
Dielectric Spectroscopy of Calcium Titanate Processed by Spark Plasma Sintering


Abstract
1. Introduction
2. Experimental
2.1. Sample Preparation
2.2. Characterization Techniques
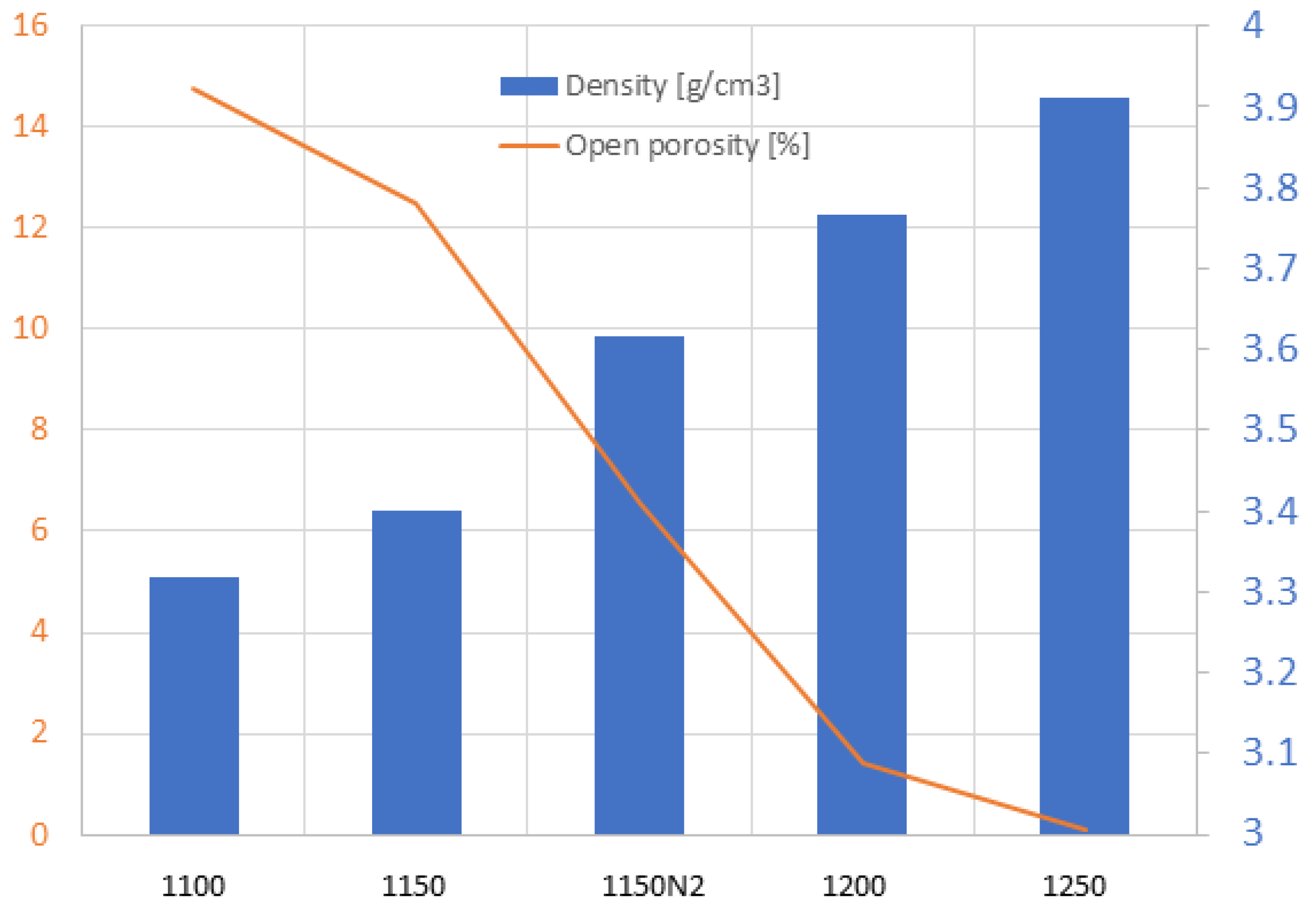
2.2.1. Porosity and Microstructure
2.2.2. Phase and Chemical Composition
2.2.3. Dielectric Parameters
2.2.4. Reflectance and Band Gap
3. Results and Discussion
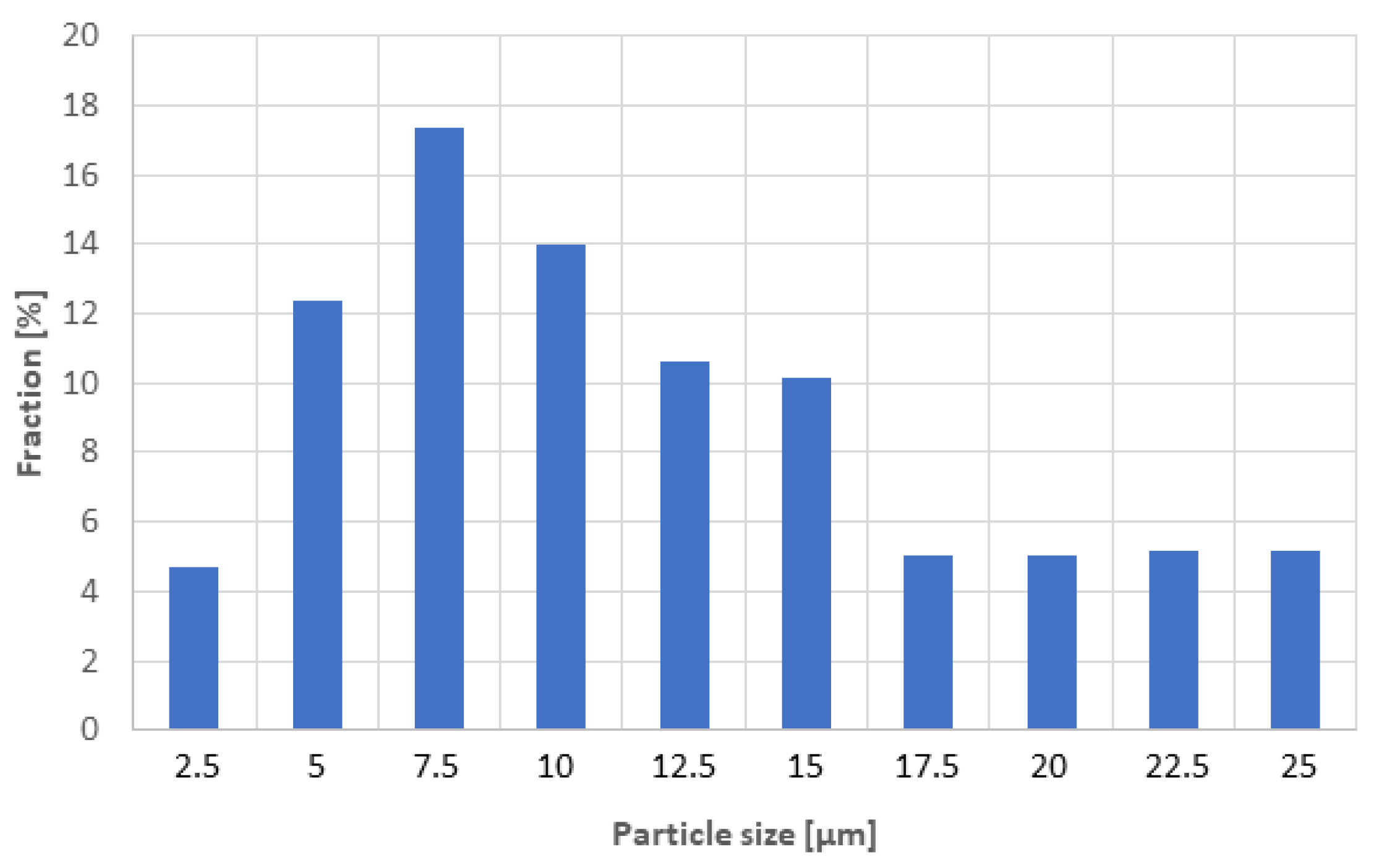
3.1. Powder and Its Processing
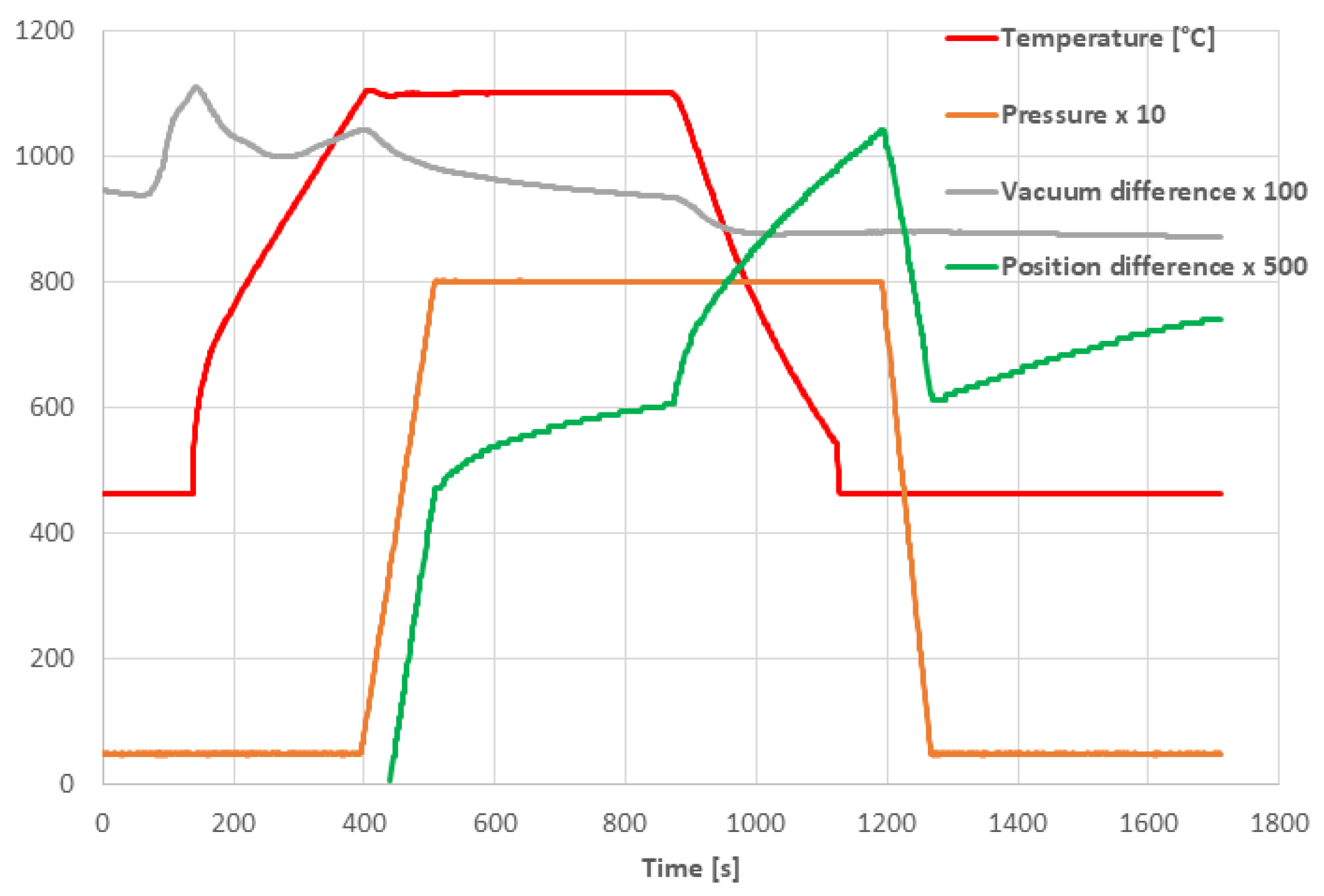
3.2. SPS Processing Parameters
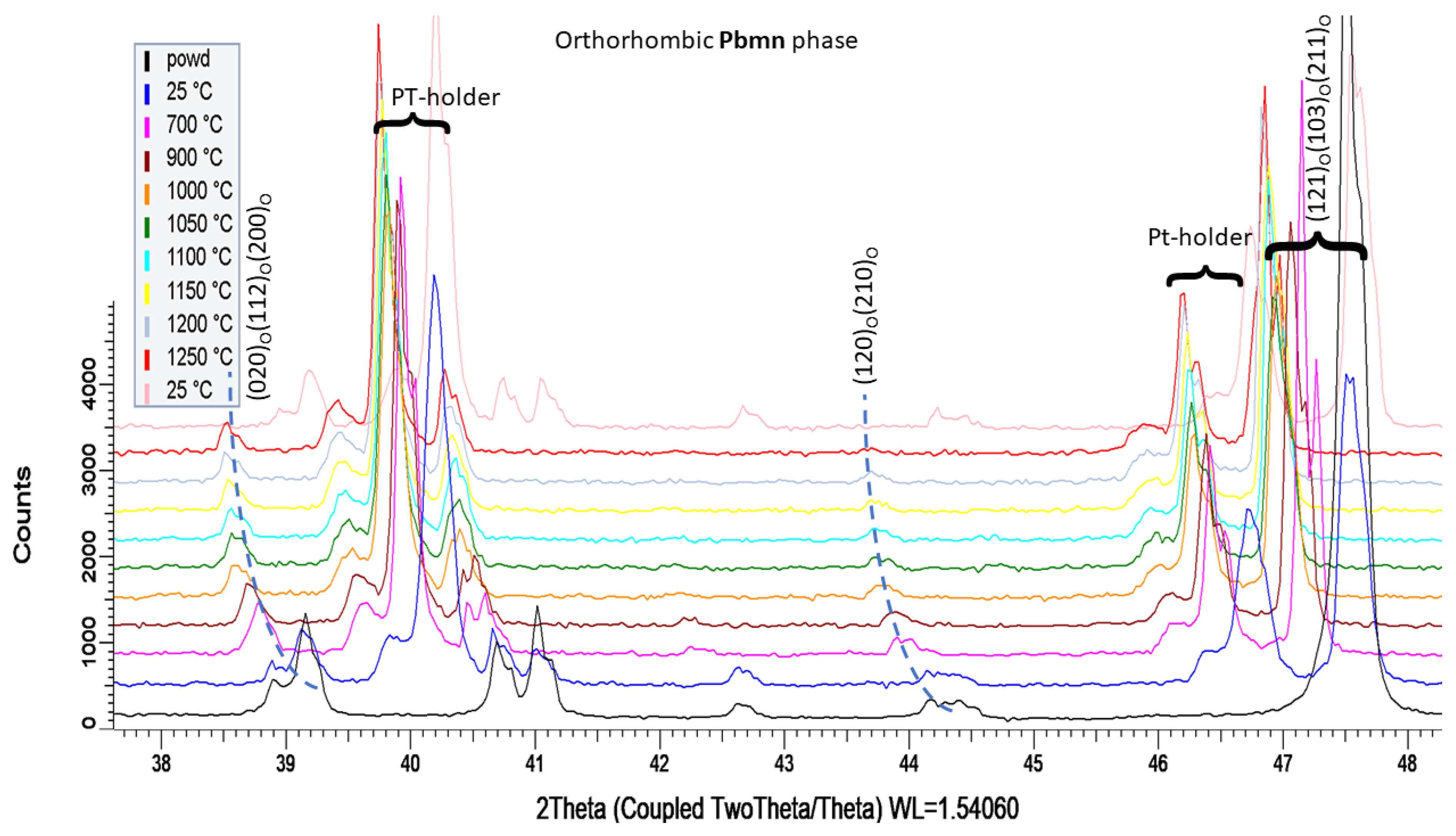
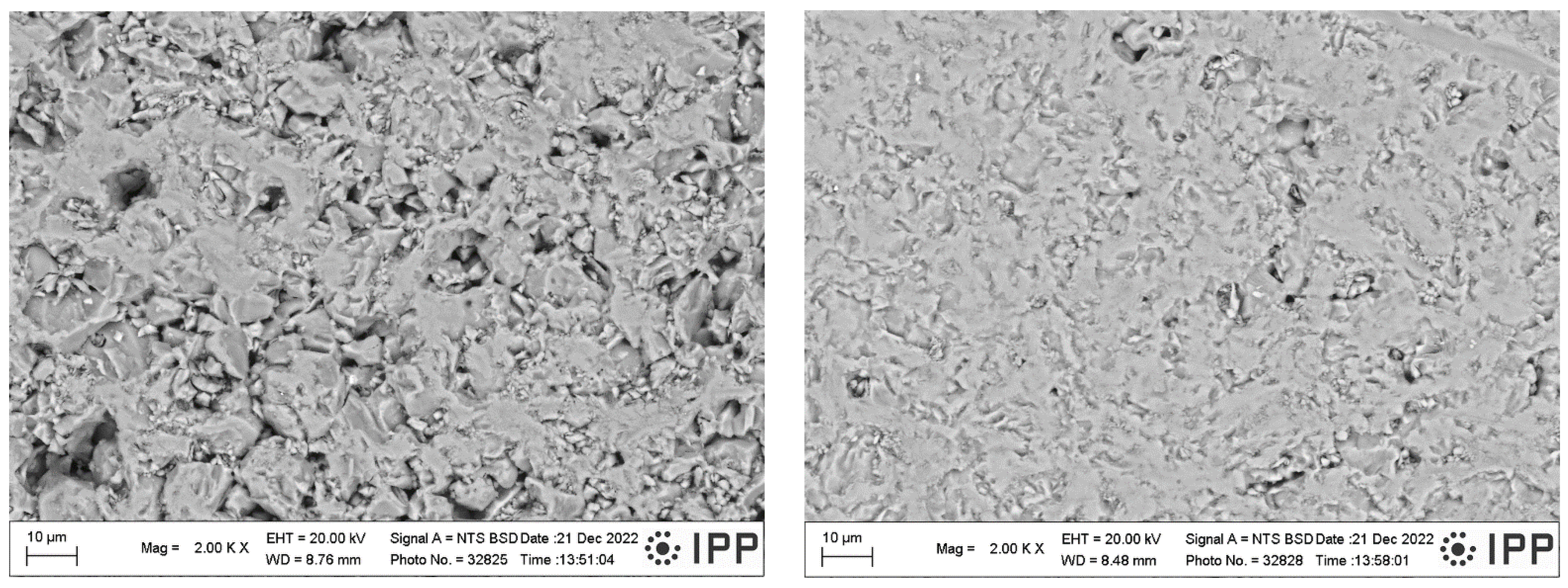
3.3. Phase Analysis and Microstructure
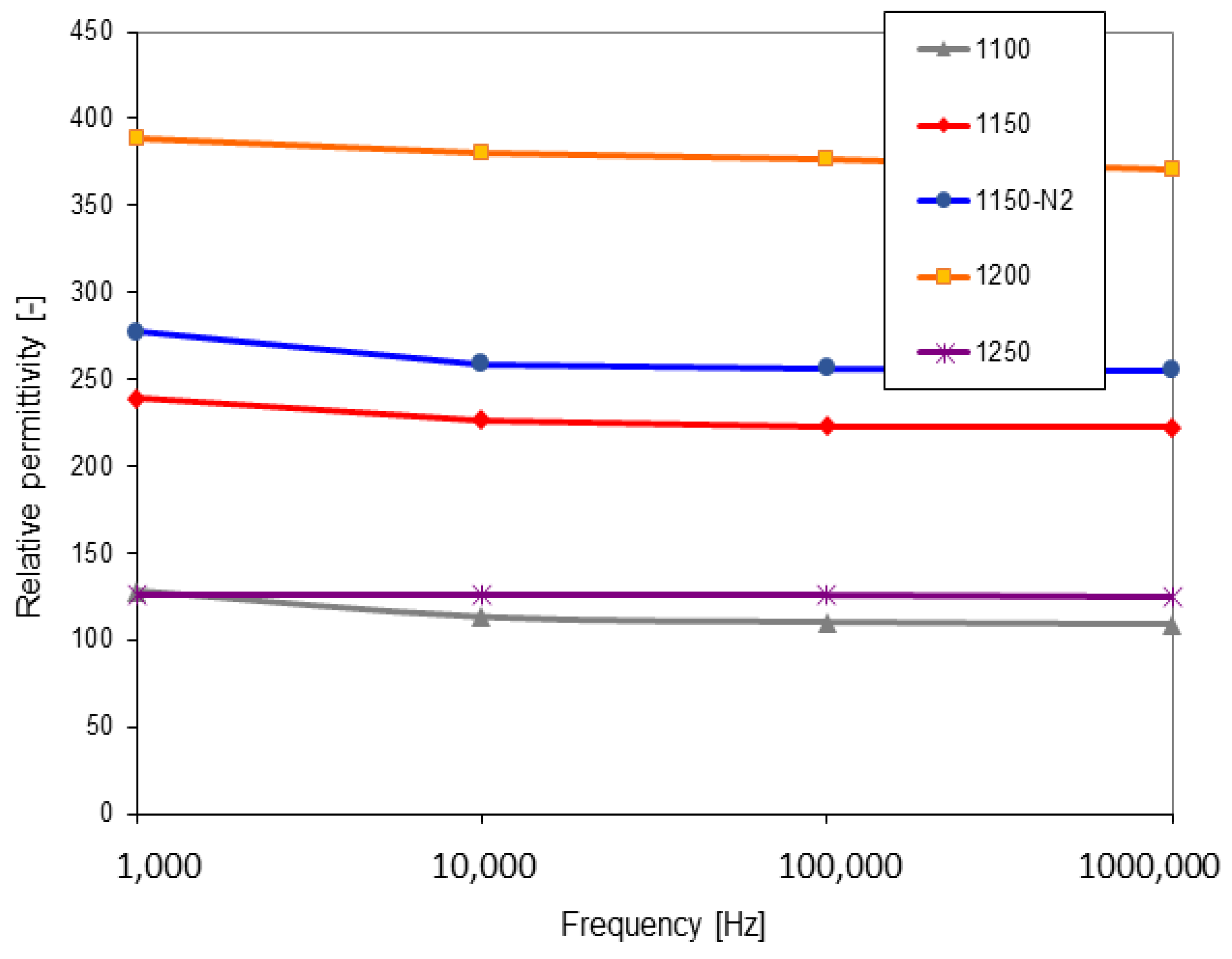
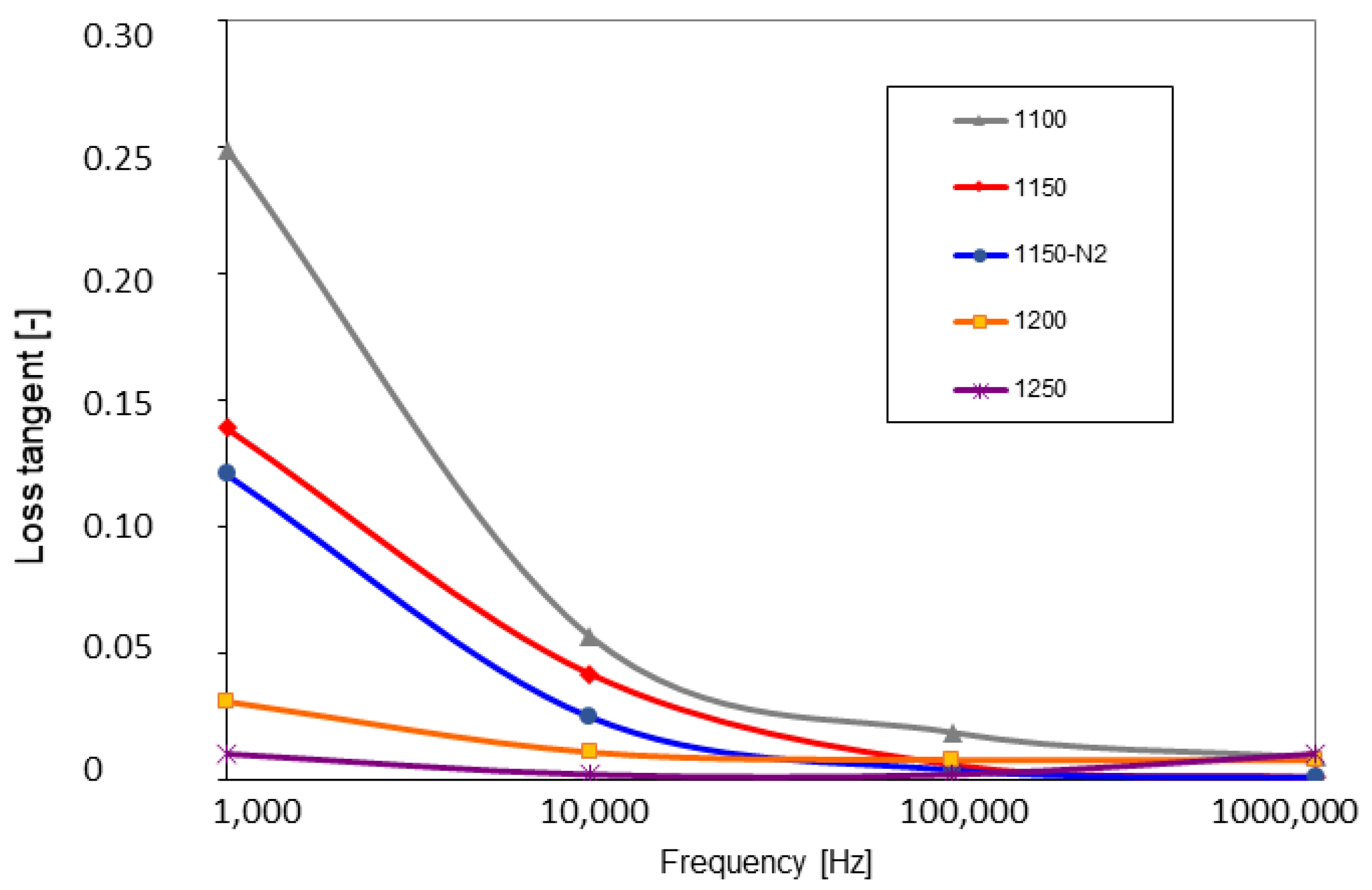
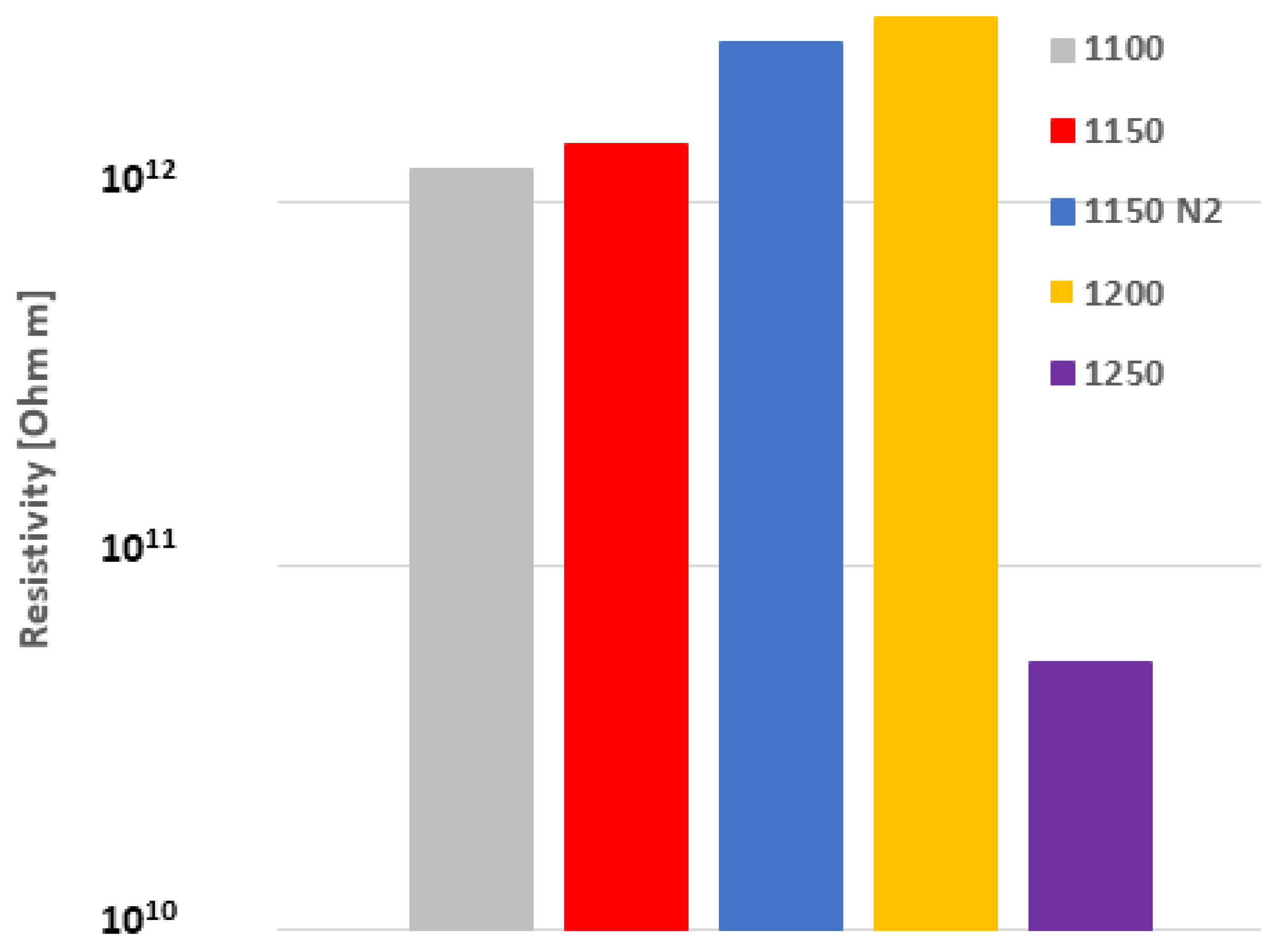
3.4. Dielectric Properties
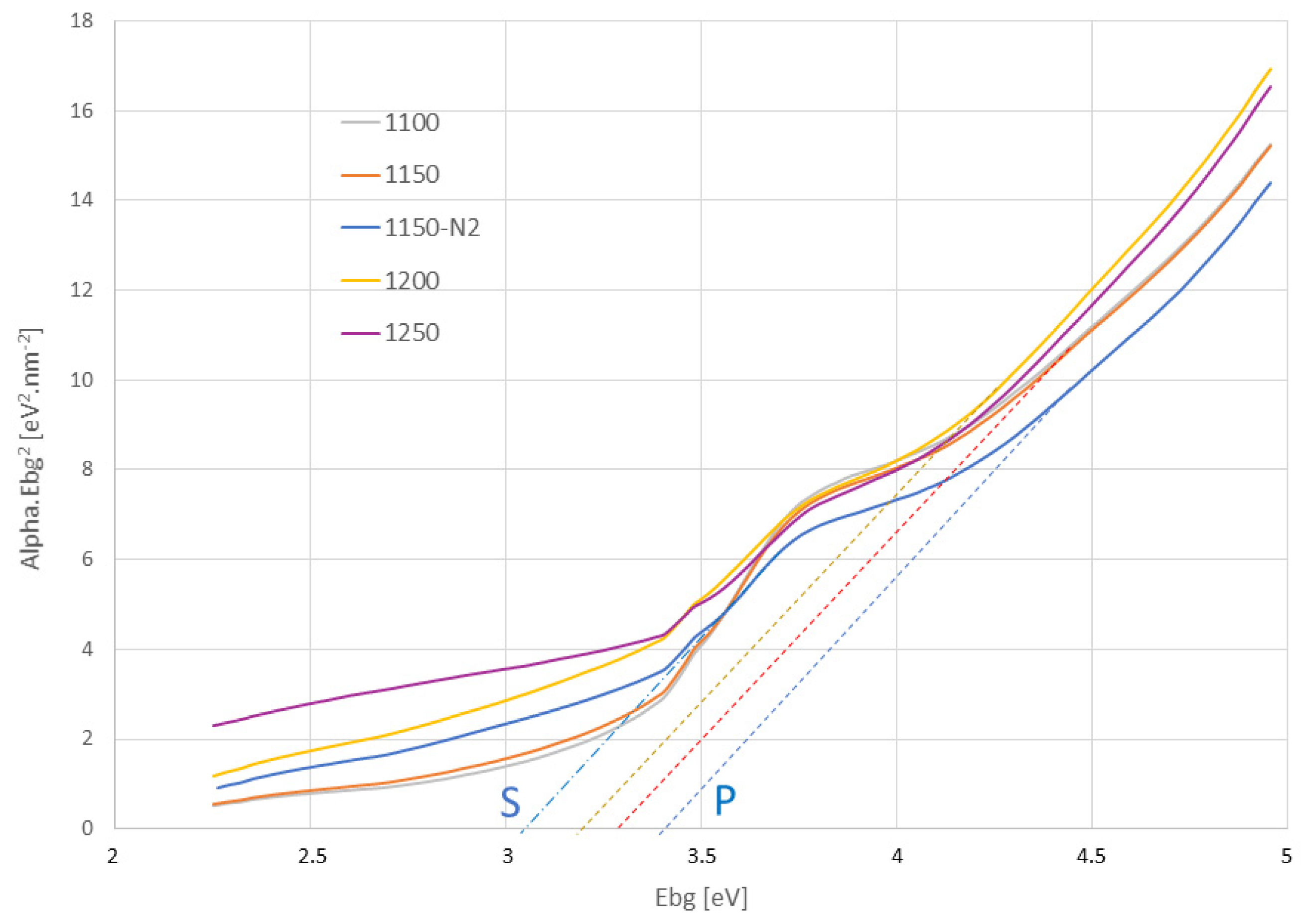
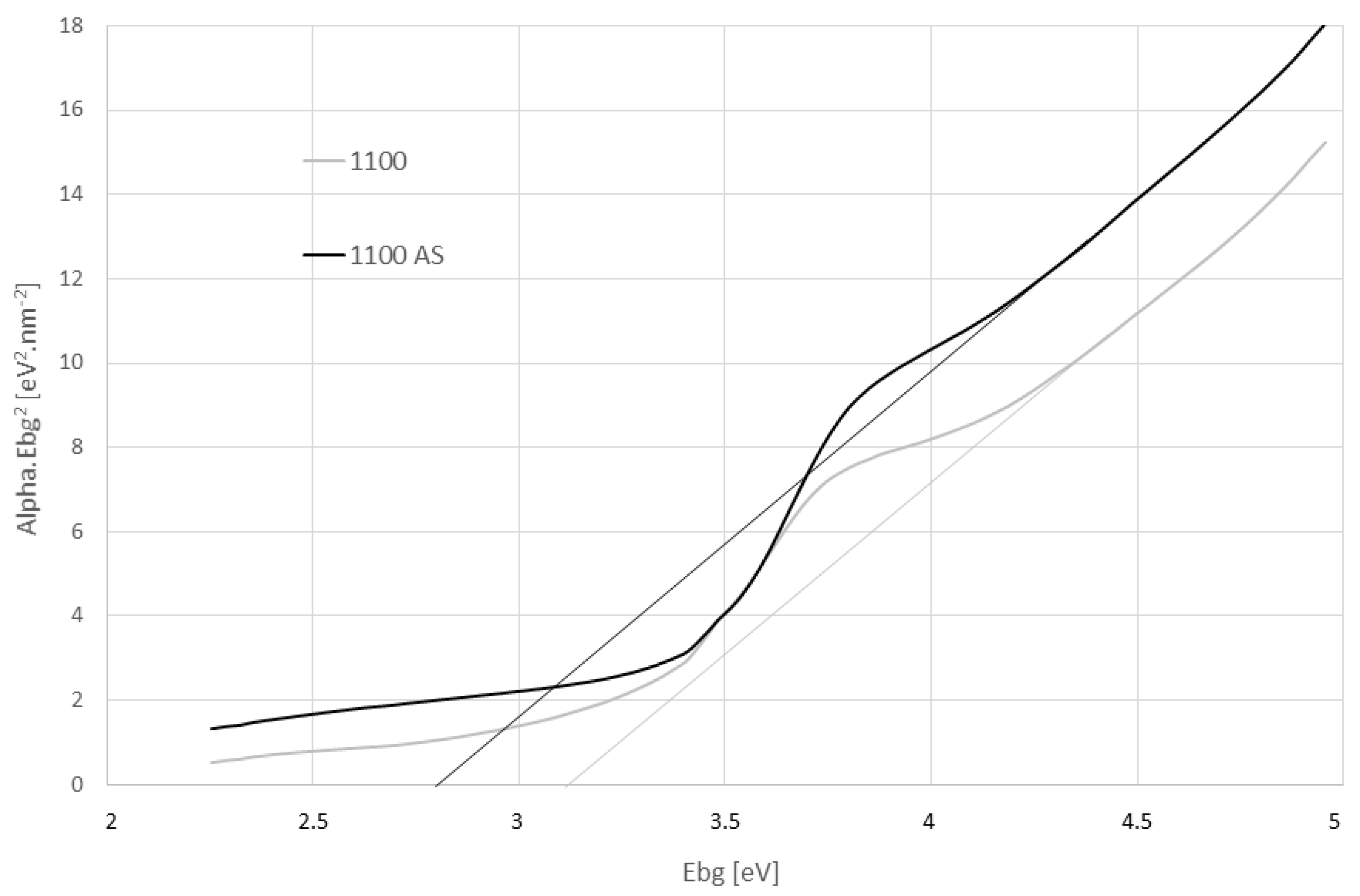
3.5. Reflectance and Optical Band Gap
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kennedy, B.; Howard, C.; Chakoumakos, B. Phase transitions in perovskite at elevated temperatures—A powder neutron diffraction study. J. Phys. Condens. Matter 1999, 11, 1479–1486. [Google Scholar] [CrossRef]
- Redfern, S.A.T. High-temperature structural phase transitions in perovskite (CaTiO3). J. Phys. Condens. Matter 1996, 8, 8267–8275. [Google Scholar] [CrossRef]
- Mallik, P.; Biswal, G.; Patnaik, S.; Senapati, S. Characterisation of sol-gel synthesis of phase pure CaTiO3 nano powders after drying. IOP Conf. Ser. Mat. Sci. Eng. 2015, 75, 012005. [Google Scholar] [CrossRef]
- Holliday, S.; Stanishevsky, A. Crystallization of CaTiO3 by sol-gel synthesis and rapid thermal processing. Surf. Coat. Technol. 2004, 188–189, 741–744. [Google Scholar] [CrossRef]
- Wang, D.; Guo, Z.; Chen, Y.; Hao, J.; Liu, W. In situ hydrothermal synthesis of nanolamellate CaTiO3 with controllable structures and wettability. Inorg. Chem. 2007, 46, 7707–7709. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Zhao, G.; Bao, Q.; Gu, X. Solvothermal preparation of CaTiO3 prism and CaTi2O4(OH)2 nanosheet by a facile surfactant-free method. Mater. Sci. (Medzg.) 2015, 21, 583–585. [Google Scholar] [CrossRef][Green Version]
- Jin, E.; Yu, J.; Wen, T.; Tian, C.; Liu, Z.; Ma, B.; Yuan, L. The effect of the MgO aggregate size on the densification of sintered MgO. Ceram. Silikáty 2020, 64, 84–91. [Google Scholar] [CrossRef]
- Robles, J.H.; Gonzalez, C.R.; de la Torre, S.D.; Cobas, L.F.; Casillas, P.G.; Montes, H.C. Dielectric properties of bismuth titanate densified by spark plasma sintering and pressureless sintering. J. Alloys Compd. 2012, 536S, 511–515. [Google Scholar] [CrossRef]
- Yoon, S.; Dornseiffer, J.; Xiong, Y.; Grüner, D.; Shen, Z.; Iwaya, S.; Pithan, C.; Waser, R. Spark Plasma Sintering of Nanocrystalline BaTiO3 powders: Consolidation Behavior and Dielectric Characteristics. J. Eur. Ceram. Soc. 2011, 31, 1723–1731. [Google Scholar] [CrossRef]
- Su, B.; He, J.; Cheng, B.; Button, T.; Liu, J.; Shen, Z.; Nygren, M. Dielectric properties of spark plasma sintered (SPS) barium strontium titanate (BST) ceramics. Integr. Ferroelectr. Int. J. 2004, 61, 74–81. [Google Scholar] [CrossRef]
- Ctibor, P.; Kubatík, T.; Chráska, P. Spark plasma sintering of multilayer ceramics—Case study of Al2O3–Mg(Ca)TiO3 sandwich. Key Eng. Mater. 2014, 606, 205–208. [Google Scholar] [CrossRef]
- Anselmi-Tamburini, U.; Garay, J.; Munir, Z. Fast low-temperature consolidation of bulk nanometric ceramic materials. Scr. Mater. 2006, 54, 823–828. [Google Scholar] [CrossRef]
- Pramanik, S.; Ravikumar, K.; Kalsar, R.; Suwas, S.; Basu, B. On the orientation relationships in phase transformation of CaTiO3. Ceram. Int. 2019, 45, 12509–12515. [Google Scholar] [CrossRef]
- Riaz, A.; Witte, K.; Bodnar, W.; Hantusch, M.; Schell, N.; Springer, A.; Burkel, E. Structural changes and pseudo-piezoelectric behaviour of field assisted sintered calcium titanate. Materialia 2021, 15, 100998. [Google Scholar] [CrossRef]
- Ctibor, P.; Sedláček, J.; Neufuss, K.; Chráska, P. Dielectric relaxation in calcium titanate-containing ceramics prepared by plasma spraying. Ceram. Int. 2003, 29, 955–960. [Google Scholar] [CrossRef]
- Sedláček, J.; Ctibor, P.; Kotlan, J.; Pala, Z. Dielectric properties of CaTiO3 coatings prepared by plasma spraying. Surf. Eng. 2013, 29, 384–389. [Google Scholar] [CrossRef]
- Dubey, A.; Mallik, P.; Kundu, S.; Basu, B. Dielectric and electrical conductivity properties of multi-stage spark plasma sintered HA–CaTiO3 composites and comparison with conventionally sintered materials. J. Eur. Ceram. Soc. 2013, 33, 3445–3453. [Google Scholar] [CrossRef]
- Mousavi, S.J.; Digest, J. The effect of sintering time on the electrical properties of CaTiO3 ceramics. Nanomater. Biostruct. 2014, 9, 1059–1063. [Google Scholar]
- Zhou, H.; Liu, X.; Zhu, X.; Chen, X. CaTiO3 linear dielectric ceramics with greatly enhanced dielectric strength and energy storage density. J. Am. Ceram. Soc. 2018, 101, 1999–2008. [Google Scholar] [CrossRef]
- Jakic, J.; Stefanic, G.; Labor, M.; Martinac, V. Microstructural characteristics of low-temperature (1400 °C) sintered MgO obtained from seawater. Sci. Sinter. 2017, 49, 61–71. [Google Scholar] [CrossRef]
- Rietveld, H. Line profiles of neutron powder-diffraction peaks for structure refinement. Acta Crystallogr. 1967, 22, 151–152. [Google Scholar] [CrossRef]
- Hill, R.; Howard, C. Quantitative phase analysis from neutron powder diffraction data using the Rietveld method. J. Appl. Crystallogr. 1987, 20, 467–474. [Google Scholar] [CrossRef]
- Dollase, W.A. Correction of Intensities for Preferred Orientation in Powder Diffractometry: Application of the March Model. J. Appl. Crystallogr. 1986, 19, 267–272. [Google Scholar] [CrossRef]
- Omar, K.; Ooi, M.J. Investigation on dielectric constant of zinc oxide. Mod. Appl. Sci. 2009, 3, 110–116. [Google Scholar] [CrossRef]
- Tauc, J.; Grigorovici, R.; Vancu, A. Optical properties and electronic structure of amorphous germanium. J. Phys. Stat. Solidi 1966, 15, 627–633. [Google Scholar] [CrossRef]
- Ctibor, P.; Sedlacek, J.; Ryukhtin, V.; Cinert, J.; Lukac, F. Barium titanate nanometric polycrystalline ceramics fired by spark plasma sintering. Ceram. Int. 2016, 42, 15989–15993. [Google Scholar] [CrossRef]
- Şavklıyıldız, İ.; Okur, Ç.; Akdoğan, E.K. Flash sintering and dielectric properties of K0.5Na0.5NbO3. J. Am. Ceram. Soc. 2022, 105, 469–480. [Google Scholar] [CrossRef]
- Cherdchom, S.; Rattanaphan, T.; Chanadee, T. Calcium titanate from food waste: Combustion synthesis, sintering, characterization, and properties. Adv. Mat. Sci. Eng. 2019, 2019, 9639016. [Google Scholar] [CrossRef]
- Vasconcelos, S.J.T.; Silva, M.A.S.; de Oliveira, R.G.M.; Bezerra, M.H., Jr.; de Andrade, H.D.; Queiroz Junio, I.S.; Singh, C.; Sombra, A.S.B. High thermal stability and colossal permittivity of novel solid solution LaFeO3/CaTiO3. Mat. Chem. Phys. 2021, 257, 123239. [Google Scholar] [CrossRef]
- Krause, A. Ultrathin CaTiO3 Capacitors: Physics and Application. Ph.D. Thesis, Fakultät Elektrotechnik und Informationstechnik der Technischen Universität, Dresden, Germany, 2014. [Google Scholar]
- Cavalcante, L.; Marques, V.; Sczancoski, J.; Escote, M.; Joya, M.; Varela, J.; Santos, M.; Pizani, P.; Longo, E. Synthesis, structural refinement and optical behavior of CaTiO3 powders: A comparative study of processing in different furnaces. Chem. Eng. J. 2008, 143, 299–306. [Google Scholar] [CrossRef]
- Oliveira, L.; Savioli, J.; Moura, A.; Nogueira, I.; Li, M.; Longo, E.; Varela, J.; Rosa, L. Investigation of structural and optical properties of CaTiO3 powders doped with Mg2+ and Eu3+ ions. J. Alloys Compd. 2015, 647, 265–275. [Google Scholar] [CrossRef]
- Capizzi, M.; Frova, A. Optical gap of strontium titanate (deviation from Urbach tail behavior). Phys. Rev. Lett. 1970, 25, 1298–1304. [Google Scholar] [CrossRef]
- El Kamel, F.; Bonin, P. Proton related defects in a-BaTiO3:H films based MIM capacitors. Solid State Ion. 2009, 180, 853–859. [Google Scholar] [CrossRef]
- Krause, A.; Weber, W.M.; Pohl, D.; Rellinghaus, B.; Kersch, A.; Mikolajick, T. Investigation of band gap and permittivity of the perovskite CaTiO3 in ultrathin layers. J. Phys. D Appl. Phys. 2015, 48, 415304. [Google Scholar] [CrossRef]
- Pontes, F.M.; Pinheiro, C.D.; Longo, E.; Leite, E.R.; de Lazaro, S.R.; Varela, J.A.; Pizani, P.S.; Boschi, T.M.; Lanciotti, F. The role of network modifiers in the creation of photoluminescence in CaTiO3. Mater. Chem. Phys. 2002, 78, 227–233. [Google Scholar] [CrossRef]
- Cavalcante, L.; Simoes, A.; Espinosa, J.; Santos, L.; Longo, E.; Varela, J.; Pizani, P. Study of structural evolution and photoluminescent properties at room temperature of Ca(Zr,Ti)O3 powders. J. Alloys Compd. 2008, 464, 340–346. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | CaTiO3 | TiO2 | CaCO3 | C |
|---|---|---|---|---|
| 1100 | 98.98 | 0.14 | 0.88 | 0 |
| 1150 | 99.28 | 0.15 | 0.57 | 0 |
| 1150-N2 | 99.03 | 0.13 | 0.84 | 0 |
| 1200 | 99.50 | 0.11 | 0.39 | 0 |
| 1250 | 93.16 | 0.33 | 0.65 | 5.86 |
| Sample | a | b | c |
|---|---|---|---|
| 1100 | 5.44313 | 7.64322 | 5.38133 |
| 1150 | 5.44397 | 7.64443 | 5.38159 |
| 1200 | 5.44345 | 7.64503 | 5.38076 |
| 1250 | 5.44333 | 7.64473 | 5.38103 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ctibor, P.; Sedláček, J.; Straka, L.; Lukáč, F.; Neufuss, K. Dielectric Spectroscopy of Calcium Titanate Processed by Spark Plasma Sintering. Materials 2023, 16, 975. https://doi.org/10.3390/ma16030975
Ctibor P, Sedláček J, Straka L, Lukáč F, Neufuss K. Dielectric Spectroscopy of Calcium Titanate Processed by Spark Plasma Sintering. Materials. 2023; 16(3):975. https://doi.org/10.3390/ma16030975
Chicago/Turabian StyleCtibor, Pavel, Josef Sedláček, Libor Straka, František Lukáč, and Karel Neufuss. 2023. "Dielectric Spectroscopy of Calcium Titanate Processed by Spark Plasma Sintering" Materials 16, no. 3: 975. https://doi.org/10.3390/ma16030975
APA StyleCtibor, P., Sedláček, J., Straka, L., Lukáč, F., & Neufuss, K. (2023). Dielectric Spectroscopy of Calcium Titanate Processed by Spark Plasma Sintering. Materials, 16(3), 975. https://doi.org/10.3390/ma16030975