1. Introduction
Surface as well as interface phenomena, taking place in the ABO
3 perovskites, including the essence of their (001), (011) and (111) surface and interface electronic properties, are very real problems in present-day physics [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26]. BaTiO
3, CaTiO
3, PbTiO
3, SrTiO
3, BaZrO
3, CaZrO
3, PbZrO
3 and SrZrO
3 perovskites are the members of the well-known ABO
3 (A = Ba, Ca, Pb, Sr and B = Ti or Zr) perovskite family [
27,
28,
29]. They all have a colossal amount of technologically essential applications [
30,
31,
32,
33]. For example, BaTiO
3 is an important ABO
3 perovskite ceramic material [
34]. It has outstanding dielectric as well as ferroelectric and piezoelectric properties [
34]. CaTiO
3 is extensively employed in electronic ceramic materials [
35]. Cubic CTO is also used as a keystone element of Synroc [
35]. PbTiO
3 is one of the worldwide most extensively used piezoelectric and ferroelectric materials for technologically important industrial applications [
36,
37,
38]. SrTiO
3 is a perovskite material with a wide-ranging spectrum of functional properties as well as physical phenomena [
39]. STO is an adaptable substrate for complex oxide electronics engineering [
40]. STO possesses excellent superconducting properties [
41] as well as impurity- and vacancy-based magnetism [
42]. BaZrO
3 perovskite has several industrially important applications in many quite different technology areas [
43]. BZO is extensively used, for example, in solid-oxide fuel cells [
44] as well as in wireless systems for communications [
45]. CaZrO
3 is used as an ionic conductor to manufacture solid electrodes for applications in various fuel cells [
46]. Moreover, CZO is used also as a key element for different sensor types [
47,
48]. PbZrO
3 is a very fascinating perovskite, since it is the end point of the Pb(Zr,Ti)O
3 alloy system, which is very interesting for numerous important applications in industry [
49]. Finally, SrZrO
3 has a lot of technological applications [
50,
51,
52,
53]. For example, in the violet–blue light emission, laser host materials as well as capacitors and oxygen sensors [
50,
51,
52,
53]. For that reason, it is clear, that during the last twenty-five years, BTO, CTO, PTO, STO, BZO, CZO, PZO and SZO perovskite (001) surfaces have been comprehensively explored theoretically as well as experimentally around the globe [
1,
2,
3,
6,
7,
11,
13,
14,
16,
18,
21,
23,
24,
54,
55,
56,
57,
58,
59,
60,
61,
62,
63,
64,
65,
66,
67,
68,
69,
70,
71,
72,
73,
74,
75,
76,
77,
78,
79,
80,
81]. It is worth noting that it is relatively easy to compute the neutral ABO
3 perovskite (001) surfaces, since they consist of alternating neutral AO and BO
2 slabs [
82,
83,
84,
85]. In contrast, it is very difficult to compute the polar ABO
3 perovskite (011) and (111) surfaces, since they consist of charged planes of ABO and O
2 as well as AO
3 and B, respectively. This is main reason why the ABO
3 perovskite charged and polar (011) [
86,
87,
88,
89,
90,
91,
92,
93,
94,
95] and (111) [
96,
97,
98,
99,
100,
101,
102,
103,
104,
105,
106,
107] surfaces are considerably less studied than their neutral and relatively simple (001) surfaces [
1,
2,
3,
6,
7,
11,
13,
14,
16,
18,
21,
23,
24,
54,
55,
56,
57,
58,
59,
60,
61,
62,
63,
64,
65,
66,
67,
68,
69,
70,
71,
72,
73,
74,
75,
76,
77,
78,
79,
80,
81,
82,
83,
84,
85].
For example, the first B3PW calculations, dealing with polar and charged BaTiO
3 and PbTiO
3 (011) surface structures were performed by Eglitis and Vanderbilt in 2007 [
1]. Two years later, Zhang et al. [
86,
87], at ab initio level, computed the electronic and structural characteristics of five different terminations of cubic PTO (110) polar surface [
86,
87]. The first ab initio computations for the polar SrTiO
3 (011) surface were carried out by Bottin et al. [
88]. They computed the atomic as well as electronic structure of a few (1 × 1) SrTiO
3 (011) surface terminations [
88]. The year after that, Heifets et al. [
89] carried out first-principles Hartree–Fock computations for four terminations (Sr, TiO as well as two different O terminations) of the polar STO (011) surface [
89]. As the next, Eglitis and Vanderbilt [
2] performed hybrid DFT computations for three different terminations (TiO, Sr and O) of the polar and charged STO (011) surface [
2]. Two years later, Enterkin et al. [
90] described the results for the 3 × 1 extended STO (011) surface structure derived experimentally via transmission electron diffraction [
90]. Experimental results, dealing with polar STO (011) surfaces, were also confirmed theoretically using modern ab initio DFT computations as well as scanning tunneling microscopy images [
90]. Finally, five years ago, Fleischer et al. [
91] experimentally investigated the STO (011) surface using reflectance anisotropy spectroscopy (RAS). World-first ab initio calculations for polar CTO (011) surfaces were performed by Zhang et al. [
92]. They constructed four different CTO polar (011) surface terminations and computed the cleavage as well as (011) surface energies [
92]. Zhang et al. [
92] also computed the CTO (011) surface grand potential as well as the (011) surface electronic and atomic structure [
92]. One year later, Eglitis and Vanderbilt, using a B3PW hybrid exchange–correlation functional, investigated three different terminations (TiO, Ca and O) of polar CTO (011) surfaces [
3]. They [
3] computed the polar CTO (011) surface atomic relaxations, energetics as well as chemical bonding properties for three different (011) surface terminations [
3].
Two world-first ab initio simulations for the polar BaZrO
3 (011) surfaces were performed independently by Heifets et al. [
93] and by Eglitis [
82] in 2007. Heifets et al. [
93] studied the charge redistribution, atomic and electronic structure of several different terminations of BZO (011) surfaces [
93]. According to the B3PW computations performed by Eglitis [
82], three different terminations of the BZO (011) surface exhibit quite different surface energies. They always (for all three terminations) are considerably larger [
82] than for the neutral BZO (001) surfaces. Eglitis and Rohlfing [
94] computed the SZO and PZO neutral (001) and polar (011) surface rumplings, relaxations, energetics, charge redistributions as well as the Γ-Γ band gaps [
94]. Four years later, Chen et al. [
95] investigated the electronic properties and stabilities of SZO (110) (1 × 1) five different polar terminations [
95]. Finally, the only existing B3LYP calculations, dealing with polar CZO (110) surfaces, were recently performed by Eglitis and co-workers [
10].
A quarter of century ago, Hagendorf et al. [
96], experimentally investigated the polar BTO (111) surface using scanning tunneling microscopy (STM), X-ray photoelectron spectroscopy (XPS) and low-energy electron diffraction (LEED) methods [
96]. Recently, Chun et al. [
97], explored the BTO surface (111) termination, using the theoretical ab initio DFT calculations and experimental XPS analysis [
97]. First-in-the-world ab initio linearized augmented-plane-wave method (LAPW) calculations for periodic (111) BTO slabs were performed by Cohen [
98]. Cohen found [
98] that the polar (111) BTO slab is considerably less stable than the BTO neutral (001) slab. In 2015, Eglitis [
5,
60,
99] performed very comprehensive B3LYP calculations for BTO, PTO, CTO, STO, PZO and SZO (111) surfaces. Eglitis found [
5] that the polar BTO, PTO, CTO, STO, PZO and SZO (111) surfaces are considerably less stable than the respective neutral (001) and even polar (011) surfaces [
5,
60,
99]. Twenty-five years ago, Haruyama et al. [
100] experimentally studied the polar STO (111) surface by means of photoemission spectroscopy [
100]. In 1999, Pojani et al. [
101], by means of simple semi-empirical HF method, computed the polar STO (111) and (110) surfaces [
101]. Recently, Torrelles et al. [
102] experimentally detected the surface structure of Ti-terminated STO (111) single crystals [
102]. Finally, Marks et al. [
103] described the reconstructions of the polar STO (111) surface by means of experimental-transmission electron diffraction, scanning tunneling microscopy as well as theoretical first-principles DFT calculations [
103]. Pang et al. [
104] performed very comprehensive first-principles computations for four different (1 × 1) polar terminations of PTO (111) surfaces. The electronic and structural properties as well as stabilities of four different polar (1 × 1) PTO (111) terminations were calculated at ab initio level [
104]. Liu et al. [
105] constructed the stoichiometric as well as nonstoichiometric terminations for polar CTO (111) surfaces [
105]. They computed the polar CTO (111) surface electronic structure, grand potential as well as the relevant surface and cleavage energies [
105]. Kim et al. [
106] explored twenty-two low-indexed BZO (001), (011) as well as (111) surface terminations in order to investigate the Gibbs free energy for their surfaces [
106]. Finally, Eglitis [
107] performed B3LYP computations for BaO
3- and Zr-terminated polar BZO (111) surface relaxations and energetics.
The objective of our review paper was to carry out necessary additional ab initio computations in order to finalize our more-than-20-year-long research work, devoted to ABO3 perovskite surfaces. Namely, we report in this place our B3PW and B3LYP computation results for BTO, CTO, PTO, STO, BZO, CZO, PZO, SZO neutral (001) as well as polar (011) and (111) surfaces. We meticulously analyzed B3PW and B3LYP computation results and detected systematic tendencies, typical for all eight of our ab initio computed ABO3 perovskite surfaces. Finally, we systematized these common systematic trends in a system, effortlessly approachable worldwide for a comprehensive audience of scientists.
2. Computational Details and Surface Models
We performed very comprehensive hybrid density functional theory (DFT) calculations for eight different ABO
3 perovskite (001), (011) and (111) surfaces by means of the CRYSTAL [
108] computer program. The CRYSTAL computer program [
108] utilizes Gaussian-type well-localized basis sets (BSs). The BSs for BTO, PTO and STO perovskites were evolved by Piskunov et al. [
109]. Almost all computations in this review were executed by means of the B3PW [
110,
111] or B3LYP [
112] hybrid exchange–correlation functionals. It is worth noting that the hybrid exchange–correlation functionals, like B3PW or B3LYP, enable us to reach an outstanding agreement with the experiment [
10,
75] for the Γ-Γ band gaps of different ABO
3 perovskites. We executed the reciprocal-space integration for the ABO
3 perovskite bulk and their surfaces by examining the Brillouin zone, utilizing the 8 × 8 × 8 and 8 × 8 × 1 times, respectively, enlarged Pack Monkhorst grid [
113]. The trump card of the CRYSTAL computer program [
108] is its ability to compute isolated, two-dimensional slabs, without any unnatural periodicity in the direction
z, perpendicular to the slab surface. We performed B3PW and B3LYP computations for all eight ABO
3-type perovskites and their surfaces in high symmetry, cubic structure
) [
114,
115,
116].
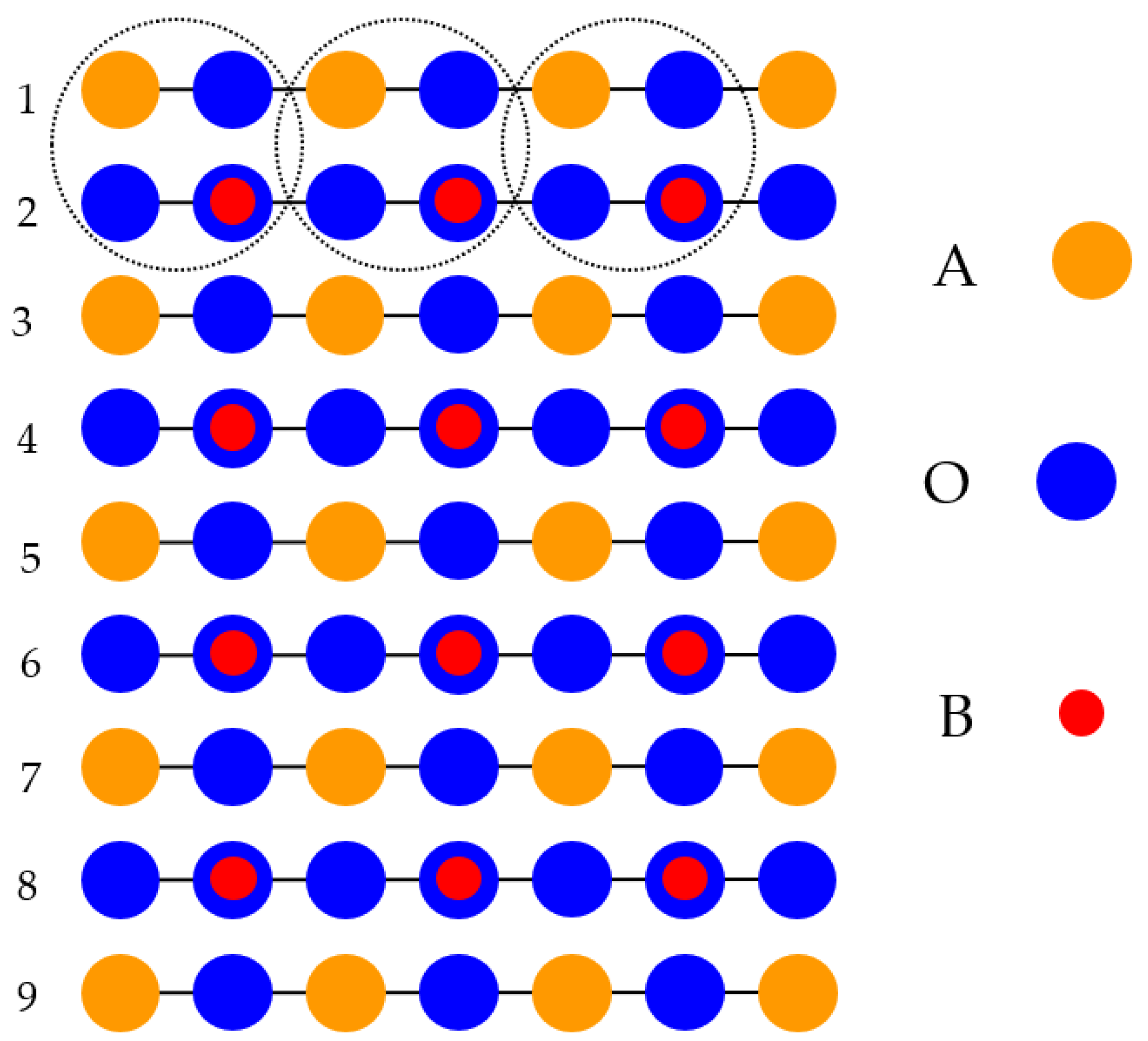
With the goal of simulating the neutral BO
2-terminated (001) surfaces of ABO
3-type perovskites [
114], we selected symmetrical slabs. These slabs, in our computations, consisted of nine neutral and alternating BO
2 as well as AO layers (
Figure 1 and
Figure 2). The first slab was terminated by the BO
2 planes and was composed of a supercell which accommodated 23 atoms (
Figure 1). The second slab was terminated by the AO planes and was composed of a supercell which accommodated 22 atoms (
Figure 2). Both these (001) surface slabs are nonstoichiometric. They have unit cell formulas equal to A
4B
5O
14 and A
5B
4O
13, respectively (
Figure 1 and
Figure 2).
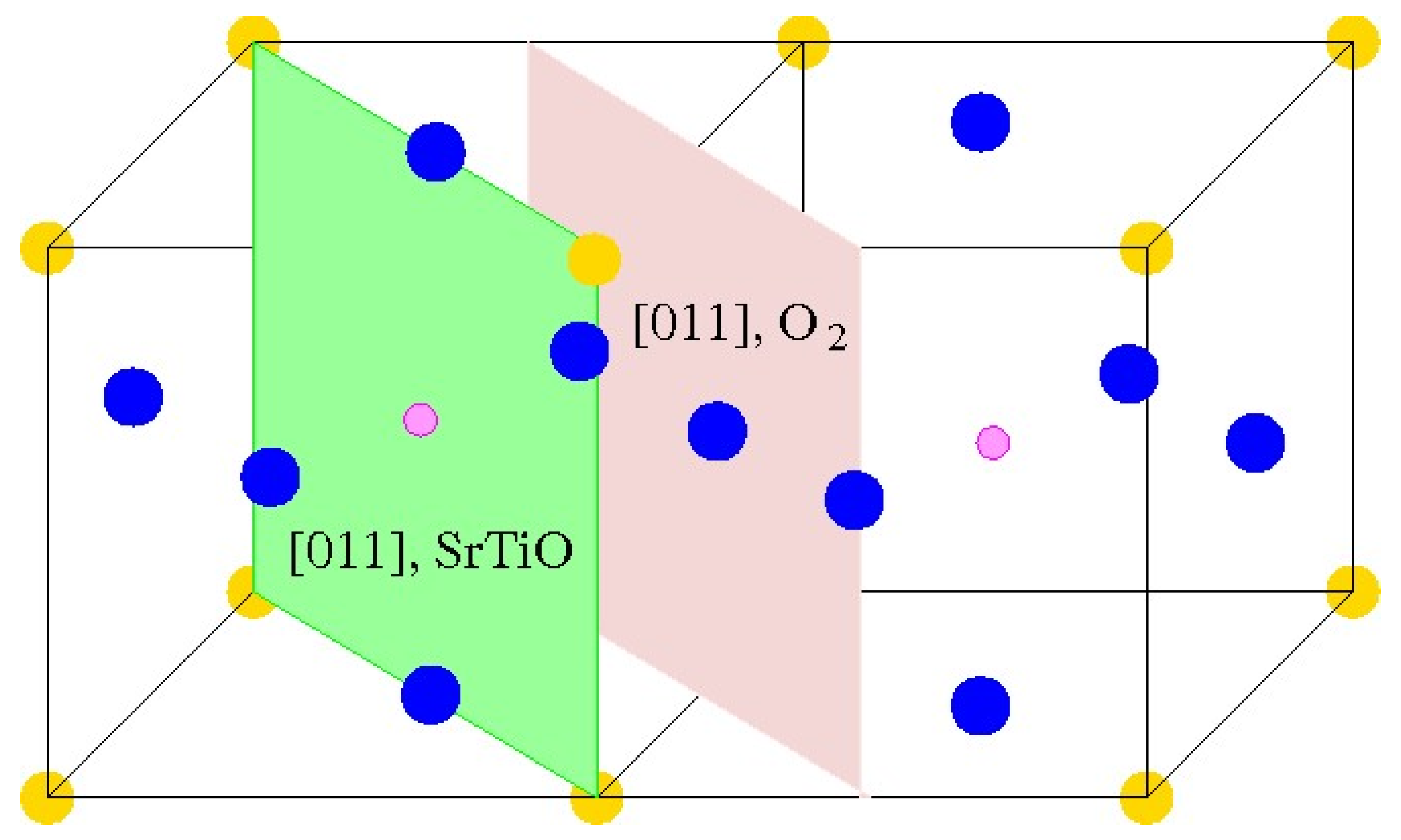
Just opposite to the (001) cleavage (
Figure 1 and
Figure 2) of ABO
3, which produce nonpolar AO and BO
2 terminations, direct cleavage of ABO
3-type perovskites, in order to generate (011) surfaces, leads to the production of polar O
2 as well as ABO surfaces (
Figure 3). The ABO
3 crystal (
Figure 3), alongside the [011] crystalic direction, is composed of cyclic planes of O
2 and ABO units (
Figure 3). These two alternating O
2 and ABO planes (
Figure 3) have ionic charges of −4
e and +4
e, assuming following constituents as O
2−, B
4+ and A
2+. Therefore, modeling of the ABO
3 (011) surfaces (
Figure 3) precisely, as they are obtained from the pristine crystal cleavage, leads to the following two problematic situations: An infinite macroscopic dipole moment, which is perpendicular to the ABO
3 perovskite (011) surface (
Figure 4), when the slab is terminated by different O
2 as well as ABO planes (
Figure 4) (stoichiometric slab). Infinite charge, in case when the slab is terminated by the same planes (O
2-O
2) (
Figure 5) or ABO-ABO (
Figure 6) (nonstoichiometric slab). Such ABO
3 perovskite (011) surface terminations (
Figure 5 and
Figure 6) make the (011) surface unstable [
117,
118].
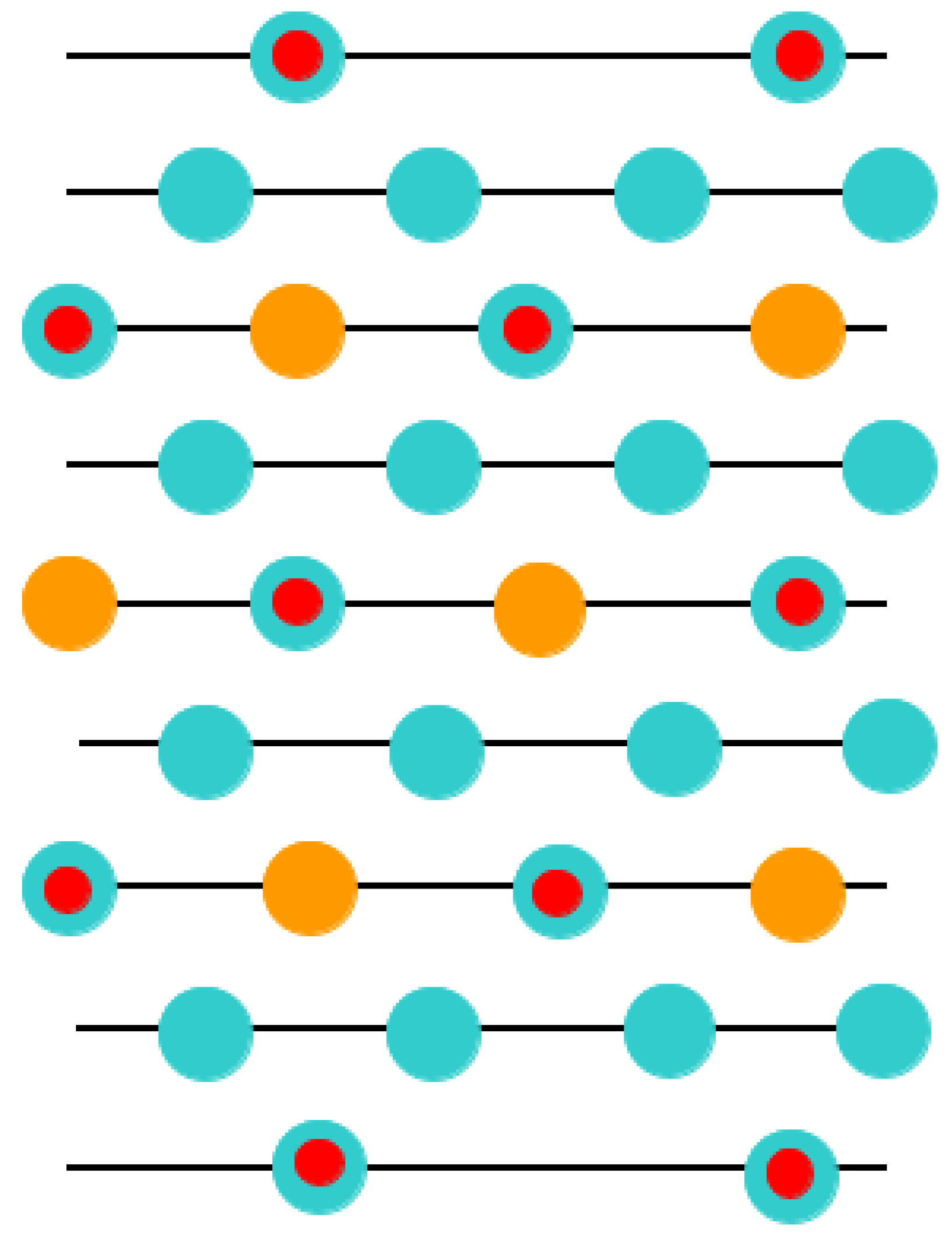
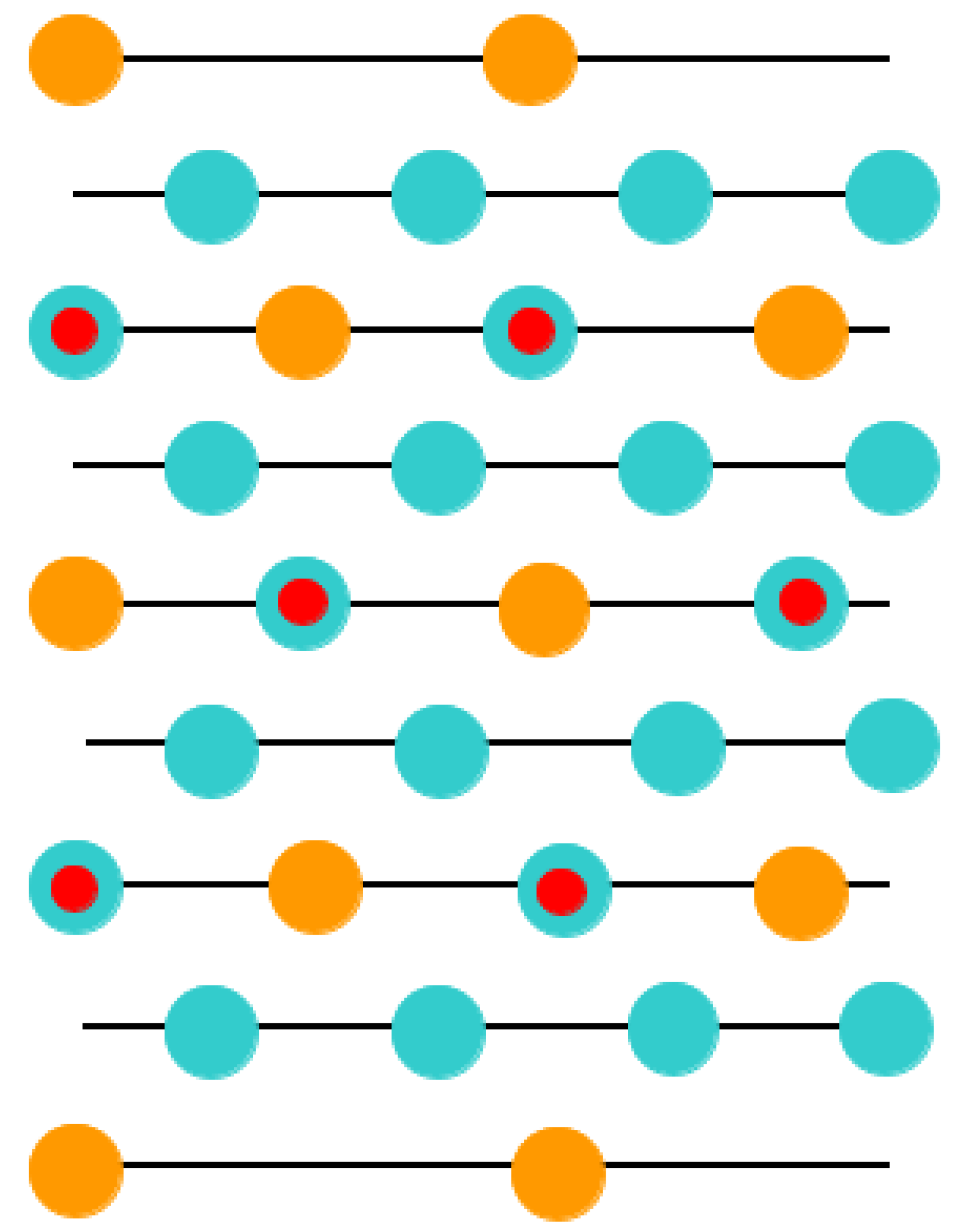
This was the key reason why in our ABO
3-type perovskite (011) surface computations, with the aim of obtaining the neutral (011) slab, we deleted some atoms (
Figure 7,
Figure 8 and
Figure 9). Namely, we deleted the O atom (
Figure 9) from the upper as well lower layers of the nine-layer O-O-terminated symmetric nonstoichiometric (011) slab. Thus, we obtain a neutral O-terminated ABO
3 perovskite (011) slab without any dipole moment perpendicular to the slab surface (
Figure 9). Similarly, we deleted both B and O atoms (
Figure 8) or an A atom (
Figure 7) from the upper and lower layers of the ABO-terminated symmetric nonstoichiometric ABO
3 perovskite (011) slabs. Thus, we obtain neutral A-terminated (
Figure 8) or BO-terminated (
Figure 7) ABO
3 perovskite (011) slabs without any dipole moment perpendicular to their (011) surfaces. Consequently, in our computations, the BO-terminated symmetric, nonstoichiometric (
Figure 7) nine-layer (011) slab consisted of a supercell enclosing 21 atoms. The A- (
Figure 8) and O- (
Figure 9) terminated nonstoichiometric and symmetric ABO
3 perovskite nine-layer (011) slabs consisted of supercells enclosing 19 and 20 atoms, respectively.
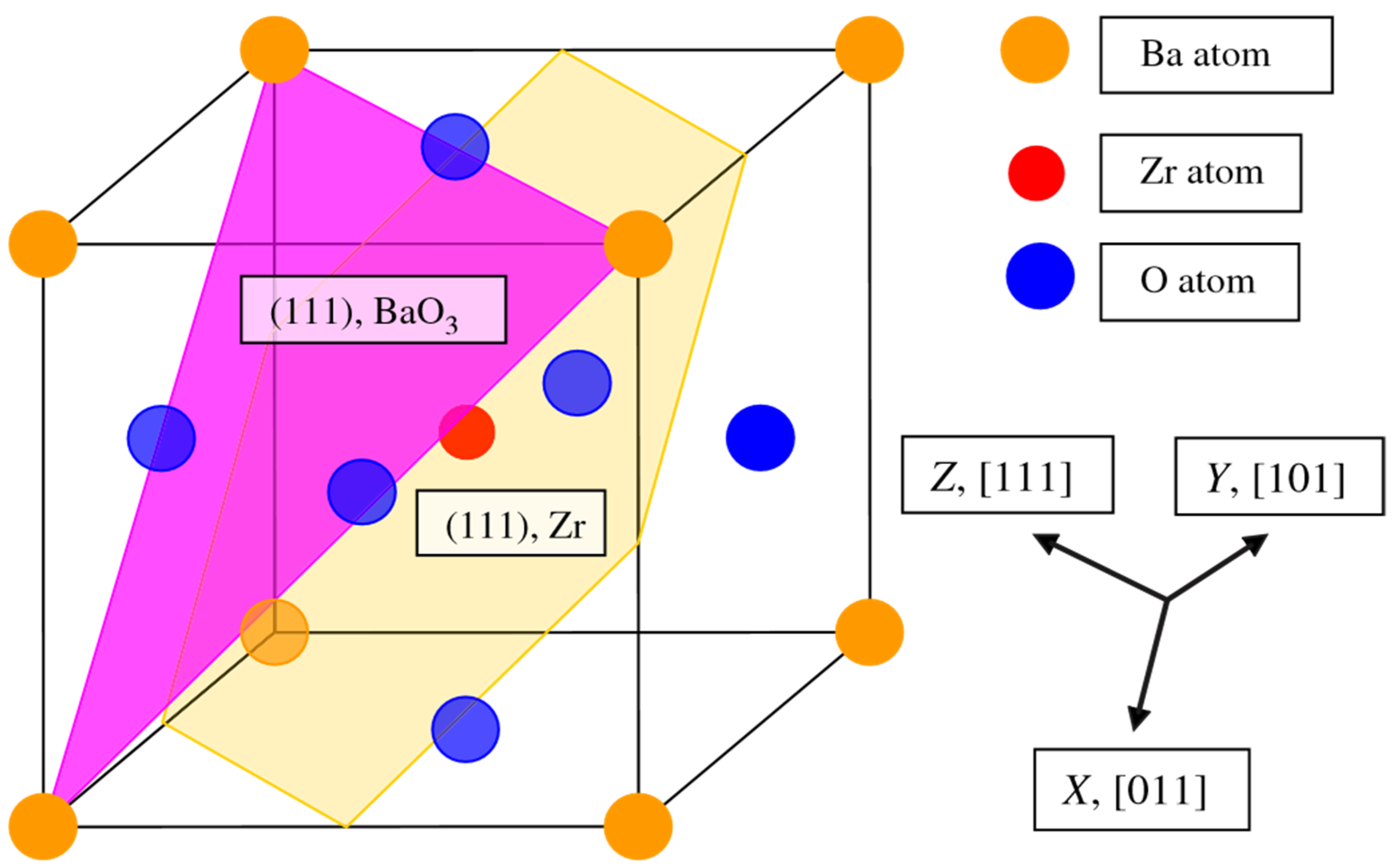
As a further action, the ABO
3 perovskite polar (111) surfaces will be described by us using BZO as an example (
Figure 10 and
Figure 11) [
107]. In order to compute the polar BZO perovskite (111) surfaces, we employed symmetrical, nonstoichiometric (111) slabs containing nine alternating Zr and BaO
3 layers (
Figure 10 and
Figure 11). One of two BZO (111) slabs (
Figure 11a) is terminated by Zr planes from both sides. It consists of a supercell accommodating 21 atoms (
Figure 11a). The second (111) slab (
Figure 11b) is terminated from both sides by BaO
3 planes. It consists of a supercell accommodating 24 atoms (
Figure 11b). Both these Zr- and BaO
3-terminated BZO (111) slabs are symmetrical and nonstoichiometric (
Figure 11). They have the unit-cell formulas Ba
4Zr
5O
12 and Ba
5Zr
4O
15, respectively (
Figure 11). As we know from studies dealing, for example, with polar STO and CTO (111) surfaces [
99,
101,
119], a strong electron redistribution happens for such (111) terminations (
Figure 11) canceling the polarity. Therefore, such calculations are possible for the Zr- or BaO
3-terminated BZO (111) surface [
99,
101,
119]. It is worth noting that we used the basis sets for neutral Ba, Zr and O atoms in all our B3LYP computations dealing with polar BaZrO
3 perovskite (111) surfaces [
5,
99,
107].
With the ultimate goal of computing the ABO
3-type perovskite, for example, the PbZrO
3 (001) surface energy, we started our B3LYP computations with the cleavage energy calculations for unrelaxed PbO- as well as ZrO
2-terminated (001) surfaces [
1,
2,
3,
94]. Surfaces with both PbO and ZrO
2 (001) terminations at the same time emerge under the (001) cleavage of the PZO crystal [
1,
2,
3,
94]. We suppose that the PZO perovskite cleavage energy is uniformly shared between the created (001) surfaces (
Figure 1 and
Figure 2) [
1,
2,
3]. In our B3LYP computations, the nine-layer PbO-terminated PZO (001) slab with 22 atoms as well as the nine-layer ZrO
2-terminated PZO (001) slab, containing 23 atoms, together contain nine bulk unit cells or 45 atoms atoms, thus:
where ϑ means PbO or ZrO
2;
Eslabunr(ϑ) is the SrO- or ZrO
2-terminated PZO (001) slab energies without relaxation;
Ebulk is the PZO bulk unit cell, containing five atoms, total energy; and the factor of ¼ means that we created four surfaces due the PZO crystal (001) cleavage [
1,
2,
3]. After this, we can compute the relaxation energies for both PbO- and ZrO
2-terminated PZO (001) slabs [
1,
2,
3,
75,
82], using the following equation:
where
Eslabrel(ϑ) is the (001) slab total energy after geometry relaxation [
1,
2,
3,
75,
82]. The surface energy is thereby described as a sum of the relevant relaxation as well as cleavage energies:
With goal of computing the PZO (011) surface energies for the ZrO- and Pb-terminated (011) surfaces, we think about the cleavage of eight PZO bulk unit cells, in order to obtain the ZrO- and Pb-terminated (011) slabs, which contain 21 and 19 atoms. Namely, we split the cleavage energy uniformly among these two surfaces and derive:
where ϑ indicates Pb or ZrO;
Eslabunr(ϑ) is our computed total energy for the unrelaxed Pb- or ZrO-terminated PZO (011) slabs; and
Ebulk is our computed PbZrO
3 perovskite total energy per five-atom bulk unit cell.
In the end, when we cut the PZO perovskite crystal in other way, we obtain two equal O-terminated PZO (011) surface slabs. Each of them contains 20 atoms [
1,
2,
3,
82]. This permits us to make our computations less complex, taking into account that the unit cell of the nine-plane O-terminated PZO (011) slab includes four PZO bulk unit cells [
1,
2,
3,
82]. Thereby, the O-terminated PZO perovskite (011) surface energy is described as follows:
where
Esurf(O) is the O-terminated PZO (011) surface energy, and
Eslabrel(O) is the relaxed O-terminated PZO (011) slab total energy. In the end, the ABO
3 perovskite polar (111) surface energy computation details are described by us in Refs. [
5,
99,
107].
4. Conclusions
We performed B3PW and B3LYP computations for BTO, CTO, PTO, STO, BZO, CZO, PZO and SZO perovskite neutral (001) along with polar (011) and (111) surfaces. For the neutral AO- as well as BO
2-terminated (001) surfaces, in most cases, all upper-layer atoms relax inwards, although the second-layer atoms shift outwards. There are only three exceptions to this systematic trend (
Table 4 and
Table 5). Namely, upward relaxation of TiO
2-terminated PTO (001) surface upper-layer O atom by (+0.31% of
a0) (
Table 5). On the (001) BO
2-terminated surface, the second-layer metal atoms, as a rule, exhibit larger atomic relaxations than the second-layer O atoms. For most ABO
3 perovskites, the (001) surface rumpling
s is bigger for the AO- than BO
2-terminated surfaces. In contrast, the surface energies, for both (001) terminations, are practically identical. Nevertheless, for the BTO, PTO and PZO perovskites, the BO
2-terminated (001) surface has a slightly smaller surface energy, and therefore, it is more stable. In contrast, for CTO, STO, CZO and SZO perovskites, the AO-terminated (001) surface has a slightly smaller surface energy, and therefore, it is slightly more stable. Conversely, different (011) surface terminations exhibit quite different surface energies for the O-terminated, A-terminated, and BO-terminated surfaces. Our computed ABO
3 perovskite (111) surface energies are always significantly larger than the neutral (001) and polar (011) surface energies. Our computed ABO
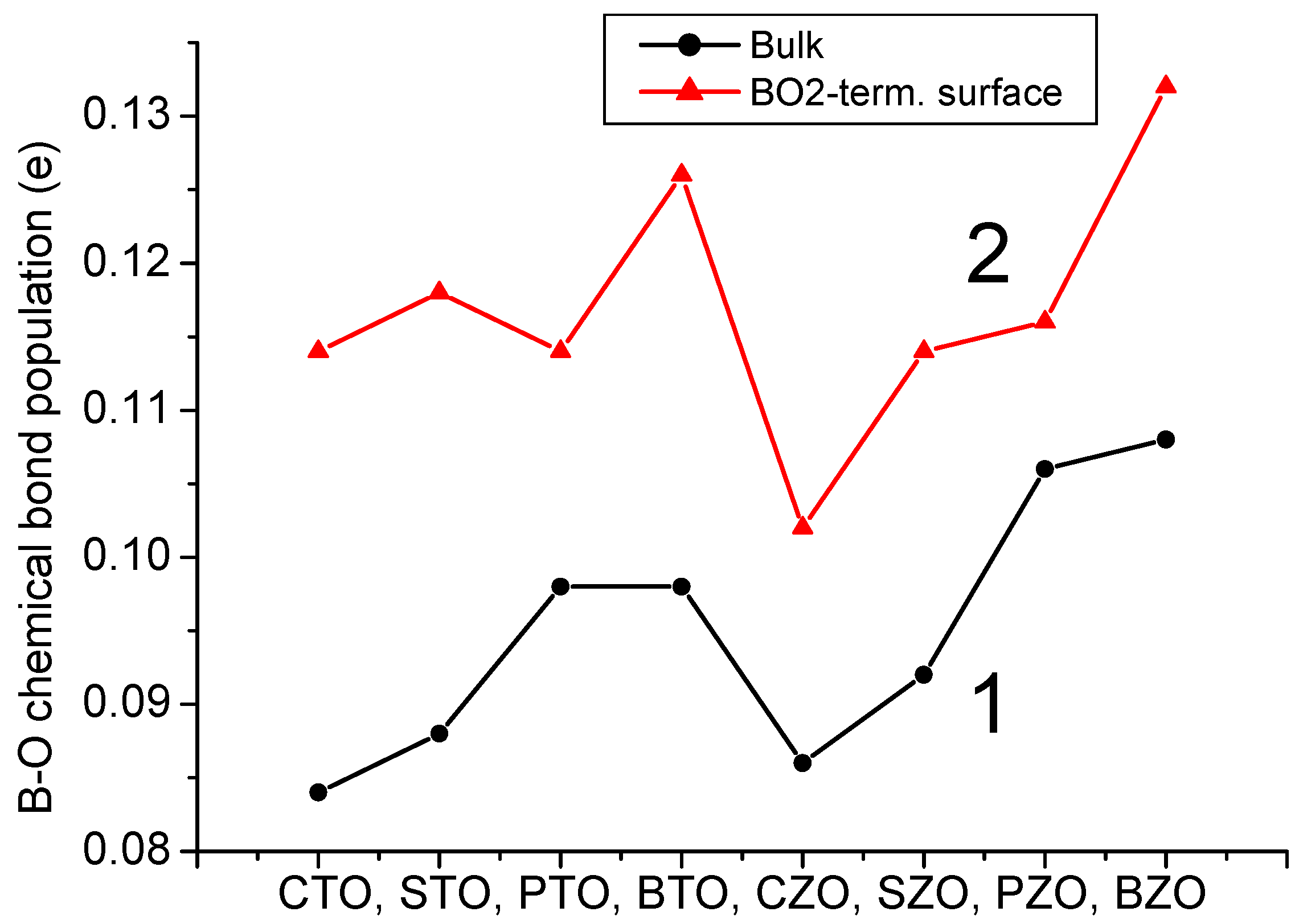
3 perovskite bulk B-O chemical bond covalency increase near their neutral (001) and especially polar (011) surfaces. It is worth noting that for the (011) surfaces, in the plane B(I)-O(I) chemical bond population for all eight of our computed ABO
3 perovskites is in the range of 0.128
e for the CTO perovskite to 0.152
e for the BZO perovskite (
Table 13 and
Figure 24). Finally, as we can see from
Table 13 and
Figure 24, the ultimately largest B-O chemical bond population, according to our B3LYP or B3PW computations for eight ABO
3 perovskites, is for the BO-terminated (011) surface B(I)-O(II) chemical bond population, in the direction perpendicular to the BO-terminated (011) surface. It is in the range of 0.186
e for the CTO perovskite to 0.252
e for the PZO and BZO perovskites (
Figure 24 and
Table 13).






























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}