google Scholar] [CrossRef]
- Pozzo, M.; Davies, C.; Gubbins, D.; Alfè, D. Transport Properties for Liquid Silicon-Oxygen-Iron Mixtures at Earth’s Core Conditions. Phys. Rev. B 2013, 87, 14110. [Google Scholar] [CrossRef]
- Morard, G.; Andrault, D.; Antonangeli, D.; Bouchet, J. Properties of Iron Alloys under the Earth’s Core Conditions. Comptes Rendus Geosci. 2014, 346, 130–139. [Google Scholar] [CrossRef]
- Chamati, H.; Papanicolaou, N.I.; Mishin, Y.; Papaconstantopoulos, D.A. Embedded-Atom Potential for Fe and Its Application to Self-Diffusion on Fe(100). Surf. Sci. 2006, 600, 1793–1803. [Google Scholar] [CrossRef]
- Stillinger, F.H.; Weber, T.A. Computer Simulation of Local Order in Condensed Phases of Silicon. Phys. Rev. B 1985, 31, 5262–5271. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, Z.; Song, Y.; Zhao, L.; Bhatia, B.; Bagnall, K.R.; Wang, E.N. Thermal Expansion Coefficient of Monolayer Molybdenum Disulfide Using Micro-Raman Spectroscopy. Nano Lett. 2019, 19, 4745–4751. [Google Scholar] [CrossRef]
- White, G.K.; Andrikidis, C. Thermal Expansion of Chromium at High Temperature. Phys. Rev. B 1996, 53, 8145. [Google Scholar] [CrossRef]
- Houze, J.; Kim, S.; Moitra, A.; Jelinek, B.; Groh, S.; Horstemeyer, M.F.; Acar, E.; Rais-Rohani, M.; Kim, S.-G. A Multi-Objective Optimization Procedure to Develop Modified-Embedded-Atom-Method Potentials: An Application to Magnesium. arXiv 2007, arXiv:0708.0075. [Google Scholar]
- Callister, W.D., Jr. Materials Science and Engineering: An Introduction, 7th ed.; Wiley Publishers: New York, NY, USA, 2007. [Google Scholar]
- Kim, S.-G.; Horstemeyer, M.F.; Baskes, M.I.; Rais-Rohani, M.; Kim, S.; Jelinek, B.; Houze, J.; Moitra, A.; Liyanage, L. Semi-Empirical Potential Methods for Atomistic Simulations of Metals and Their Construction Procedures. J. Eng. Mater. Technol. 2009, 131, 041210. [Google Scholar] [CrossRef]
- Hestenes, M.R.; Stiefel, E. Methods of conjugate gradients for solving linear systems. J. Res. Natl. Bur. Stand. 1952, 49, 409–435. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Reference | System | Systems Extracted |
|---|---|---|---|
| MEAM | Chen et al. [77] | Fe-C-Cr-Mo | Mo |
| Fe-Mo | |||
| Cr-Mo | |||
| Choi et al. [81] | Co-Cr-Fe-Mn-Ni | Fe | |
| Cr | |||
| Fe-Cr | |||
| Jelinek et al. [82] | Al-Si-Mg-Cu-Fe | Si | |
| Fe-Si | |||
| Baskes [79] | Si-Mo | Si-Mo | |
| DFT | Ren et al. [80] | Cr-Si | Cr-Si |
| Cohesive Energy (eV) | Lattice (Å) | Elastic Constants (GPA) | Shear Modulus (GPA) | Young Modulus (GPa) | Bulk Modulus (GPA) | Poisson’s Ratio | Zener’s Ratio (Anisotropic) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Ec | alat | C11 | C12 | C44 | G | E | B | v | ar | |
| Fe (bcc) | ||||||||||
| MEAM | −4.29 | 2.863 | 242.17 | 138.41 | 121.46 | 51.88 | 141.49 | 173.00 | 0.36 | 2.34 |
| MEAM [77] | −4.29 | 2.860 | 302.69 | 112.78 | 120.45 | |||||
| MEAM [81] | −4.29 | 2.860 | 226.00 | 140.00 | 116.01 | |||||
| MEAM [93] | 243.00 | 138.00 | 121.90 | |||||||
| Exp. [95] | 243.10 | 138.10 | 121.90 | |||||||
| Cr (bcc) | ||||||||||
| MEAM | −4.10 | 2.880 | 344.45 | 112.89 | 130.47 | 115.78 | 288.71 | 190.08 | 0.25 | 1.13 |
| MEAM [81] | 344.50 | 112.90 | 130.50 | |||||||
| MEAM [77] | −4.10 | 2.885 | 350.00 | 67.00 | 100.00 | |||||
| MEAM [93] | 390.90 | 89.70 | 103.40 | |||||||
| Exp. [95] | 391.00 | 89.60 | 103.20 | |||||||
| Si (dia) | ||||||||||
| MEAM | −4.63 | 5.431 | 163.78 | 64.54 | 76.46 | 49.62 | 127.29 | 97.62 | 0.28 | 1.54 |
| MEAM [96] | 164 | 65 | 76 | |||||||
| DFT [97] | −4.63 | 5.429 | 171.5 | 67.1 | 81.1 | 101.9 | ||||
| DFT [98] | 154.6 | 58.1 | 74.4 | 122.8 | 0.27 | |||||
| Exp. [96] | 165.8 | 63.5 | 79.6 | |||||||
| Mo (bcc) | ||||||||||
| MEAM | −6.81 | 3.146 | 460.18 | 167.82 | 110.56 | 146.18 | 370.49 | 265.27 | 0.27 | 0.76 |
| MEAM [77] | −6.81 | 3.149 | 459.26 | 167.86 | 110.79 | |||||
| MEAM [93] | 464.90 | 165.50 | 108.80 | |||||||
| Exp. [99] | 464.70 | 161.50 | 108.90 | |||||||
| Ec | alat | C11 | C12 | C44 | G | E | B | v | |
|---|---|---|---|---|---|---|---|---|---|
| Str. Type | eV | Å | (GPa) | GPa | GPa | GPa | |||
| Fe Cr | |||||||||
| B2 | −4.31 | 2.800 | 348.63 | 115.85 | 88.38 | 116.39 | 290.84 | 193.45 | 0.25 |
| DFT [100] | 350.00 | 150.00 | 124.00 | 116.00 | 295.00 | 219.00 | 0.27 | ||
| Fe Si | |||||||||
| B20 | −6.83 | 4.208 | 317.42 | 296.31 | 13.40 | 10.56 | 31.31 | 303.35 | 0.48 |
| DFT [82] | 226.50 | ||||||||
| DFT LDA [100] | 4.83 | 440.00 | 150.00 | 190.00 | 235.00 | ||||
| DFT [100] | 4.48 | 385.00 | 120.00 | 160.00 | 210.00 | ||||
| B2 | −4.45 | 2.803 | 543.04 | 29.93 | −30.40 | 256.56 | 539.91 | 200.97 | 0.05 |
| DFT [82] | 87.00 | 231.90 | |||||||
| MEAM [82] | 36.20 | 177.70 | |||||||
| DFT LDA [101] | 2.70 | 510.00 | 160 | 135.00 | 285.00 | ||||
| DFT [101] | 2.77 | 435.00 | 125 | 95.00 | 230.00 | ||||
| B1 | −5.05 | 4.240 | 682.15 | 7.18 | 52.96 | 337.48 | 682.00 | 232.17 | 0.01 |
| DFT [82] | −70.00 | 100.90 | |||||||
| MEAM [82] | 65.00 | 157.90 | |||||||
| Fe Mo | |||||||||
| B1 | −4.96 | 4.702 | 286.15 | 109.27 | 92.77 | 88.44 | 225.76 | 168.23 | 0.28 |
| MoFe2 P63/mmc | −4.81 | 3.09/7.82 † | 64 | 164 | 26 | −390.86 | 2818.71 | 92.01 | −0.52 |
| DFT [102] | 4.71/7.64 † | 441.40 | 161.50 | 110.60 | 126.60 | 244.90 | |||
| Cr Mo | |||||||||
| B2 | −4.96 | 2.901 | 396.04 | 204.62 | −21.93 | 95.71 | 256.64 | 268.43 | 0.34 |
| Cr Si | |||||||||
| P213 | −9.23 | 4.550 | 306.00 | 316.05 | 29.85 | −5.03 | −15.16 | 312.70 | 0.51 |
| DFT [103] | −8.13 | 4.590 | 390.50 | 112.30 | 125.70 | 130.90 | 323.80 | 205.00 | 0.24 |
| Exp. [104] | 107.70 | 260.90 | 150 | ||||||
| MoSi | |||||||||
| MoSi2 C11b | −5.66 | 8.80/3.46 † | 242.73 | 156.82 | 42.31 | 84.83 | s225.37 | 218.93 | 0.33 |
| MEAM [79] | −5.92 | 8.42/3.59 † | 252.00 | 145.00 | 26.00 | 75.0 | 202.00 | 0.34 | |
| DFT [105] | −23.19 | 7.87/3.22 † | 203.70 | ||||||
| DFT [106] | 7.79/3.18 † | 406.4 | 111.5 | 202.1 | 211.6 | ||||
| Exp [107] | 7.85/3.20 † | 410.00 | 114.90 | 195.00 | |||||
| Exp. [108] | 19.14 * | 2.446 | 401.00 | 102.00 | 208.00 | 222.0 | |||
| Cr-Si | Ec | alat | C11 | C12 | C44 | G | E | B | v |
|---|---|---|---|---|---|---|---|---|---|
| P213 | eV | Å | GPa | GPa | GPa | GPa | |||
| MEAM-A | −9.23 | 4.550 | 306.00 | 316.05 | 29.85 | −5.03 | −15.16 | 312.70 | 0.51 |
| MEAM-B | −7.95 | 4.601 | 394.56 | 112.28 | 127.34 | 141.14 | 344.82 | 206.37 | 0.22 |
| DFT [103] | −8.13 | 4.590 | 390.50 | 112.30 | 125.70 | 130.90 | 323.80 | 205.00 | 0.24 |
| Exp. [104] | 107.70 | 260.90 | 150 | ||||||
| MoSi2 | Ec | clat/alat | C11 | C12 | C44 | G | E | B | v |
|---|---|---|---|---|---|---|---|---|---|
| C11b | eV | Å | GPa | GPa | |||||
| MEAM-A | −5.66 | 8.80/3.46 | 242.73 | 156.82 | 42.31 | 84.83 | 225.37 | 218.93 | 0.33 |
| MEAM-B1 | −5.65 | 9.55/3.11 | 396.52 | 116.16 | 173.55 | 158.70 | 385.09 | 223.86 | 0.28 |
| MEAM-B2 | −5.645 | 8.98/3.22 | 266.36 | 129.58 | 178.34 | 214.12 | 489.19 | 227.93 | 0.24 |
| MEAM [79] | −5.92 | 8.42/3.59 | 252.0 | 145 | 26.00 | 75.00 | 202.00 | 0.34 | |
| DFT [105] | −23.19 | 7.87/3.22 | 203.70 | ||||||
| DFT [106] | 7.79/3.18 | 406.4 | 111.5 | 202.1 | 211.6 | ||||
| Exp. [107] | 7.85/3.20 | 410.0 | 114.9 | 195.00 | |||||
| Exp. [108] | −19.14 * | 2.446 | 401.0 | 102.00 | 208.00 | 222.00 | |||
| FeSi | Ec | alat | C11 | C12 | C44 | G | E | B | v |
|---|---|---|---|---|---|---|---|---|---|
| eV | Å | GPa | GPa | GPa | GPa | ||||
| B20 | |||||||||
| MEAM-A | −6.83 | 4.208 | 317.42 | 296.31 | 13.40 | 10.56 | 31.31 | 303.35 | 0.48 |
| MEAM-B | −6.41 | 4.02 | 328.21 | 201.23 | 279.08 | 12.28 | 35. 61 | 298.09 | 0.45 |
| DFT [82] | 226.50 | ||||||||
| DFT LDA [101] | 4.38 | 440.00 | 150 | 190.00 | 235.00 | ||||
| DFT [101] | 4.48 | 385.00 | 120 | 160.00 | 210.00 | ||||
| Exp. [108] | 4.483 | 345.0 | 106 | 138 | |||||
| B2 | |||||||||
| MEAM-A | −4.45 | 2.803 | 543.04 | 29.93 | −30.40 | 256.56 | 539.91 | 200.97 | 0.05 |
| MEAM-B | −4.93 | 2.63 | 377.02 | 146.0 | 100.0 | 254.0 | 548.64 | 207.57 | 0.06 |
| DFT [82] | 87.00 | 231.90 | |||||||
| MEAM [82] | 36.20 | 177.70 | |||||||
| DFT LDA [101] | 2.70 | 510.00 | 160.0 | 135.00 | 285.00 | ||||
| DFT [101] | 2.77 | 435.00 | 125.0 | 95.00 | 230.00 | ||||
| DFT LDA [113] | 2.72 | 460.0 | 173.0 | 114.3 | 269.5 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paul, S.; Schwen, D.; Short, M.P.; Momeni, K. A Modified Embedded-Atom Method Potential for a Quaternary Fe-Cr-Si-Mo Solid Solution Alloy. Materials 2023, 16, 2825. https://doi.org/10.3390/ma16072825
Paul S, Schwen D, Short MP, Momeni K. A Modified Embedded-Atom Method Potential for a Quaternary Fe-Cr-Si-Mo Solid Solution Alloy. Materials. 2023; 16(7):2825. https://doi.org/10.3390/ma16072825
Chicago/Turabian StylePaul, Shiddartha, Daniel Schwen, Michael P. Short, and Kasra Momeni. 2023. "A Modified Embedded-Atom Method Potential for a Quaternary Fe-Cr-Si-Mo Solid Solution Alloy" Materials 16, no. 7: 2825. https://doi.org/10.3390/ma16072825
APA StylePaul, S., Schwen, D., Short, M. P., & Momeni, K. (2023). A Modified Embedded-Atom Method Potential for a Quaternary Fe-Cr-Si-Mo Solid Solution Alloy. Materials, 16(7), 2825. https://doi.org/10.3390/ma16072825