



Modeling of Charge Injection, Recombination, and Diffusion in Complete Perovskite Solar Cells on Short Time Scales



Abstract
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Sample Preparation
3.2. Transient Absorption
3.3. Numerical Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- National Renewable Energy Laboratory Best Research Cell Efficiencies Chart. Available online: https://www.nrel.gov/pv/cell-efficiency.html (accessed on 6 June 2023).
- Nishida, J.; Chang, P.T.S.; Ye, J.Y.; Sharma, P.; Wharton, D.M.; Johnson, S.C.; Shaheen, S.E.; Raschke, M.B. Nanoscale Heterogeneity of Ultrafast Many-Body Carrier Dynamics in Triple Cation Perovskites. Nat. Commun. 2022, 13, 6582. [Google Scholar] [CrossRef] [PubMed]
- Marchioro, A.; Teuscher, J.; Friedrich, D.; Kunst, M.; Van De Krol, R.; Moehl, T.; Grätzel, M.; Moser, J.E. Unravelling the Mechanism of Photoinduced Charge Transfer Processes in Lead Iodide Perovskite Solar Cells. Nat. Photonics 2014, 8, 250–255. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, M.; Moore, D.T.; Yan, Y.; Miller, E.M.; Zhu, K.; Beard, M.C. Top and Bottom Surfaces Limit Carrier Lifetime in Lead Iodide Perovskite Films. Nat. Energy 2017, 2, 16207. [Google Scholar] [CrossRef]
- Brauer, J.C.; Tsokkou, D.; Sanchez, S.; Droseros, N.; Hua, X.; Stolterfoht, M.; Neher, D.; Roose, B.; Steiner, U.; De Angelis, F. Comparing the Excited-State Properties of a Mixed-Cation—Mixed-Halide Perovskite to Methylammonium Lead Iodide. J. Chem. Phys. 2020, 152, 104703. [Google Scholar] [CrossRef]
- Shen, Q.; Ogomi, Y.; Chang, J.; Tsukamoto, S.; Kukihara, K.; Oshima, T.; Osada, N.; Yoshino, K.; Katayama, K.; Toyoda, T.; et al. Charge Transfer and Recombination at the Metal Oxide/CH3NH3PbClI2/Spiro-OMeTAD Interfaces: Uncovering the Detailed Mechanism behind High Efficiency Solar Cells. Phys. Chem. Chem. Phys. 2014, 16, 19984–19992. [Google Scholar] [CrossRef]
- Ponseca, C.S.; Hutter, E.M.; Piatkowski, P.; Cohen, B.; Pascher, T.; Douhal, A.; Yartsev, A.; Sundstrom, V.; Savenije, T.J. Mechanism of Charge Transfer and Recombination Dynamics in Organo Metal Halide Perovskites and Organic Electrodes, PCBM, and Spiro-OMeTAD: Role of Dark Carriers. J. Am. Chem. Soc. 2015, 137, 16043–16048. [Google Scholar] [CrossRef]
- Navedu, M.; Apurba, D.; Sumanta, P.; Anunay, S. Ultrafast Carrier Dynamics of Metal Halide Perovskite Nanocrystals and Perovskite-Composites. Nanoscale 2019, 11, 9796–9818. [Google Scholar] [CrossRef]
- Jiang, Q.; Tong, J.; Xian, Y.; Kerner, R.A.; Dunfield, S.P.; Xiao, C.; Scheidt, R.A.; Kuciauskas, D.; Wang, X.; Hautzinger, M.P.; et al. Surface Reaction for Efficient and Stable Inverted Perovskite Solar Cells. Nature 2022, 611, 278–283. [Google Scholar] [CrossRef]
- Giordano, F.; Abate, A.; Pablo, J.; Baena, C.; Saliba, M.; Matsui, T.; Im, S.H.; Zakeeruddin, S.M.; Nazeeruddin, M.K.; Hagfeldt, A.; et al. Enhanced Electronic Properties in Mesoporous TiO2 via Lithium Doping for High-Efficiency Perovskite Solar Cells. Nat. Commun. 2016, 7, 10379. [Google Scholar] [CrossRef]
- Kirchartz, T. Picturing Charge Carrier Diffusion. Nat. Mater. 2022, 21, 1344–1345. [Google Scholar] [CrossRef]
- Stranks, S.D.; Eperon, G.E.; Grancini, G.; Menelaou, C.; Alcocer, M.J.P.; Leijtens, T.; Herz, L.M.; Petrozza, A.; Snaith, H.J. Electron-Hole Diffusion Lengths Exceeding 1 Micrometer in an Organometal Trihalide Perovskite Absorber. Science 2013, 342, 341–344. [Google Scholar] [CrossRef]
- Xing, G.; Mathews, N.; Sun, S.; Lim, S.S.; Lam, Y.M.; Graẗzel, M.; Mhaisalkar, S.; Sum, T.C. Long-Range Balanced Electron-and Hole-Transport Lengths in Organic-Inorganic CH3NH3PbI3. Science 2013, 342, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Herz, L.M. Charge-Carrier Dynamics in Organic-Inorganic Metal Halide Perovskites. Annu. Rev. Phys. Chem. 2016, 67, 65–89. [Google Scholar] [CrossRef] [PubMed]
- Crothers, T.W.; Milot, R.L.; Patel, J.B.; Parrott, E.S.; Schlipf, J.; Müller-Buschbaum, P.; Johnston, M.B.; Herz, L.M. Photon Reabsorption Masks Intrinsic Bimolecular Charge-Carrier Recombination in CH3NH3PbI3 Perovskite. Nano Lett. 2017, 17, 5782–5789. [Google Scholar] [CrossRef] [PubMed]
- Leng, J.; Liu, J.; Zhang, J.; Jin, S. Decoupling Interfacial Charge Transfer from Bulk Diffusion Unravels Its Intrinsic Role for Efficient Charge Extraction in Perovskite Solar Cells. J. Phys. Chem. Lett. 2016, 7, 5056–5061. [Google Scholar] [CrossRef] [PubMed]
- Serpetzoglou, E.; Konidakis, I.; Kakavelakis, G.; Maksudov, T.; Kymakis, E.; Stratakis, E. Improved Carrier Transport in Perovskite Solar Cells Probed by Femtosecond Transient Absorption Spectroscopy. ACS Appl. Mater. Interfaces 2017, 9, 43910–43919. [Google Scholar] [CrossRef] [PubMed]
- Pydzińska, K.; Karolczak, J.; Kosta, I.; Tena-Zaera, R.; Todinova, A.; Idígoras, J.; Anta, J.A.; Ziółek, M. Determination of Interfacial Charge-Transfer Rate Constants in Perovskite Solar Cells. ChemSusChem 2016, 9, 1647–1659. [Google Scholar] [CrossRef]
- Scholz, M.; Flender, O.; Oum, K.; Lenzer, T. Pronounced Exciton Dynamics in the Vacancy-Ordered Bismuth Halide Perovskite (CH3NH3)3Bi2I9 Observed by Ultrafast UV-Vis-NIR Transient Absorption Spectroscopy. J. Phys. Chem. C 2017, 121, 12110–12116. [Google Scholar] [CrossRef]
- Wang, L.; McCleese, C.; Kovalsky, A.; Zhao, Y.; Burda, C. Femtosecond Time-Resolved Transient Absorption Spectroscopy of CH3NH3PbI3 Perovskite Films: Evidence for Passivation Effect of PbI2. J. Am. Chem. Soc. 2014, 110, 12205–12208. [Google Scholar] [CrossRef]
- Pydzińska-Białek, K.; Szeremeta, J.; Wojciechowski, K.; Ziółek, M. Insights into the Femtosecond to Nanosecond Charge Carrier Kinetics in Perovskite Materials for Solar Cells. J. Phys. Chem. C 2019, 123, 110–119. [Google Scholar] [CrossRef]
- Pydzińska, K.; Drushliak, V.; Coy, E.; Załęski, K.; Flach, J.; Idígoras, J.; Contreras-Bernal, L.; Hagfeldt, A.; Anta, J.A.; Ziółek, M. Understanding the Interfaces between Triple Cation Perovskite and Electron or Hole Transporting Material. ACS Appl. Mater. Interfaces 2020, 12, 30399–30410. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Sarmiento, J.S.; Fei, C.; Wang, H. Charge Transfer and Diffusion at the Perovskite/PCBM Interface Probed by Transient Absorption and Reflection. J. Phys. Chem. C 2019, 123, 22095–22103. [Google Scholar] [CrossRef]
- Droseros, N.; Dänekamp, B.; Tsokkou, D.; Boix, P.P.; Banerji, N. Charge Injection and Trapping at Perovskite Interfaces with Organic Hole Transporting Materials of Different Ionization Energies. APL Mater. 2019, 7, 041115. [Google Scholar] [CrossRef]
- Shen, W.; Wu, Z.; Yang, G.; Kong, Y.; Li, W.; Liang, G.; Huang, F.; Cheng, Y.; Zhong, J. Differentiated Functions of Potassium Interface Passivation and Doping on Charge-Carrier Dynamics in Perovskite Solar Cells. J. Phys. Chem. Lett. 2022, 13, 3188–3193. [Google Scholar] [CrossRef]
- Li, C.; Wang, A.; Deng, X.; Wang, S.; Yuan, Y.; Ding, L.; Hao, F. Insights into Ultrafast Carrier Dynamics in Perovskite Thin Films and Solar Cells. ACS Photonics 2020, 7, 1893–1907. [Google Scholar] [CrossRef]
- Yan, L. Ultrafast Photoinduced Carrier Dynamics in Single Crystalline Perovskite Films. J. Mater. Chem. C 2023, 11, 3736–3742. [Google Scholar] [CrossRef]
- Pydzińska-Białek, K.; Glinka, A.; Drushliak, V.; Nowaczyk, G.; Florczak, P.; Ziółek, M. Impact of Improvements in Mesoporous Titania Layers on Ultrafast Electron Transfer Dynamics in Perovskite and Dye-Sensitized Solar Cells. Phys. Chem. Chem. Phys. 2020, 22, 21947–21960. [Google Scholar] [CrossRef]
- Handa, T.; Tex, D.M.; Shimazaki, A.; Wakamiya, A.; Kanemitsu, Y. Charge Injection Mechanism at Heterointerfaces in CH3NH3PbI3 Perovskite Solar Cells Revealed by Simultaneous Time-Resolved Photoluminescence and Photocurrent Measurements. J. Phys. Chem. Lett. 2017, 8, 954–960. [Google Scholar] [CrossRef]
- Li, X.; Bi, D.; Yi, C.; Décoppet, J.-D.; Luo, J.; Zakeeruddin, S.M.; Hagfeldt, A.; Grätzel, M. A Vacuum Flash-Assisted Solution Process for High-Efficiency Large-Area Perovskite Solar Cells. Science 2016, 353, 58–62. [Google Scholar] [CrossRef]
- Quere, B.; Pydzińska-Białek, K.; Karolczak, J.; Nowaczyk, G.; Coy, E.; Ziółek, M. Understanding the Effect of Different Synthesis Conditions on the Physicochemical Properties of Mixed-Ion Perovskite Solar Cells. Chem. A Eur. J. 2019, 25, 5978–5986. [Google Scholar] [CrossRef]
- Pydzińska-Białek, K.; Nowaczyk, G.; Ziółek, M. Complete Perovskite Solar Cells with Gold Electrodes Studied in the Visible and Near-Infrared Ranges. Chem. Mater. 2022, 34, 6355–6366. [Google Scholar] [CrossRef]
- Brauer, J.C.; Lee, Y.H.; Nazeeruddin, M.K.; Banerji, N. Charge Transfer Dynamics from Organometal Halide Perovskite to Polymeric Hole Transport Materials in Hybrid Solar Cells. J. Phys. Chem. Lett. 2015, 6, 3675–3681. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liu, J.; Isikgor, F.H.; Wang, M.; Khan, J.I.; Wolf, S. De Probing Ultrafast Interfacial Carrier Dynamics in Metal Halide Perovskite Films and Devices by Transient Reflection Spectroscopy. ACS Appl. Mater. Interfaces 2022, 14, 34281–34290. [Google Scholar] [CrossRef] [PubMed]
- Staub, F.; Anusca, I.; Lupascu, D.C.; Rau, U.; Kirchartz, T. Effect of Reabsorption and Photon Recycling on Photoluminescence Spectra and Transients in Lead-Halide Perovskite Crystals. J. Phys. Mater. 2020, 3, 025003. [Google Scholar] [CrossRef]
- Kovalenko, S.A.; Schanz, R.; Farztdinov, V.M.; Hennig, H.; Ernsting, N.P. Femtosecond Relaxation of Photoexcited Para-Nitroaniline: Solvation, Charge Transfer, Internal Conversion and Cooling. Chem. Phys. Lett. 2000, 323, 312–322. [Google Scholar] [CrossRef]
- Dong, C.; Wang, Z.K.; Liao, L.S. Progress of Triple Cation Organometal Halide Perovskite Solar Cells. Energy Technol. 2020, 8, 1–14. [Google Scholar] [CrossRef]
- Yaiche, A.; Sha, H.; Cacovich, S.; Messou, D.; Bercegol, A.; Rousset, J.; Schulz, P.; Bouttemy, M.; Lombez, L. Light-Induced Passivation in Triple Cation Mixed Halide Perovskites: Interplay between Transport Properties and Surface Chemistry. ACS Appl. Mater. Interfaces 2020, 12, 34784–34794. [Google Scholar] [CrossRef]
- Krückemeier, L.; Krogmeier, B.; Liu, Z.; Rau, U.; Kirchartz, T. Understanding Transient Photoluminescence in Halide Perovskite Layer Stacks and Solar Cells. Adv. Energy Mater. 2021, 11, 2003489. [Google Scholar] [CrossRef]
- Krogmeier, B.; Staub, F.; Grabowski, D.; Rau, U.; Kirchartz, T. Quantitative Analysis of the Transient Photoluminescence of CH3NH3PbI3/PC61BM Heterojunctions by Numerical Simulations. Sustain. Energy Fuels 2018, 2, 1027–1034. [Google Scholar] [CrossRef]
- Burkhard, B.G.F.; Hoke, E.T.; Mcgehee, M.D. Accounting for Interference, Scattering, and Electrode Absorption to Make Accurate Internal Quantum Efficiency Measurements in Organic and Other Thin Solar Cells. Adv. Mater. 2010, 94305, 3293–3297. [Google Scholar] [CrossRef]
- Kar, S.; Kaushal, K.; Yantara, N.; Mhaisalkar, S.G. Optical Simulations in Perovskite Devices: A Critical Analysis. ACS Photonics 2022, 9, 3196–3214. [Google Scholar] [CrossRef]
- Richardson, G.; Foster, J.M. How Transport Layer Properties Affect Perovskite Solar Cell Performance: Insights from a Coupled Charge Transport / Ion Migration Model. Energy Environ. Sci. 2019, 12, 396–409. [Google Scholar] [CrossRef]
- Singh, A.; Kaiser, W.; Gagliardi, A. Role of Cation-Mediated Recombination in Perovskite Solar Cells. Sol. Energy Mater. Sol. Cells 2021, 221, 110912. [Google Scholar] [CrossRef]
- Saliba, M.; Wol, C.M.; Stolterfoht, M.; Phung, N.; Albrecht, S.; Neher, D.; Abate, A. How to Make over 20% Efficient Perovskite Solar Cells in Regular (n–i–p) and Inverted (p–i–n) Architectures. Chem. Mater. 2018, 30, 4193–4201. [Google Scholar] [CrossRef]
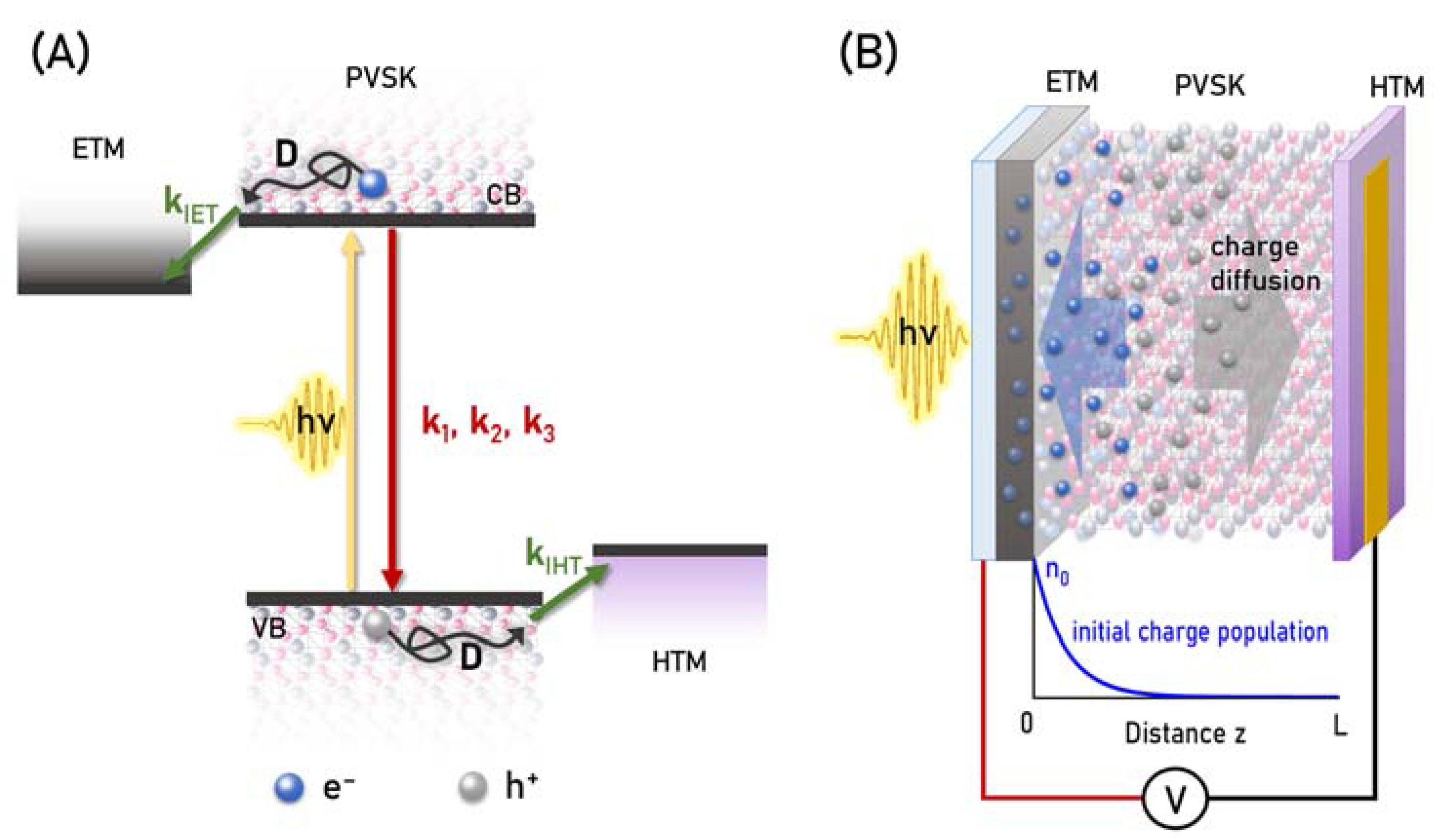
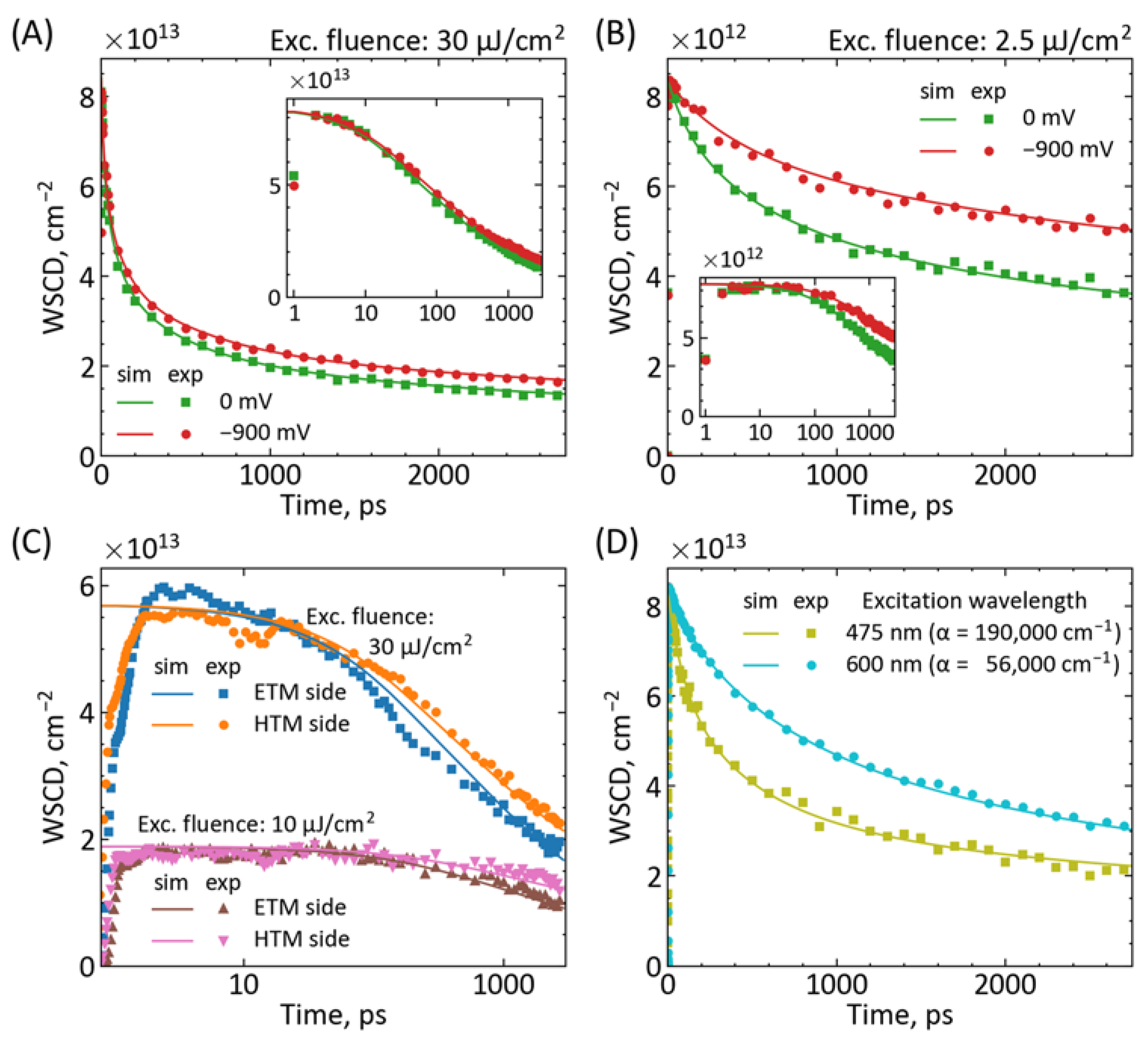
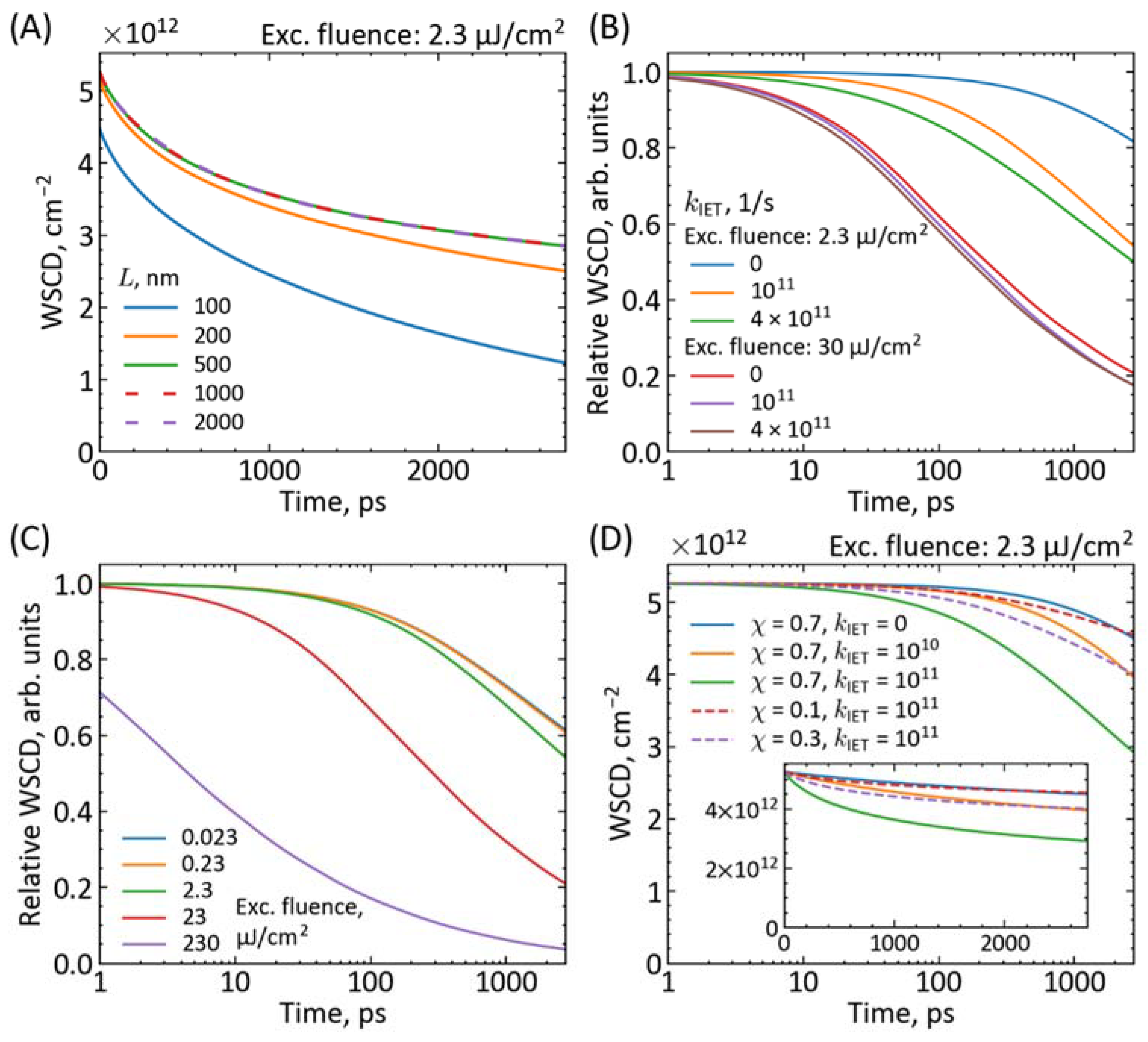

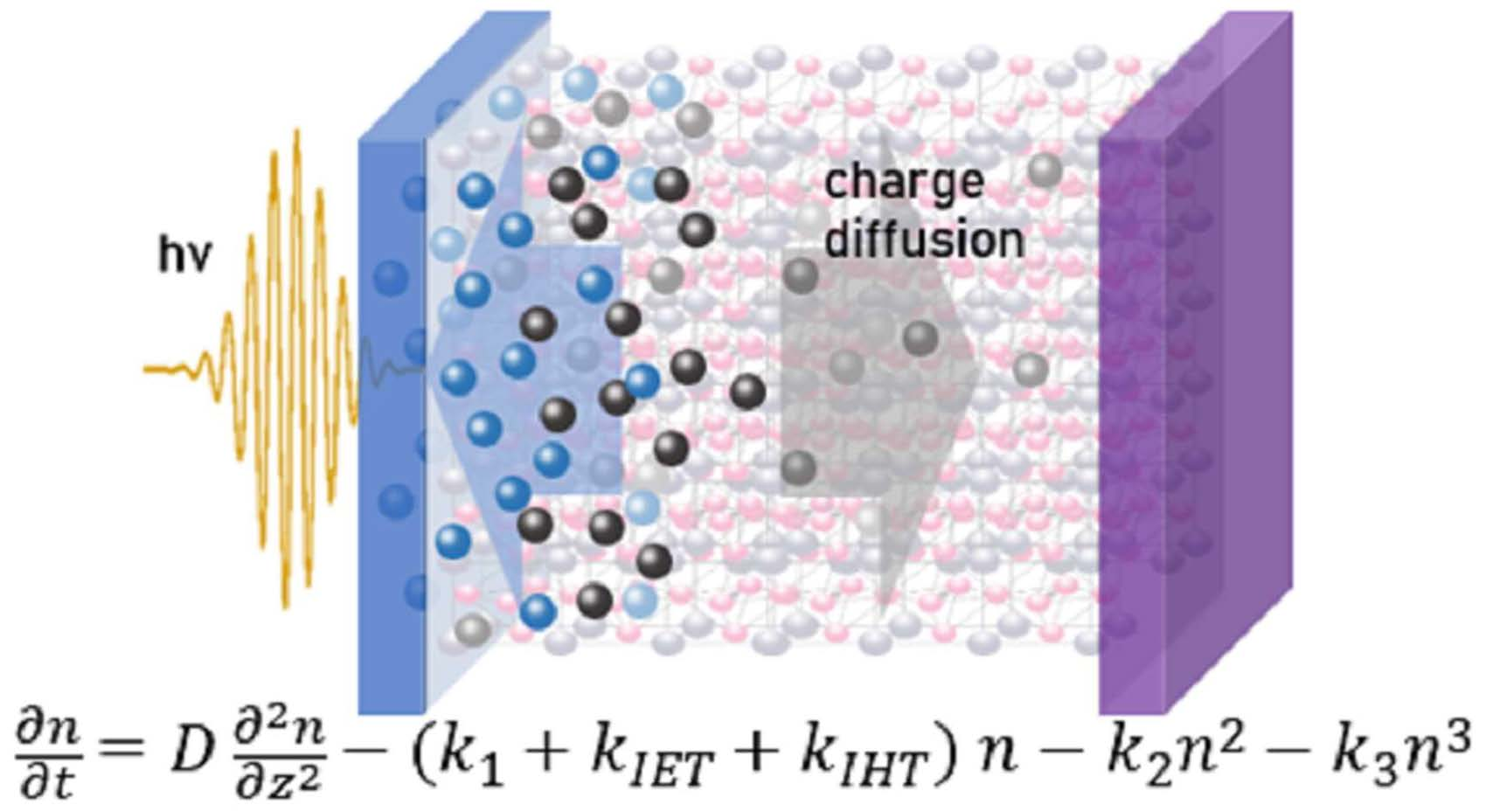{kind=link}
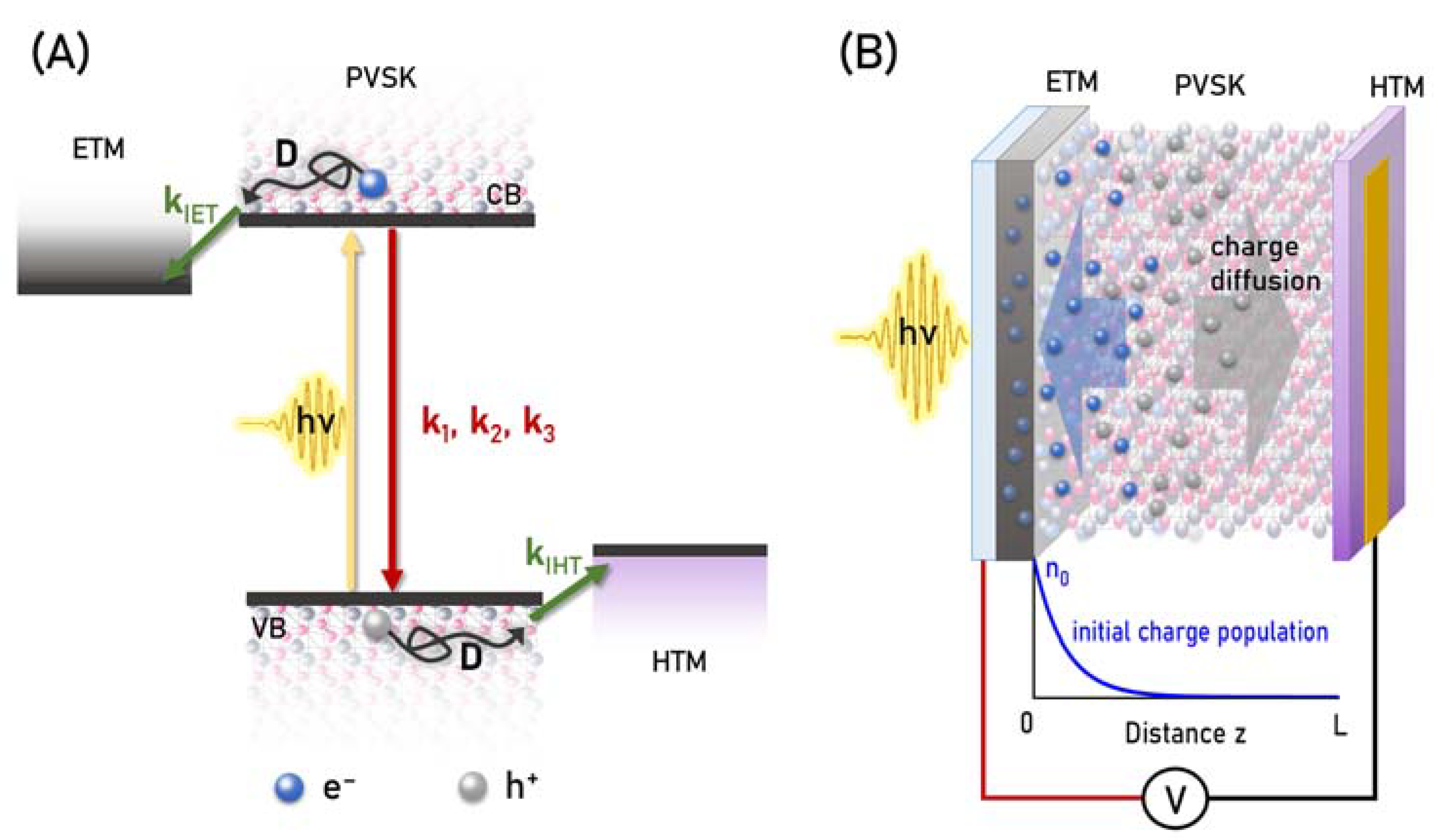{kind=link}
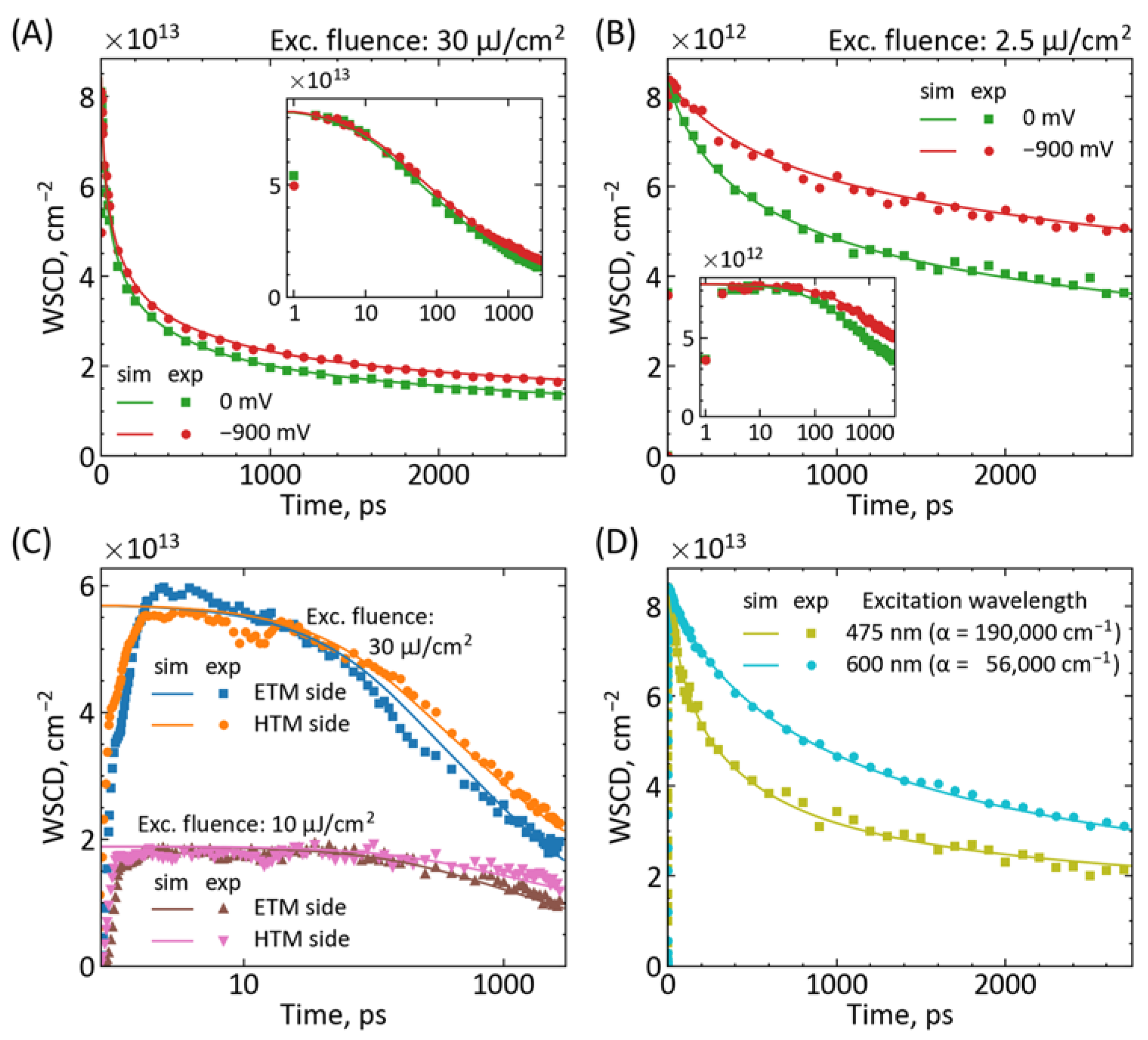{kind=link}
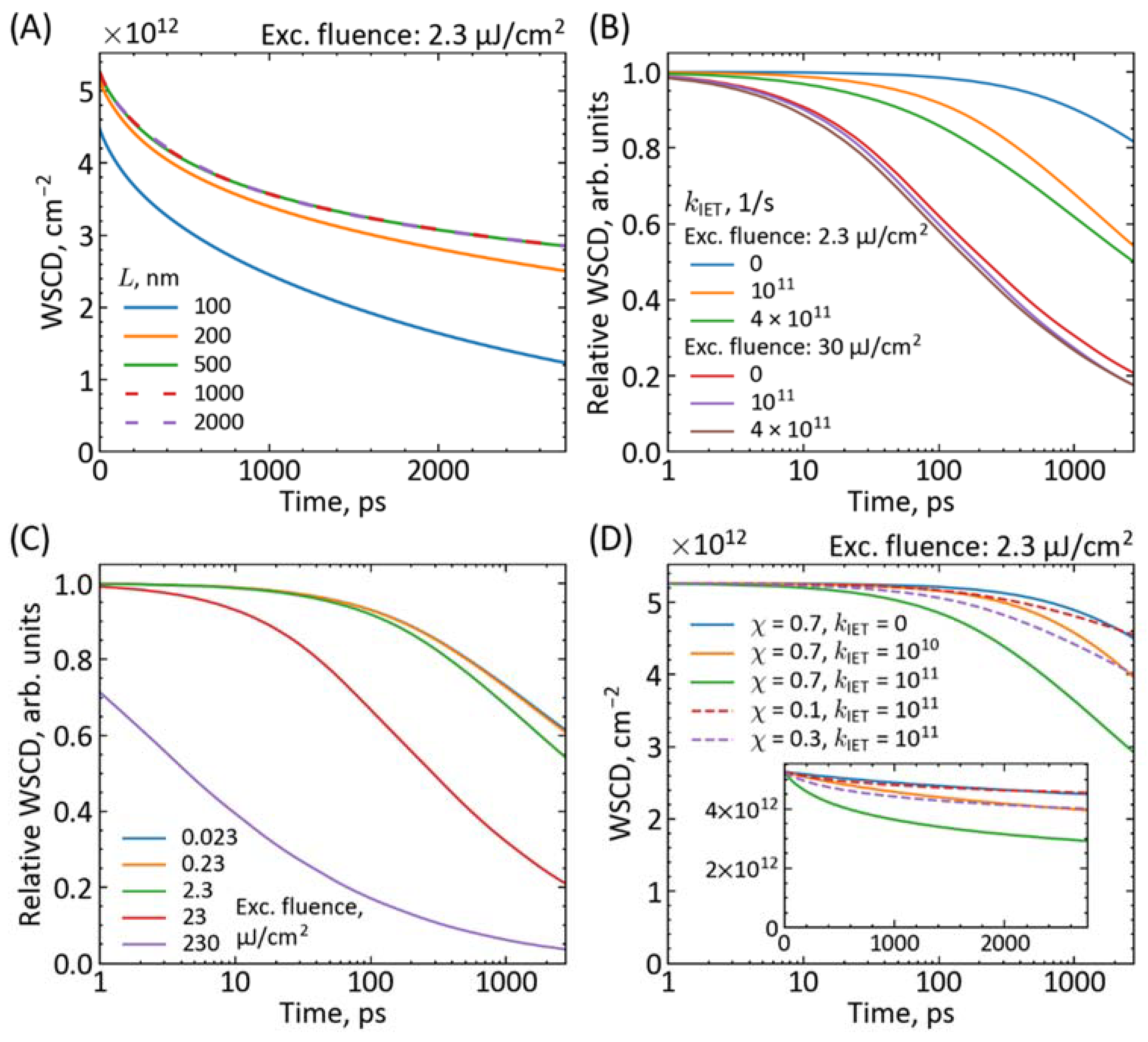{kind=link}
| Parameter | Figure 2A | Figure 2B | Figure 2C | Figure 2D |
|---|---|---|---|---|
| Pump (side, fluence) | ETM side, 30 μJ/cm2 | ETM side, 2.5 μJ/cm2 | Both sides, 10 and 30 μJ/cm2 | HTM side, 30 μJ/cm2 |
| L, nm | 500 | 500 | 600 | 500 |
| D, cm2 s−1 | 3 × 10−2 | 3 × 10−2 | 1 × 10−2 | 2 × 10−2 |
| k1, s−1 | 3 × 105 | 3 × 105 | 3 × 105 | 3 × 105 |
| k2, cm3 s−1 | 0.9 × 10−10 | 3 × 10−10 | 0.8 × 10−10 | 3 × 10−10 |
| k3, cm6 s−1 | 2.5 × 10−28 | 7 × 10−28 | 0.4 × 10−28 | 0.2 × 10−28 |
| kIET, s−1 | 3 × 1011 or 0 | 1 × 1011 or 0 | 1 × 1011 | 1 × 1011 |
| kIHT, s−1 | - | - | 1 × 1011 | - |
| Jsc, mA cm−2 | 20 | 15 | 24 | 20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szulc, K.; Pydzińska-Białek, K.; Ziółek, M. Modeling of Charge Injection, Recombination, and Diffusion in Complete Perovskite Solar Cells on Short Time Scales. Materials 2023, 16, 7110. https://doi.org/10.3390/ma16227110
Szulc K, Pydzińska-Białek K, Ziółek M. Modeling of Charge Injection, Recombination, and Diffusion in Complete Perovskite Solar Cells on Short Time Scales. Materials. 2023; 16(22):7110. https://doi.org/10.3390/ma16227110
Chicago/Turabian StyleSzulc, Krzysztof, Katarzyna Pydzińska-Białek, and Marcin Ziółek. 2023. "Modeling of Charge Injection, Recombination, and Diffusion in Complete Perovskite Solar Cells on Short Time Scales" Materials 16, no. 22: 7110. https://doi.org/10.3390/ma16227110
APA StyleSzulc, K., Pydzińska-Białek, K., & Ziółek, M. (2023). Modeling of Charge Injection, Recombination, and Diffusion in Complete Perovskite Solar Cells on Short Time Scales. Materials, 16(22), 7110. https://doi.org/10.3390/ma16227110