

Surface Modification of an Absorbable Bimodal Fe-Mn-Ag Alloy by Nitrogen Plasma Immersion Ion Implantation

 ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Alloy Fabrication and Surface Preparation
2.2. Mechanical Testing
2.3. Plasma Surface Modification
2.4. Surface Characterizations
2.5. Electrochemical Measurements
2.6. Static Degradation Immersion Tests
2.7. Cytotoxicity Tests
2.8. Statistical Analysis
3. Results and Discussion
3.1. Microstructure and Mechanical Properties
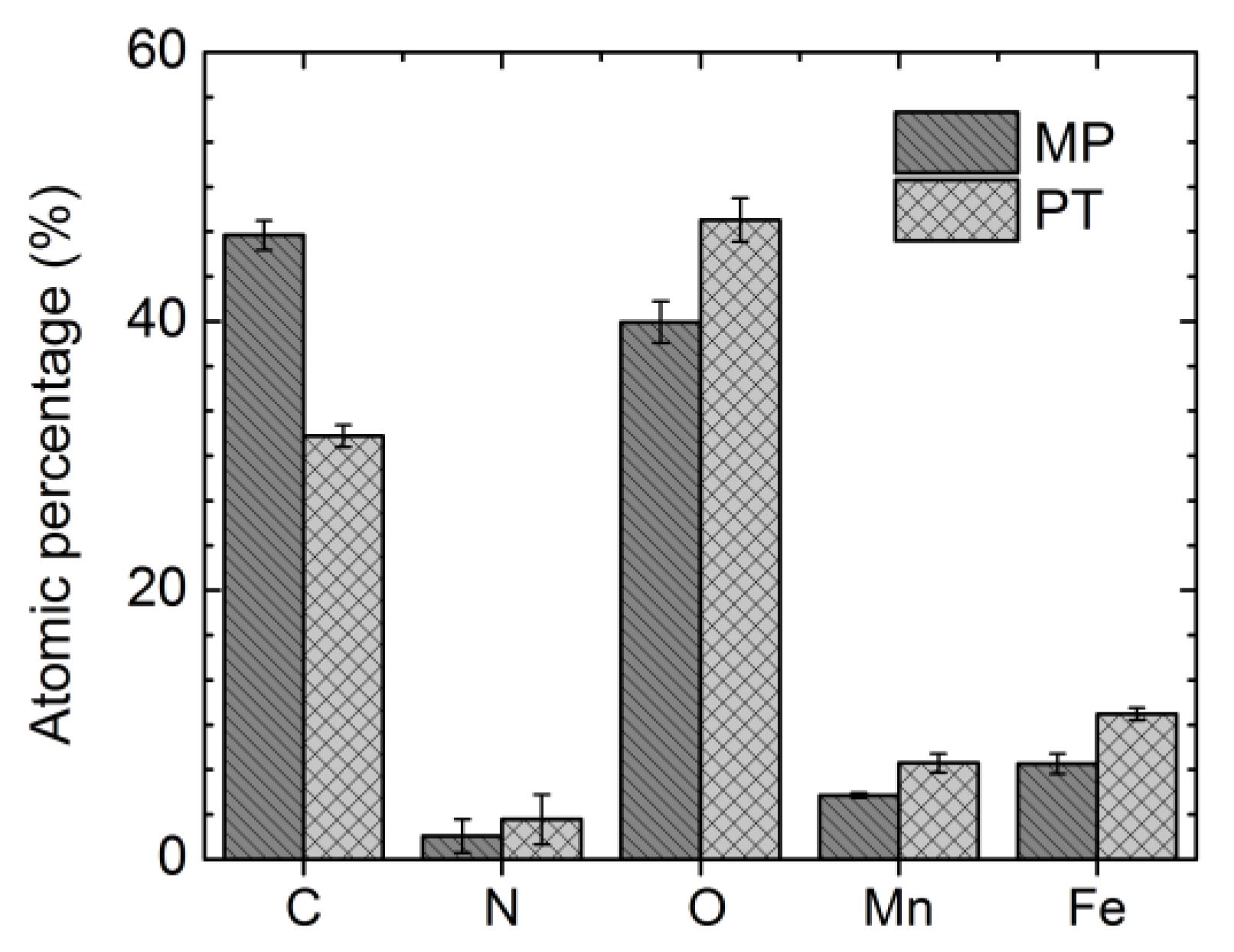
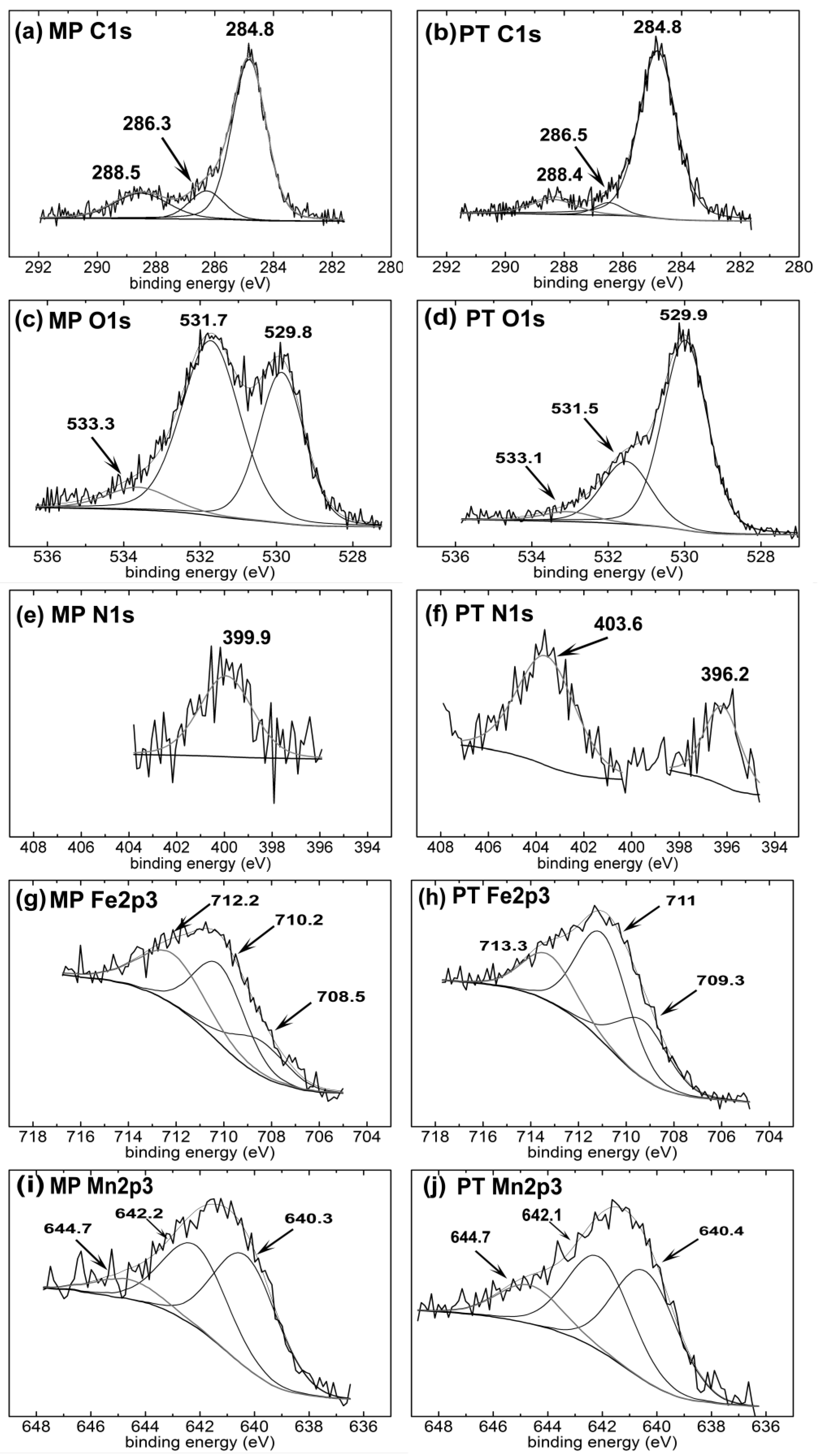
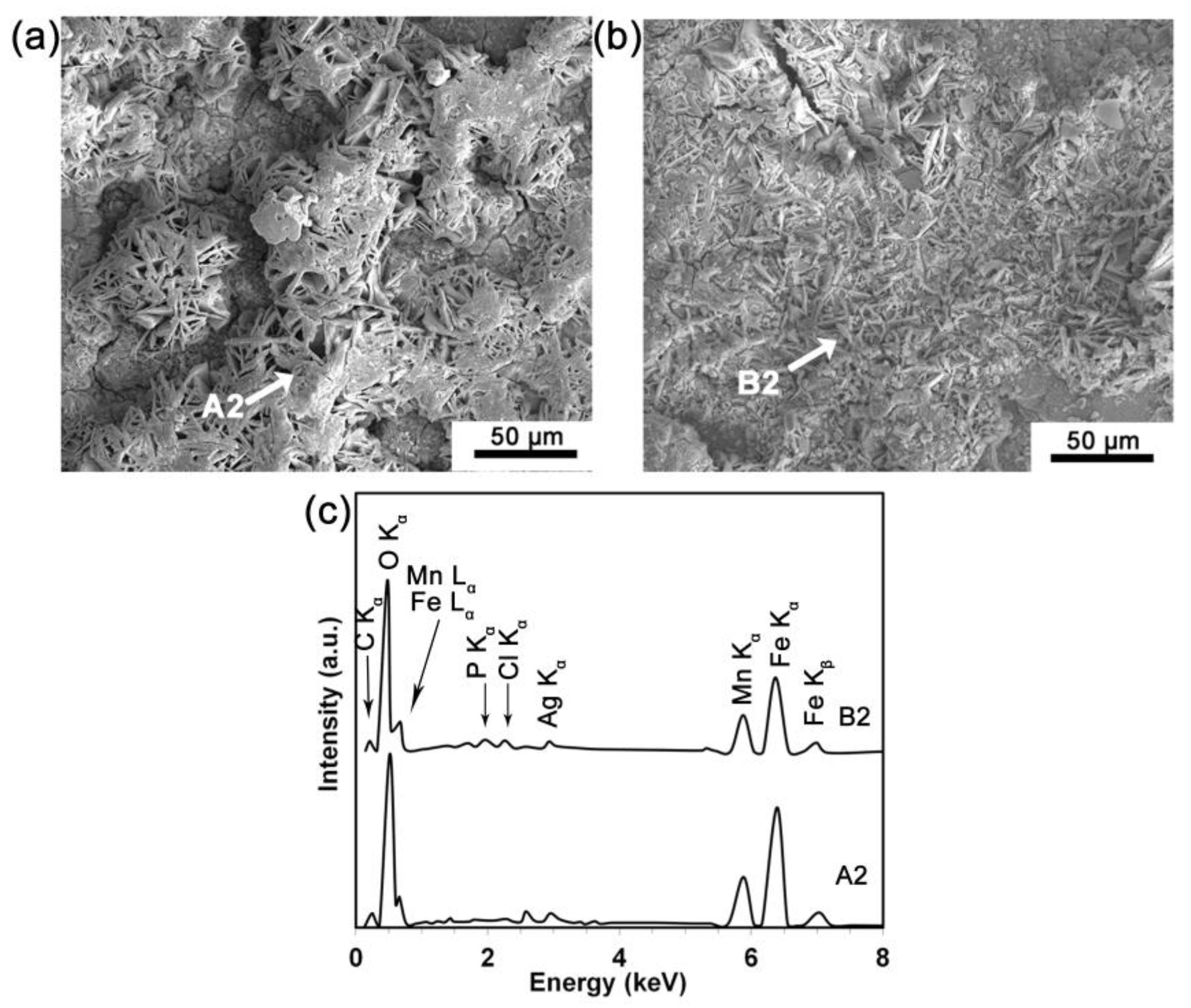
3.2. Surface Chemical Composition
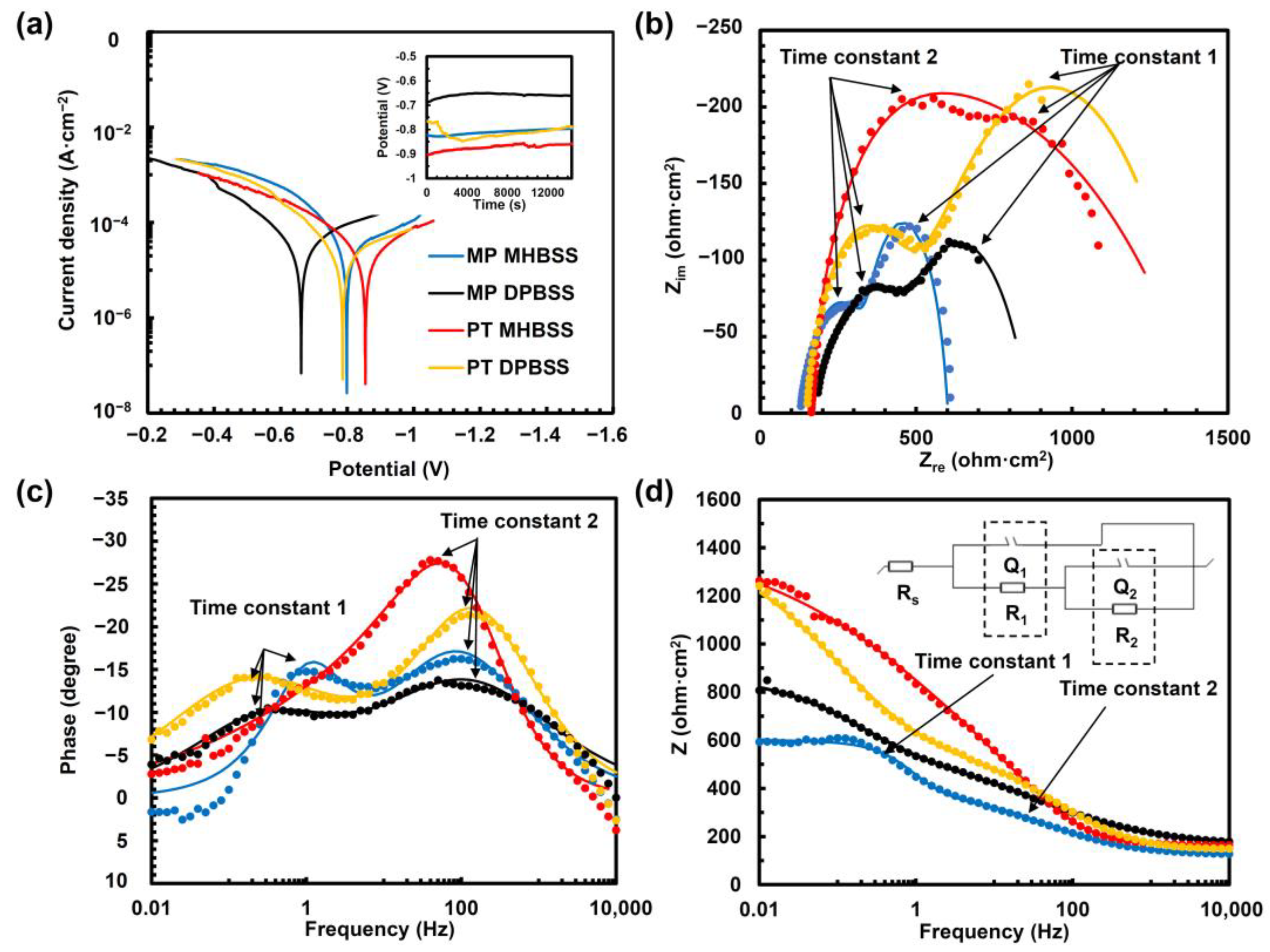
3.3. Electrochemical Corrosion Behavior
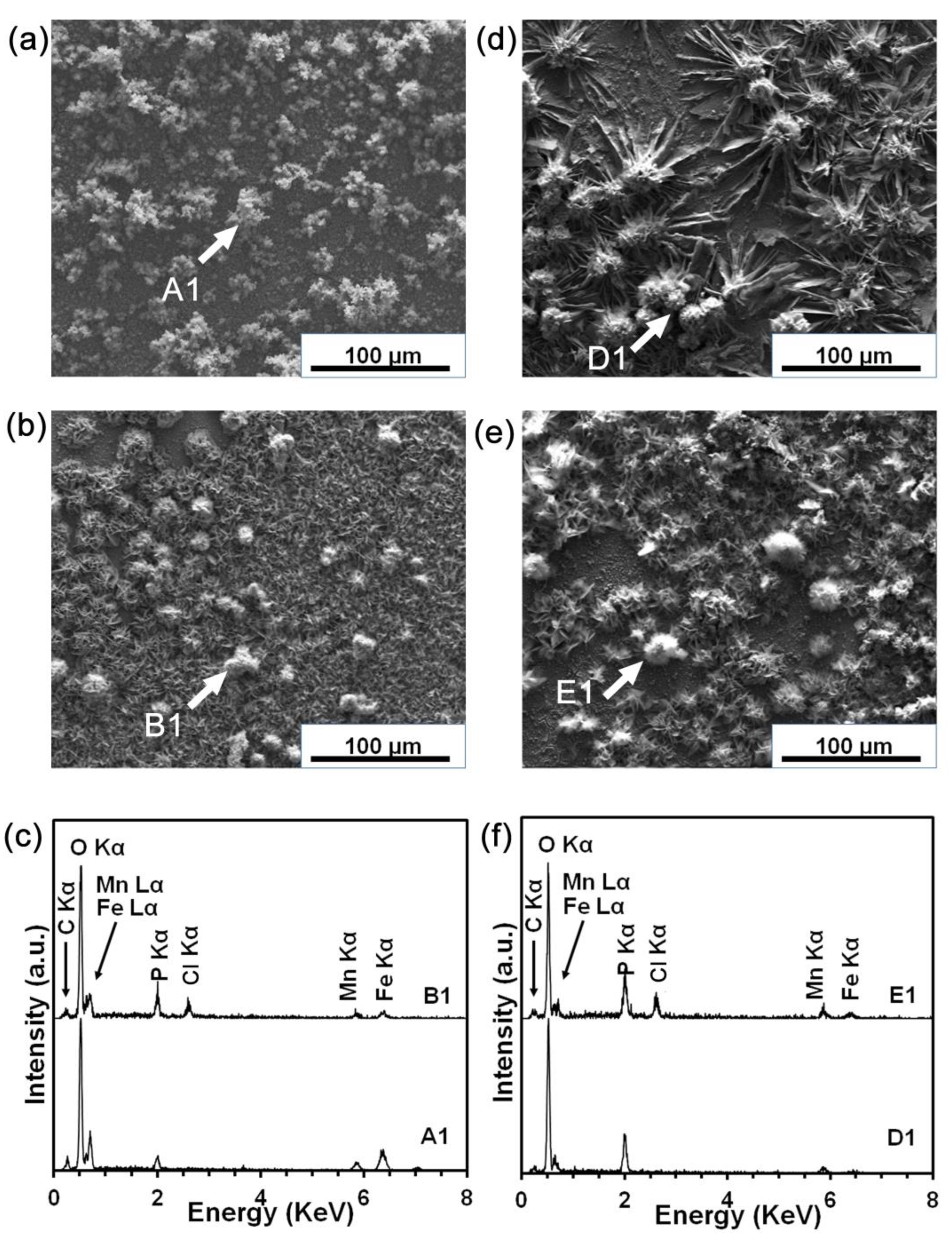
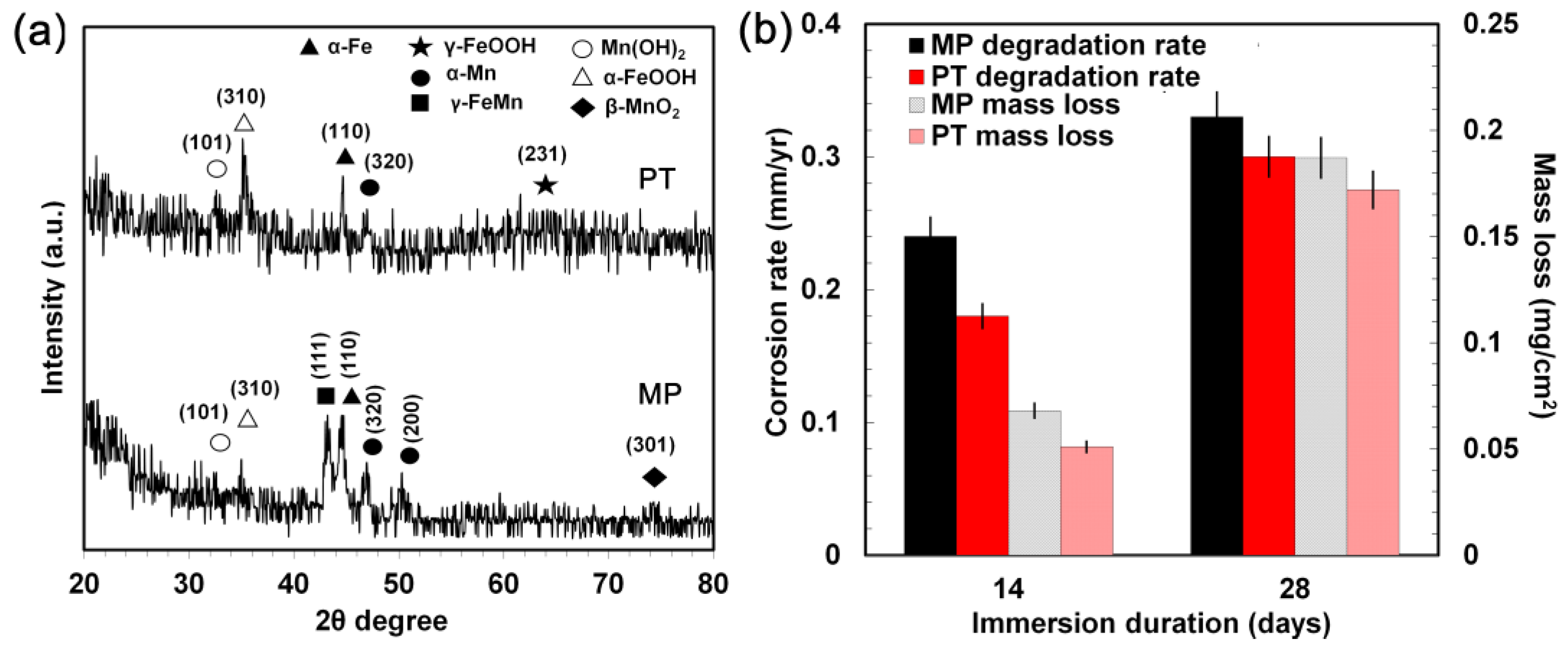
3.4. Static Degradation Tests Results
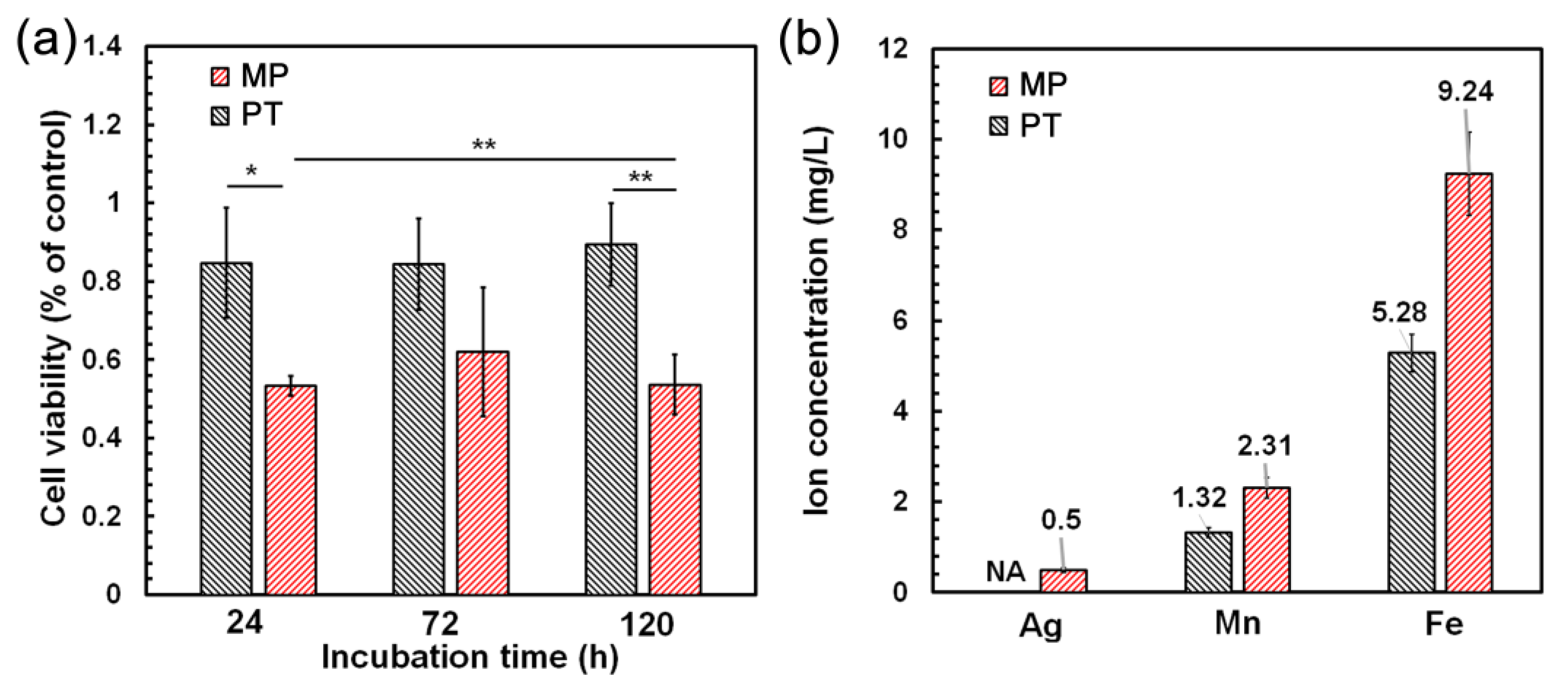
3.5. Cell Viability
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hermawan, H.; Purnama, A.; Dube, D.; Couet, J.; Mantovani, D. Fe-Mn alloys for metallic biodegradable stents: Degradation and cell viability studies. Acta Biomat. 2010, 6, 1852–1860. [Google Scholar] [CrossRef] [PubMed]
- Hermawan, H. Biodegradable Metals, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Schinhammer, M.; Hanzi, A.C.; Loffler, J.F.; Uggowitzer, P.J. Design strategy for biodegradable Fe-based alloys for medical applications. Acta Biomat. 2010, 6, 1705–1713. [Google Scholar] [CrossRef] [PubMed]
- Witte, F. The history of biodegradable magnesium implants: A review. Acta Biomat. 2010, 6, 1680–1692. [Google Scholar] [CrossRef]
- Zheng, Y.F.; Gu, X.N.; Witte, F. Biodegradable metals. Mat. Sci. Eng. R Rep. 2014, 77, 1–34. [Google Scholar] [CrossRef]
- Li, H.; Zheng, Y.; Qin, L. Progress of biodegradable metals. Progr. Nat. Sci. Mat. Intern. 2014, 24, 414–422. [Google Scholar] [CrossRef]
- Vojtech, D.; Kubasek, J.; Serak, J.; Novak, P. Mechanical and corrosion properties of newly developed biodegradable Zn-based alloys for bone fixation. Acta Biomat. 2011, 7, 3515–3522. [Google Scholar] [CrossRef] [PubMed]
- Sotoudeh Bagha, P.; Khaleghpanah, S.; Sheibani, S.; Khakbiz, M.; Zakeri, A. Characterization of nanostructured biodegradable Zn-Mn alloy synthesized by mechanical alloying. J. Alloys Compd. 2018, 735, 1319–1327. [Google Scholar] [CrossRef]
- Sikora-Jasinska, M.; Paternoster, C.; Mostaed, E.; Tolouei, R.; Casati, R.; Vedani, M.; Mantovani, D. Synthesis, mechanical properties and corrosion behavior of powder metallurgy processed Fe/Mg2Si composites for biodegradable implant applications. Mat. Sci. Eng. C 2017, 81, 511–521. [Google Scholar] [CrossRef]
- Li, H.; Lin, G.; Wang, P.; Huang, J.; Wen, C. Nutrient alloying elements in biodegradable metals: A review. J. Mat. Chem. B 2021, 9, 9806–9825. [Google Scholar] [CrossRef]
- Otto, M.; Pilz, S.; Gebert, A.; Kühn, U.; Hufenbach, J. Effect of Build Orientation on the Microstructure, Mechanical and Corrosion Properties of a Biodegradable High Manganese Steel Processed by Laser Powder Bed Fusion. Metals 2021, 11, 944. [Google Scholar] [CrossRef]
- Prokoshkin, S.; Pustov, Y.; Zhukova, Y.; Kadirov, P.; Karavaeva, M.; Prosviryakov, A.; Dubinskiy, S. Effect of Thermomechanical Treatment on Structure and Functional Fatigue Characteristics of Biodegradable Fe-30Mn-5Si (wt %) Shape Memory Alloy. Materials 2021, 14, 3327. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wang, Z.; Zang, S.; Yu, X.; Yang, H.; Chang, S. Research Progress on Surface Treatments of Biodegradable Mg Alloys: A Review. ACS Omega 2020, 5, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Huang, N.; Xu, L.; Zhang, Y.; Liu, H.; Sun, H.; Leng, Y. Biocompatibility of pure iron: In vitro assessment of degradation kinetics and cytotoxicity on endothelial cells. Mat. Sci. Eng. C 2009, 29, 1589–1592. [Google Scholar] [CrossRef]
- Gasior, G.; Szczepanski, J.; Radtke, A. Biodegradable Iron-Based Materials-What Was Done and What More Can Be Done? Materials 2021, 14, 3381. [Google Scholar] [CrossRef] [PubMed]
- Venezuela, J.; Dargusch, M.S. Addressing the slow corrosion rate of biodegradable Fe-Mn: Current approaches and future trends. Curr. Op. Sol. State Mat. Sci. 2020, 24, 100822. [Google Scholar] [CrossRef]
- Hermawan, H. Updates on the research and development of absorbable metals for biomedical applications. Prog. Biomater. 2018, 7, 93–110. [Google Scholar] [CrossRef]
- Shuai, C.; Li, S.; Yang, W.; Yang, Y.; Deng, Y.; Gao, C. MnO2 catalysis of oxygen reduction to accelerate the degradation of Fe-C composites for biomedical applications. Corr. Sci. 2020, 170, 108679. [Google Scholar] [CrossRef]
- Abdalla, M.; Joplin, A.; Elahinia, M.; Ibrahim, H. Corrosion Modeling of Magnesium and Its Alloys for Biomedical Applications: Review. Corr. Mat. Degrad. 2020, 1, 219–248. [Google Scholar] [CrossRef]
- Sotoudeh Bagha, P.; Khakbiz, M.; Sheibani, S.; Hermawan, H. Design and characterization of nano and bimodal structured biodegradable Fe-Mn-Ag alloy with accelerated corrosion rate. J. Alloys Compd. 2018, 767, 955–965. [Google Scholar] [CrossRef]
- Huang, T.; Cheng, J.; Zheng, Y.F. In vitro degradation and biocompatibility of Fe-Pd and Fe-Pt composites fabricated by spark plasma sintering. Mat. Sci. Eng. C 2014, 35, 43–53. [Google Scholar] [CrossRef]
- Mandal, S.; Ummadi, R.; Bose, M.; Balla, V.K.; Roy, M. Fe–Mn–Cu alloy as biodegradable material with enhanced antimicrobial properties. Mat. Lett. 2019, 237, 323–327. [Google Scholar] [CrossRef]
- Thull, R. Surface treatment. In Metals for Biomedical Devices; Woodhead Publishing Series in Biomaterials; Woodhead Publishing: Cambridge, UK, 2010; pp. 251–259. [Google Scholar]
- Lu, T.; Qiao, Y.; Liu, X. Surface modification of biomaterials using plasma immersion ion implantation and deposition. Interf. Focus 2012, 2, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Cisternas, M.; Bhuyan, H.; Retamal, M.J.; Casanova-Morales, N.; Favre, M.; Volkmann, U.G.; Saikia, P.; Diaz-Droguett, D.E.; Mandl, S.; Manova, D.; et al. Study of nitrogen implantation in Ti surface using plasma immersion ion implantation & deposition technique as biocompatible substrate for artificial membranes. Mater. Sci. Eng. C 2020, 113, 111002. [Google Scholar]
- Catanio Bortolan, C.; Paternoster, C.; Turgeon, S.; Paoletti, C.; Cabibbo, M.; Lecis, N.; Mantovani, D. Plasma-immersion ion implantation surface oxidation on a cobalt-chromium alloy for biomedical applications. Biointerphases 2020, 15, 041004. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Castanon, E.; Gigax, J.G.; Kim, H.; Balerio, R.; Fan, J.; Garner, F.A.; Shao, L. Nitrogen ion implantation into pure iron for formation of surface nitride layer. Nucl. Instr. Met. Phy. Res. B Beam Int. Mat. At. 2019, 451, 10–13. [Google Scholar] [CrossRef]
- Bao, J.J.; Leng, Y.X.; Su, Y.Y.; Chen, J.Y.; Zhang, P.C.; Bai, B.; Huang, N. High frequency and low voltage plasma immersion ion implantation of nitrogen on industrial pure iron at different Rf power. Surf. Coat. Technol. 2011, 206, 943–946. [Google Scholar] [CrossRef]
- Bagha, P.S.; Khakbiz, M.; Sheibani, S.; Ebrahimi-Barough, S.; Hermawan, H. In Vitro Degradation, Hemocompatibility, and Cytocompatibility of Nanostructured Absorbable Fe-Mn-Ag Alloys for Biomedical Application. ACS Biomat. Sci. Eng. 2020, 6, 2094–2106. [Google Scholar] [CrossRef]
- Zhu, S.; Huang, N.; Xu, L.; Zhang, Y.; Liu, H.; Lei, Y.; Sun, H.; Yao, Y. Biocompatibility of Fe–O films synthesized by plasma immersion ion implantation and deposition. Surf. Coat. Technol. 2009, 203, 1523–1529. [Google Scholar] [CrossRef]
- Zhao, Y.; Wong, S.M.; Wong, H.M.; Wu, S.; Hu, T.; Yeung, K.W.; Chu, P.K. Effects of carbon and nitrogen plasma immersion ion implantation on in vitro and in vivo biocompatibility of titanium alloy. ACS Appl. Mat. Interf. 2013, 5, 1510–1516. [Google Scholar] [CrossRef]
- Nguyen, H.G.T.; Horn, J.C.; Bleakney, M.; Siderius, D.W.; Espinal, L. Understanding material characteristics through signature traits from helium pycnometry. Langmuir 2019, 35, 2115–2122. [Google Scholar] [CrossRef]
- Tolouei, R.; Harrison, J.; Paternoster, C.; Turgeon, S.; Chevallier, P.; Mantovani, D. The use of multiple pseudo-physiological solutions to simulate the degradation behavior of pure iron as a metallic resorbable implant: A surface-characterization study. Phys. Chem. Chem. Phys. 2016, 18, 19637–19646. [Google Scholar] [CrossRef] [PubMed]
- ASTM G59-97(2014); Standard Test Method for Conducting Potentiodynamic Polarization Resistance Measurements. ASTM: West Conshohocken, PA, USA, 2014.
- ASTM G31-72(2004); Standard Practice for Laboratory Immersion Corrosion Testing of Metals. ASTM: West Conshohocken, PA, USA, 2004.
- ISO 10993-12:2012; Biological Evaluation of Medical Devices—Part 12: Sample Preparation and Reference Materials. ISO: Geneva, Switzerland, 2012.
- Safaie, N.; Khakbiz, M.; Sheibani, S.; Bagha, P.S. Synthesizing of Nanostructured Fe-Mn Alloys by Mechanical Alloying Process. Proc. Mat. Sci. 2015, 11, 381–385. [Google Scholar] [CrossRef]
- ASTM E140-12b (2013); Standard Hardness Conversion Tables for Metals Relationship among Brinell Hardness, Vickers Hardness, Rockwell Hardness, Superficial Hardness, Knoop Hardness, Scleroscope Hardness, and Leeb Hardness. ASTM: West Conshohocken, PA, USA, 2013.
- Nieh, T.G.; Wadsworth, J. Hall-petch relation in nanocrystalline solids. Scr. Metall. Et Mater. 1991, 25, 955–958. [Google Scholar] [CrossRef]
- Wang, Y.M.; Ma, E. Three strategies to achieve uniform tensile deformation in a nanostructured metal. Acta Mater. 2004, 52, 1699–1709. [Google Scholar] [CrossRef]
- Alizadeh, R.; Mahmudi, R.; Langdon, T.G. Superplasticity of a fine-grained Mg–9Gd–4Y–0.4Zr alloy evaluated using shear punch testing. J. Mat. Res. Technol. 2014, 3, 228–232. [Google Scholar] [CrossRef]
- ASTM F138-19 (2020); Standard Specification for Wrought 18Chromium-14Nickel-2.5Molybdenum Stainless Steel Bar and Wire for Surgical Implants (UNS S31673). ASTM: West Conshohocken, PA, USA, 2013.
- Li, Z.; Tang, G.; Ma, X.; Sun, M.; Wang, L. XPS Study on Chemical State and Phase Structure of PBII Nitriding M50 Steel. IEEE Trans. Plasma Sci. 2010, 38, 3079–3082. [Google Scholar] [CrossRef]
- Graat, P.C.J.; Somers, M.A.J.; Mittemeijer, E.J. The initial oxidation of ε-Fe2N1−x: An XPS investigation. Appl. Surf. Sci. 1998, 136, 238–259. [Google Scholar] [CrossRef]
- Swift, P. Adventitious carbon?the panacea for energy referencing? Surf. Interface Anal. 1982, 4, 47–51. [Google Scholar] [CrossRef]
- Miller, D.J.; Biesinger, M.C.; McIntyre, N.S. Interactions of CO2 and CO at fractional atmosphere pressures with iron and iron oxide surfaces: One possible mechanism for surface contamination? Surf. Interface Anal. 2002, 33, 299–305. [Google Scholar] [CrossRef]
- Hueso, J.L.; Espinós, J.P.; Caballero, A.; Cotrino, J.; González-Elipe, A.R. XPS investigation of the reaction of carbon with NO, O2, N2 and H2O plasmas. Carbon 2007, 45, 89–96. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R.; Freitas, M.M.A.; Órfão, J.J.M. Modification of the surface chemistry of activated carbons. Carbon 1999, 37, 1379–1389. [Google Scholar] [CrossRef]
- Liu, Z.; Duchon, T.; Wang, H.; Grinter, D.C.; Waluyo, I.; Zhou, J.; Liu, Q.; Jeong, B.; Crumlin, E.J.; Matolin, V.; et al. Ambient pressure XPS and IRRAS investigation of ethanol steam reforming on Ni-CeO2(111) catalysts: An in situ study of C-C and O-H bond scission. Phys. Chem. Chem. Phys. 2016, 18, 16621–16628. [Google Scholar] [CrossRef] [PubMed]
- Mullet, M.; Khare, V.; Ruby, C. XPS study of Fe(II)–Fe(III) (oxy)hydroxycarbonate green rust compounds. Surf. Interface Anal. 2008, 40, 323–328. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Yakupova, I.V.; Mamchenko, A.V.; Savchenko, O.V.; Chernova, N.N.; Kosygina, I.M. Investigation of the structure of the surface of sorbents–catalysts modified with MnO2 by the method of X-ray photoelectronic spectroscopy. J. Water Chem. Technol. 2016, 38, 134–142. [Google Scholar] [CrossRef]
- Heuer, J.K.; Stubbins, J.F. An XPS characterization of FeCO3 films from CO2 corrosion. Corr. Sci. 1999, 41, 1231–1243. [Google Scholar] [CrossRef]
- Diekmann, W.; Panzner, G.; Grabke, H.J. The bonding state of nitrogen segregated on Fe(100) and on iron nitrides Fe4N and Fe2N. Surf. Sci. 1989, 218, 507–518. [Google Scholar] [CrossRef]
- Liao, H.M.; Sodhi, R.N.S.; Coyle, T.W. Surface composition of AlN powders studied by X-ray photoelectron spectroscopy and bremsstrahlung-excited Auger electron spectroscopy. J. Vac. Sci. Technol. A 1993, 11, 2681–2686. [Google Scholar] [CrossRef]
- Wu, S.X.; Xia, Y.Q.; Yu, X.L.; Liu, Y.J.; Li, S.W. Magnetic properties of MnxTi1−xN thin films grown by plasma-assisted molecular beam epitaxy. J. Appl. Phys. 2007, 102, 063911. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, W.T.; Tian, H.W.; Yu, S.S.; Xu, W.; Meng, S.H.; He, X.D.; Han, J.C.; Sun, C.Q.; Tay, B.K. Growth, structural, and magnetic properties of iron nitride thin films deposited by dc magnetron sputtering. Appl. Surf. Sci. 2003, 220, 30–39. [Google Scholar] [CrossRef]
- Rohith Vinod, K.; Saravanan, P.; Sakar, M.; Balakumar, S. Insights into the nitridation of zero-valent iron nanoparticles for the facile synthesis of iron nitride nanoparticles. RSC Adv. 2016, 6, 45850–45857. [Google Scholar] [CrossRef]
- Jie, J.; Shao, T. Graded Microstructure and Mechanical Performance of Ti/N-Implanted M50 Steel with Polyenergy. Materials 2017, 10, 1204. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Chao, S.; Wang, P.; Bai, Z.; Yan, H.; Wang, K.; Yang, L. Fe–N/C catalysts synthesized by heat-treatment of iron triazine carboxylic acid derivative complex for oxygen reduction reaction. RSC Adv. 2014, 4, 12168–12174. [Google Scholar] [CrossRef]
- Grosvenor, A.P.; Kobe, B.A.; Biesinger, M.C.; McIntyre, N.S. Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds. Surf. Interface Anal. 2004, 36, 1564–1574. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, W.T.; Tian, H.W.; Yu, S.S.; Wang, L.L. Effect of substrate temperature and bias voltage on DC magnetron sputtered Fe–N thin films. J. Magnet. Magn. Mat. 2004, 283, 282–290. [Google Scholar] [CrossRef]
- Ram Mohan Rao, K.; Mukherjee, S.; Raole, P.M.; Manna, I. Low energy isothermal plasma-immersion ion implantation of nitrogen for enhanced hardness of AISI 52100 ball bearing steel. Surf. Coat. Technol. 2002, 150, 80–87. [Google Scholar] [CrossRef]
- Miola, E.J.; de Souza, S.D.; Nascente, P.A.P.; Olzon-Dionysio, M.; Olivieri, C.A.; Spinelli, D. Surface characterisation of plasma-nitrided iron by X-ray photoelectron spectroscopy. Appl. Surf. Sci. 1999, 144–145, 272–277. [Google Scholar] [CrossRef]
- Jutte, R.H.; Kooi, B.J.; Somers, M.A.J.; Mittemeijer, E.J. On the oxidation ofα-Fe andε-Fe2N1−z: I. Oxidation kinetics and microstructural evolution of the oxide and nitride layers. Oxid. Met. 1997, 48, 87–109. [Google Scholar] [CrossRef]
- Ilton, E.S.; Post, J.E.; Heaney, P.J.; Ling, F.T.; Kerisit, S.N. XPS determination of Mn oxidation states in Mn (hydr)oxides. Appl. Surf. Sci. 2016, 366, 475–485. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, L.; Li, X.; Hu, P.; Li, S. Growth and magnetic property of ζ-phase Mn2N1±x thin films by plasma-assisted molecular beam epitaxy. J. Appl. Phy. 2010, 107, 103914. [Google Scholar] [CrossRef]
- Yang, R.; Yang, L. Experimental Study of Static Contact-angle on Peak-like Microstructural Surfaces Produced by PIII Technology. J. Therm. Sci. 2018, 27, 241–248. [Google Scholar] [CrossRef]
- Yao, Z.Q.; Yang, P.; Huang, N.; Sun, H.; Wang, J. Structural, mechanical and hydrophobic properties of fluorine-doped diamond-like carbon films synthesized by plasma immersion ion implantation and deposition (PIII–D). Appl. Surf. Sci. 2004, 230, 172–178. [Google Scholar] [CrossRef]
- Suegama, P.H.; de Melo, H.G.; Benedetti, A.V.; Aoki, I.V. Influence of cerium (IV) ions on the mechanism of organosilane polymerization and on the improvement of its barrier properties. Electrochim. Acta 2009, 54, 2655–2662. [Google Scholar] [CrossRef]
- Hedberg, Y.; Karlsson, M.E.; Blomberg, E.; Odnevall Wallinder, I.; Hedberg, J. Correlation between surface physicochemical properties and the release of iron from stainless steel AISI 304 in biological media. Coll. Surf. B Biointerf. 2014, 122, 216–222. [Google Scholar] [CrossRef]
- Groth, T.; Altankov, G. Studies on cell-biomaterial interaction: Role of tyrosine phosphorylation during fibroblast spreading on surfaces varying in wettability. Biomaterials 1996, 17, 1227–1234. [Google Scholar] [CrossRef]
- Mueller, P.P.; May, T.; Perz, A.; Hauser, H.; Peuster, M. Control of smooth muscle cell proliferation by ferrous iron. Biomaterials 2006, 27, 2193–2200. [Google Scholar] [CrossRef]
- Nygren, H. Initial reactions of whole blood with hydrophilic and hydrophobic titanium surfaces. Coll. Surf. B Biointerf. 1996, 6, 329–333. [Google Scholar] [CrossRef]
- Hansen, A.W.; Führ, L.T.; Antonini, L.M.; Villarinho, D.J.; Marino, C.E.B.; Malfatti, C.d.F. The Electrochemical Behavior of the NiTi Alloy in Different Simulated Body Fluids. Mat. Res. 2015, 18, 184–190. [Google Scholar] [CrossRef]
- Jirásková, Y.; Brauer, G.; Schneeweiss, O.; Blawert, C.; Anwand, W.; Coleman, P.G. The migration of defects and nitrogen atoms in nitrided surface layers of austenitic stainless steel followed by microscopic methods. Appl. Surf. Sci. 2002, 194, 145–149. [Google Scholar] [CrossRef]
- Kim, Y.-S.; Kim, D.-W.; Lee, I.-S.; Yoon, S.; Kim, D.; Jun, S.; Cha, B.-C. Effect of N+ Implantation on Surface Characteristics of 316L Stainless Steels for Bipolar Plate in PEMFC. Coatings 2020, 10, 604. [Google Scholar] [CrossRef]
- Ravi Kumar, B.; Singh, R.; Mahato, B.; De, P.K.; Bandyopadhyay, N.R.; Bhattacharya, D.K. Effect of texture on corrosion behavior of AISI 304L stainless steel. Mat. Charact. 2005, 54, 141–147. [Google Scholar] [CrossRef]
- Guleryuz, L.F.; Ipek, R.; Arıtman, I.; Karaoglu, S. Microstructure and mechanical properties of Zn-Mg alloys as implant materials manufactured by powder metallurgy method. AIP Conf. Proc. 2017, 1809, 020020. [Google Scholar]
- Lee, J.-S.; Fushimi, K.; Nakanishi, T.; Hasegawa, Y.; Park, Y.-S. Corrosion behaviour of ferrite and austenite phases on super duplex stainless steel in a modified green-death solution. Corr. Sci. 2014, 89, 111–117. [Google Scholar] [CrossRef]
- Tao, X.; Liu, X.; Matthews, A.; Leyland, A. The influence of stacking fault energy on plasticity mechanisms in triode-plasma nitrided austenitic stainless steels: Implications for the structure and stability of nitrogen-expanded austenite. Acta Mat. 2019, 164, 60–75. [Google Scholar] [CrossRef]
- Ram Mohan Rao, K.; Mukherjee, S.; Roy, S.K.; Richter, E.; Möller, W.; Manna, I. Plasma immersion ion implantation of nitrogen on austenitic stainless steel at variable energy for enhanced corrosion resistance. Surf. Coat. Technol. 2007, 201, 4919–4921. [Google Scholar] [CrossRef]
- Fosbøl, P.L.; Thomsen, K.; Stenby, E.H. Review and recommended thermodynamic properties of FeCO3. Corr. Eng. Sci. Technol. 2013, 45, 115–135. [Google Scholar] [CrossRef]
- Mouzou, E.; Paternoster, C.; Tolouei, R.; Purnama, A.; Chevallier, P.; Dube, D.; Prima, F.; Mantovani, D. In vitro degradation behavior of Fe-20 Mn-1.2C alloy in three different pseudo-physiological solutions. Mat. Sci. Eng. C 2016, 61, 564–573. [Google Scholar] [CrossRef]
- Martini, E.M.A.; Muller, I.L. Characterization of the film formed on iron in borate solution by electrochemical impedance spectroscopy. Corr. Sci. 2000, 42, 443–454. [Google Scholar] [CrossRef]
- Zhao, C.; Pan, F.; Zhao, S.; Pan, H.; Song, K.; Tang, A. Preparation and characterization of as-extruded Mg–Sn alloys for orthopedic applications. Mat. Des. 2015, 70, 60–67. [Google Scholar] [CrossRef]
- Naganuma, H.; Nakatani, R.; Endo, Y.; Kawamura, Y.; Yamamoto, M. Magnetic and electrical properties of iron nitride films containing both amorphous matrices and nanocrystalline grains. Sci. Technol. Adv. Mat. 2016, 5, 101–106. [Google Scholar] [CrossRef]
- Schibicheski Kurelo, B.C.E.; de Souza, G.B.; Serbena, F.C.; de Oliveira, W.R.; Marino, C.E.B.; Taminato, L.A. Performance of nitrogen ion-implanted supermartensitic stainless steel in chlorine- and hydrogen-rich environments. Surf. Coat. Technol. 2018, 351, 29–41. [Google Scholar] [CrossRef]
- Durai, G.; Kuppusami, P.; Maiyalagan, T.; Ahila, M.; Vinoth kumar, P. Supercapacitive properties of manganese nitride thin film electrodes prepared by reactive magnetron sputtering: Effect of different electrolytes. Ceram. Intern. 2019, 45, 17120–17127. [Google Scholar] [CrossRef]
- Zatkalikova, V.; Halanda, J.; Vana, D.; Uhricik, M.; Markovicova, L.; Strbak, M.; Kucharikova, L. Corrosion Resistance of AISI 316L Stainless Steel Biomaterial after Plasma Immersion Ion Implantation of Nitrogen. Materials 2021, 14, 6790. [Google Scholar] [CrossRef] [PubMed]
- Luiz, L.A.; Kurelo, B.C.E.S.; Souza, G.B.d.; Andrade, J.d.; Marino, C.E.B. Effect of nitrogen plasma immersion ion implantation on the corrosion protection mechanisms of different stainless steels. Mater. Today Commun. 2021, 28, 102655. [Google Scholar] [CrossRef]
- Casero, E.; Parra-Alfambra, A.M.; Petit-Domínguez, M.D.; Pariente, F.; Lorenzo, E.; Alonso, C. Differentiation between graphene oxide and reduced graphene by electrochemical impedance spectroscopy (EIS). Electrochem. Commun. 2012, 20, 63–66. [Google Scholar] [CrossRef]
- Yohai, L.; Valcarce, M.B.; Vázquez, M. Testing phosphate ions as corrosion inhibitors for construction steel in mortars. Electrochim. Acta 2016, 202, 316–324. [Google Scholar] [CrossRef]
- Grabke, H.J. High Nitrogen Steels. The Role of Nitrogen in the Corrosion of Iron and Steels. ISIJ Intern. 1996, 36, 777–786. [Google Scholar] [CrossRef]
- Tian, X.; Chu, P.K. Electrochemical corrosion properties of AISI304 steel treated by low-temperature plasma immersion ion implantation. Scr. Mat. 2000, 43, 417–422. [Google Scholar] [CrossRef]
- Baranwal, P.K.; Rajaraman, P.V. Electrochemical investigation on effect of sodium thiosulfate (Na2S2O3) and ammonium chloride (NH4Cl) on carbon steel corrosion. J. Mat. Res. Technol. 2019, 8, 1366–1378. [Google Scholar] [CrossRef]
- Gambaro, S.; Paternoster, C.; Occhionero, B.; Fiocchi, J.; Biffi, C.A.; Tuissi, A.; Mantovani, D. Mechanical and degradation behavior of three Fe-Mn-C alloys for potential biomedical applications. Mat. Today Comm. 2021, 27, 102250. [Google Scholar] [CrossRef]
- Saravanan, P.; Raja, V.S.; Mukherjee, S. Effect of plasma immersion ion implantation of nitrogen on the wear and corrosion behavior of 316LVM stainless steel. Surf. Coat. Technol. 2007, 201, 8131–8135. [Google Scholar] [CrossRef]
- De Andrade, L.M.; Paternoster, C.; Montaño-Machado, V.; Barucca, G.; Sikora-Jasinska, M.; Tolouei, R.; Turgeon, S.; Mantovani, D. Surface modification of L605 by oxygen plasma immersion ion implantation for biomedical applications. MRS Comm. 2018, 8, 1404–1412. [Google Scholar] [CrossRef]
- Manova, D.; Höche, T.; Mändl, S.; Neumann, H. Development of CrN precipitates during the initial stages of PIII nitriding of stainless steel thin films. Nucl. Instr. Meth. Phys. Res. Sec. B Beam Inter. Mat. At. 2009, 267, 1536–1539. [Google Scholar] [CrossRef]
- Choe, H.-C. Effects of nitrogen ion implantation on the corrosion characteristics of Cu-electroless plated and sintered stainless steel. Surf. Coat. Technol. 1999, 112, 299–306. [Google Scholar] [CrossRef]
- Feng, Q.; Zhang, D.; Xin, C.; Liu, X.; Lin, W.; Zhang, W.; Chen, S.; Sun, K. Characterization and in vivo evaluation of a bio-corrodible nitrided iron stent. J. Mat. Sci. Mat. Med. 2013, 24, 713–724. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, G.; Jiang, J.; Wong, H.M.; Yeung, K.W.K.; Chu, P.K. Improved corrosion resistance and cytocompatibility of magnesium alloy by two-stage cooling in thermal treatment. Corr. Sci. 2012, 59, 360–365. [Google Scholar] [CrossRef]
- Yamamoto, A.; Kohyama, Y.; Kuroda, D.; Hanawa, T. Cytocompatibility evaluation of Ni-free stainless steel manufactured by nitrogen adsorption treatment. Mat. Sci. Eng. C 2004, 24, 737–743. [Google Scholar] [CrossRef]
- Wan, P.; Ren, Y.; Zhang, B.; Yang, K. Effect of nitrogen on blood compatibility of nickel-free high nitrogen stainless steel for biomaterial. Mat. Sci. Eng. C 2010, 30, 1183–1189. [Google Scholar] [CrossRef]
- Buhagiar, J.; Bell, T.; Sammons, R.; Dong, H. Evaluation of the biocompatibility of S-phase layers on medical grade austenitic stainless steels. J. Mater. Sci. Mat. Med. 2011, 22, 1269–1278. [Google Scholar] [CrossRef]
- Ali, S.; Abdul Rani, A.M.; Mufti, R.A.; Hastuty, S.; Hussain, M.; Shehzad, N.; Baig, Z.; Abdu Aliyu, A.A. An Efficient Approach for Nitrogen Diffusion and Surface Nitriding of Boron-Titanium Modified Stainless Steel Alloy for Biomedical Applications. Metals 2019, 9, 755. [Google Scholar] [CrossRef]
- Braz, J.; Martins, G.M.; Sabino, V.; Vitoriano, J.O.; Barboza, C.A.G.; Soares, A.; Rocha, H.A.O.; Oliveira, M.F.; Alves Junior, C.; Moura, C.E.B. Plasma nitriding under low temperature improves the endothelial cell biocompatibility of 316L stainless steel. Biotechn. Lett. 2019, 41, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-H.; Liu, C.-F.; Wang, S.; Chen, C.-S.; Chang, J.-H. Nitrogen plasma immersion ion implantation treatment of Ti6Al7Nb alloy for bone-implant applications: Enhanced in vitro biological responses and in vivo initial bone-implant contact. Surf. Coat. Technol. 2021, 405, 126551. [Google Scholar] [CrossRef]
- Lau, K.; Heu, C.; Moore, M.J.; Zhang, A.; Akhavan, B.; Wise, S.G.; Bilek, M.M.M.; Lord, M.S.; Rnjak-Kovacina, J. Effect of plasma ion immersion implantation on physiochemical and biological properties of silk towards creating a versatile biomaterial platform. Mater. Today Adv. 2022, 13, 100212. [Google Scholar] [CrossRef]
- Guo, S.; Liu, N.; Liu, K.; Li, Y.; Zhang, W.; Zhu, B.; Gu, B.; Wen, N. Effects of carbon and nitrogen plasma immersion ion implantation on bioactivity of zirconia. RSC Adv. 2020, 10, 35917–35929. [Google Scholar] [CrossRef]
- Huang, T.; Cheng, Y.; Zheng, Y. In vitro studies on silver implanted pure iron by metal vapor vacuum arc technique. Coll. Surf. B Biointerf. 2016, 142, 20–29. [Google Scholar] [CrossRef]
- Huang, T.; Cheng, J.; Bian, D.; Zheng, Y. Fe-Au and Fe-Ag composites as candidates for biodegradable stent materials. J. Biomed. Mat. Res. B Appl. Biomat. 2016, 104, 225–240. [Google Scholar] [CrossRef]
- Schinhammer, M.; Gerber, I.; Hanzi, A.C.; Uggowitzer, P.J. On the cytocompatibility of biodegradable Fe-based alloys. Mater. Sci. Eng. C 2013, 33, 782–789. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, Y.F. Effects of alloying elements (Mn, Co, Al, W, Sn, B, C and S) on biodegradability and in vitro biocompatibility of pure iron. Acta Biomat. 2011, 7, 1407–1420. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ion or Compound (mg/L) | Blood Plasma | MHBSS | DPBSS |
|---|---|---|---|
| Na+ | 3000–3400 | 2795 | 3519 |
| K+ | 130–210 | 172 | 162 |
| Cl− | 3400–3750 | 3542 | 4947 |
| HCO3− | 1100–2400 | 1654 | - |
| H2PO4−/HPO42− | 270–450 | 48 | 920 |
| Ca2+ | 84–110 | 35 | - |
| Mg2+ | 15–30 | 14 | - |
| SO42− | 5–15 | 78 | - |
| D-Glucose | 600–1100 | 720 | - |
| Albumin | 35,000–50,000 | - | - |
| Composition (Wt.%) | Micro- Hardness (Hv), Phase | Hardness (HRB) | Yield Shear Strength (MPa) | Ultimate Shear Strength (MPa) | Ultimate Shear Strain (%) |
|---|---|---|---|---|---|
| Fe-30Mn-1Ag | 103 ± 13, α-Fe | 96.5 ± 3.1 | 361.5 ± 22.4 | 420.3 ± 35.1 | 74 ± 12 |
| 556 ± 77, γ-FeMn | |||||
| 515 ± 91, α-Mn |
| Sample | Element | Peak1 (eV) | FWHM (eV) | Area (%) | Peak2 (eV) | FWHM (eV) | Area (%) | Peak3 (eV) | FWHM (eV) | Area (%) | Gauss (%) | χ2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MP | C | 284.8 | 1.37 | 69.9 | 286.3 | 1.37 | 12.3 | 288.5 | 2.2 | 17.8 | 80 | 1.3 |
| O | 529.8 | 1.4 | 37.1 | 531.7 | 1.8 | 54.7 | 533.3 | 2 | 8.2 | 80 | 1.3 | |
| N | 399.9 | 2.6 | 100 | - | - | - | - | - | - | 80 | 1.4 | |
| Fe | 708.5 | 2.7 | 20.1 | 710.2 | 2.5 | 42 | 712.2 | 3.2 | 37.9 | 90 | 1.2 | |
| Mn | 640.3 | 2.8 | 51.3 | 642.2 | 2.75 | 39.2 | 644.7 | 2.8 | 9.4 | 90 | 1.1 | |
| PT | C | 284.8 | 1.5 | 85.7 | 286.5 | 1.1 | 4.8 | 288.4 | 1.86 | 9.5 | 70 | 1 |
| O | 529.9 | 1.4 | 70.3 | 531.5 | 1.5 | 25.3 | 533.1 | 1.6 | 4.4 | 80 | 1.3 | |
| N | 396.2 | 1.8 | 100 | 403.6 | 2.9 | 100 | - | - | - | 80 | 0.9 | |
| Fe | 709.3 | 2.5 | 27.2 | 711 | 2.5 | 50.8 | 713.3 | 2.5 | 22.1 | 90 | 1.3 | |
| Mn | 640.4 | 2.8 | 44.3 | 642.1 | 2.8 | 39.5 | 644.7 | 2.8 | 16.2 | 90 | 1.3 |
| (A) PDP Parameters | |||||||||
| Condition (Solution) | iC (µA/cm2) | CP (mV) | βa (mV/decade) | βc (mV/decade) | CR (mm/year) | ||||
| MP + MHBSS | 26.3 ± 4.1 | −800 ± 77 | 116.7 ± 10 | 309.3 ± 27 | 3.2 ± 0.2 | ||||
| MP + DPBSS | 18 ± 16.5 | −780 ± 49 | 132.4 ± 47.4 | 186.9 ± 60.9 | 2.4 ± 1.7 | ||||
| PT + MHBSS | 16 ± 11.3 | −698 ± 96 | 141.5 ± 37 | 214.1 ± 40 | 2 ± 1.4 | ||||
| PT + DPBSS | 15.9 ± 2.2 | −781 ± 81 | 123.2 ± 15 | 253.7 ± 32 | 1.9 ± 0.6 | ||||
| (B) EIS Parameters | |||||||||
| Condition (Solution) | R1 (ohm·cm2) | Q1 (µF·cm−2·sn) | n1 | τ1 (s) | R2 (ohm·cm2) | Q2 (µF·cm−2·sn) | n2 | τ2 (s) | χʋ2 |
| MP + MHBSS | 256 ± 9.9 | 115.7 ± 13 | 0.7 ± 0.1 | 0.03 ± 0.014 | 235.1 ± 11 | 220 ± 9.2 | 1 ± 0.1 | 0.027 ± 0.003 | 0.25 |
| MP + DPBSS | 754.7 ± 81.7 | 65.2 ± 7.6 | 0.7 ± 0.2 | 0.045 ± 0.006 | 311.1 ± 89.5 | 1454 ± 148 | 0.6 ± 0.1 | 0.02 ± 0.026 | 0.12 |
| PT + MHBSS | 673.4 ± 61 | 30.1 ± 5.9 | 0.9 ± 0.1 | 0.02 ± 0.005 | 1547 ± 578 | 946 ± 79 | 0.6 ± 0.3 | 1.46 ± 0.32 | 0.74 |
| PT + DPBSS | 349.3 ± 6.4 | 35.2 ± 2.4 | 0.7 ± 0.1 | 0.012 ± 0.002 | 921.6 ± 3.5 | 1637 ± 28.7 | 0.5 ± 0.1 | 1.51 ± 0.25 | 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sotoudeh Bagha, P.; Paternoster, C.; Khakbiz, M.; Sheibani, S.; Gholami, N.; Mantovani, D. Surface Modification of an Absorbable Bimodal Fe-Mn-Ag Alloy by Nitrogen Plasma Immersion Ion Implantation. Materials 2023, 16, 1048. https://doi.org/10.3390/ma16031048
Sotoudeh Bagha P, Paternoster C, Khakbiz M, Sheibani S, Gholami N, Mantovani D. Surface Modification of an Absorbable Bimodal Fe-Mn-Ag Alloy by Nitrogen Plasma Immersion Ion Implantation. Materials. 2023; 16(3):1048. https://doi.org/10.3390/ma16031048
Chicago/Turabian StyleSotoudeh Bagha, Pedram, Carlo Paternoster, Mehrdad Khakbiz, Saeed Sheibani, Navid Gholami, and Diego Mantovani. 2023. "Surface Modification of an Absorbable Bimodal Fe-Mn-Ag Alloy by Nitrogen Plasma Immersion Ion Implantation" Materials 16, no. 3: 1048. https://doi.org/10.3390/ma16031048
APA StyleSotoudeh Bagha, P., Paternoster, C., Khakbiz, M., Sheibani, S., Gholami, N., & Mantovani, D. (2023). Surface Modification of an Absorbable Bimodal Fe-Mn-Ag Alloy by Nitrogen Plasma Immersion Ion Implantation. Materials, 16(3), 1048. https://doi.org/10.3390/ma16031048