
Morphology Determines an Efficient Coherent Electron Transport for Push–Pull Organic Semiconductors Based on Triphenylamine and Dicyanovinyl Groups


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
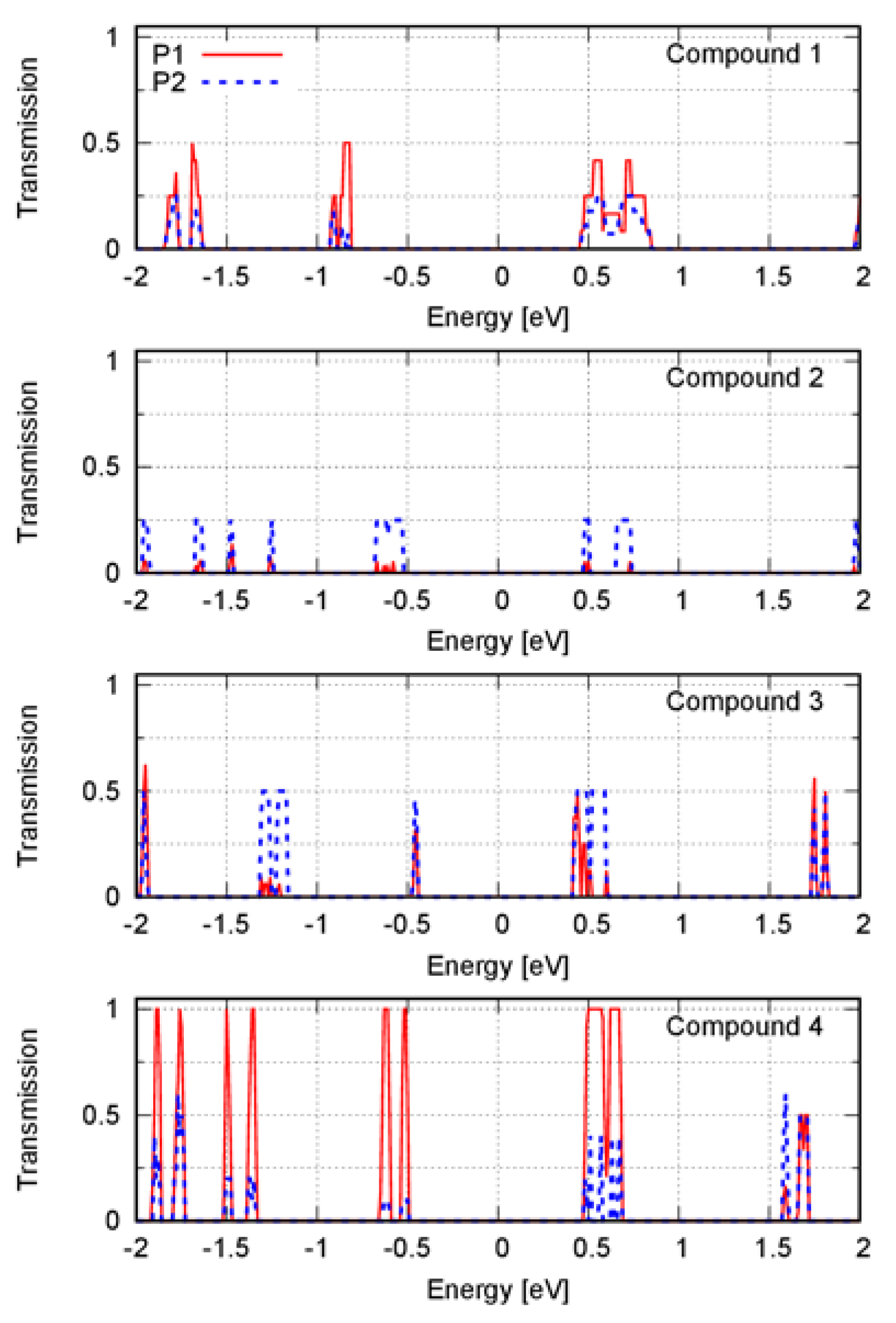
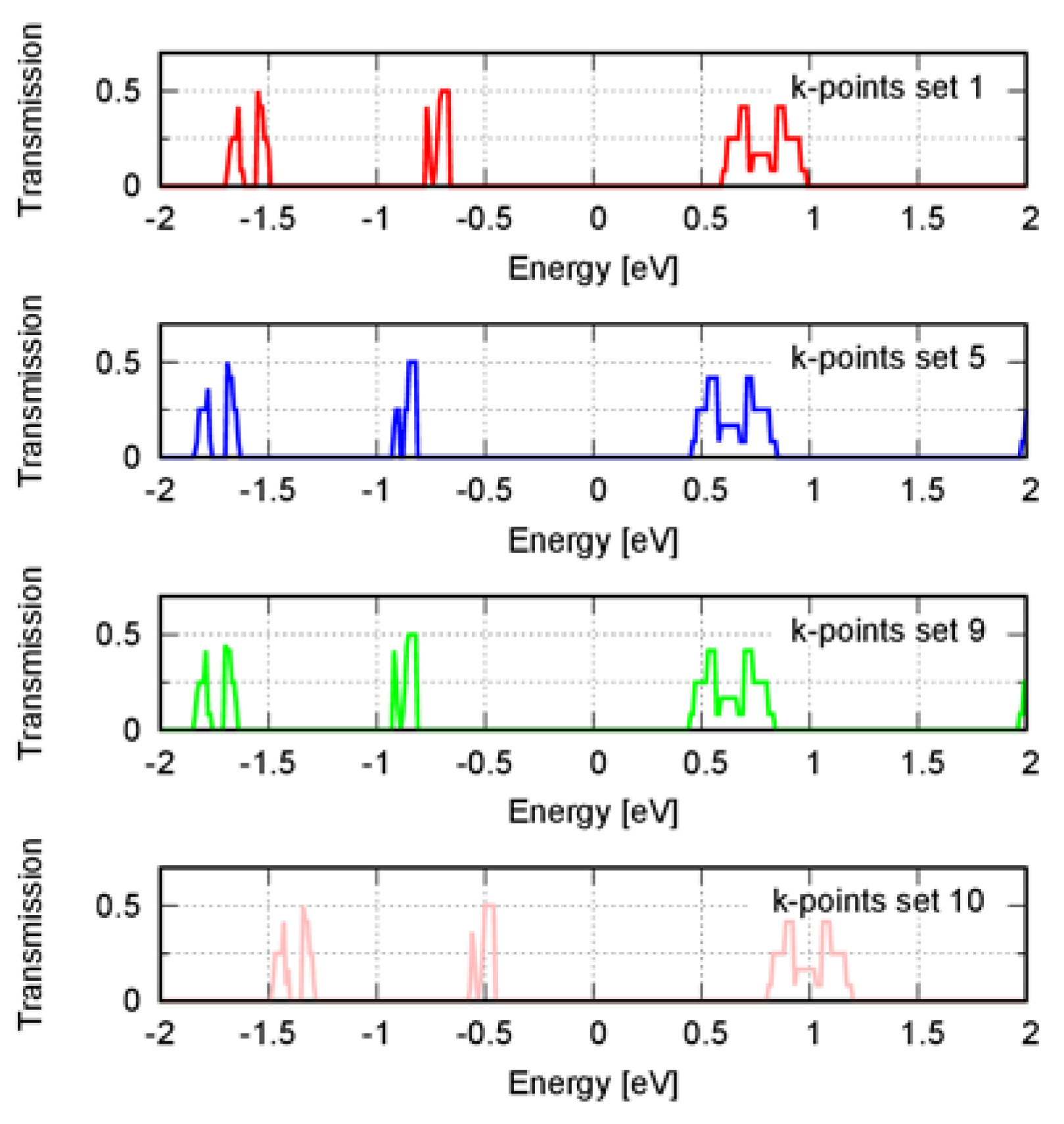
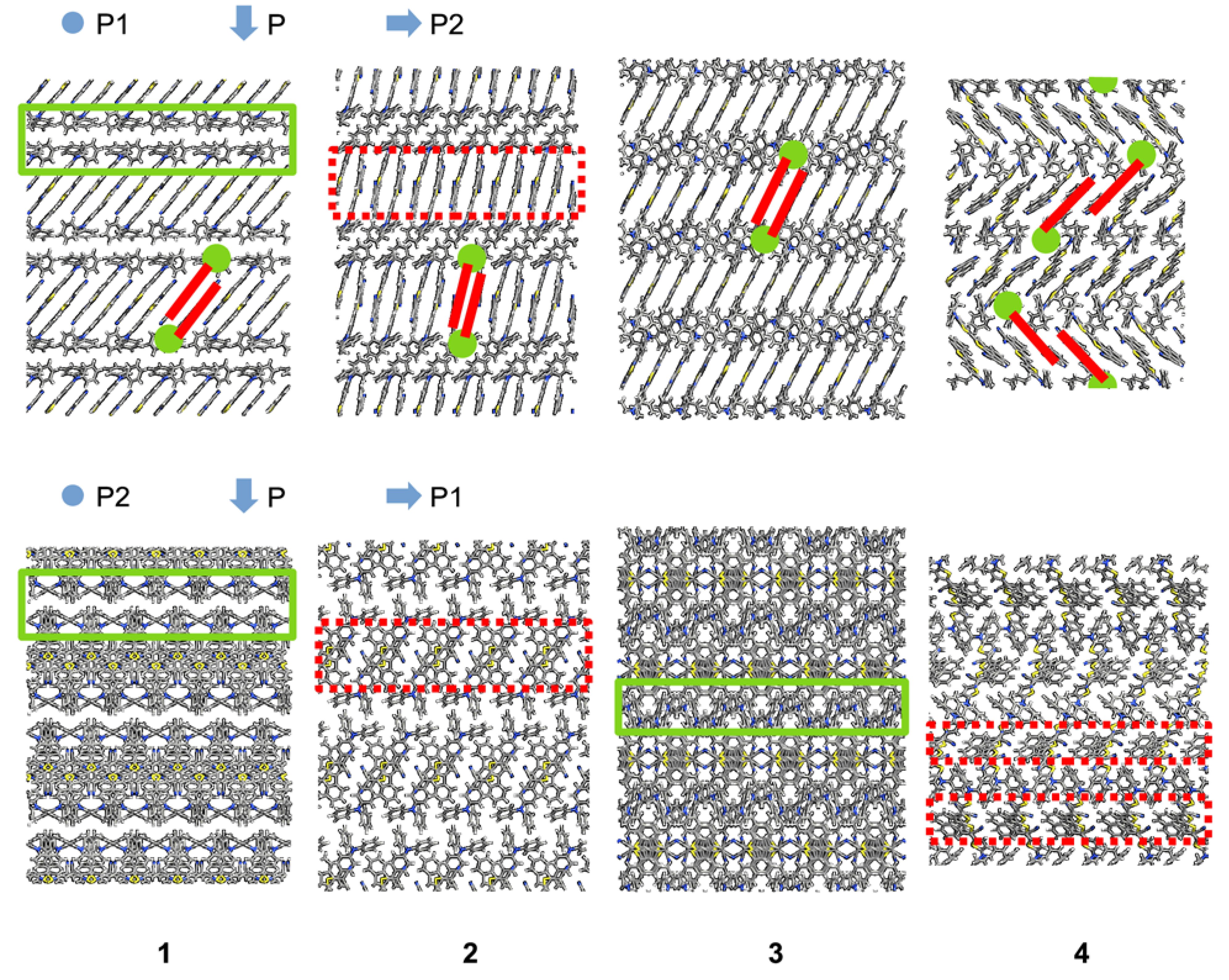
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Q.; Li, Z. Molecular Packing: Another Key Point for the Performance of Organic and Polymeric Optoelectronic Materials. Acc. Chem. Res. 2020, 53, 962–973. [Google Scholar] [CrossRef]
- Boehm, B.J.; Nguyen, H.T.L.; Huang, D.M. The Interplay of Interfaces, Supramolecular Assembly, and Electronics in Organic Semiconductors. J. Phys. Condens. Matter 2019, 31, 423001. [Google Scholar] [CrossRef]
- Lee, H.; Lee, D.; Sin, D.H.; Kim, S.W.; Jeong, M.S.; Cho, K. Effect of Donor–Acceptor Molecular Orientation on Charge Photogeneration in Organic Solar Cells. NPG Asia Mater. 2018, 10, 469–481. [Google Scholar] [CrossRef]
- Lou, S.J.; Loser, S.; Luck, K.A.; Zhou, N.; Leonardi, M.J.; Timalsina, A.; Manley, E.F.; Hayes, D.; Strzalka, J.; Hersam, M.C.; et al. Charge Generation Mechanism Tuned via Film Morphology in Small Molecule Bulk-Heterojunction Photovoltaic Materials. J. Mater. Chem. C 2020, 8, 15234–15252. [Google Scholar] [CrossRef]
- Budzinauskas, K.; Fazzi, D.; Hertel, D.; Rüth, S.; Schelter, J.; Weitkamp, P.; Diesing, S.; Meerholz, K.; van Loosdrecht, P.H.M. Impact of the Interfacial Molecular Structure Organization on the Charge Transfer State Formation and Exciton Delocalization in Merocyanine:PC 61 BM Blends. J. Phys. Chem. C 2020, 124, 21978–21984. [Google Scholar] [CrossRef]
- Kupgan, G.; Chen, X.-K.; Brédas, J.-L. Molecular Packing in the Active Layers of Organic Solar Cells Based on Non-Fullerene Acceptors: Impact of Isomerization on Charge Transport, Exciton Dissociation, and Nonradiative Recombination. ACS Appl. Energy Mater. 2021, 4, 4002–4011. [Google Scholar] [CrossRef]
- Coropceanu, V.; Chen, X.-K.; Wang, T.; Zheng, Z.; Brédas, J.-L. Charge-Transfer Electronic States in Organic Solar Cells. Nat. Rev. Mater. 2019, 4, 689–707. [Google Scholar] [CrossRef]
- Suzuki, M.; Suzuki, K.; Won, T.; Yamada, H. Impact of Substituents on the Performance of Small-Molecule Semiconductors in Organic Photovoltaic Devices via Regulating Morphology. J. Mater. Chem. C 2022, 10, 1162–1195. [Google Scholar] [CrossRef]
- Memon, W.A.; Zhou, R.; Zhang, Y.; Wang, Y.; Liu, L.; Yang, C.; Zhang, J.; Liaqat, A.; Xie, L.; Wei, Z. Precise Control of Crystal Orientation of Conjugated Molecule Enables Anisotropic Charge Transport Properties. Adv. Funct. Mater. 2022, 32, 2110080. [Google Scholar] [CrossRef]
- Wang, T.; Kupgan, G.; Brédas, J.-L. Organic Photovoltaics: Relating Chemical Structure, Local Morphology, and Electronic Properties. Trends Chem. 2020, 2, 535–554. [Google Scholar] [CrossRef]
- Cavassilas, N.; Michelini, F.; Bescond, M. Modeling of Nanoscale Solar Cells: The Green’s Function Formalism. J. Renew. Sustain. Energy 2014, 6, 011203. [Google Scholar] [CrossRef]
- Aeberhard, U. Theory and Simulation of Quantum Photovoltaic Devices Based on the Non-Equilibrium Green’s Function Formalism. J. Comput. Electron. 2011, 10, 394–413. [Google Scholar] [CrossRef]
- Biele, R.; D’Agosta, R. Beyond the State of the Art: Novel Approaches for Thermal and Electrical Transport in Nanoscale Devices. Entropy 2019, 21, 752. [Google Scholar] [CrossRef]
- Ryndyk, D. Theory of Quantum Transport at Nanoscale; Springer Series in Solid-State Sciences; Springer International Publishing: Cham, Switzerland, 2016; Volume 184, pp. 17–46. ISBN 978-3-319-24086-2. [Google Scholar]
- Leliège, A.; Grolleau, J.; Allain, M.; Blanchard, P.; Demeter, D.; Rousseau, T.; Roncali, J. Small D-π-A Systems with o-Phenylene-Bridged Accepting Units as Active Materials for Organic Photovoltaics. Chem. Eur. J. 2013, 19, 9948–9960. [Google Scholar] [CrossRef]
- Smidstrup, S.; Markussen, T.; Vancraeyveld, P.; Wellendorff, J.; Schneider, J.; Gunst, T.; Verstichel, B.; Stradi, D.; Khomyakov, P.A.; Vej-Hansen, U.G.; et al. QuantumATK: An Integrated Platform of Electronic and Atomic-Scale Modelling Tools. J. Phys. Condens. Matter 2020, 32, 015901. [Google Scholar] [CrossRef]
- Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.; Frauenheim, T.; Suhai, S.; Seifert, G. Self-Consistent-Charge Density-Functional Tight-Binding Method for Simulations of Complex Materials Properties. Phys. Rev. B 1998, 58, 7260–7268. [Google Scholar] [CrossRef]
- Niehaus, T.A.; Elstner, M.; Frauenheim, T.; Suhai, S. Application of an Approximate Density-Functional Method to Sulfur Containing Compounds. J. Mol. Struct. THEOCHEM 2001, 541, 185–194. [Google Scholar] [CrossRef]
- Strange, M.; Kristensen, I.S.; Thygesen, K.S.; Jacobsen, K.W. Benchmark Density Functional Theory Calculations for Nanoscale Conductance. J. Chem. Phys. 2008, 128, 114714. [Google Scholar] [CrossRef]
- Glukhova, O.E.; Shmygin, D.S. The Electrical Conductivity of CNT/Graphene Composites: A New Method for Accelerating Transmission Function Calculations. Beilstein J. Nanotechnol. 2018, 9, 1254–1262. [Google Scholar] [CrossRef]
- Falkenberg, J.T.; Brandbyge, M. Simple and Efficient Way of Speeding up Transmission Calculations with k-Point Sampling. Beilstein J. Nanotechnol. 2015, 6, 1603–1608. [Google Scholar] [CrossRef]
- Datta, S. Electronic Transport in Mesoscopic Systems; Cambridge Studies in Semiconductor Physics and Microelectronic Engineering: 3; Cambridge University Press: Cambridge, UK, 1997; pp. 48–50. ISBN 0-521-59943-1. [Google Scholar]
- Yamada, K.; Suzuki, M.; Suenobu, T.; Nakayama, K. High Vertical Carrier Mobilities of Organic Semiconductors due to a Deposited Laid-Down Herringbone Structure Induced by a Reduced Graphene Oxide Template. ACS Appl. Mater. Interfaces 2020, 12, 9489–9497. [Google Scholar] [CrossRef]
- Lee, H.; Park, C.; Sin, D.H.; Park, J.H.; Cho, K. Recent Advances in Morphology Optimization for Organic Photovoltaics. Adv. Mater. 2018, 30, 1800453. [Google Scholar] [CrossRef]
- Wang, L.; Guo, S.; Zhou, K.; Ma, W. Control of the Molecular Orientation in Small Molecule-Based Organic Photovoltaics. Sustain. Energy Fuels 2020, 4, 4934–4955. [Google Scholar] [CrossRef]
- Han, G.; Yi, Y.; Shuai, Z. From Molecular Packing Structures to Electronic Processes: Theoretical Simulations for Organic Solar Cells. Adv. Energy Mater. 2018, 8, 1702743. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero, A.; Velasco-Medina, J.; Ortiz, A. Morphology Determines an Efficient Coherent Electron Transport for Push–Pull Organic Semiconductors Based on Triphenylamine and Dicyanovinyl Groups. Materials 2023, 16, 2442. https://doi.org/10.3390/ma16062442
Romero A, Velasco-Medina J, Ortiz A. Morphology Determines an Efficient Coherent Electron Transport for Push–Pull Organic Semiconductors Based on Triphenylamine and Dicyanovinyl Groups. Materials. 2023; 16(6):2442. https://doi.org/10.3390/ma16062442
Chicago/Turabian StyleRomero, Alexander, Jaime Velasco-Medina, and Alejandro Ortiz. 2023. "Morphology Determines an Efficient Coherent Electron Transport for Push–Pull Organic Semiconductors Based on Triphenylamine and Dicyanovinyl Groups" Materials 16, no. 6: 2442. https://doi.org/10.3390/ma16062442
APA StyleRomero, A., Velasco-Medina, J., & Ortiz, A. (2023). Morphology Determines an Efficient Coherent Electron Transport for Push–Pull Organic Semiconductors Based on Triphenylamine and Dicyanovinyl Groups. Materials, 16(6), 2442. https://doi.org/10.3390/ma16062442