
Metal Complexes in the Synthesis of Biodegradable Polymers: Achievements and Prospects

Abstract
: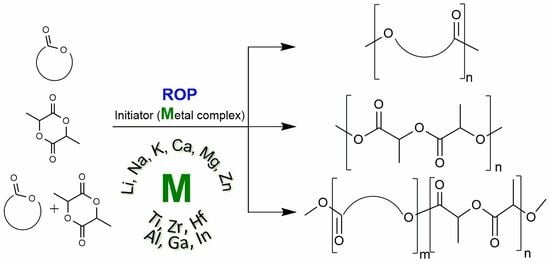
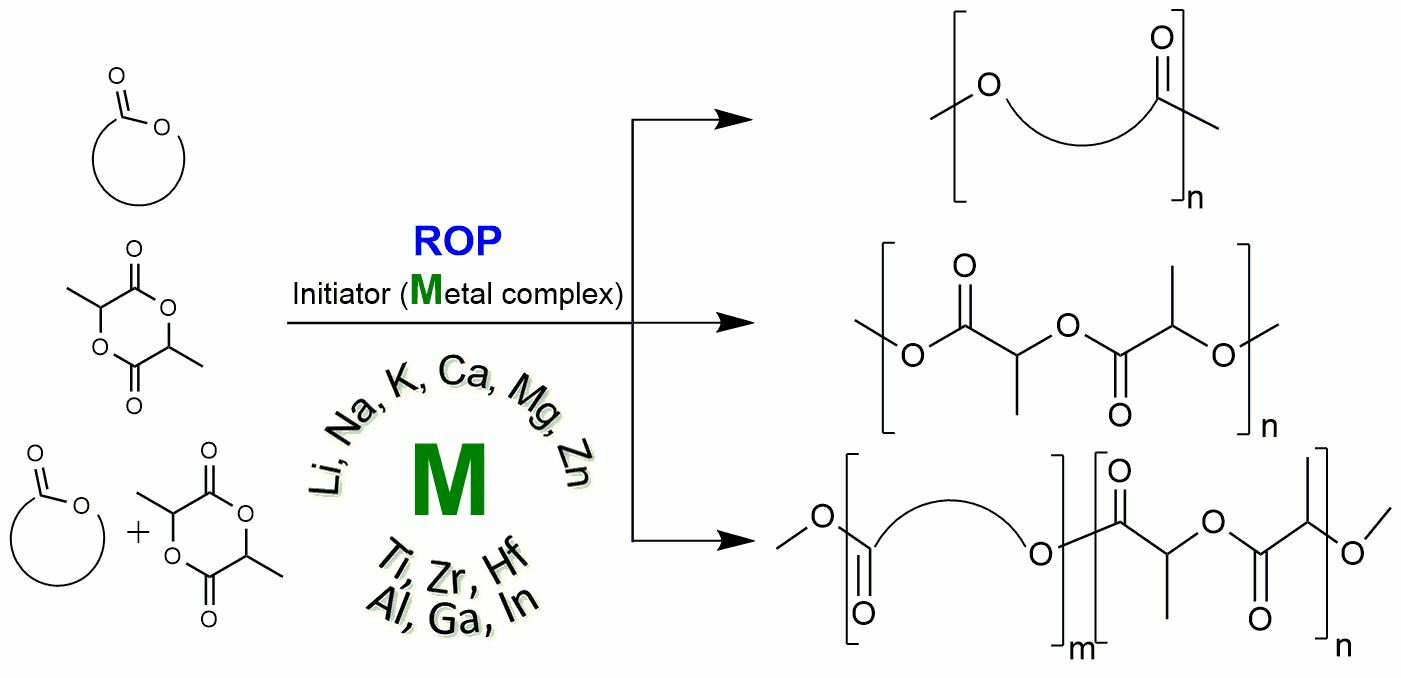
1. Introduction
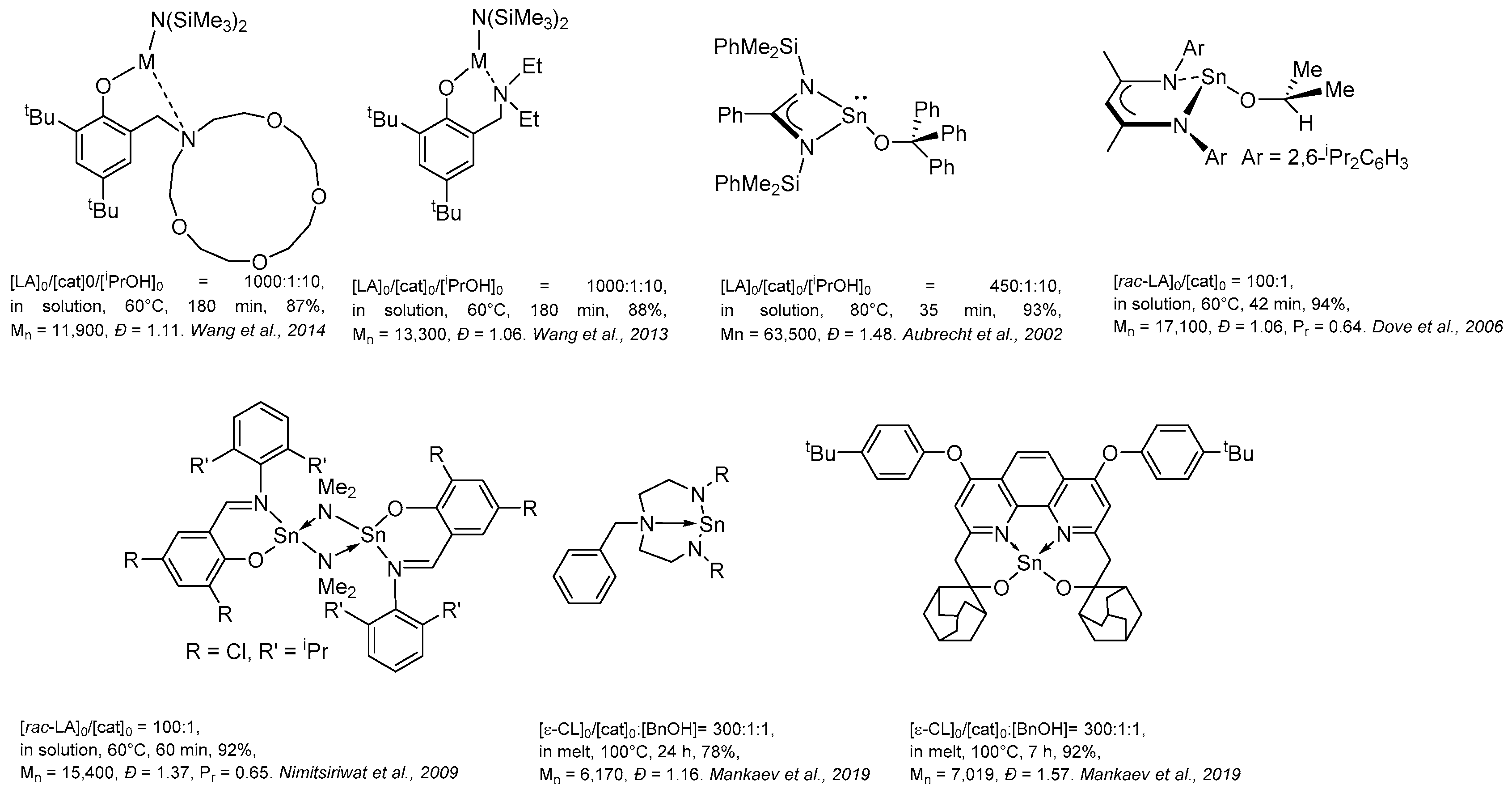
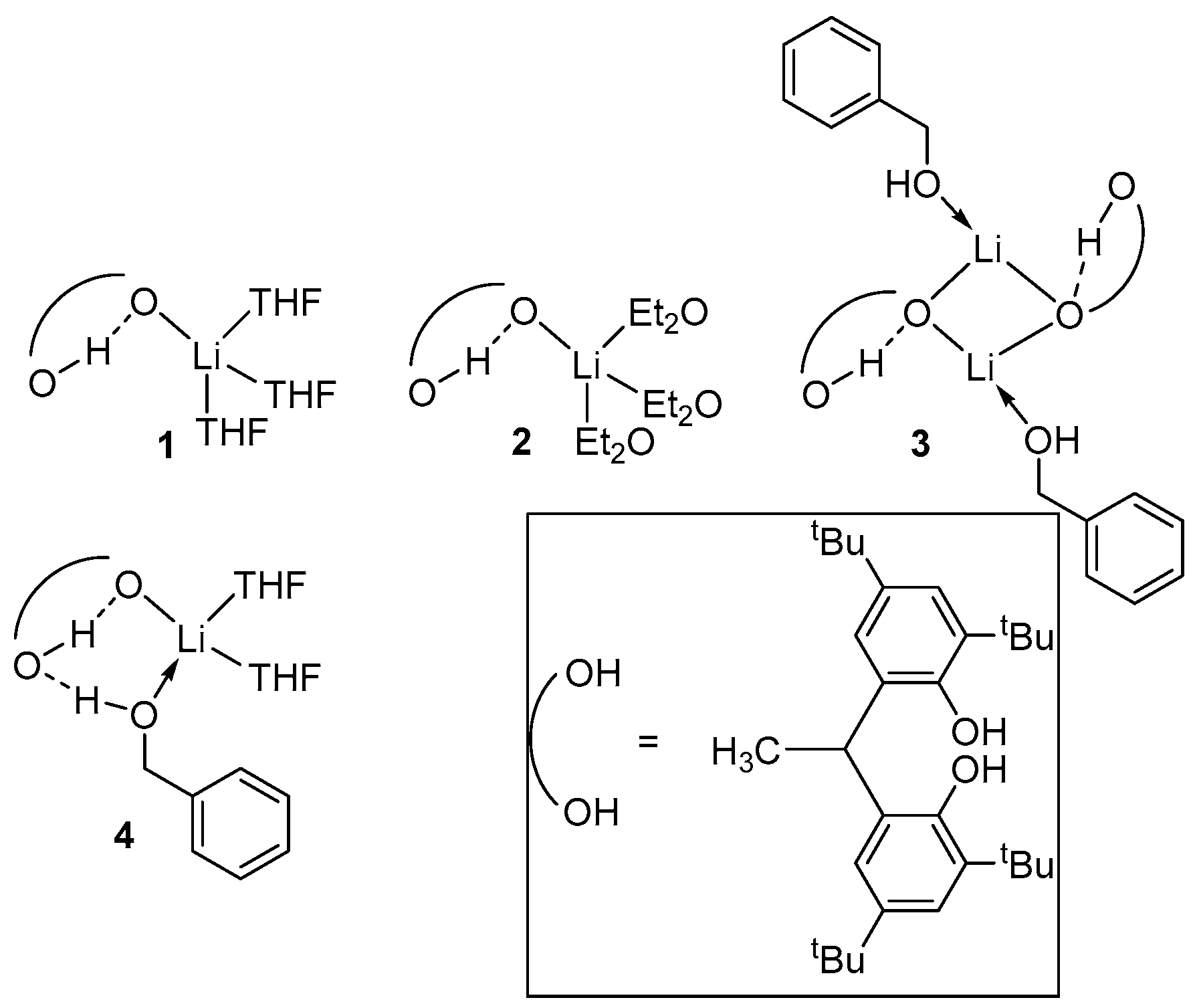
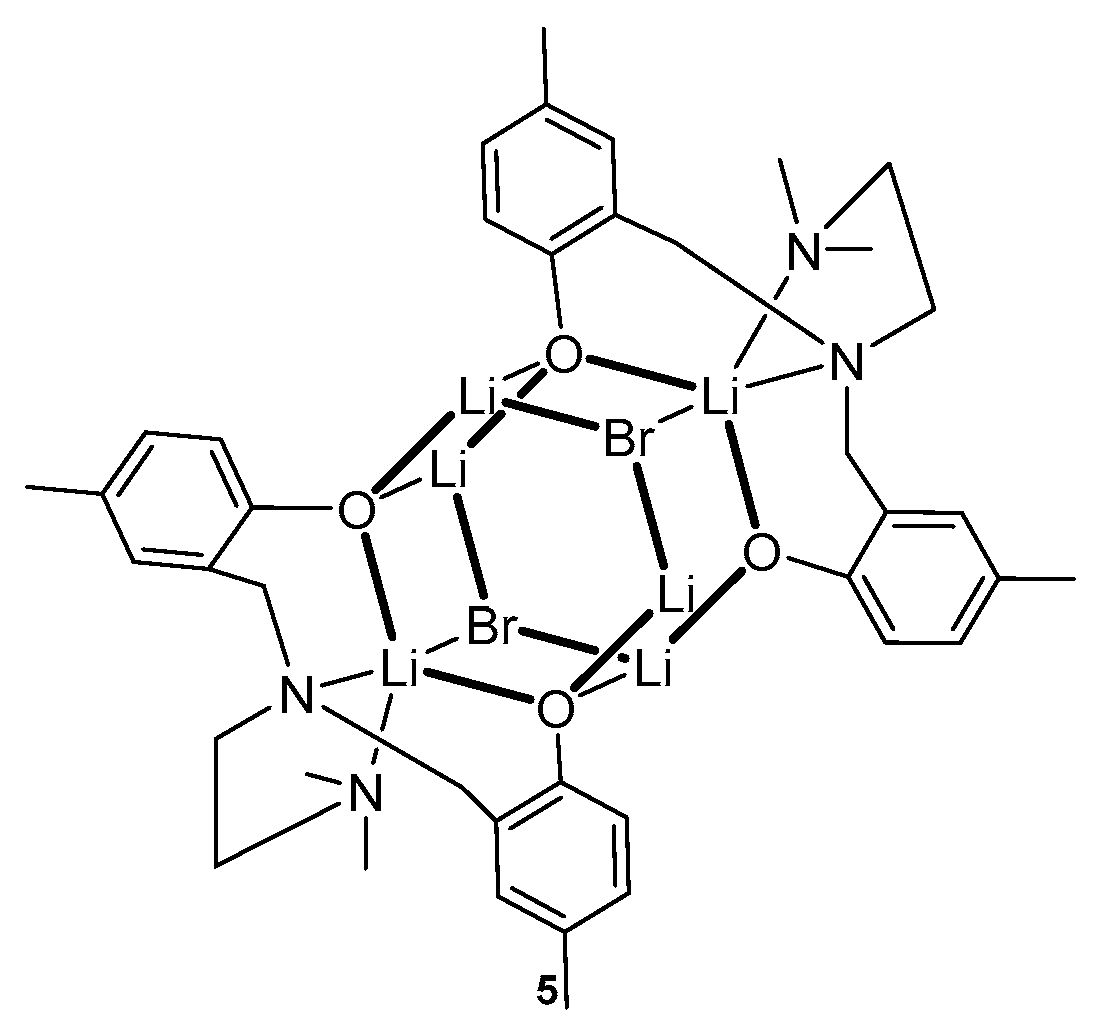
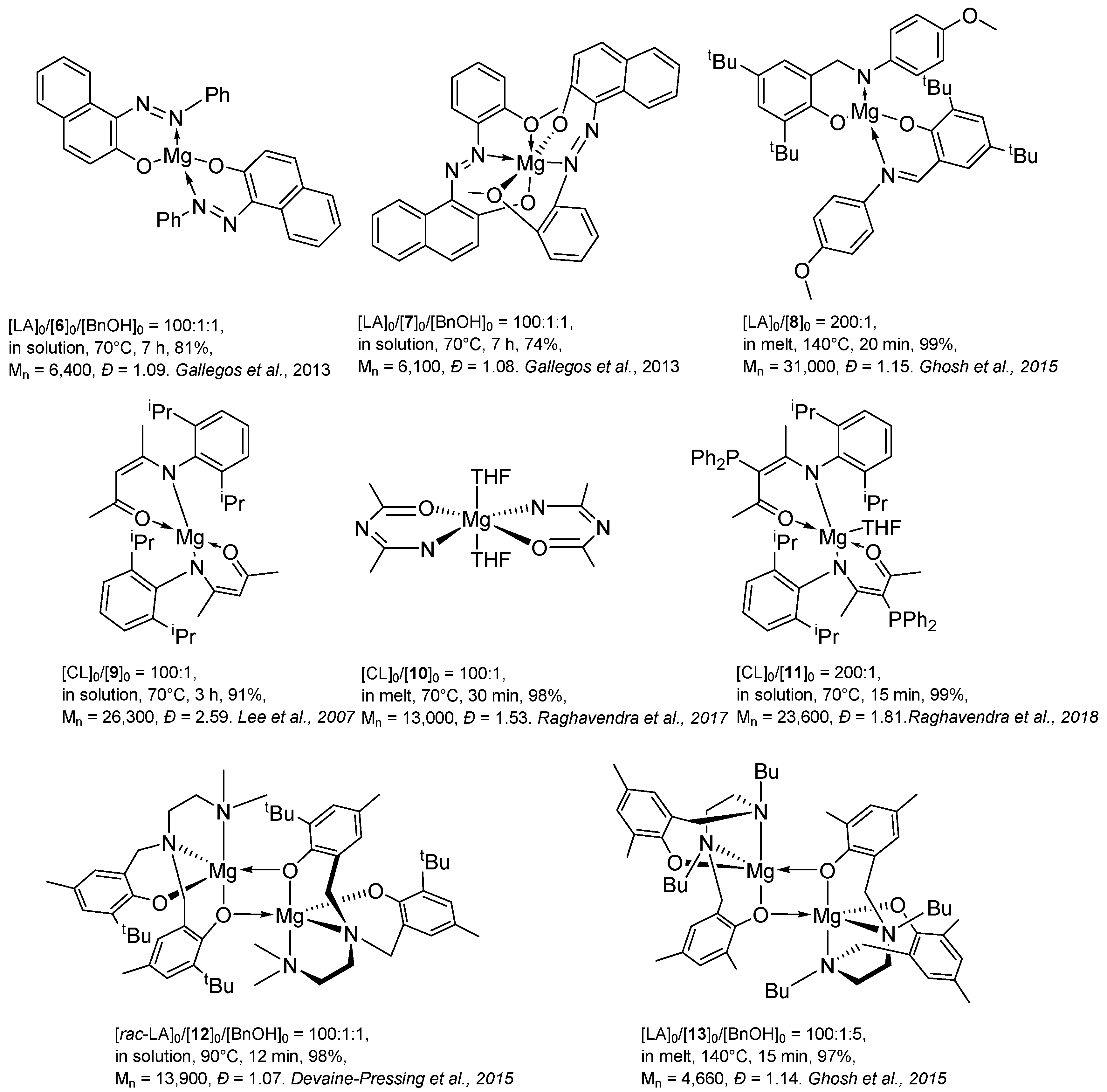
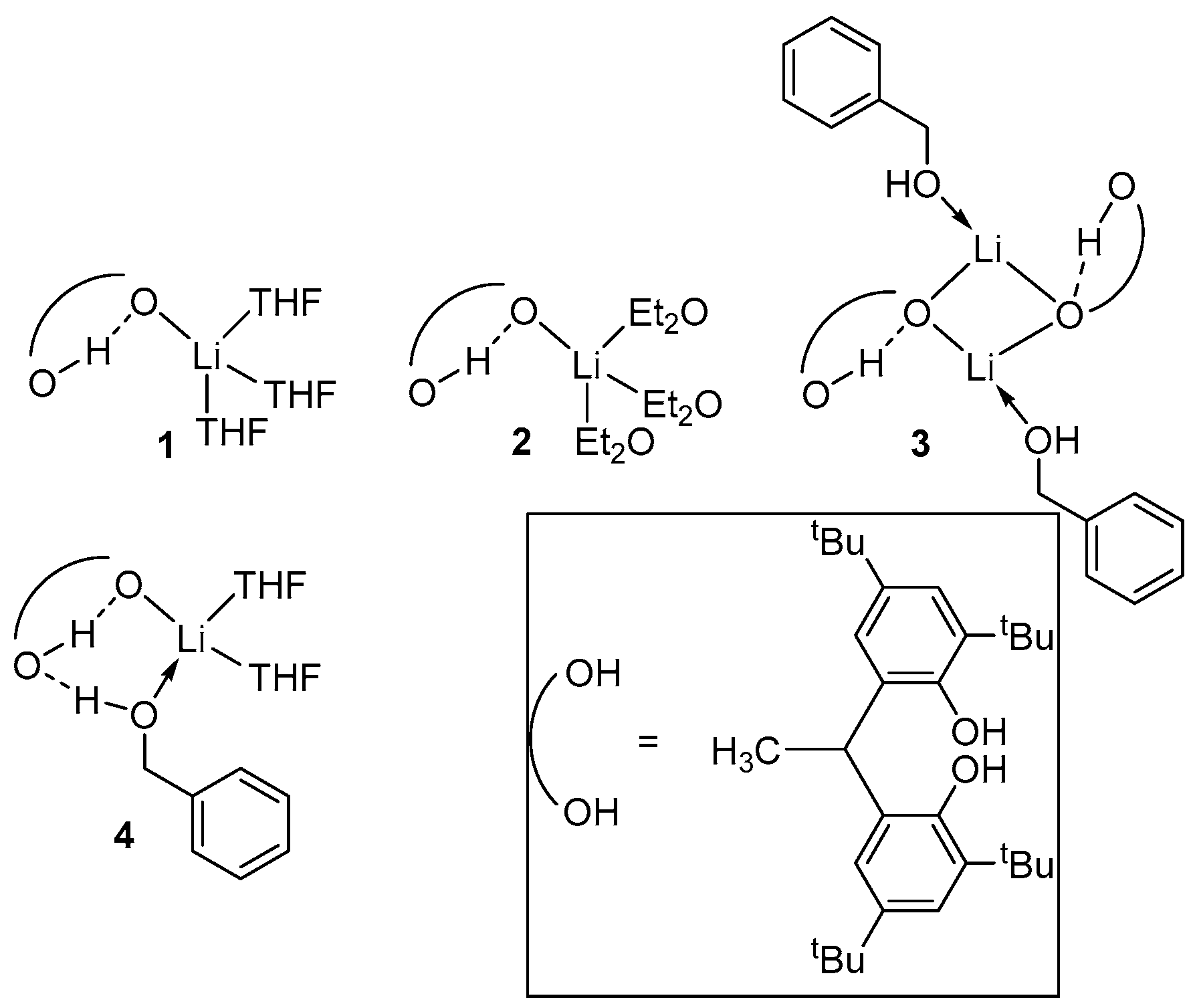
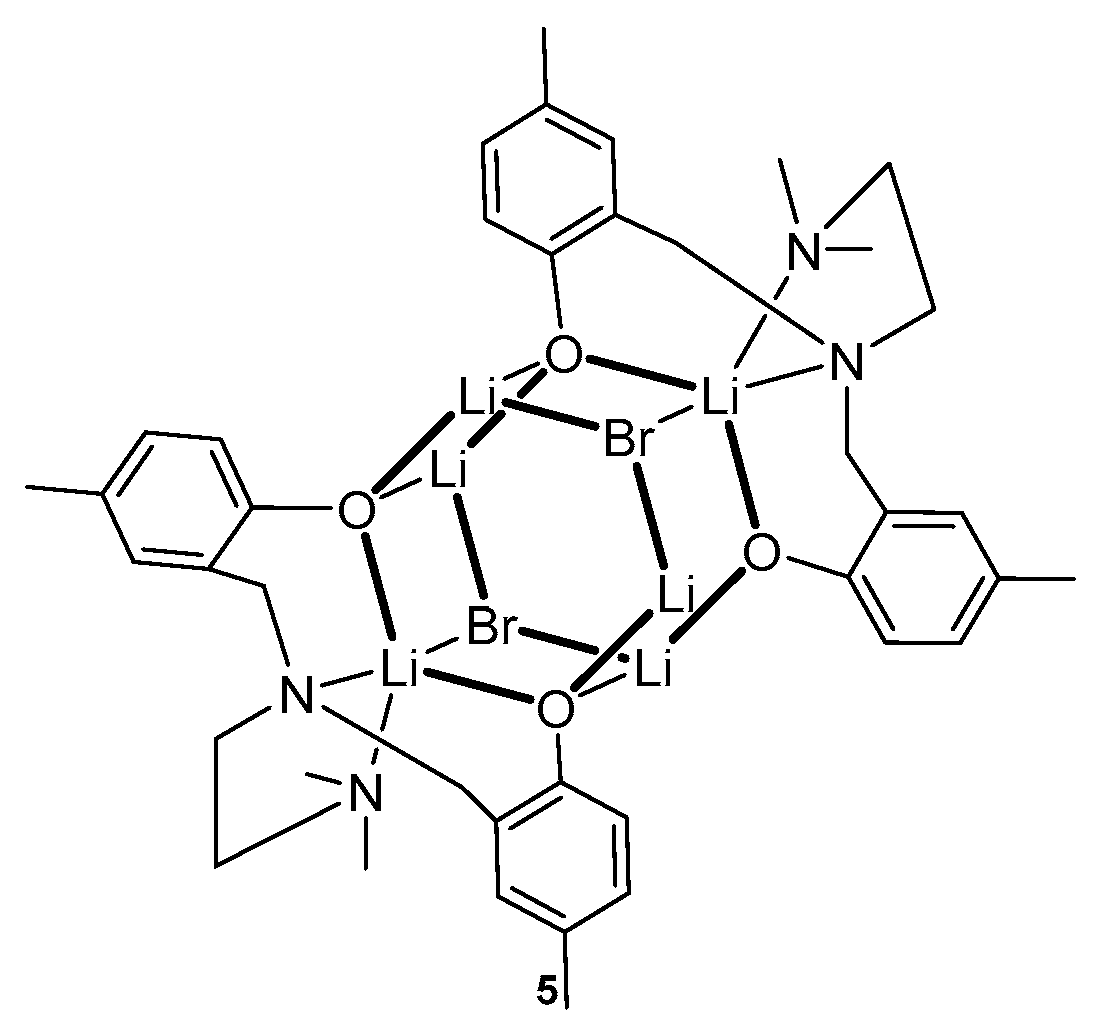
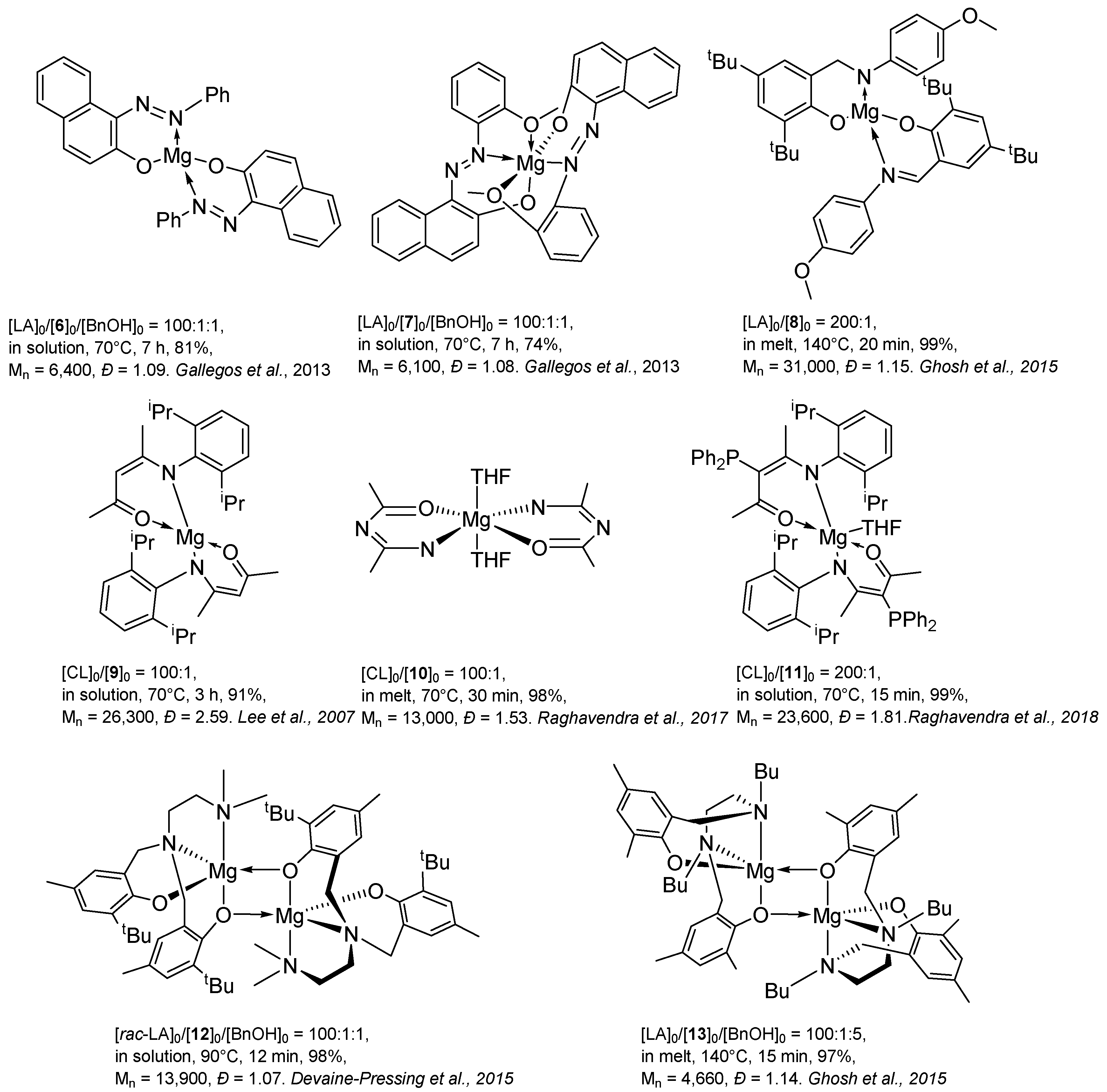
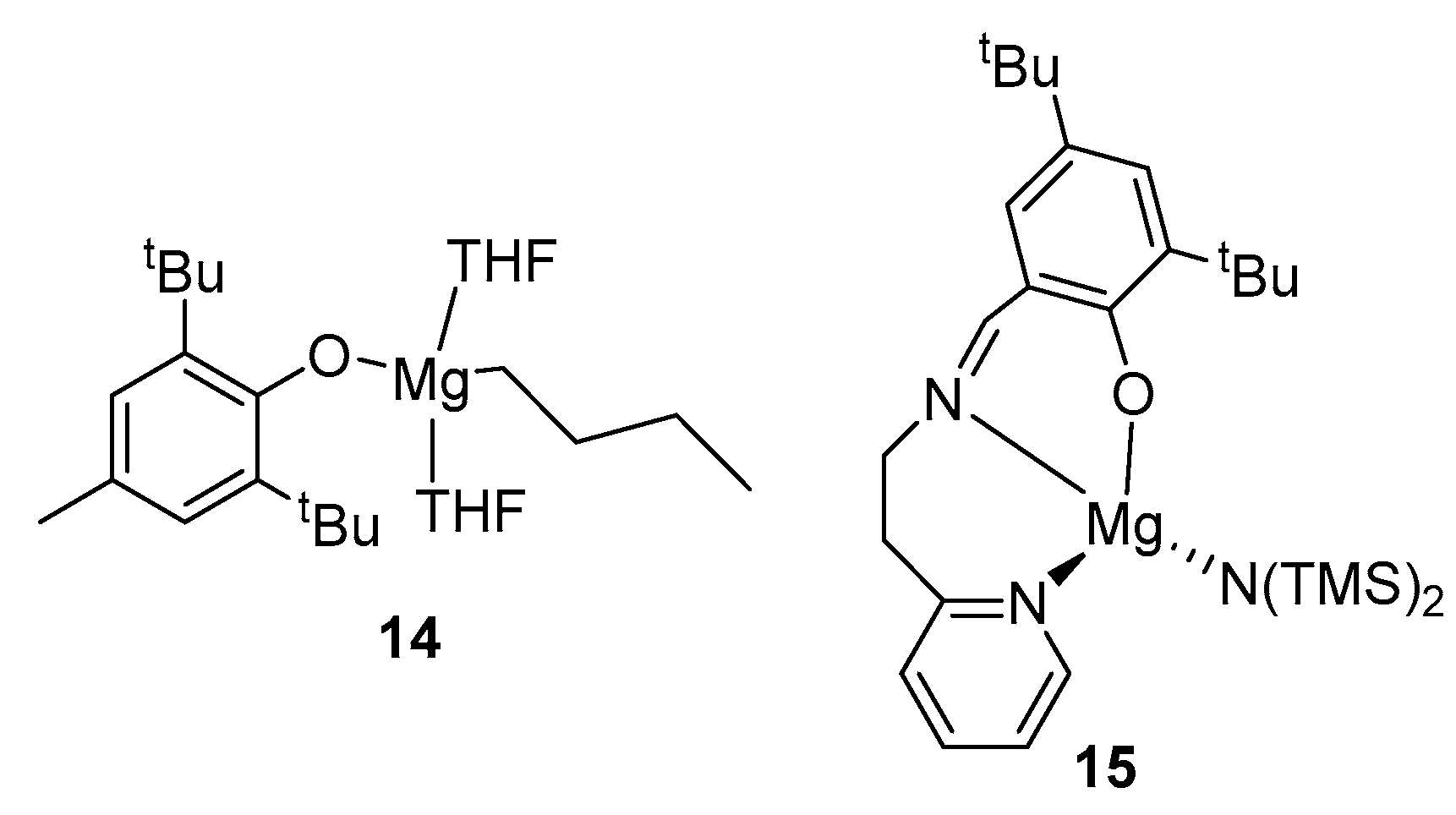
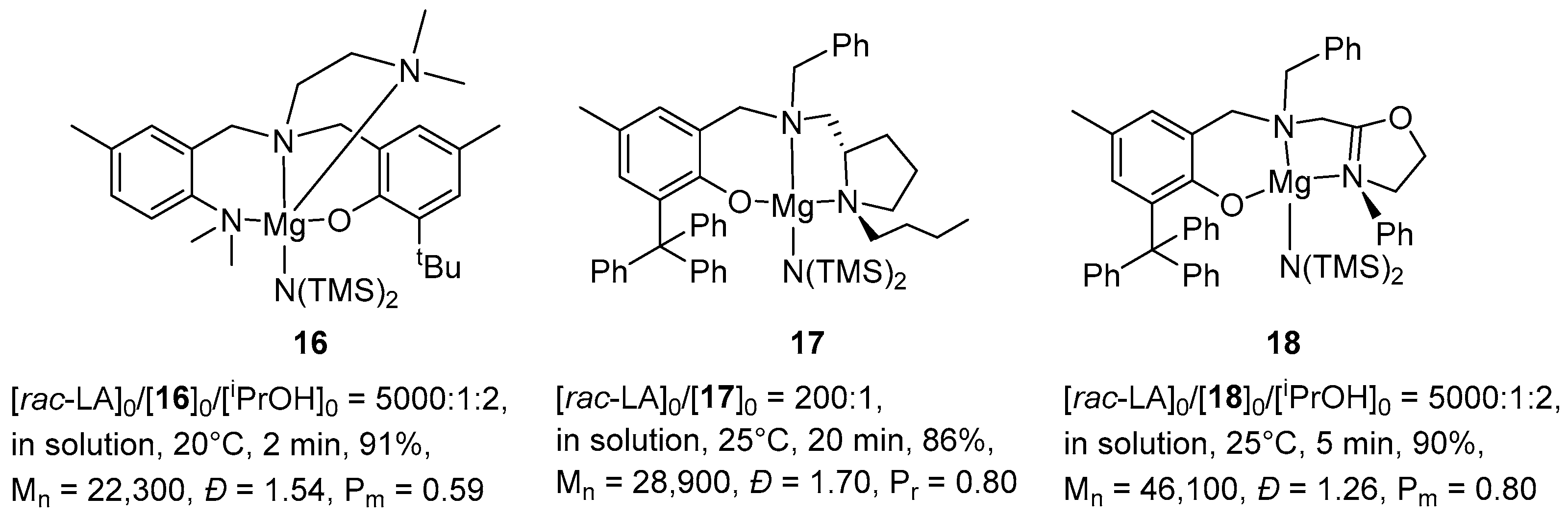
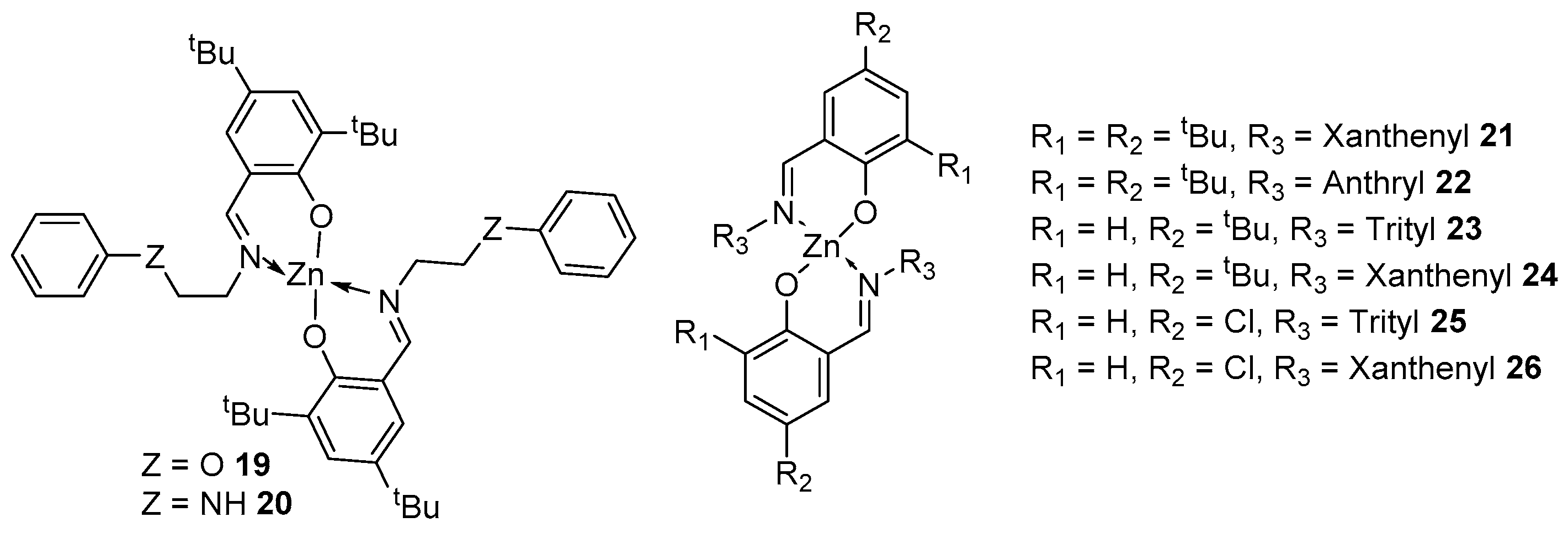
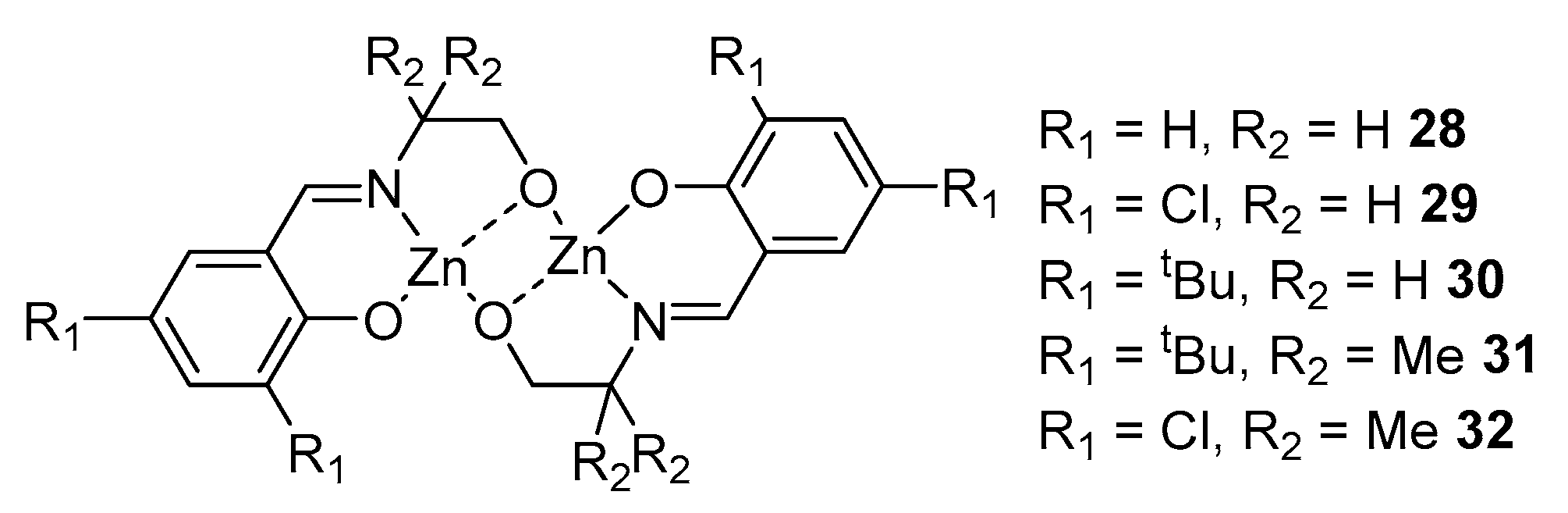
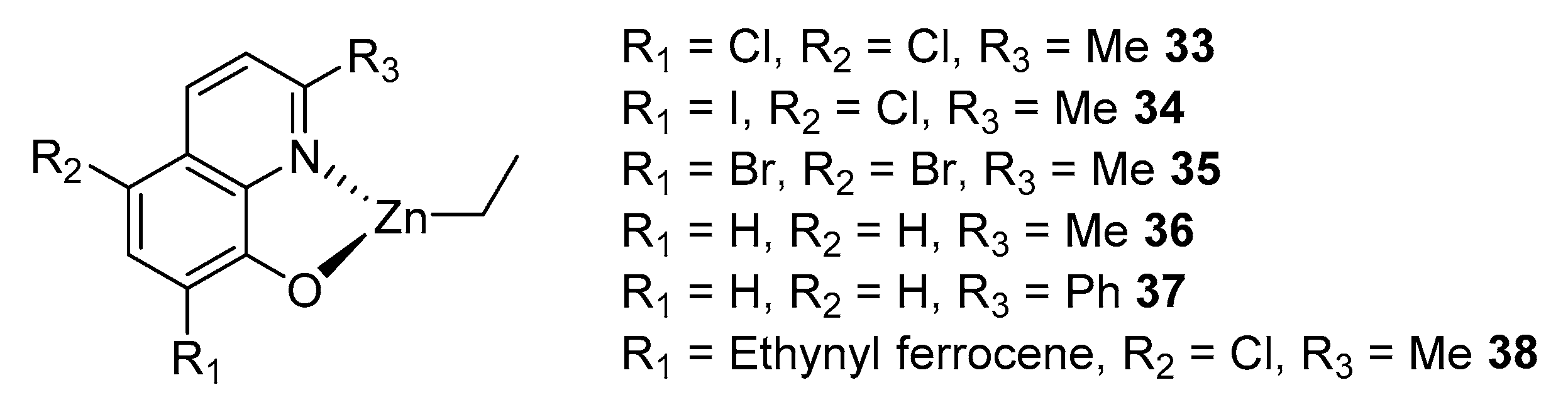
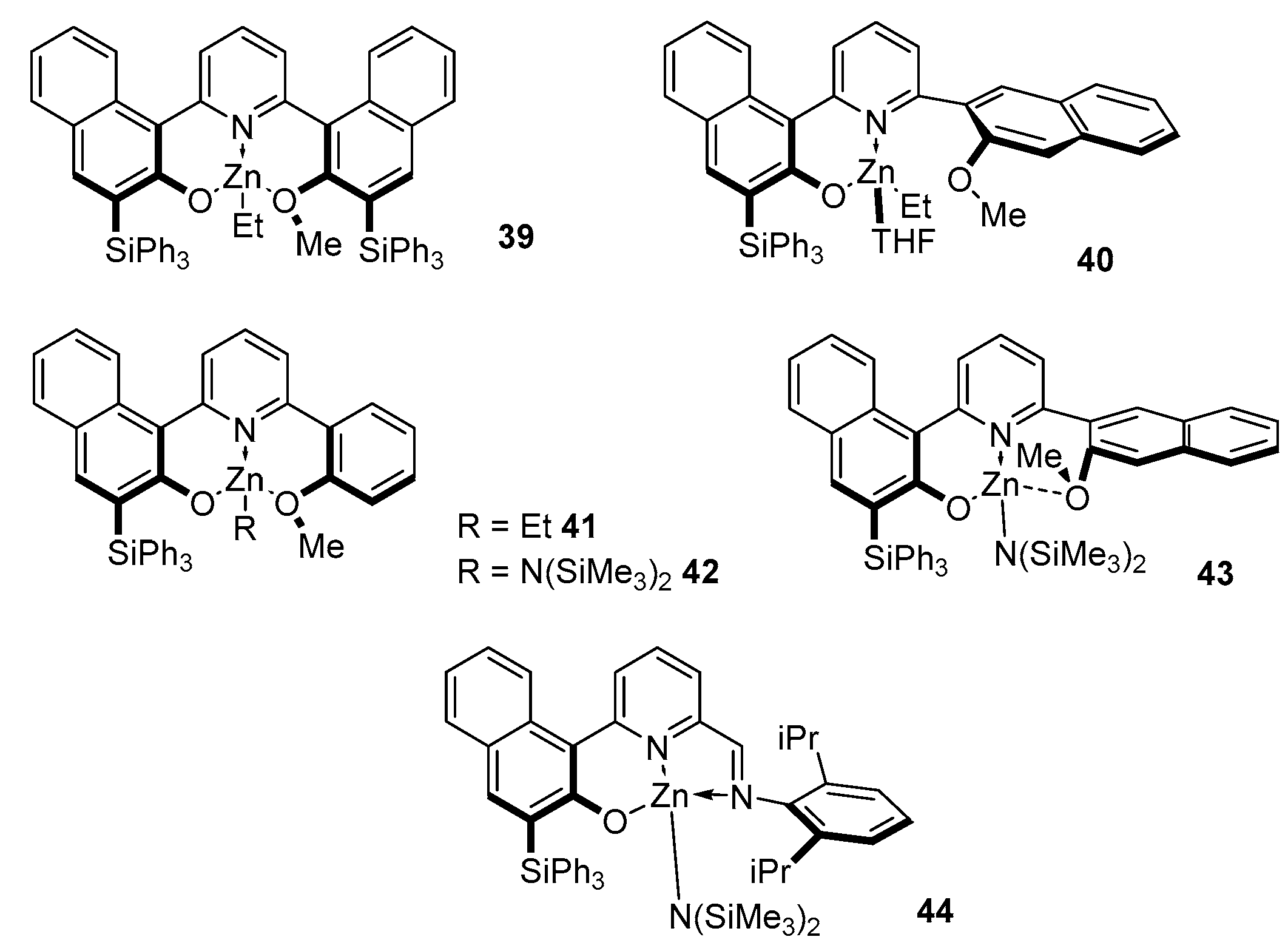
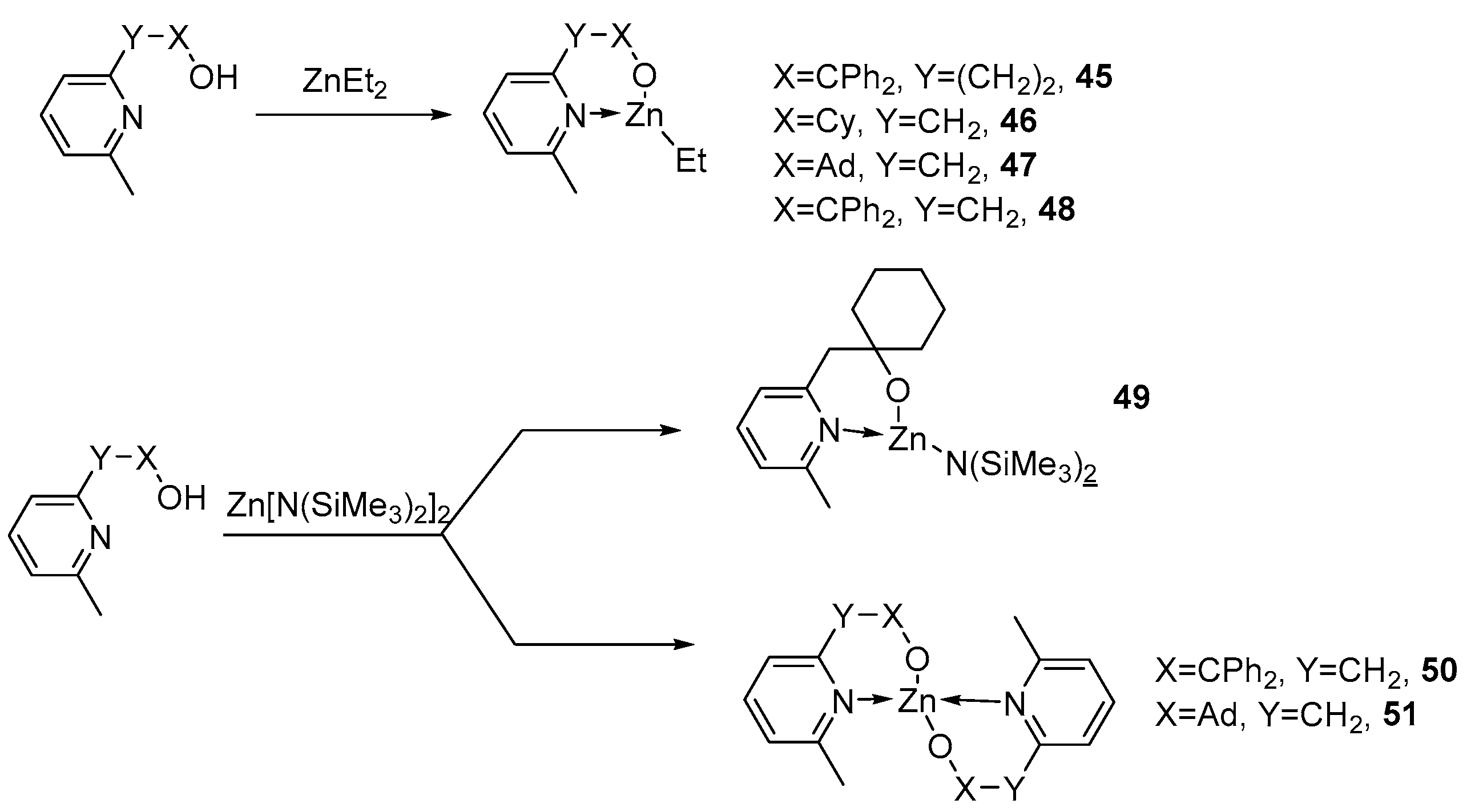
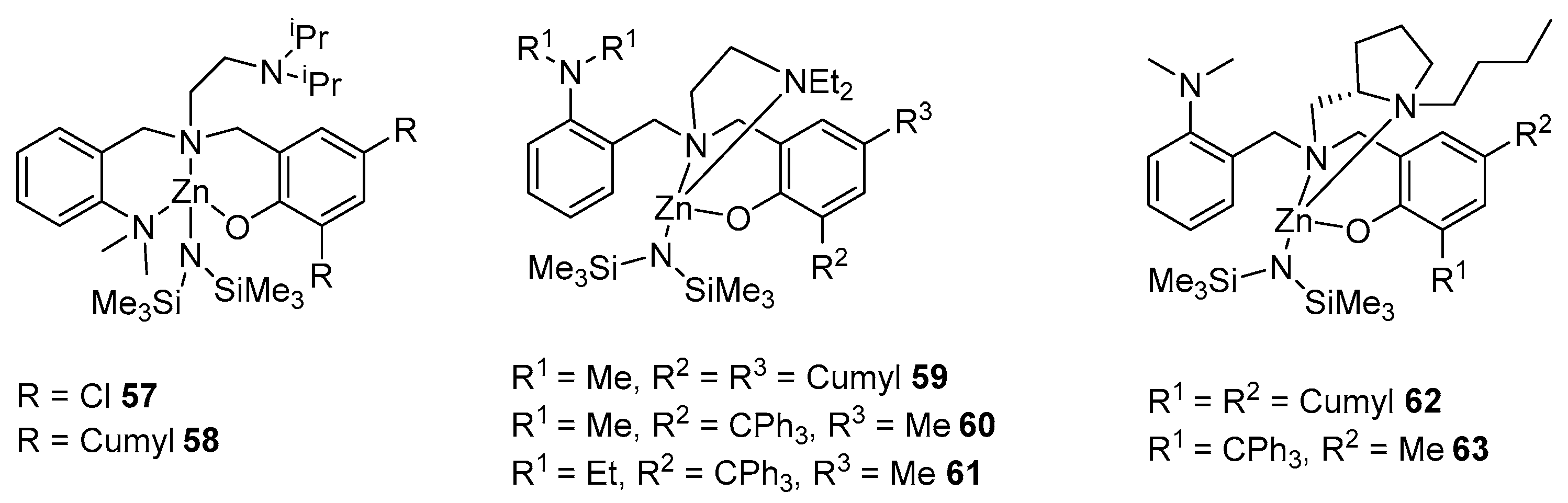
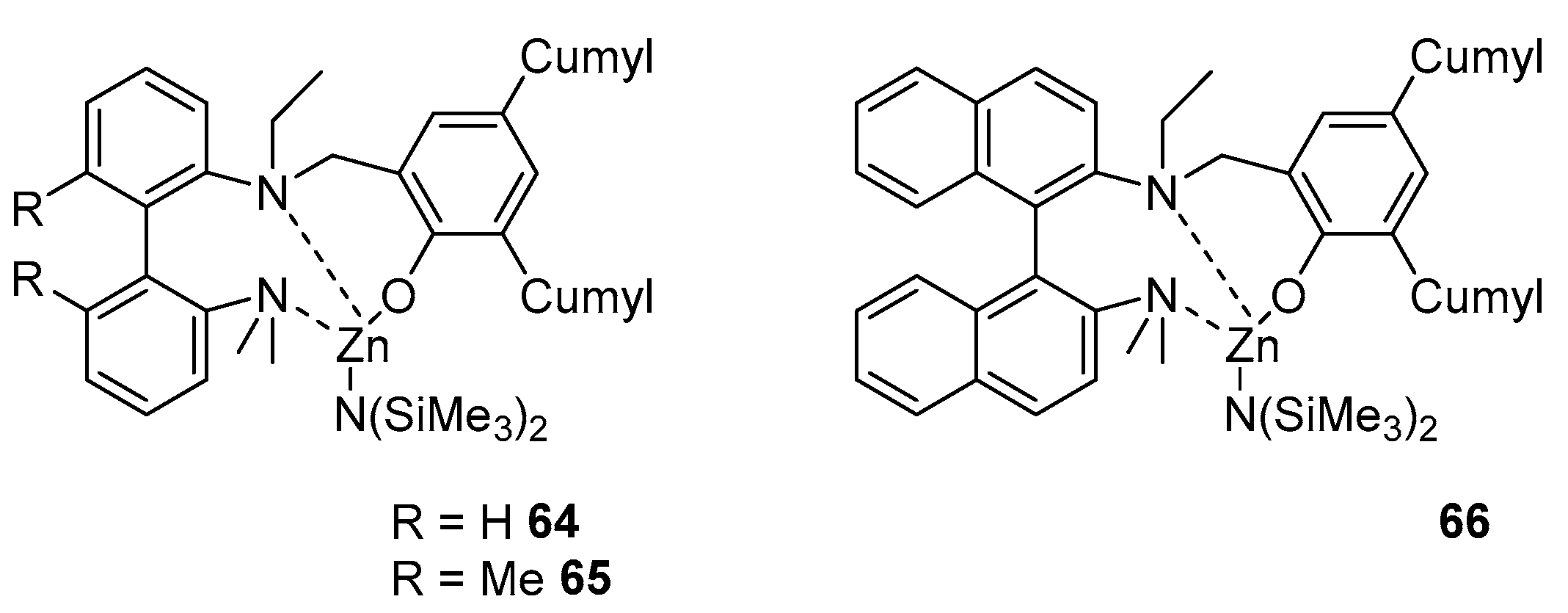
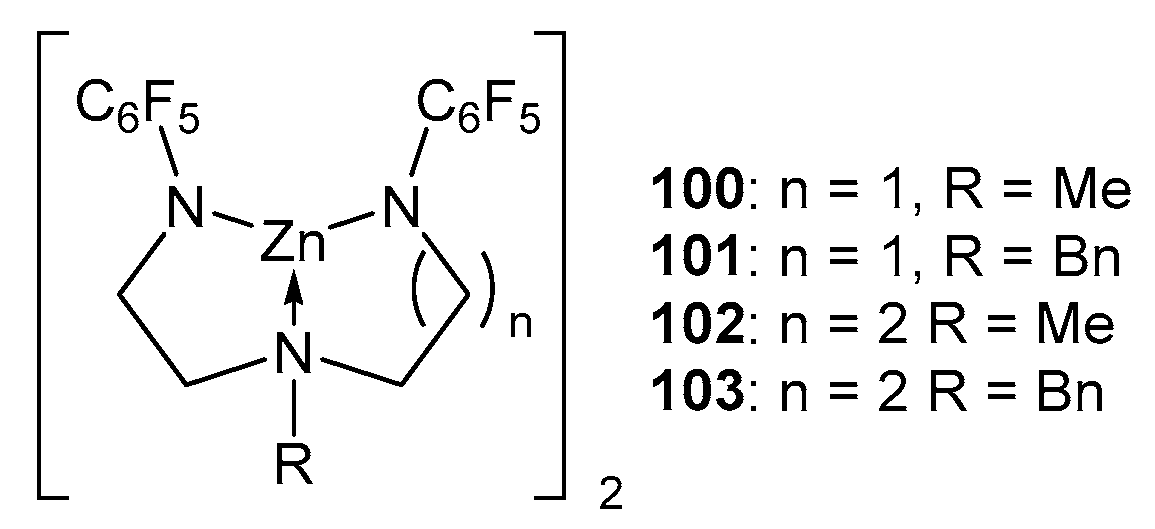
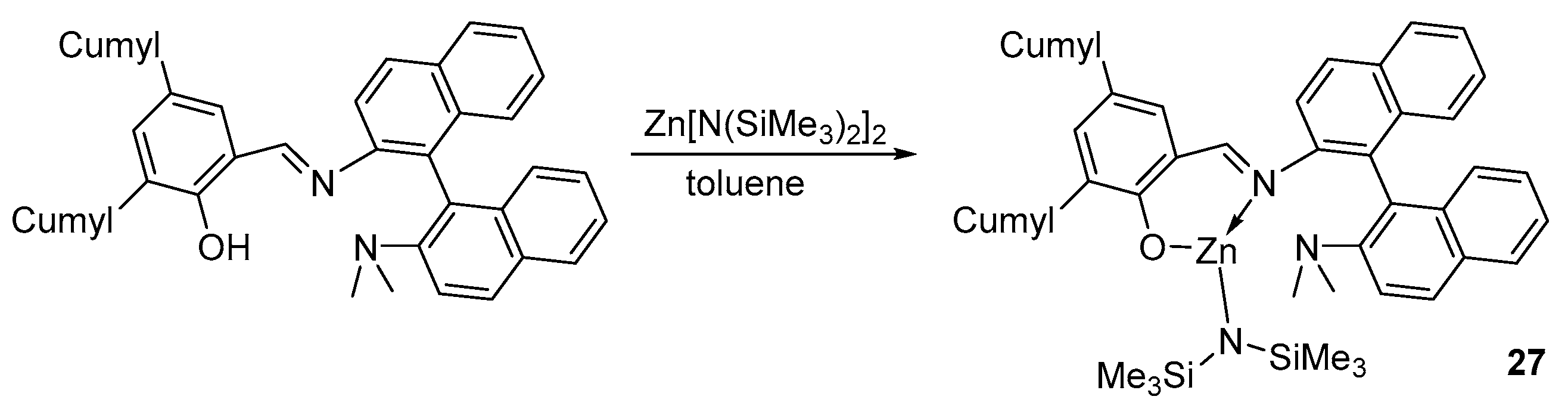
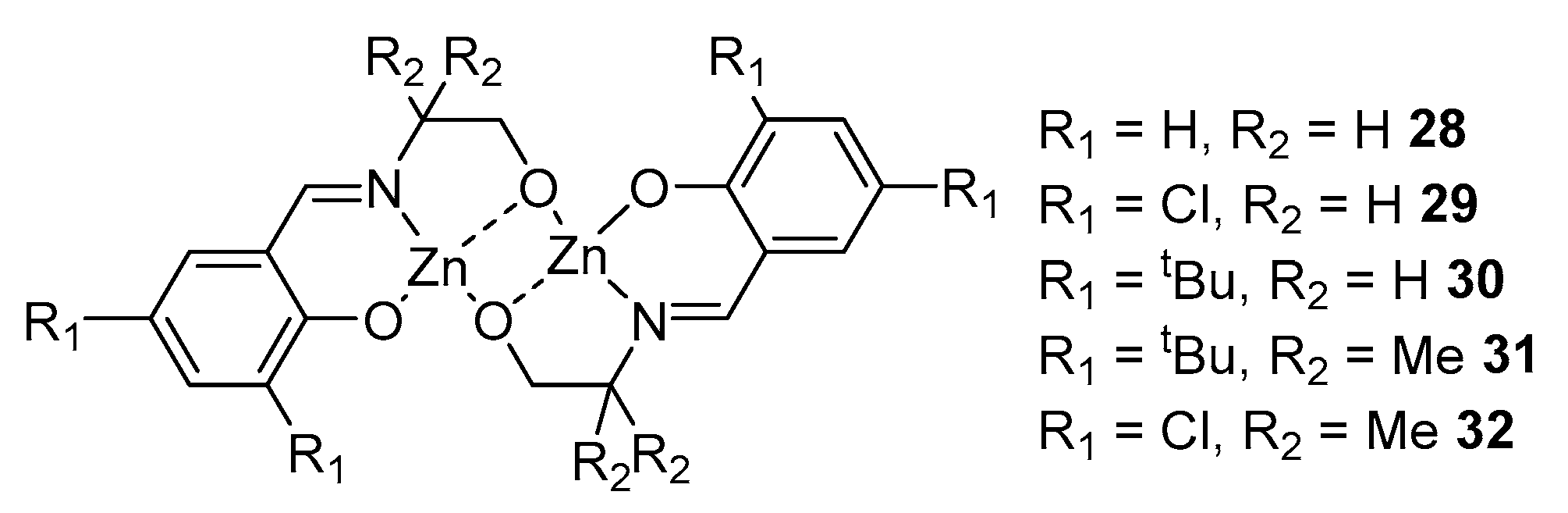
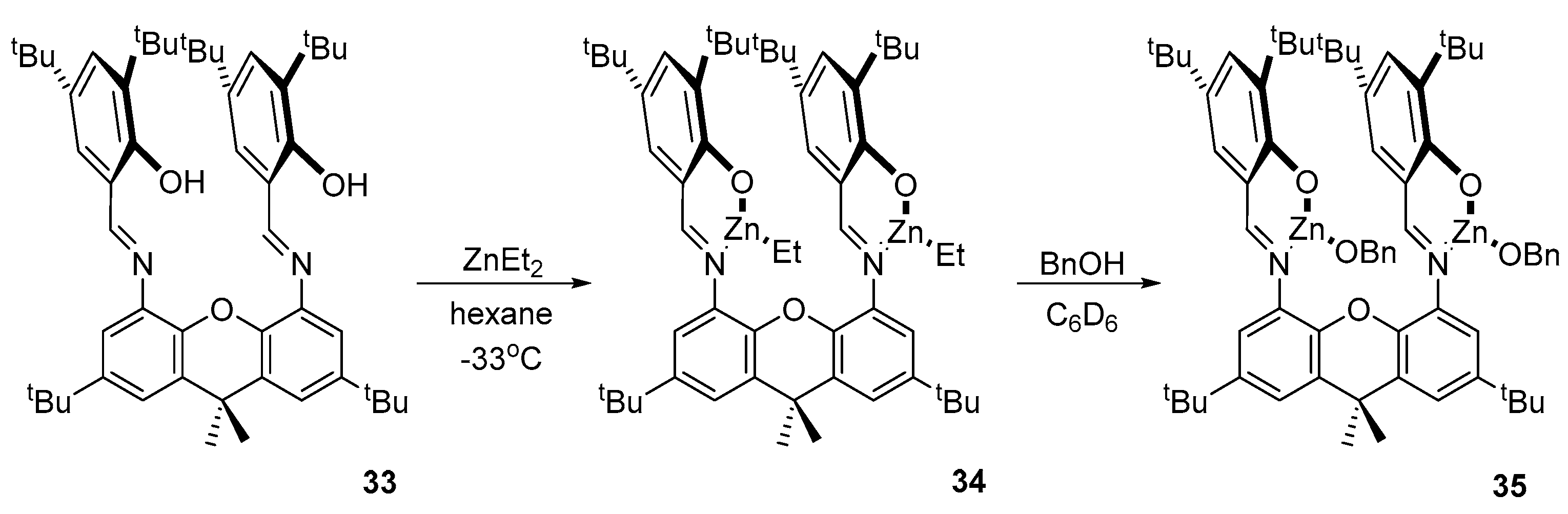
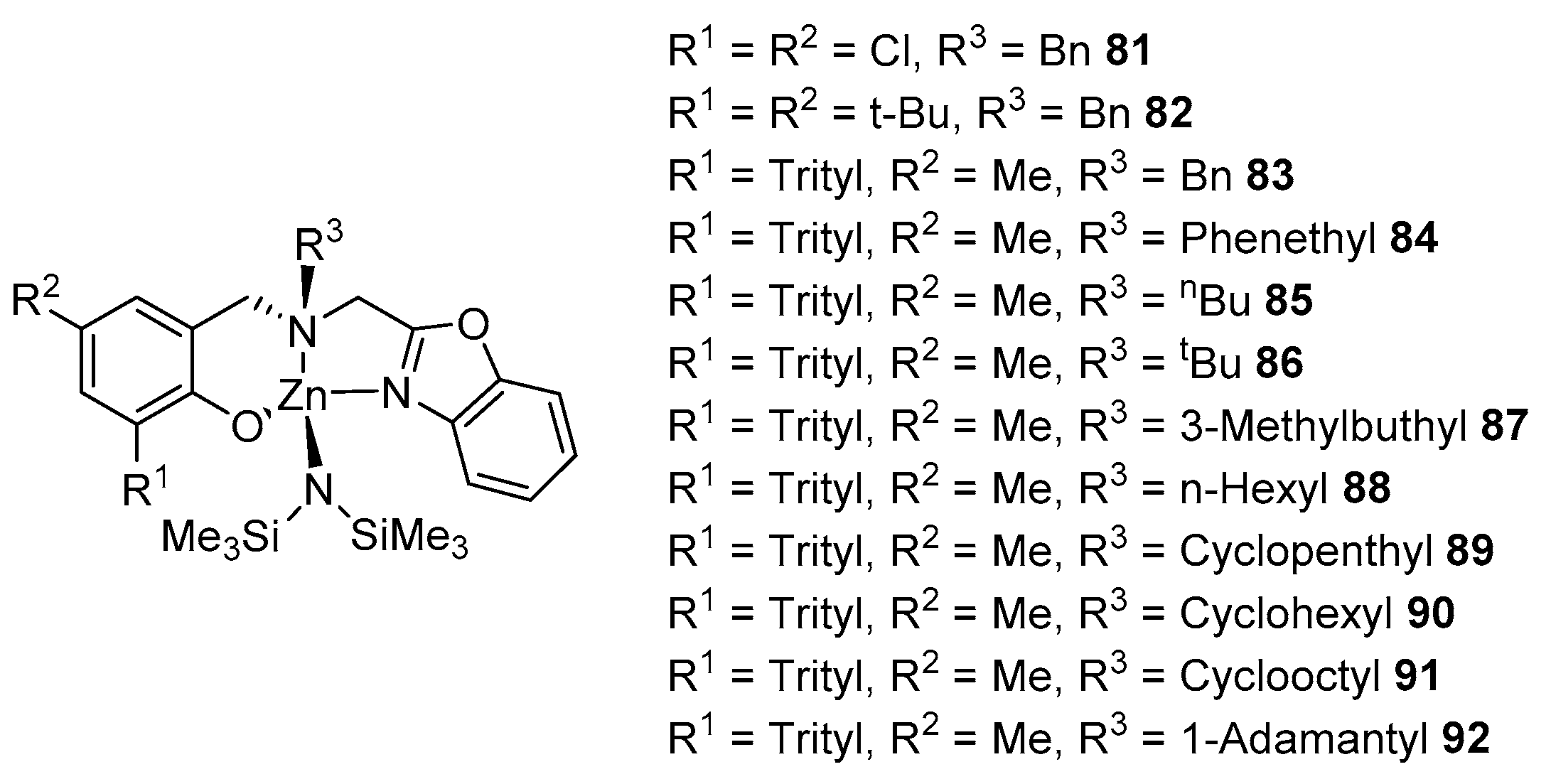
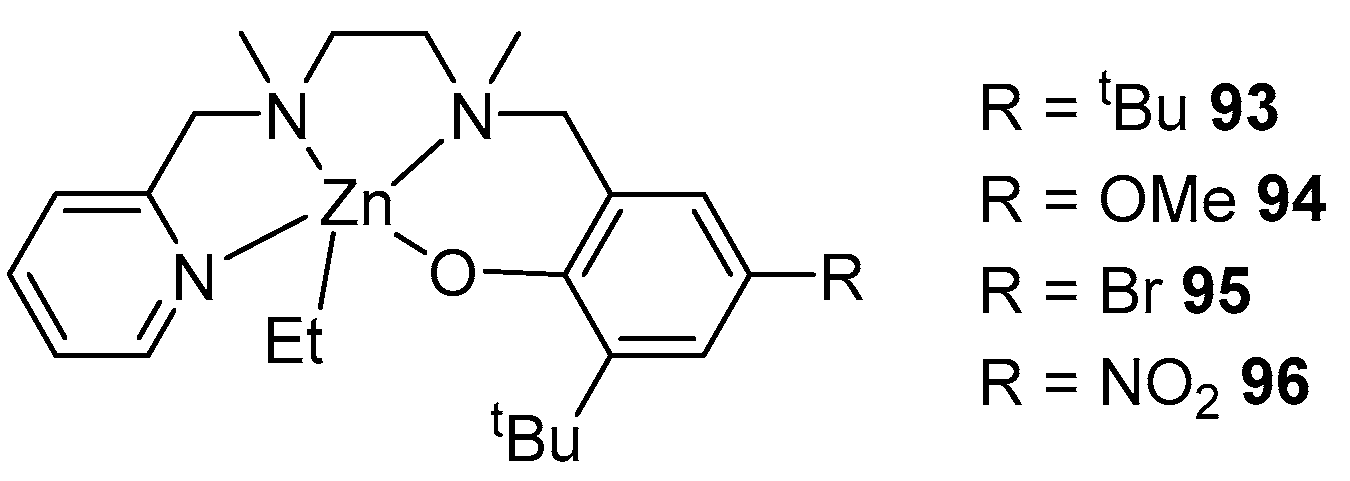
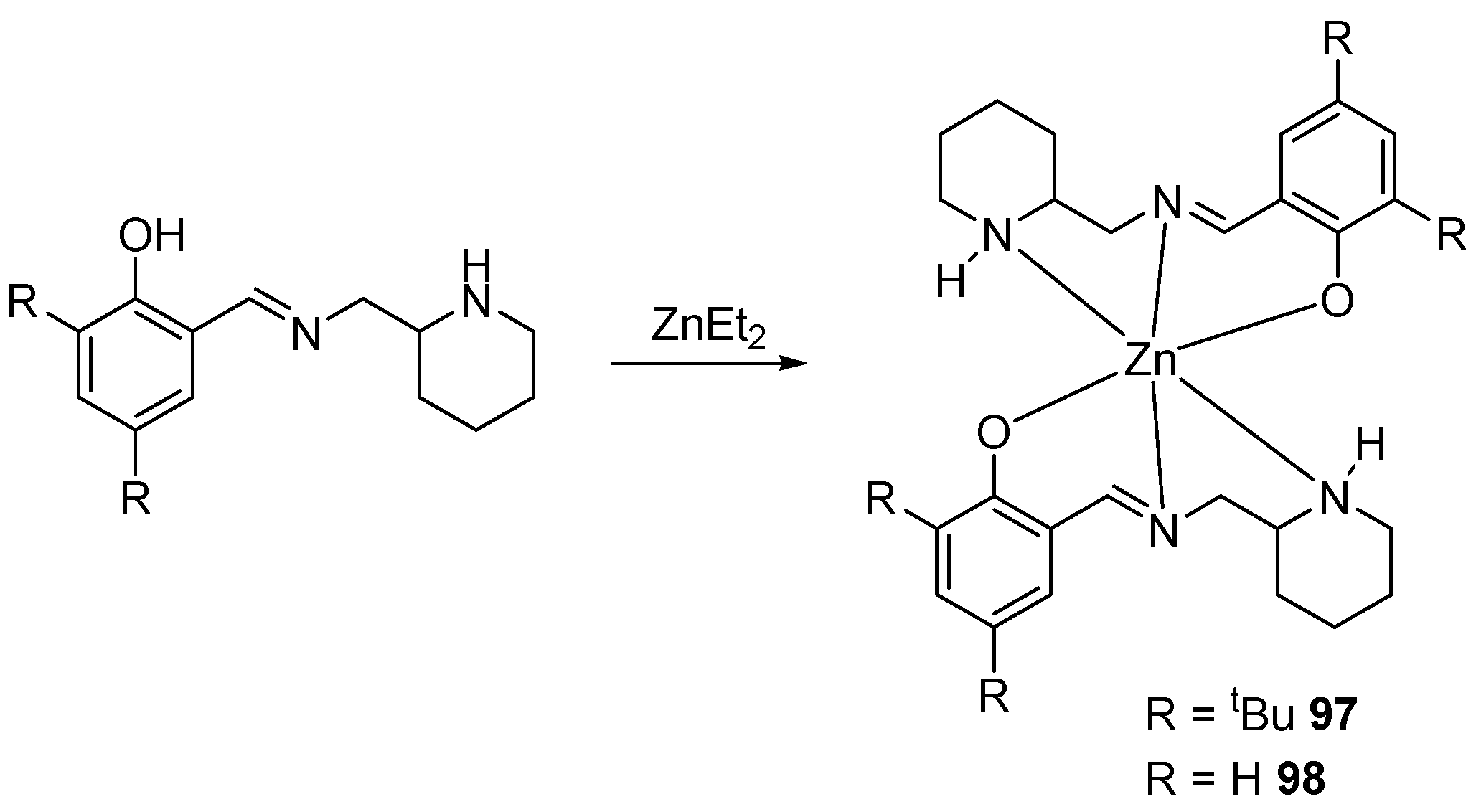
2. Group 1, 2 and 12 Metals (Li, Na, K, Ca, Mg, Zn)
3. Group 4 Metals (Ti, Zr, Hf)
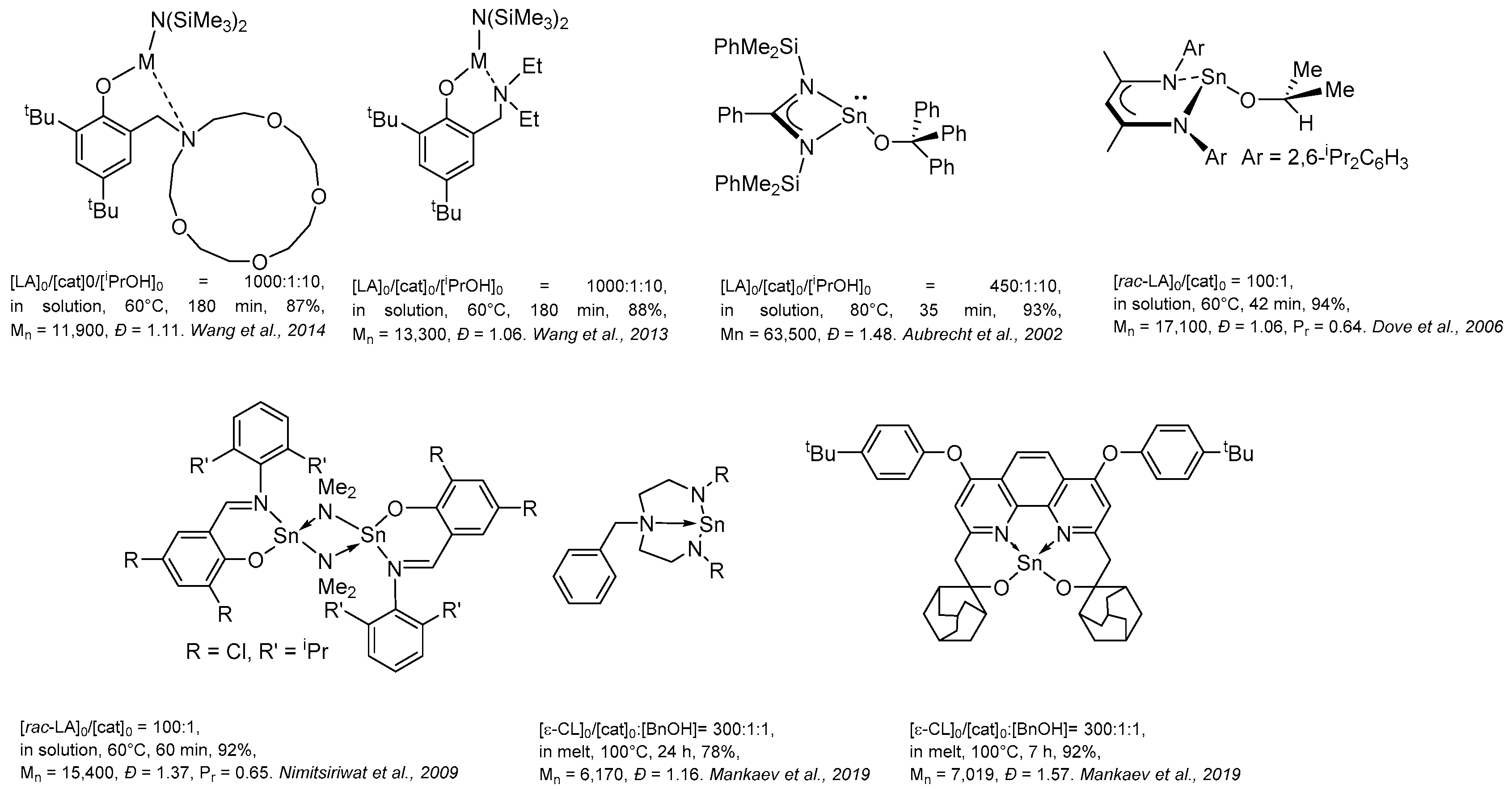
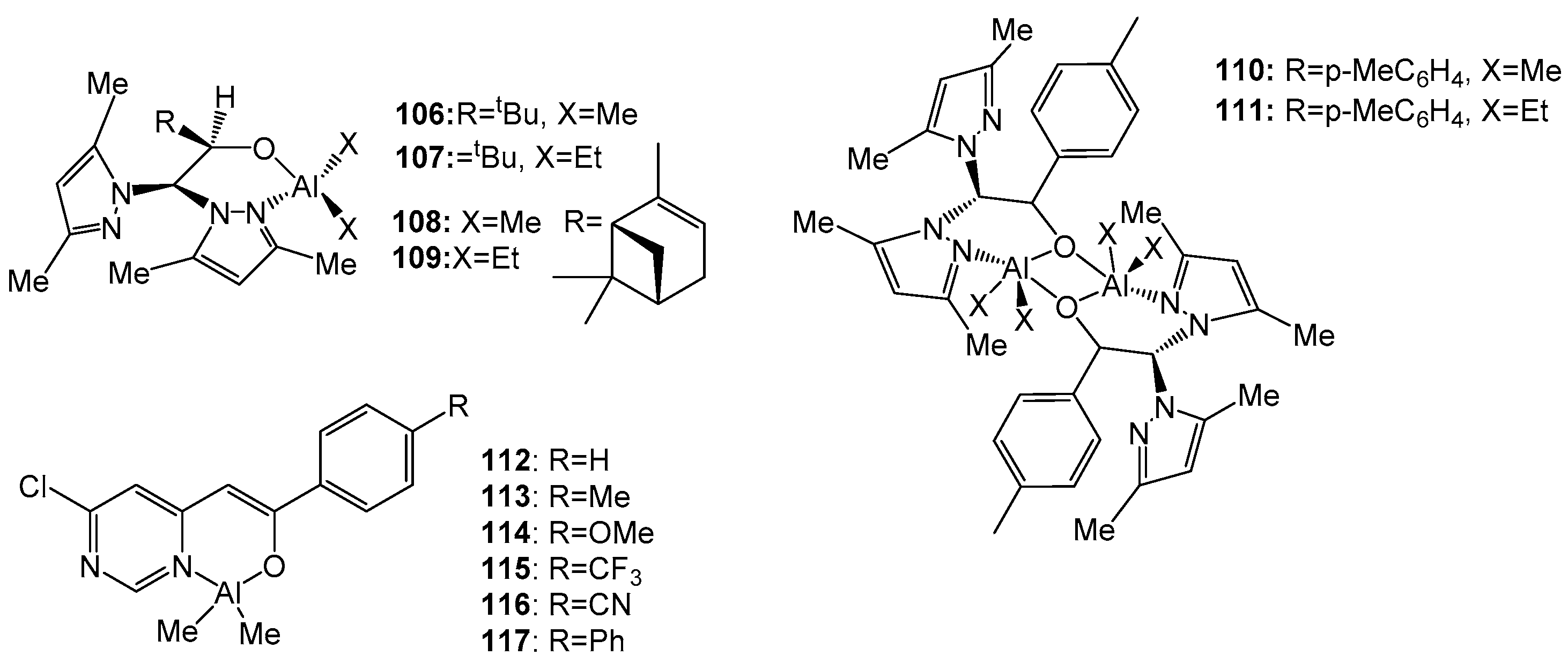
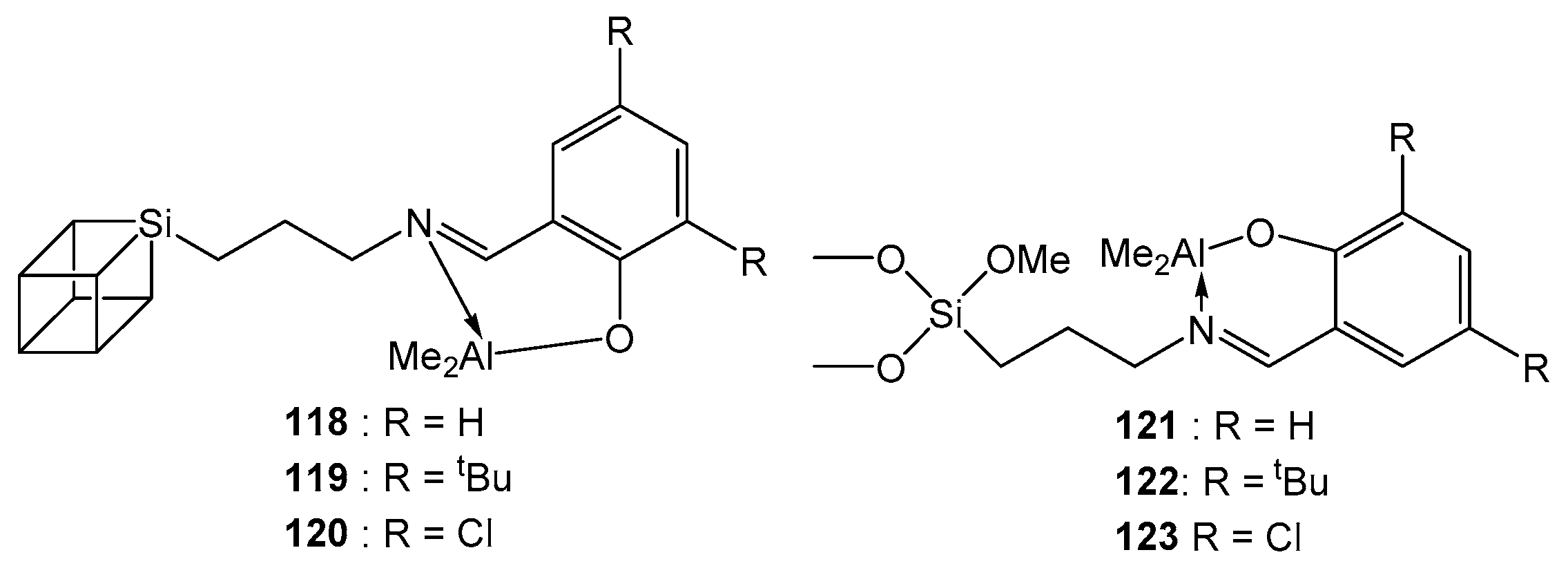
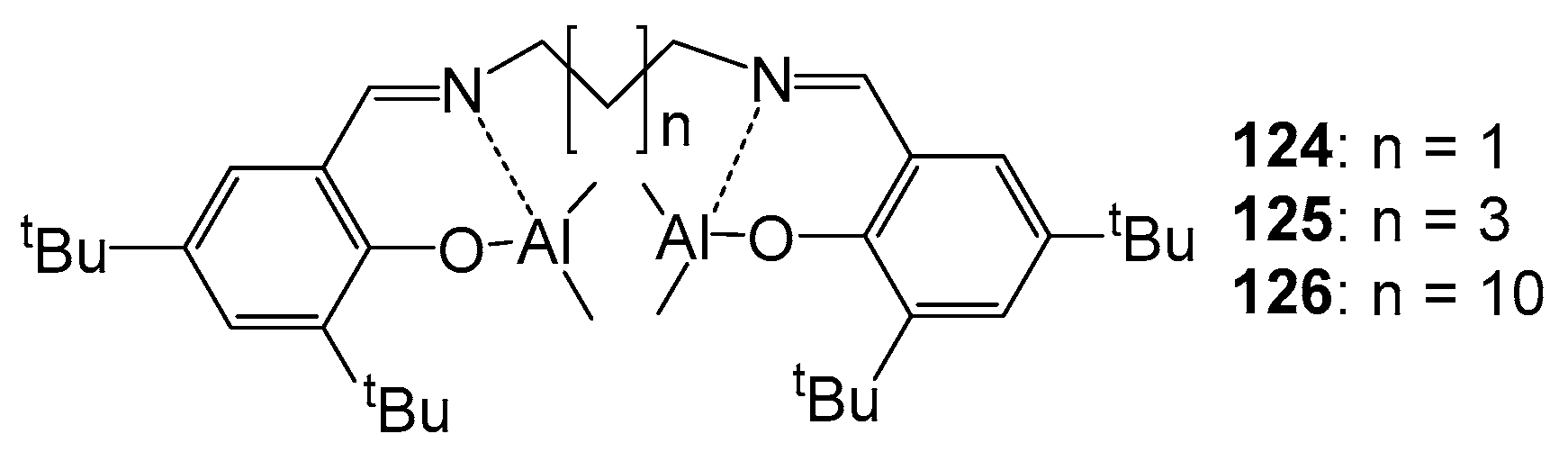
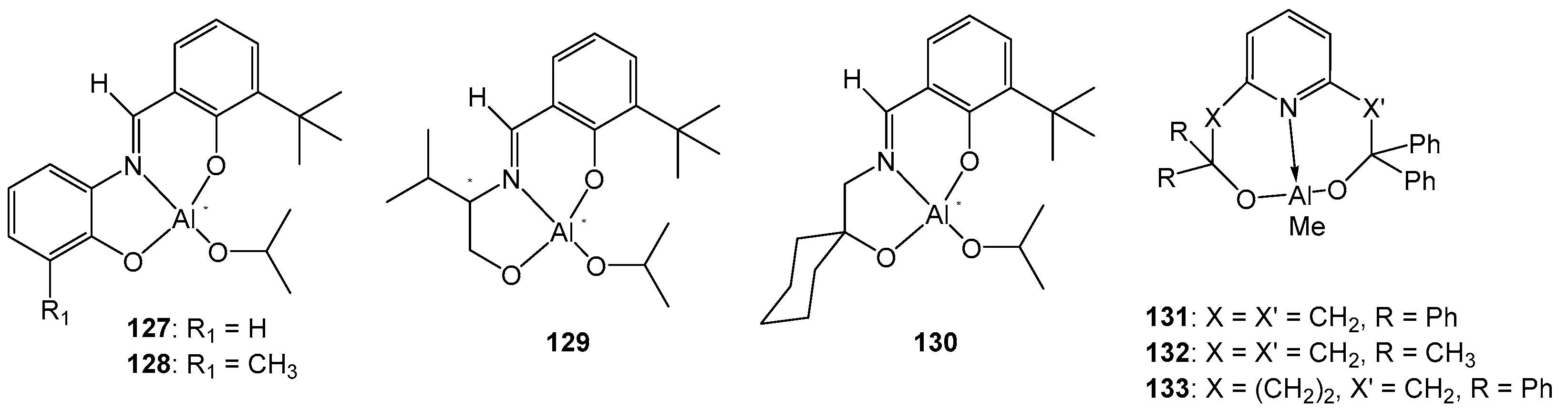
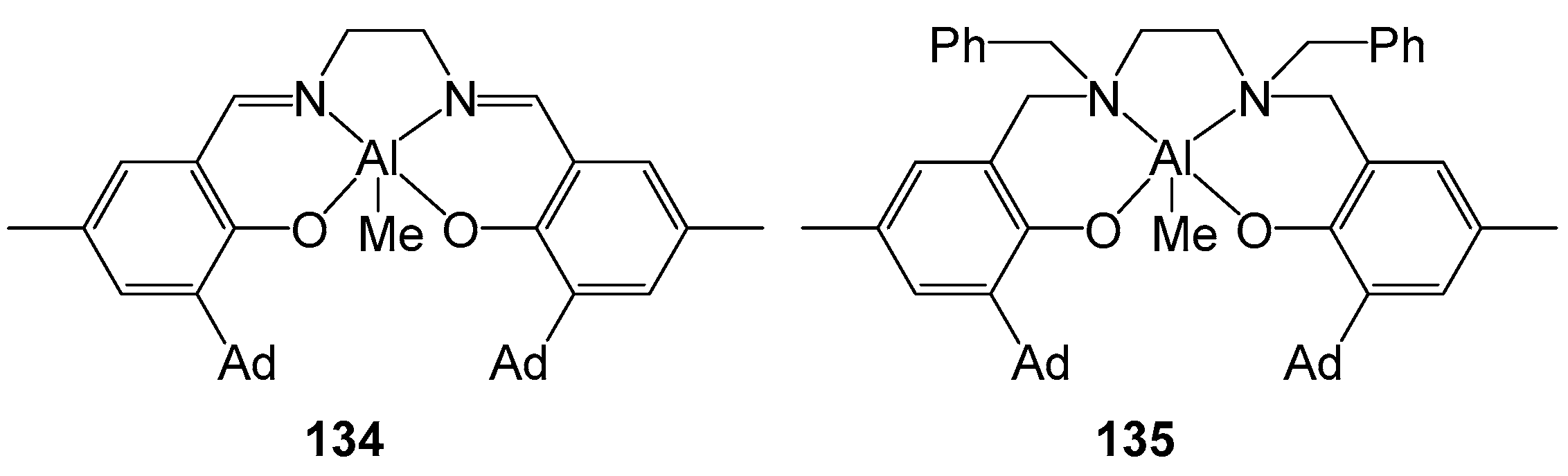
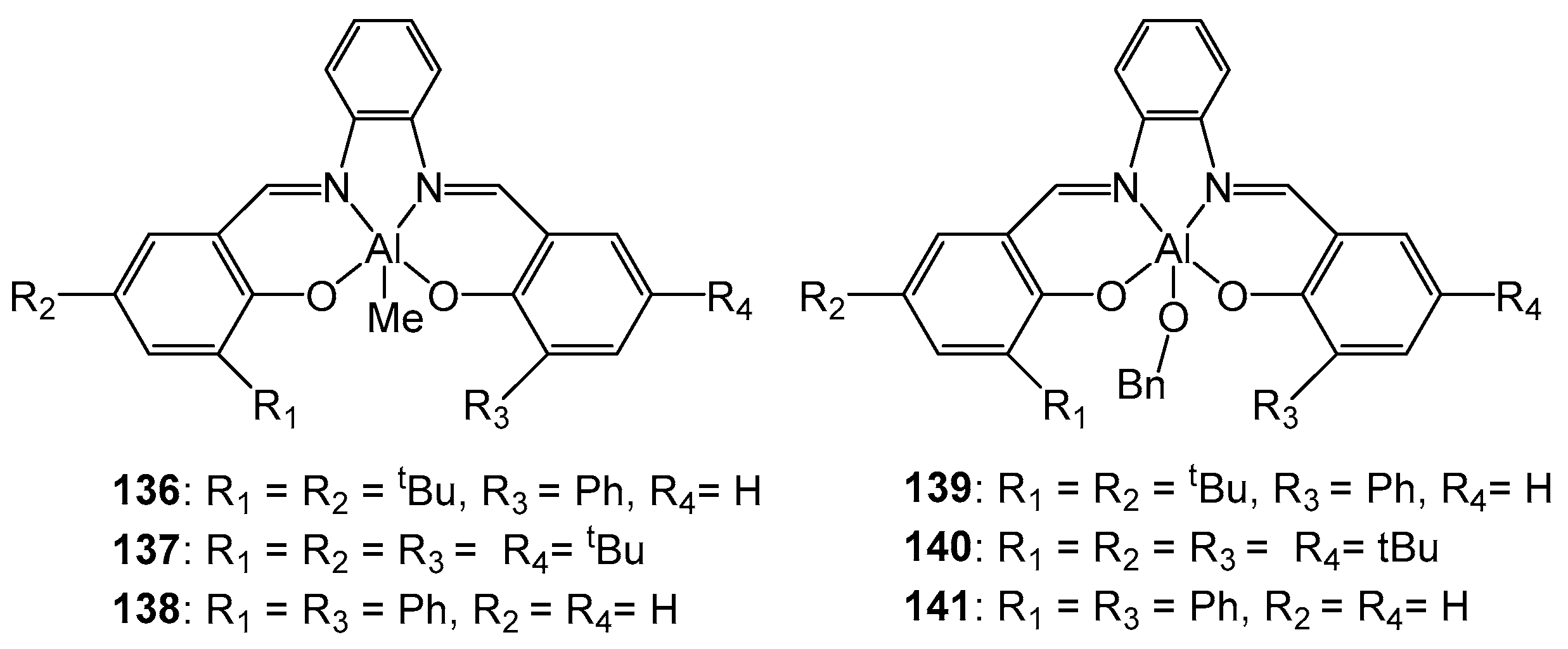
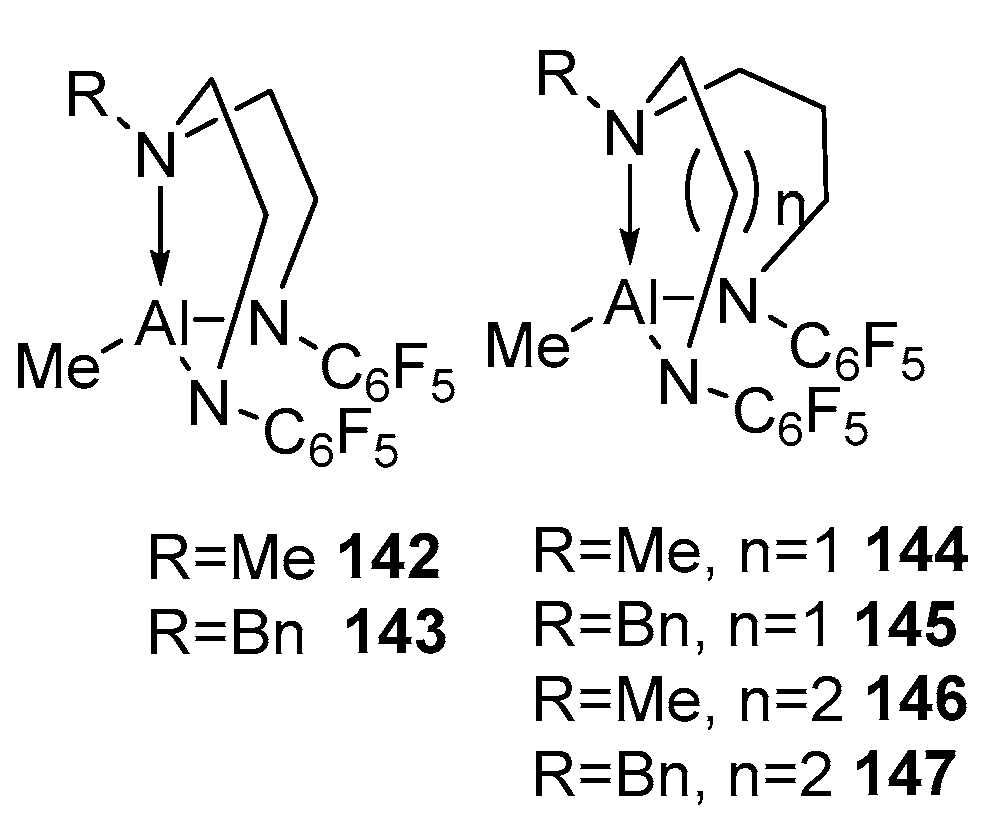
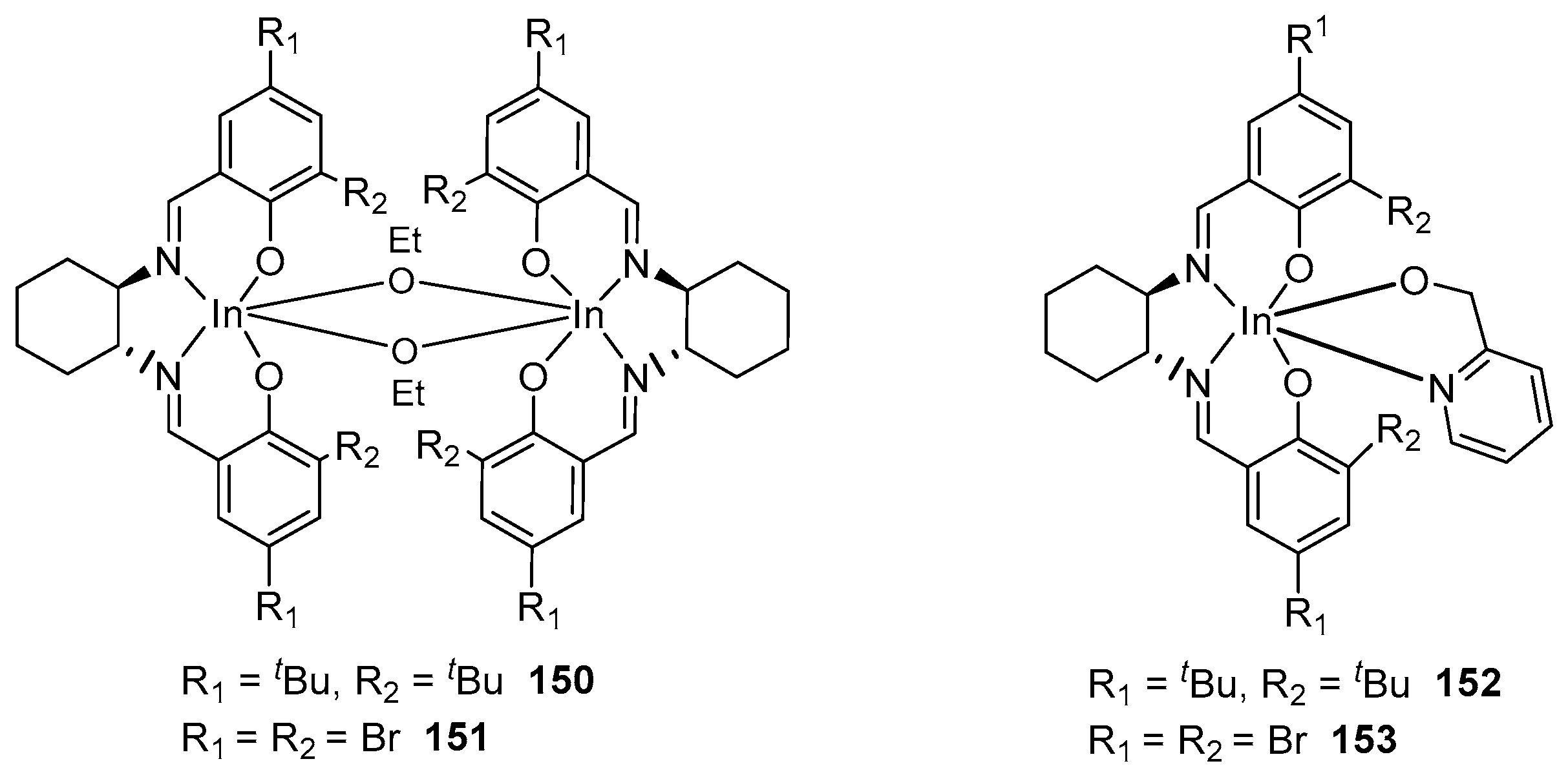
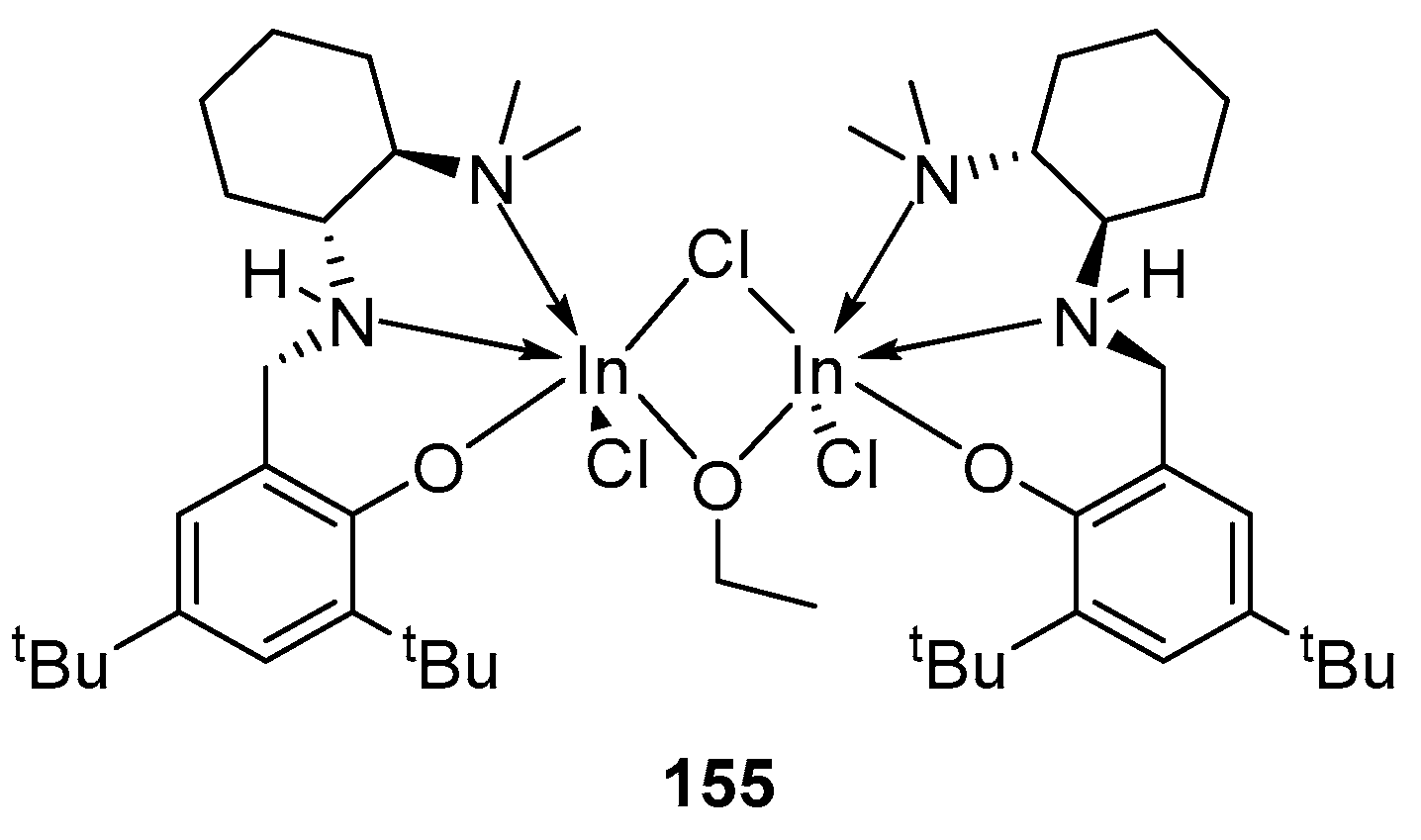
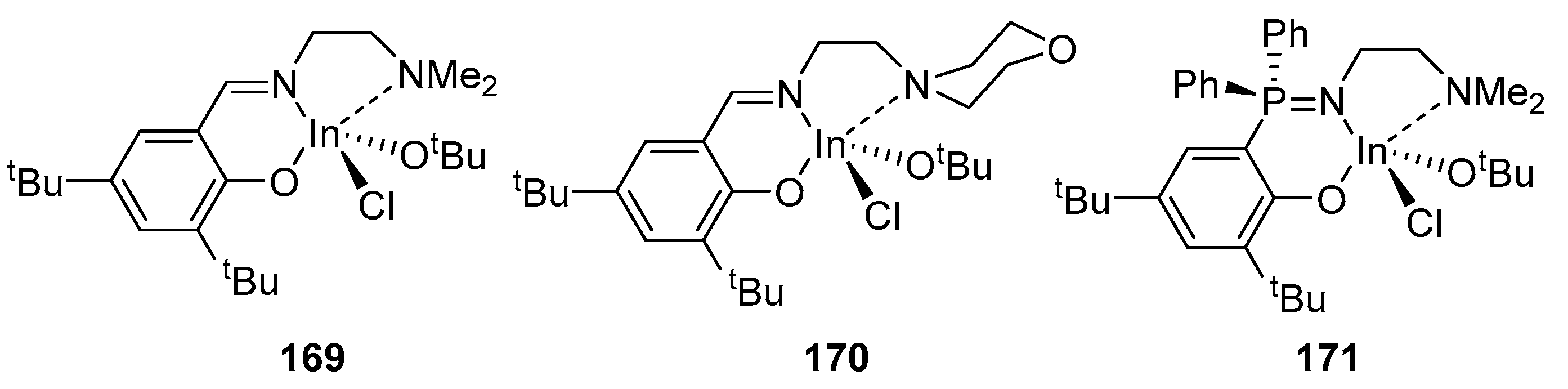
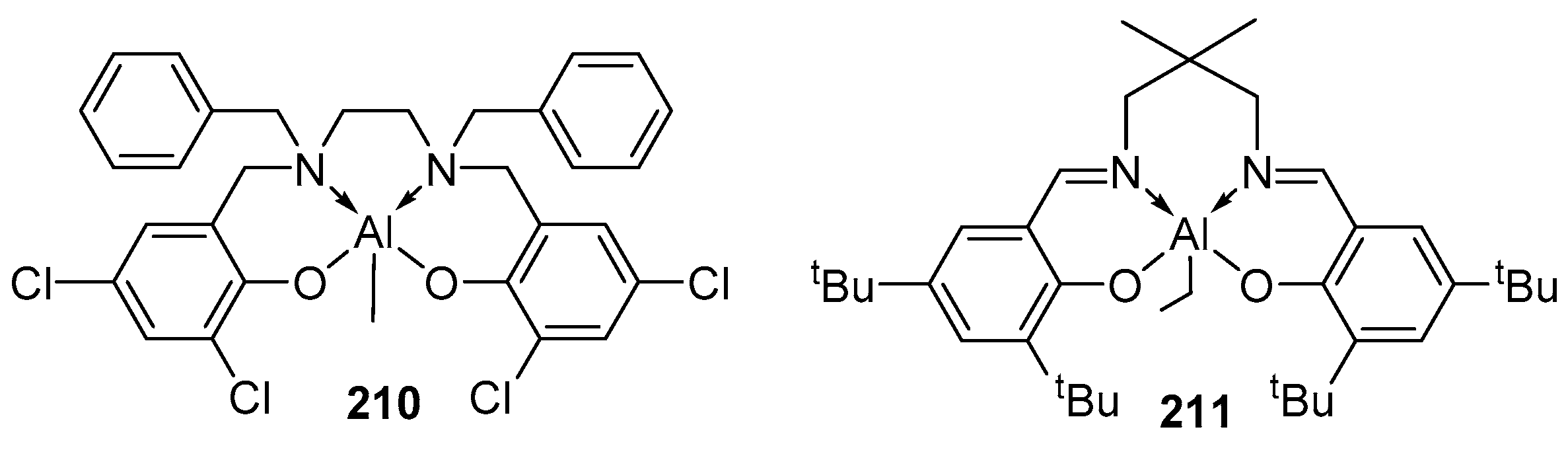
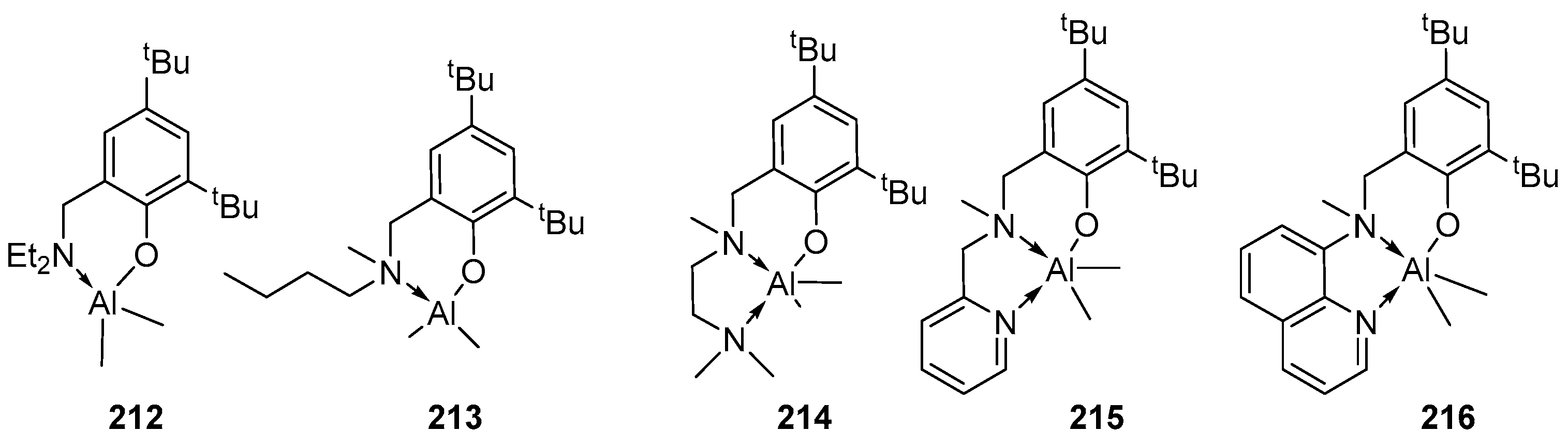
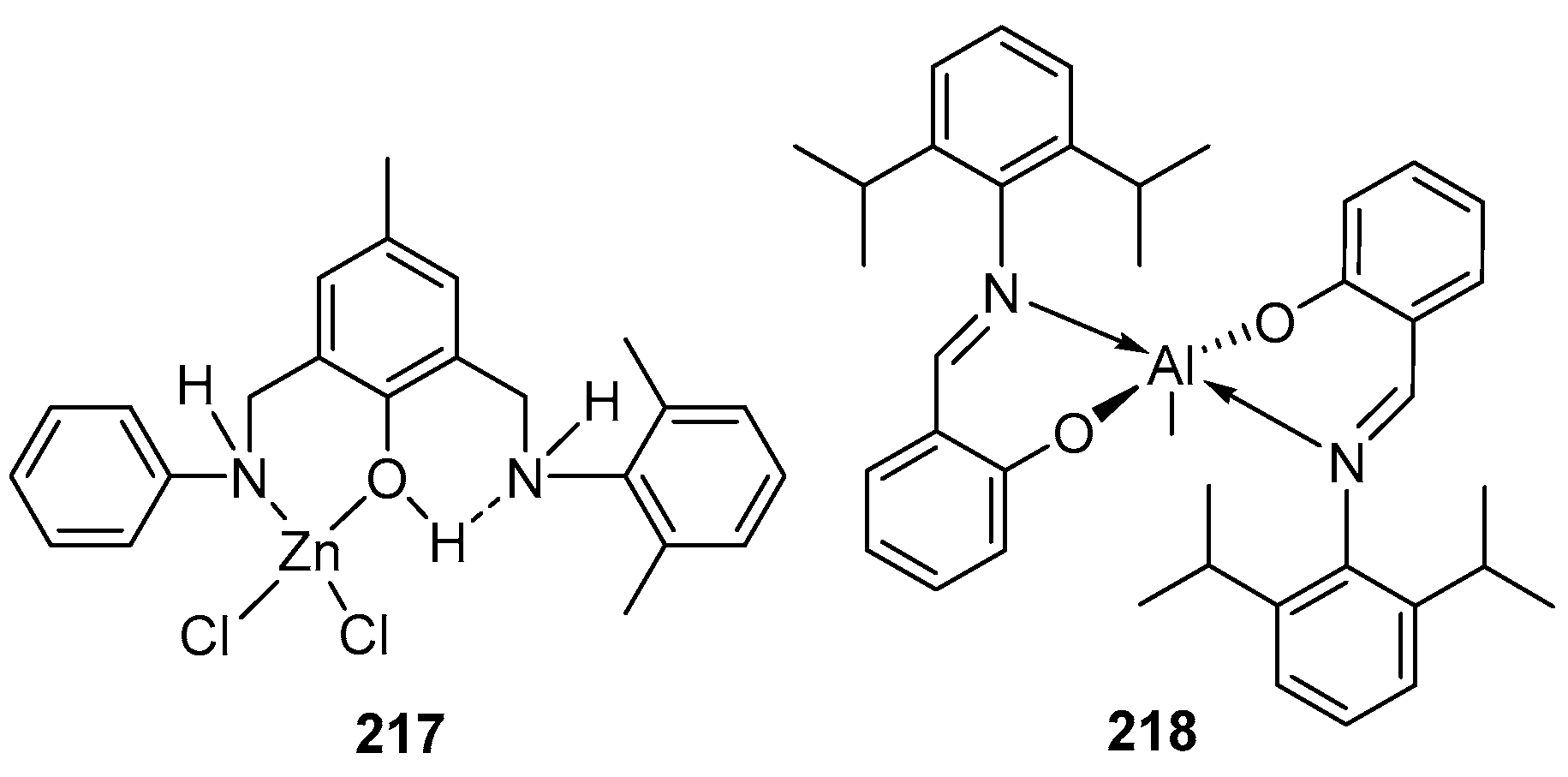
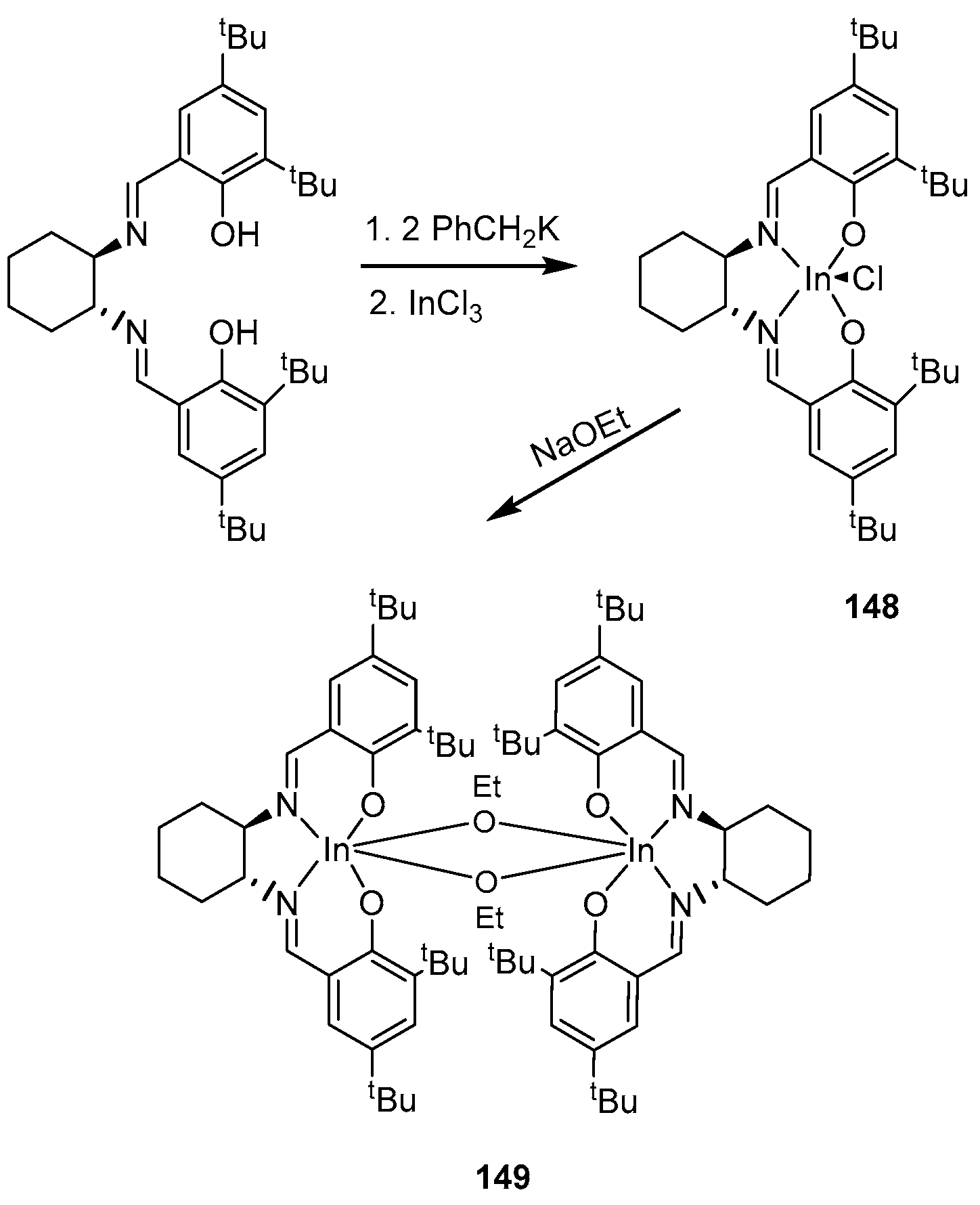
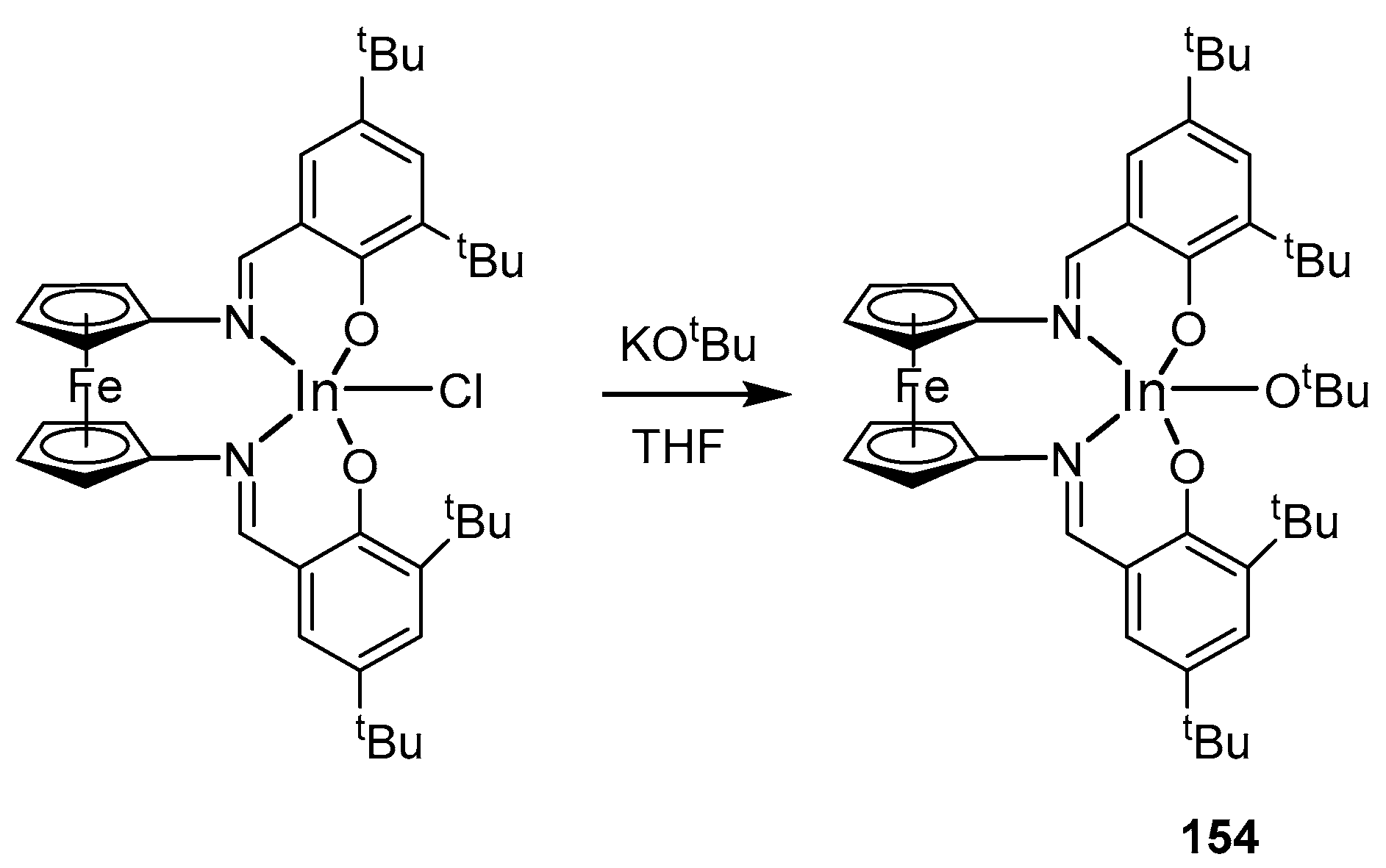
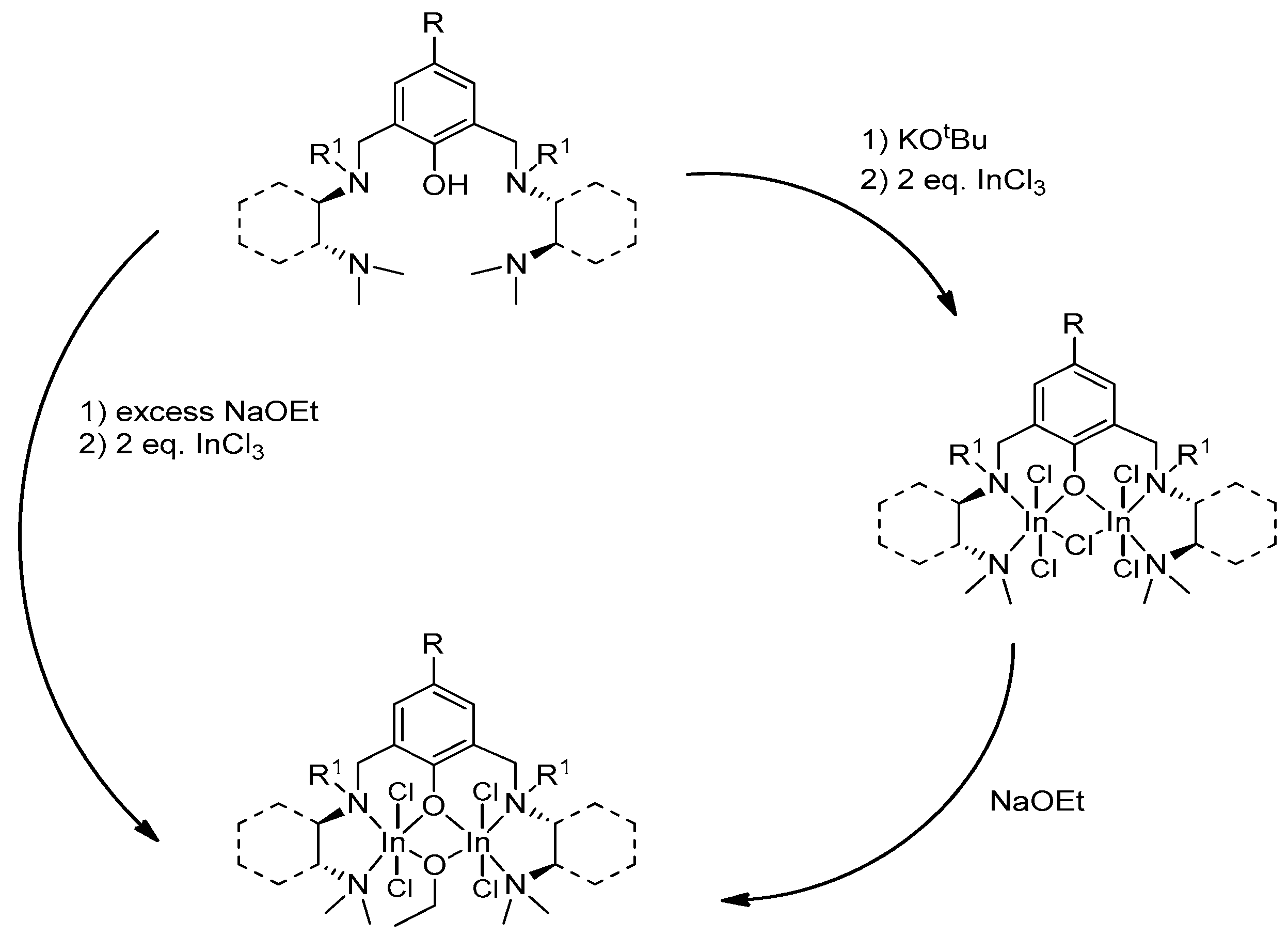
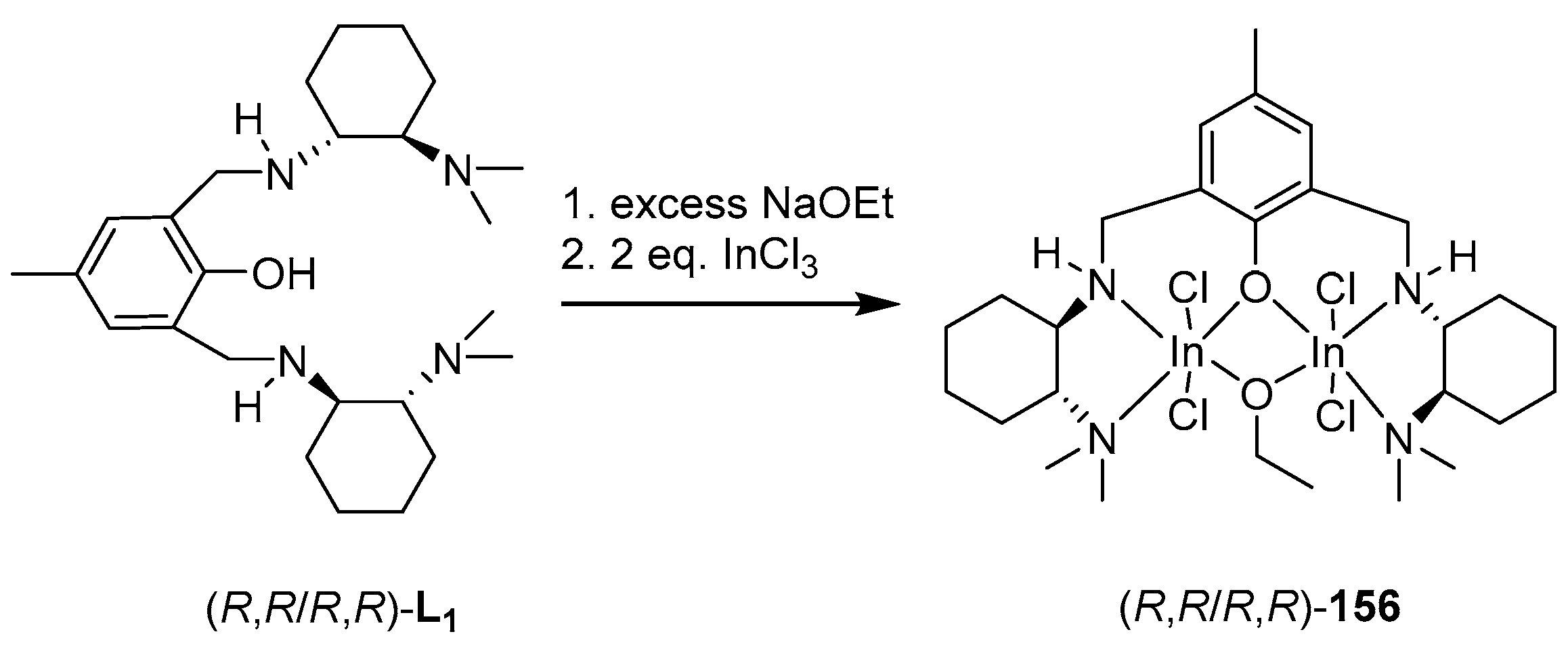
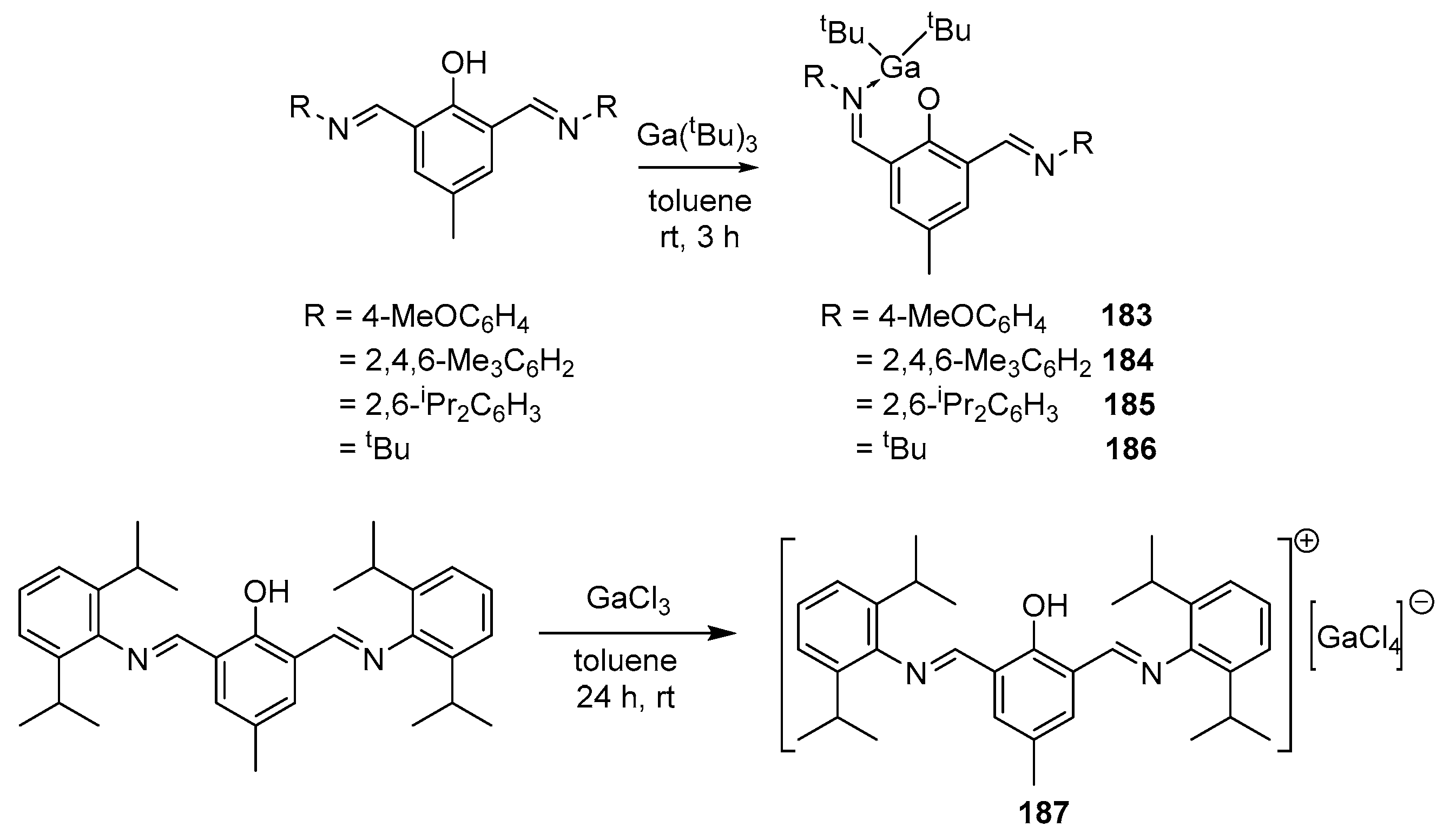
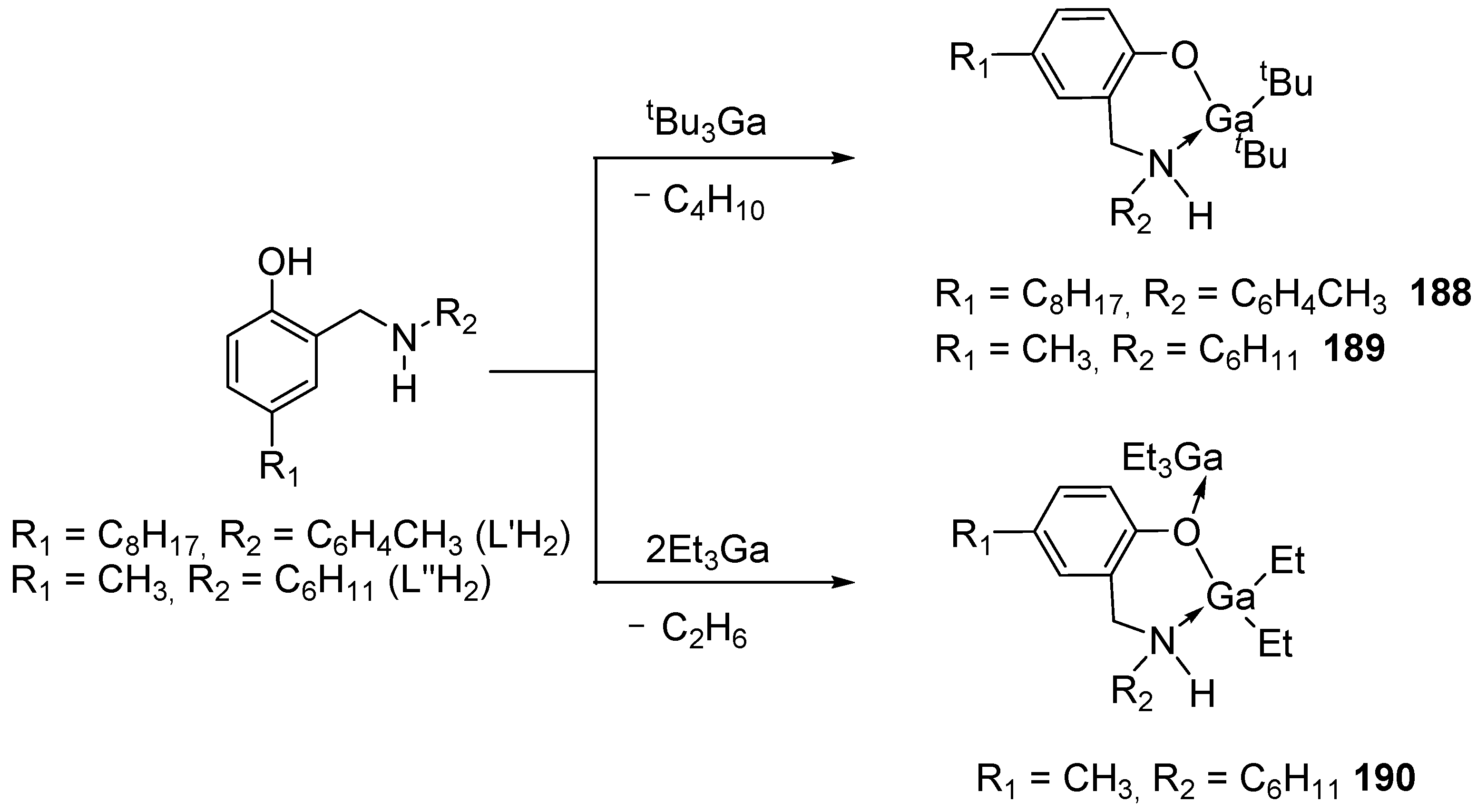
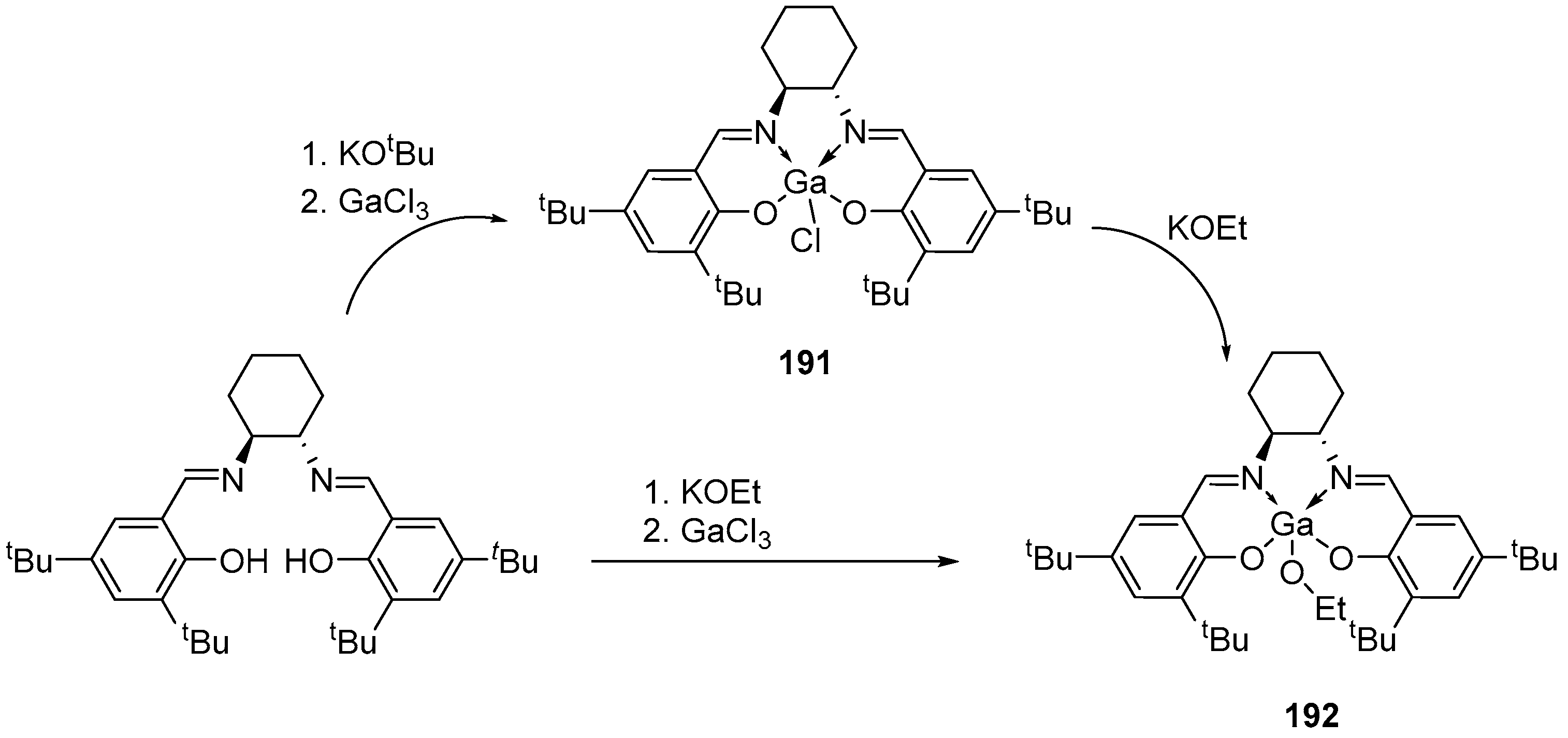
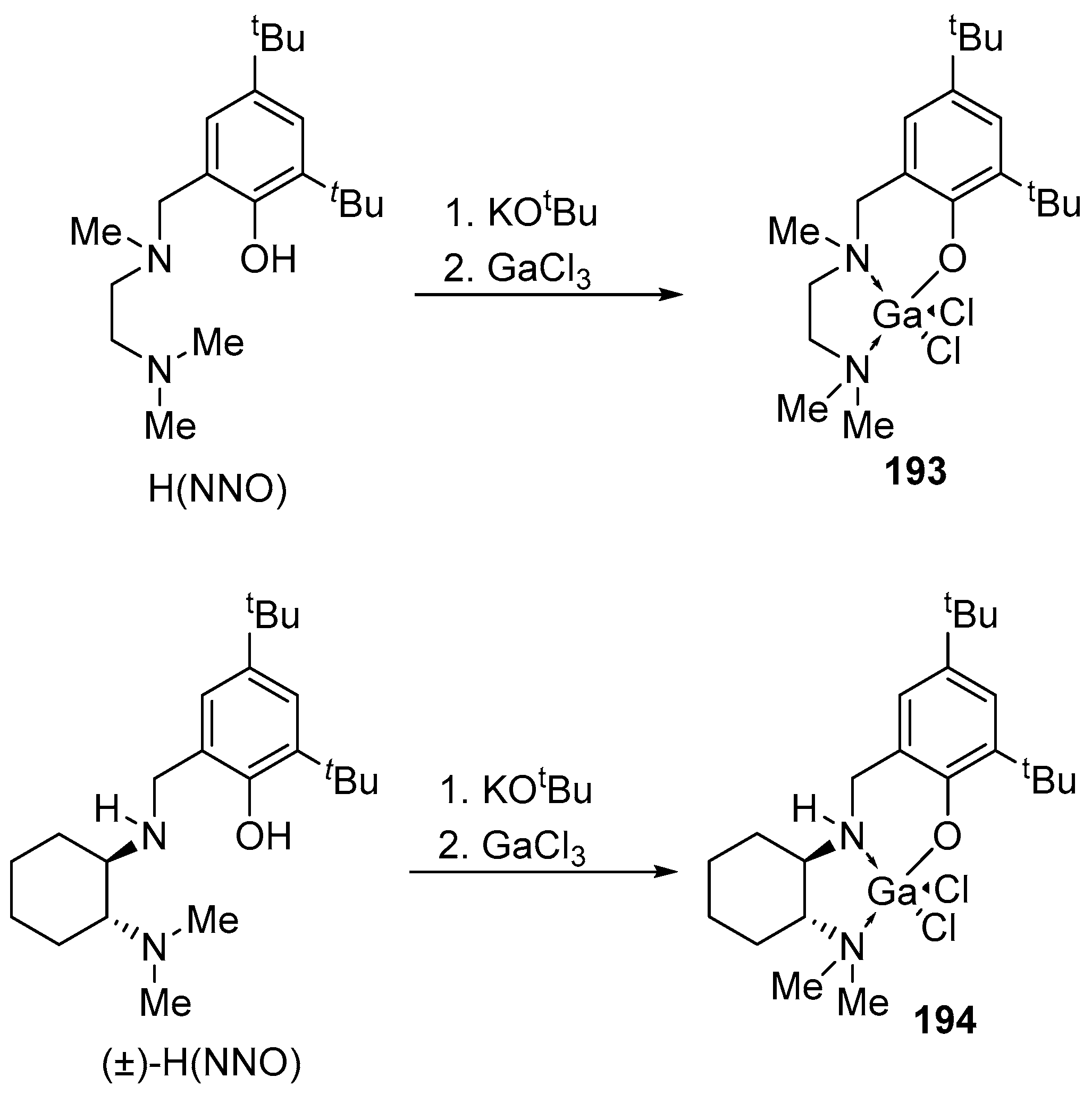
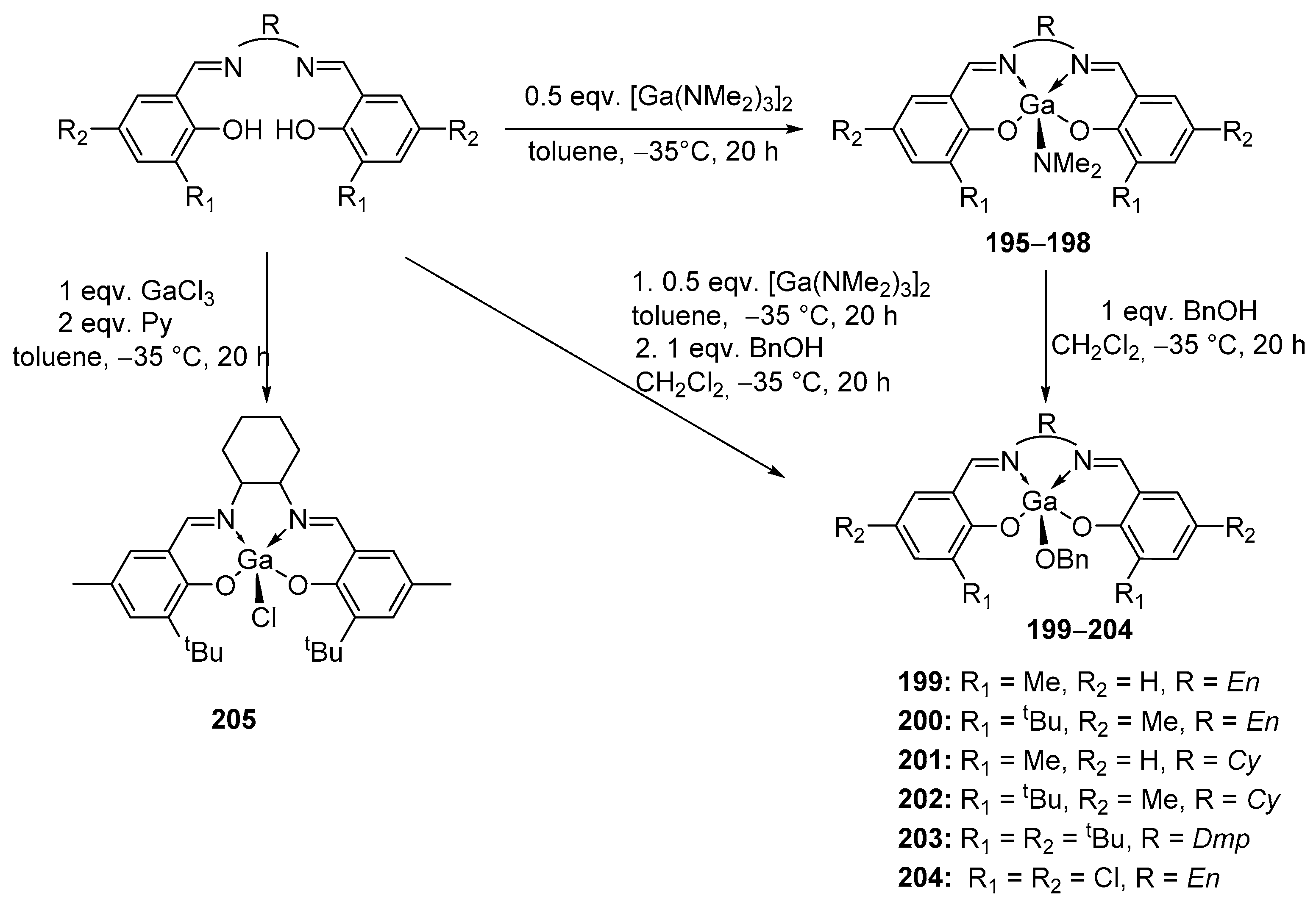
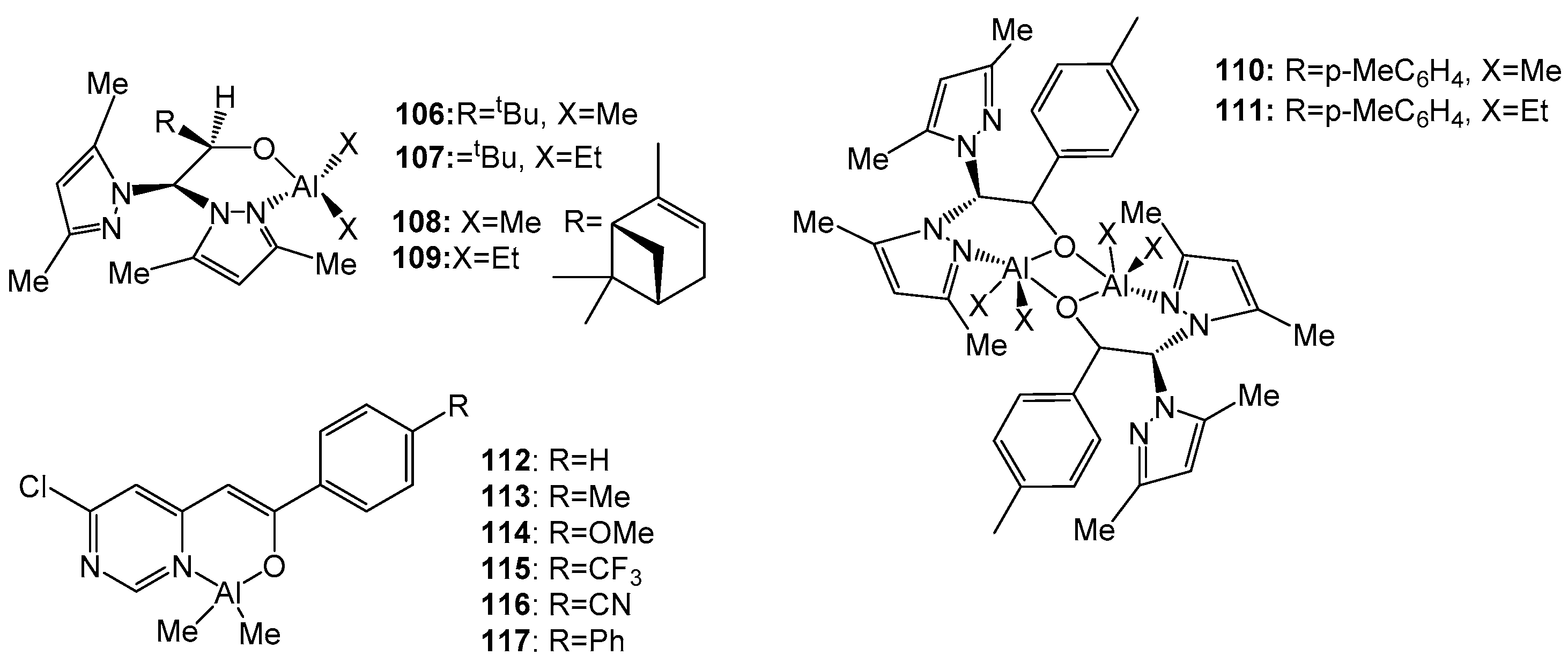
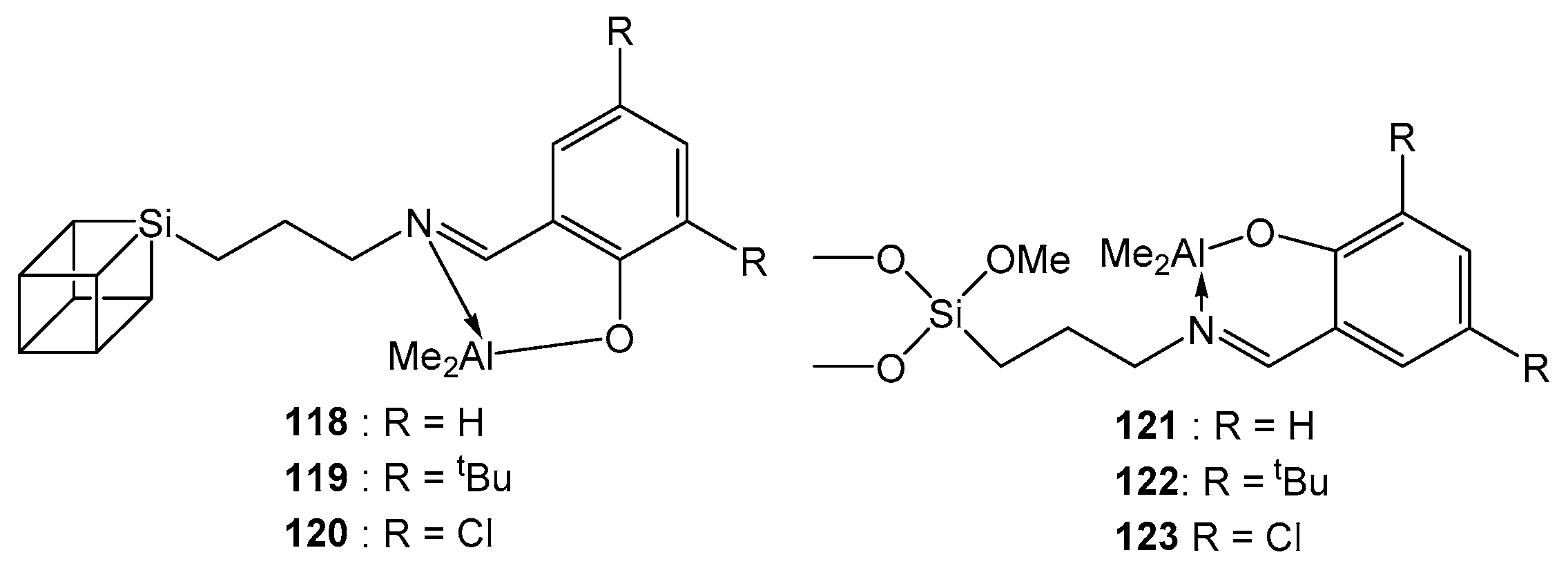
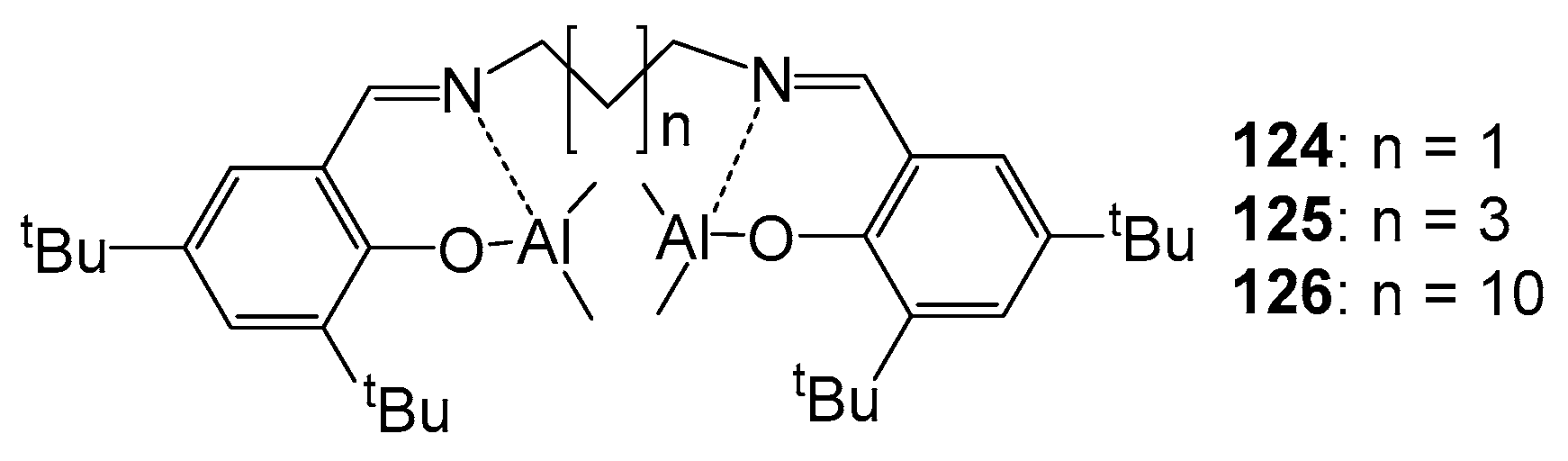
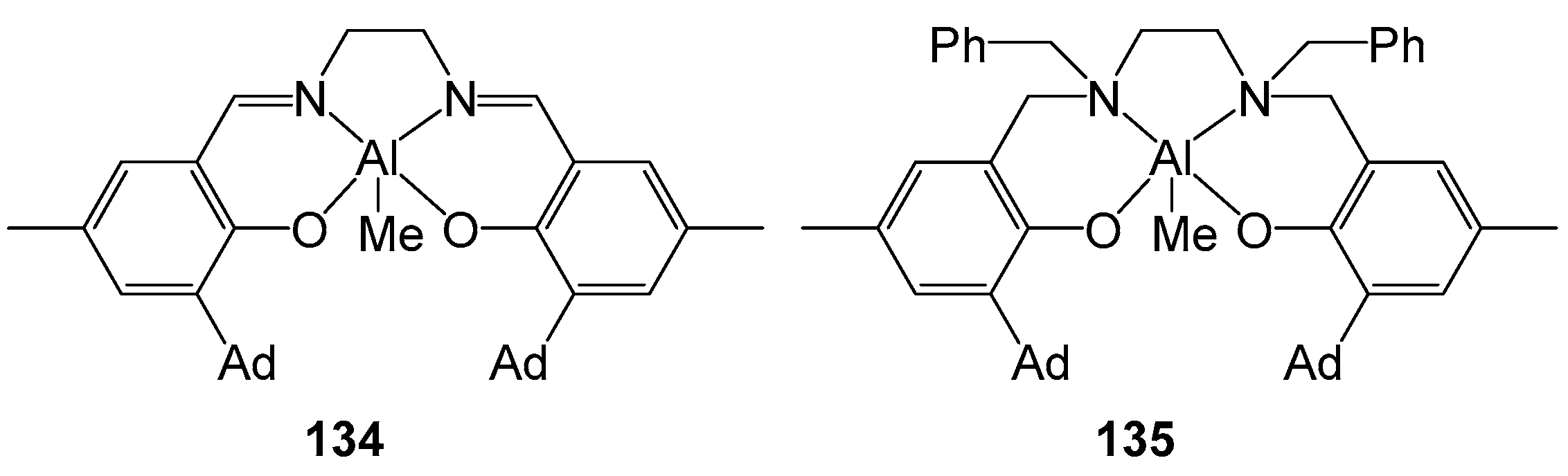
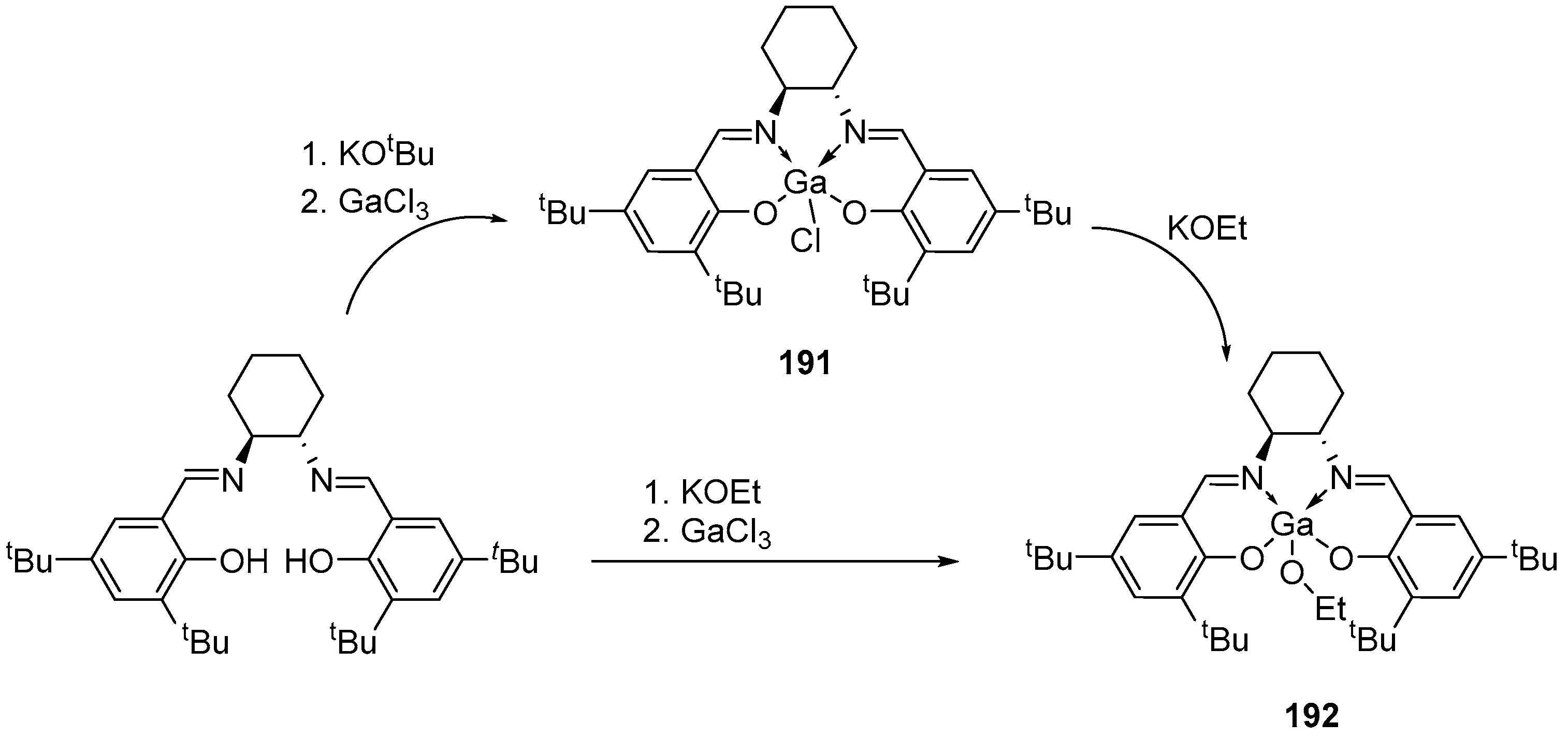
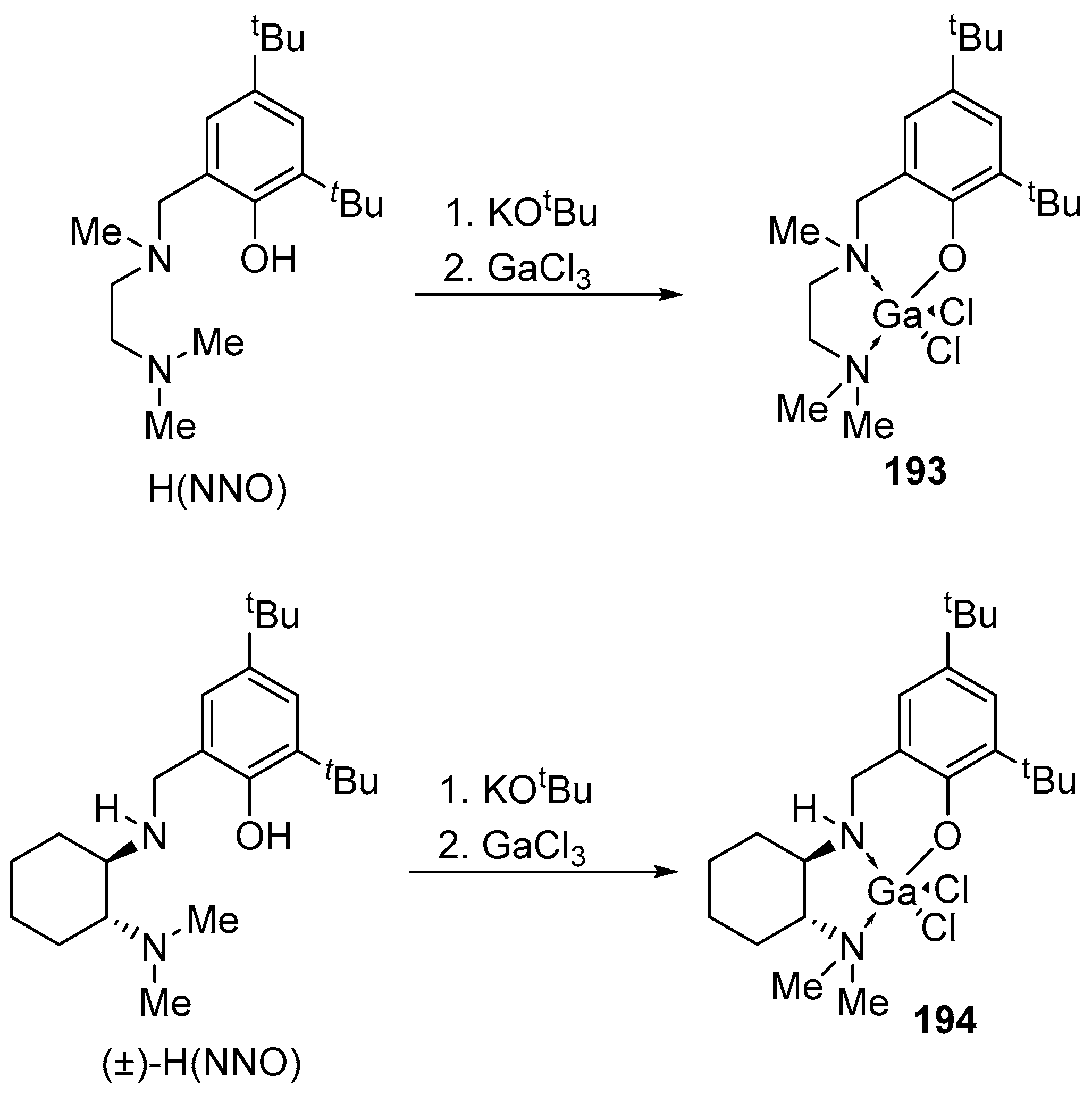
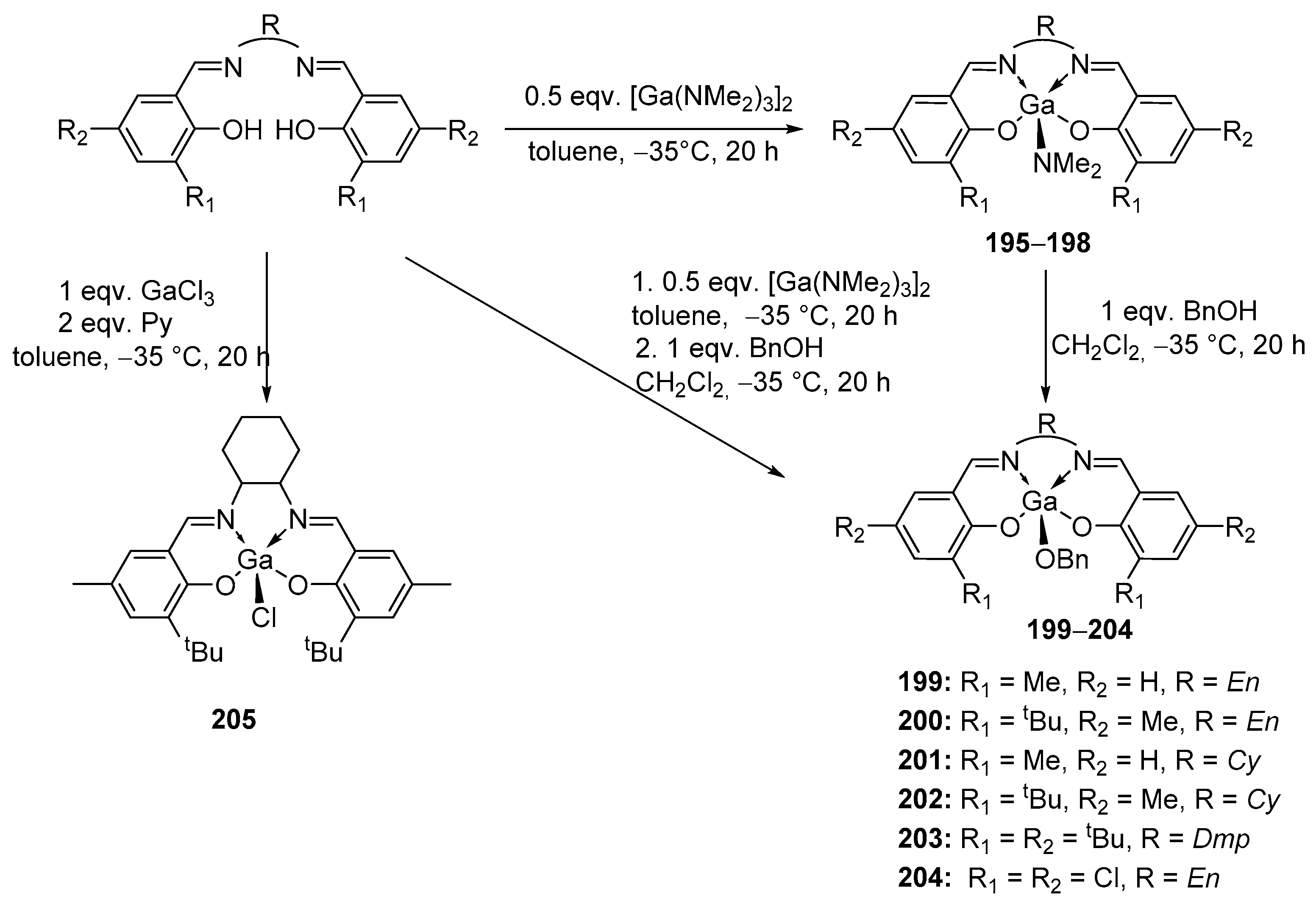
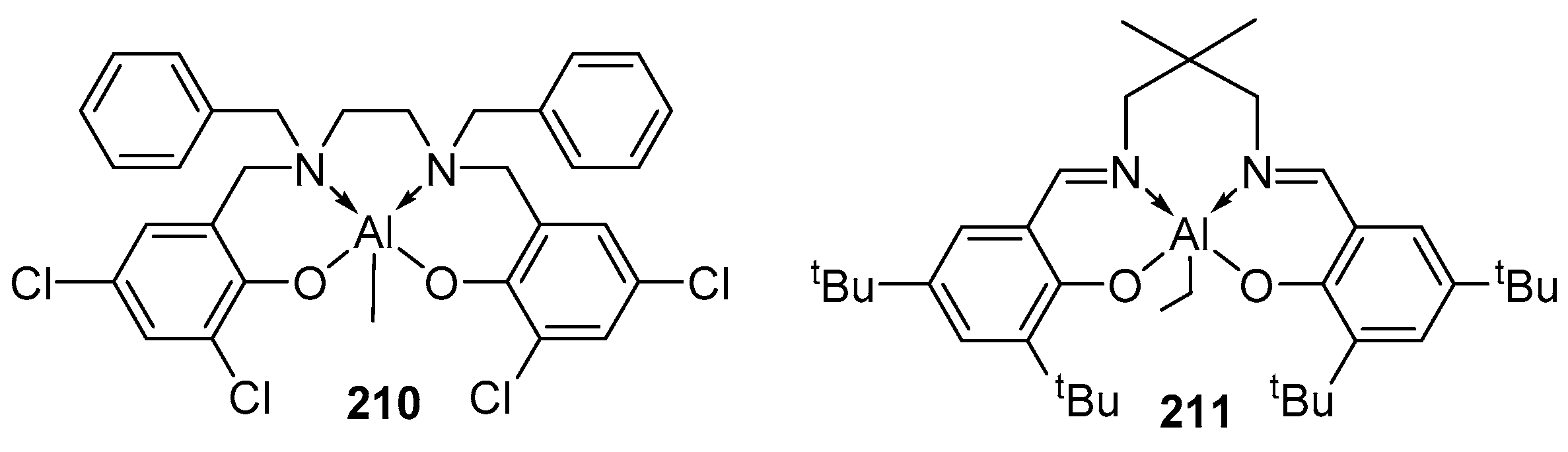
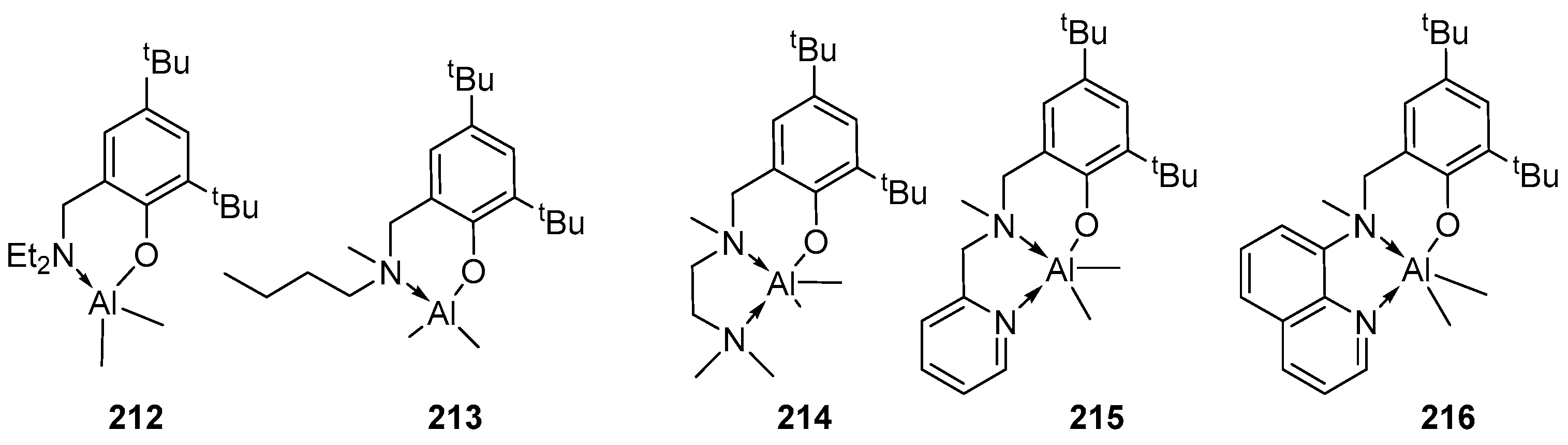
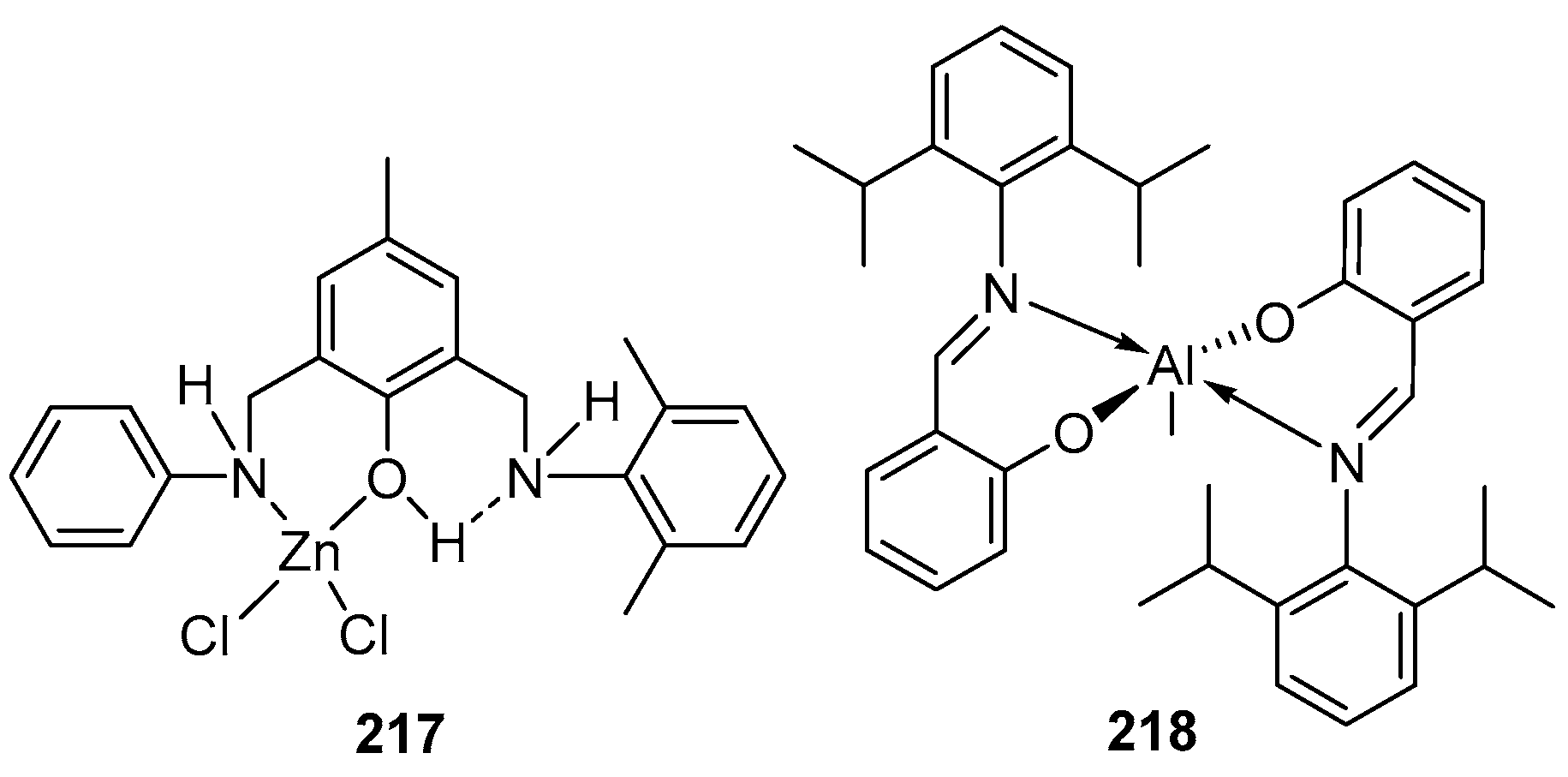
4. Group 13 Metals (Al, Ga, In)
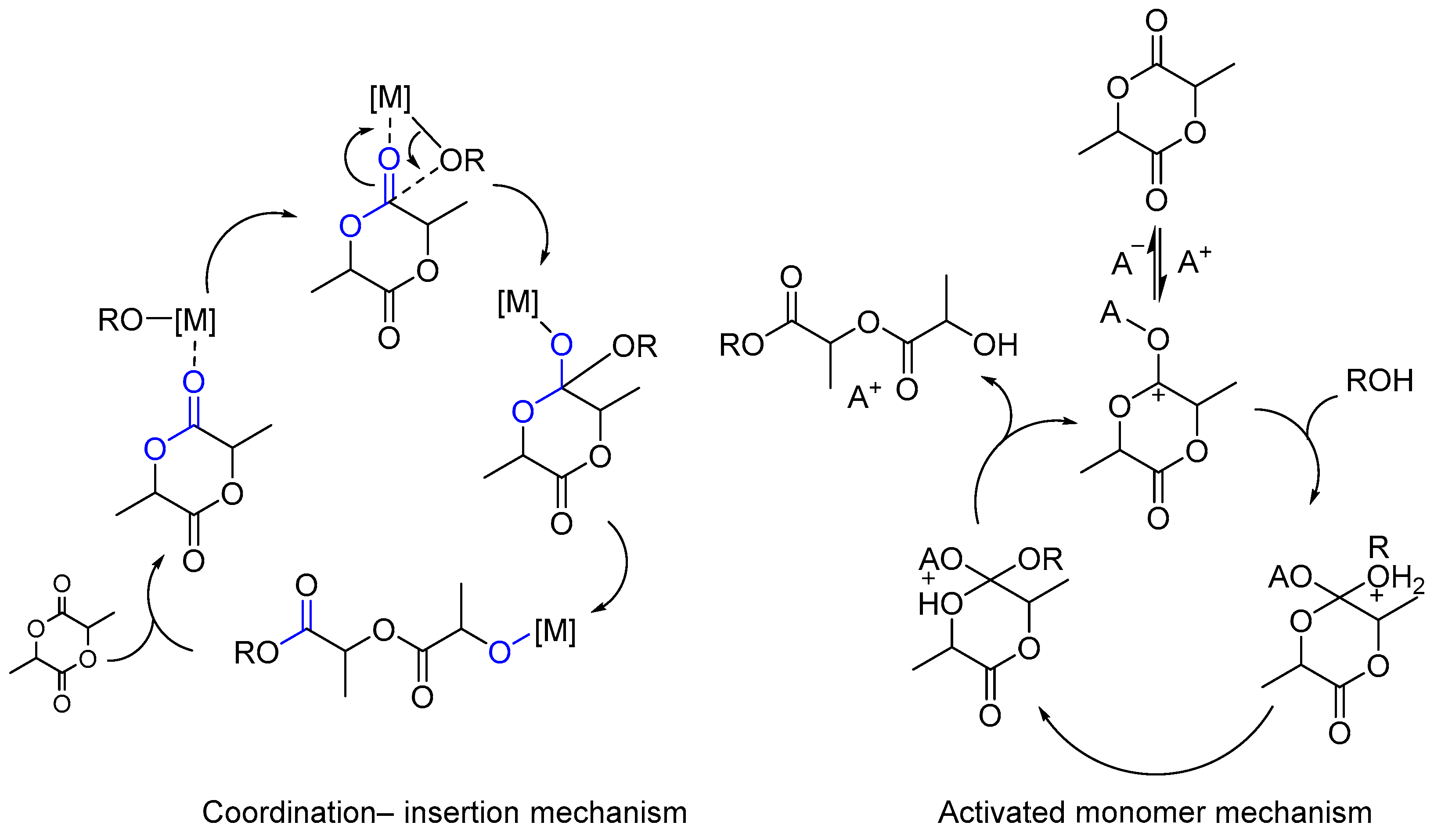
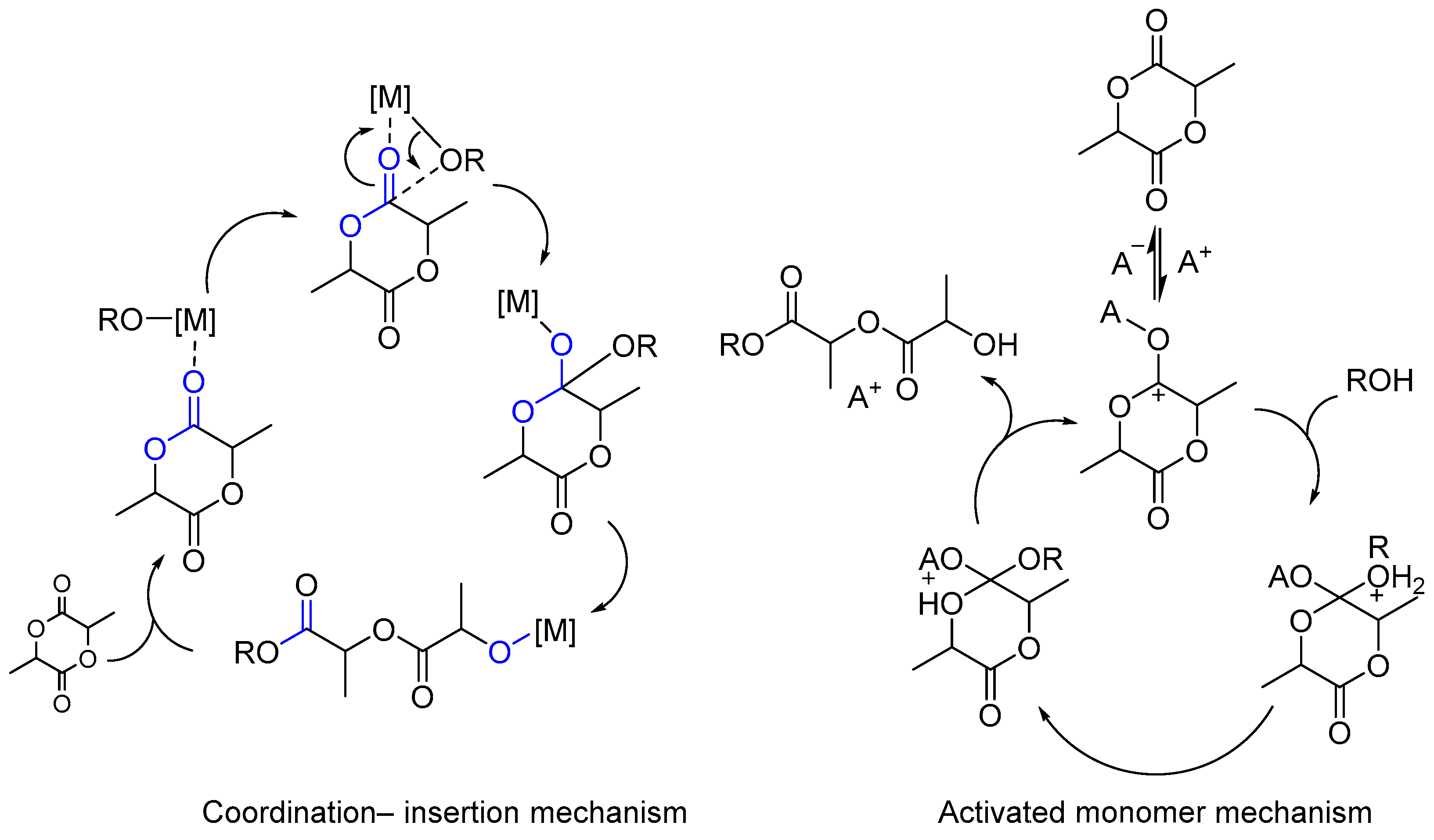
5. Metal Complexes as Initiators for Controlled Copolymerization of Lactide with Lactones and Cyclic Carbonates
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Dechy-Cabaret, O.; Martin-Vaca, B.; Bourissou, D. Controlled Ring-Opening Polymerization of Lactide and Glycolide. Chem. Rev. 2004, 104, 6147–6176. [Google Scholar] [CrossRef]
- Dove, A.P.; Gibson, V.C.; Marshall, E.L.; White, A.J.P.; Williams, D.J. Magnesium and zinc complexes of a potentially tridentate β-diketiminate ligand. Dalton Trans. 2004, 4, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.; Kumar, V.; Bhunia, H.; Upadhyay, S.N. Synthesis of Poly(Lactic Acid): A Review. J. Macromol. Sci. Part C 2005, 45, 325–349. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Patmore, N.J.; Zhou, Z. Concerning the relative importance of enantiomorphic site vs. chain end control in the stereoselective polymerization of lactides: Reactions of (R,R-salen)- and (S,S-salen)–aluminium alkoxides LAlOCH2R complexes (R = CH3 and S-CHMeCl). Chem. Commun. 2005, 1, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Nomura, N.; Ishii, R.; Yamamoto, Y.; Kondo, T. Stereoselective Ring-Opening Polymerization of a Racemic Lactide by Using Achiral Salen– and Homosalen–Aluminum Complexes. Chem. Eur. J. 2007, 13, 4433–4451. [Google Scholar] [CrossRef] [PubMed]
- Kricheldorf, H.R. Syntheses and application of polylactides. Chemosphere 2001, 43, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Degée, P.; Dubois, P.; Jérǒme, R.; Jacobsen, S.; Fritz, H.-G. New catalysis for fast bulk ring-opening polymerization of lactide monomers. Macromol. Symp. 1999, 144, 289–302. [Google Scholar] [CrossRef]
- Dong, C.-M.; Qiu, K.-Y.; Gu, Z.-W.; Feng, X.-D. Synthesis of Star-Shaped Poly(ε-caprolactone)-b-poly(dl-lactic acid-alt-glycolic acid) with Multifunctional Initiator and Stannous Octoate Catalyst. Macromolecules 2001, 34, 4691–4696. [Google Scholar] [CrossRef]
- Nakayama, Y.; Aihara, K.; Cai, Z.; Shiono, T.; Tsutsumi, C. Synthesis and Biodegradation of Poly(l-lactide-co-β-propiolactone). Int. J. Mol. Sci. 2017, 18, 1312. [Google Scholar] [CrossRef]
- Santoro, O.; Zhang, X.; Redshaw, C. Synthesis of Biodegradable Polymers: A Review on the Use of Schiff-Base Metal Complexes as Catalysts for the Ring Opening Polymerization (ROP) of Cyclic Esters. Catalysts 2020, 10, 800. [Google Scholar] [CrossRef]
- Fuoco, T.; Pappalardo, D. Aluminum Alkyl Complexes Bearing Salicylaldiminato Ligands: Versatile Initiators in the Ring-Opening Polymerization of Cyclic Esters. Catalysts 2017, 7, 64. [Google Scholar] [CrossRef]
- Wang, L.; Roşca, S.-C.; Poirier, V.; Sinbandhit, S.; Dorcet, V.; Roisnel, T.; Carpentier, J.-F.; Sarazin, Y. Stable divalent germanium, tin and lead amino(ether)-phenolate monomeric complexes: Structural features, inclusion heterobimetallic complexes, and ROP catalysis. Dalton Trans. 2014, 43, 4268–4286. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kefalidis, C.E.; Sinbandhit, S.; Dorcet, V.; Carpentier, J.-F.; Maron, L.; Sarazin, Y. Heteroleptic Tin(II) Initiators for the Ring-Opening (Co)Polymerization of Lactide and Trimethylene Carbonate: Mechanistic Insights from Experiments and Computations. Chem. Eur. J. 2013, 19, 13463–13478. [Google Scholar] [CrossRef] [PubMed]
- Aubrecht, K.B.; Hillmyer, M.A.; Tolman, W.B. Polymerization of Lactide by Monomeric Sn(II) Alkoxide Complexes. Macromolecules 2002, 35, 644–650. [Google Scholar] [CrossRef]
- Dove, A.P.; Gibson, V.C.; Marshall, E.L.; Rzepa, H.S.; White, A.J.P.; Williams, D.J. Synthetic, Structural, Mechanistic, and Computational Studies on Single-Site β-Diketiminate Tin(II) Initiators for the Polymerization of rac-Lactide. J. Am. Chem. Soc. 2006, 128, 9834–9843. [Google Scholar] [CrossRef] [PubMed]
- Nimitsiriwat, N.; Gibson, V.C.; Marshall, E.L.; Elsegood, M.R.J. Bidentate salicylaldiminato tin(ii) complexes and their use as lactide polymerisation initiators. Dalton Trans. 2009, 19, 3710–3715. [Google Scholar] [CrossRef] [PubMed]
- Mankaev, B.N.; Zaitsev, K.V.; Kuchuk, E.A.; Vershinina, M.V.; Zaitseva, G.S.; Egorov, M.P.; Karlov, S.S. New tetrylenes based on substituted diethylenetriamines: Synthesis and use as initiators for ε-caprolactone polymerization. Russ. Chem. Bull. 2019, 68, 389–393. [Google Scholar] [CrossRef]
- Mankaev, B.N.; Zaitsev, K.V.; Zaitseva, G.S.; Churakov, A.V.; Egorov, M.P.; Karlov, S.S. Sterically hindered tetrylenes based on new 1,10-phenanthroline-containing diols: Initiators for ε-caprolactone polymerization. Russ. Chem. Bull. 2019, 68, 380–388. [Google Scholar] [CrossRef]
- DeStefano, V.; Khan, S.; Tabada, A. Applications of PLA in modern medicine. Eng. Regen. 2020, 1, 76–87. [Google Scholar] [CrossRef]
- Dhanasekaran, N.P.D.; Muthuvelu, K.S.; Arumugasamy, S.K. Recent Advancement in Biomedical Applications of Polycaprolactone and Polycaprolactone-Based Materials. In Encyclopedia of Materials: Plastics and Polymers; Hashmi, M.S.J., Ed.; Elsevier: Oxford, UK, 2022; pp. 795–809. [Google Scholar]
- Stirling, E.; Champouret, Y.; Visseaux, M. Catalytic metal-based systems for controlled statistical copolymerisation of lactide with a lactone. Polym. Chem. 2018, 9, 2517–2531. [Google Scholar] [CrossRef]
- Lyubov, D.M.; Tolpygin, A.O.; Trifonov, A.A. Rare-earth metal complexes as catalysts for ring-opening polymerization of cyclic esters. Coord. Chem. Rev. 2019, 392, 83–145. [Google Scholar] [CrossRef]
- Amgoune, A.; Thomas, C.M.; Carpentier, J.-F. Controlled ring-opening polymerization of lactide by group 3 metal complexes. Pure Appl. Chem. 2007, 79, 2013–2030. [Google Scholar] [CrossRef]
- Ajellal, N.; Carpentier, J.-F.; Guillaume, C.; Guillaume, S.M.; Helou, M.; Poirier, V.; Sarazin, Y.; Trifonov, A. Metal-catalyzed immortal ring-opening polymerization of lactones, lactides and cyclic carbonates. Dalton Trans. 2010, 39, 8363–8376. [Google Scholar] [CrossRef] [PubMed]
- Singha Roy, S.; Sarkar, S.; Chakraborty, D. Living and Immortal Ring-Opening Polymerization of Cyclic Esters. Adv. Mater. Lett. 2023, 14, 23021717. [Google Scholar] [CrossRef]
- Chellali, J.E.; Alverson, A.K.; Robinson, J.R. Zinc Aryl/Alkyl β-diketiminates: Balancing Accessibility and Stability for High-Activity Ring-Opening Polymerization of rac-Lactide. ACS Catal. 2022, 12, 5585–5594. [Google Scholar] [CrossRef]
- Zhang, H.; Dong, Y.; Huang, K.; Liu, J.; Dong, B.; Wang, F. Immortal Ring Opening Polymerization of ε-caprolactone and rac-lactide by magnesium precatalysts bearing sterically congested phenoxide ligands. Eur. Polym. J. 2019, 118, 633–641. [Google Scholar] [CrossRef]
- Zang, M.; Cao, W.; Zhang, X.; Liu, S.; Li, Z. Fast and living ring-opening polymerization of ε-caprolactone by aluminum complexes bearing amino-quinoline ligands. Eur. Polym. J. 2023, 193, 112103. [Google Scholar] [CrossRef]
- Ryves, W.J.; Harwood, A.J. Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem. Biophys. Res. Commun. 2001, 280, 720–725. [Google Scholar] [CrossRef]
- Sipos, L.; Zsuga, M. Anionic Polymerization of L-Lactide Effect of Lithium and Potassium as Counterions. J. Macromol. Sci. Part A 1997, 34, 1269–1284. [Google Scholar] [CrossRef]
- Hsueh, M.-L.; Huang, B.-H.; Wu, J.; Lin, C.-C. Synthesis, Characterization, and Catalytic Studies of Lithium Complexes: Efficient Initiators for Ring-Opening Polymerization of l-Lactide. Macromolecules 2005, 38, 9482–9487. [Google Scholar] [CrossRef]
- Huang, C.-A.; Chen, C.-T. Lithium complexes supported by amine bis-phenolate ligands as efficient catalysts for ring-opening polymerization of l-lactide. Dalton Trans. 2007, 47, 5561–5566. [Google Scholar] [CrossRef] [PubMed]
- Ko, B.-T.; Lin, C.-C. Synthesis, Characterization, and Catalysis of Mixed-Ligand Lithium Aggregates, Excellent Initiators for the Ring-Opening Polymerization of l-Lactide. J. Am. Chem. Soc. 2001, 123, 7973–7977. [Google Scholar] [CrossRef]
- Huang, B.H.; Dutta, S.; Lin, C.C. 1.39-Main-Group Catalysts for Lactide Polymerization. In Comprehensive Inorganic Chemistry II, 2nd ed.; Reedijk, J., Poeppelmeier, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1217–1249. [Google Scholar]
- Gao, J.; Zhu, D.; Zhang, W.; Solan, G.A.; Ma, Y.; Sun, W.-H. Recent progress in the application of group 1, 2 & 13 metal complexes as catalysts for the ring opening polymerization of cyclic esters. Inorg. Chem. Front. 2019, 6, 2619–2652. [Google Scholar]
- Kober, E.; Petrus, R.; Kocięcka, P.; Janas, Z.; Sobota, P. Lithium diaminebis(aryloxido) complexes: Synthesis, structures and reactivity in l-lactide polymerization. Polyhedron 2015, 85, 814–823. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Lin, C.-C.; Gallucci, J.C.; Ko, B.-T. Binolate complexes of lithium, zinc, aluminium, and titanium; preparations, structures, and studies of lactide polymerization. Dalton Trans. 2003, 3, 406–412. [Google Scholar] [CrossRef]
- Ghosh, S.; Chakraborty, D.; Varghese, B. Group 1 salts of the imino(phenoxide) scaffold: Synthesis, structural characterization and studies as catalysts towards the bulk ring opening polymerization of lactides. Eur. Polym. J. 2015, 62, 51–65. [Google Scholar] [CrossRef]
- Xiong, J.; Sun, Y.; Jiang, J.; Chen, C.; Pan, X.; Wang, C.; Wu, J. Metal-size influence of alkali metal complexes for polymerization of rac-lactide. Polyhedron 2018, 141, 118–124. [Google Scholar] [CrossRef]
- Devaine-Pressing, K.; Oldenburg, F.J.; Menzel, J.P.; Springer, M.; Dawe, L.N.; Kozak, C.M. Lithium, sodium, potassium and calcium amine-bis(phenolate) complexes in the ring-opening polymerization of rac-lactide. Dalton Trans. 2020, 49, 1531–1544. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Zhou, Z. New generation polymers: The role of metal alkoxides as catalysts in the production of polyoxygenates. J. Mater. Chem. 2004, 14, 3081–3092. [Google Scholar] [CrossRef]
- Biela, T.; Kowalski, A.; Libiszowski, J.; Duda, A.; Penczek, S. Progress in Polymerization of Cyclic Esters: Mechanisms and Synthetic Applications. Macromol. Symp. 2006, 240, 47–55. [Google Scholar] [CrossRef]
- Wu, J.; Yu, T.-L.; Chen, C.-T.; Lin, C.-C. Recent developments in main group metal complexes catalyzed/initiated polymerization of lactides and related cyclic esters. Coord. Chem. Rev. 2006, 250, 602–626. [Google Scholar] [CrossRef]
- Carpentier, J.-F. Discrete Metal Catalysts for Stereoselective Ring-Opening Polymerization of Chiral Racemic β-Lactones. Macromol. Rapid Commun. 2010, 31, 1696–1705. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M. Stereocontrolled ring-opening polymerization of cyclic esters: Synthesis of new polyester microstructures. Chem. Soc. Rev. 2010, 39, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Buffet, J.-C.; Okuda, J. Initiators for the stereoselective ring-opening polymerization of meso-lactide. Polym. Chem. 2011, 2, 2758–2763. [Google Scholar] [CrossRef]
- Gallegos, C.; Tabernero, V.; García-Valle, F.M.; Mosquera, M.E.G.; Cuenca, T.; Cano, J. Synthesis and Structure of Homo- and Heterometallic Lithium–Magnesium Complexes and Their Reactivity in the ROP of rac-Lactide. Organometallics 2013, 32, 6624–6627. [Google Scholar] [CrossRef]
- Gallegos, C.; Tabernero, V.; Mosquera, M.E.G.; Cuenca, T.; Cano, J. Comparative Study of Lactide Polymerization with Lithium, Sodium, Potassium, Magnesium, Calcium, and Zinc Azonaphthoxide Complexes. Eur. J. Inorg. Chem. 2015, 2015, 5124–5132. [Google Scholar] [CrossRef]
- Ghosh, S.; Chakraborty, D.; Ramkumar, V. Imino(phenoxide) compounds of magnesium: Synthesis, structural characterization, and polymerization studies. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 1474–1491. [Google Scholar] [CrossRef]
- Lee, W.-Y.; Hsieh, H.-H.; Hsieh, C.-C.; Lee, H.M.; Lee, G.-H.; Huang, J.-H.; Wu, T.-C.; Chuang, S.-H. Four- and five-coordinate magnesium compounds containing ketiminate ligands. Synthesis and characterization of L2Mg, L2Mg(LH), and L2Mg(Py), where L=MeC(O)CHC(NAr)Me. J. Organomet. Chem. 2007, 692, 1131–1137. [Google Scholar] [CrossRef]
- Raghavendra, B.; Bakthavachalam, K.; Ramakrishna, B.; Dastagiri Reddy, N. N-Benzoylbenzamidinate Complexes of Magnesium: Catalysts for the Ring-Opening Polymerization of ε-Caprolactone and CO2/Epoxide Coupling. Organometallics 2017, 36, 4005–4012. [Google Scholar] [CrossRef]
- Raghavendra, B.; Shashank, P.V.S.; Pandey, M.K.; Reddy, N.D. CO2/Epoxide Coupling and the ROP of ε-Caprolactone: Mg and Al Complexes of γ-Phosphino-ketiminates as Dual-Purpose Catalysts. Organometallics 2018, 37, 1656–1664. [Google Scholar] [CrossRef]
- Devaine-Pressing, K.; Lehr, J.H.; Pratt, M.E.; Dawe, L.N.; Sarjeant, A.A.; Kozak, C.M. Magnesium amino-bis(phenolato) complexes for the ring-opening polymerization of rac-lactide. Dalton Trans. 2015, 44, 12365–12375. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Antharjanam, P.K.S.; Chakraborty, D. Magnesium complexes of the N, O polydentate scaffold: Synthesis, structural characterization and polymerization studies. Polymer 2015, 70, 38–51. [Google Scholar] [CrossRef]
- Nifant’ev, I.; Komarov, P.; Ovchinnikova, V.; Kiselev, A.; Minyaev, M.; Ivchenko, P. Comparative Experimental and Theoretical Study of Mg, Al and Zn Aryloxy Complexes in Copolymerization of Cyclic Esters: The Role of the Metal Coordination in Formation of Random Copolymers. Polymers 2020, 12, 2273. [Google Scholar] [CrossRef] [PubMed]
- Santulli, F.; Gravina, G.; Lamberti, M.; Tedesco, C.; Mazzeo, M. Zinc and magnesium catalysts for the synthesis for PLA and its degradation: Clues for catalyst design. Mol. Catal. 2022, 528, 112480. [Google Scholar] [CrossRef]
- Chamberlain, B.M.; Cheng, M.; Moore, D.R.; Ovitt, T.M.; Lobkovsky, E.B.; Coates, G.W. Polymerization of Lactide with Zinc and Magnesium β-Diiminate Complexes: Stereocontrol and Mechanism. J. Am. Chem. Soc. 2001, 123, 3229–3238. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, M.H.; Gallucci, J.; Phomphrai, K. Coordination Chemistry and Reactivity of Monomeric Alkoxides and Amides of Magnesium and Zinc Supported by the Diiminato Ligand CH(CMeNC6H3-2,6-iPr2)2. A Comparative Study. Inorg. Chem. 2002, 41, 2785–2794. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, M.H.; Gallucci, J.C.; Phomphrai, K. Comparative Study of the Coordination Chemistry and Lactide Polymerization of Alkoxide and Amide Complexes of Zinc and Magnesium with a β-Diiminato Ligand Bearing Ether Substituents. Inorg. Chem. 2005, 44, 8004–8010. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Barba, L.F.; Hughes, D.L.; Humphrey, S.M.; Bochmann, M. Ligand Transfer Reactions of Mixed-Metal Lanthanide/Magnesium Allyl Complexes with β-Diketimines: Synthesis, Structures, and Ring-Opening Polymerization Catalysis. Organometallics 2006, 25, 1012–1020. [Google Scholar] [CrossRef]
- Xie, H.; Mou, Z.; Liu, B.; Li, P.; Rong, W.; Li, S.; Cui, D. Phosphinimino-amino Magnesium Complexes: Synthesis and Catalysis of Heteroselective ROP of rac-Lactide. Organometallics 2014, 33, 722–730. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Eilerts, N.W.; Huffman, J.C.; Iyer, S.S.; Pacold, M.; Phomphrai, K. Molecular Design of Single-Site Metal Alkoxide Catalyst Precursors for Ring-Opening Polymerization Reactions Leading to Polyoxygenates. 1. Polylactide Formation by Achiral and Chiral Magnesium and Zinc Alkoxides, (η3-L)MOR, Where L = Trispyrazolyl- and Trisindazolylborate Ligands. J. Am. Chem. Soc. 2000, 122, 11845–11854. [Google Scholar]
- Chisholm, M.H.; Gallucci, J.; Phomphrai, K. Lactide polymerization by well-defined calcium coordination complexes: Comparisons with related magnesium and zinc chemistry. Chem. Commun. 2003, 1, 48–49. [Google Scholar] [CrossRef]
- Wu, J.-C.; Huang, B.-H.; Hsueh, M.-L.; Lai, S.-L.; Lin, C.-C. Ring-opening polymerization of lactide initiated by magnesium and zinc alkoxides. Polymer 2005, 46, 9784–9792. [Google Scholar] [CrossRef]
- Hung, W.-C.; Lin, C.-C. Preparation, Characterization, and Catalytic Studies of Magnesium Complexes Supported by NNO-Tridentate Schiff-Base Ligands. Inorg. Chem. 2009, 48, 728–734. [Google Scholar] [CrossRef]
- Huang, Y.; Hung, W.-C.; Liao, M.-Y.; Tsai, T.-E.; Peng, Y.-L.; Lin, C.-C. Ring-opening polymerization of lactides initiated by magnesium and zinc complexes based on NNO-tridentate ketiminate ligands: Activity and stereoselectivity studies. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 2318–2329. [Google Scholar] [CrossRef]
- Garcés, A.; Sánchez-Barba, L.F.; Fernández-Baeza, J.; Otero, A.; Honrado, M.; Lara-Sánchez, A.; Rodríguez, A.M. Heteroscorpionate Magnesium Alkyls Bearing Unprecedented Apical σ-C(sp3)–Mg Bonds: Heteroselective Ring-Opening Polymerization of rac-Lactide. Inorg. Chem. 2013, 52, 12691–12701. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Barba, L.F.; Garcés, A.; Fajardo, M.; Alonso-Moreno, C.; Fernández-Baeza, J.; Otero, A.; Antiñolo, A.; Tejeda, J.; Lara-Sánchez, A.; López-Solera, M.I. Well-Defined Alkyl Heteroscorpionate Magnesium Complexes as Excellent Initiators for the ROP of Cyclic Esters. Organometallics 2007, 26, 6403–6411. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Ma, H. Magnesium complexes in diverse coordination patterns supported by tetradentate aminophenolate ligands: Synthesis, characterization and application in the stereocontrolled ring-opening polymerization of rac-LA. Polyhedron 2016, 117, 569–578. [Google Scholar] [CrossRef]
- Wang, H.; Guo, J.; Yang, Y.; Ma, H. Diastereoselective synthesis of chiral aminophenolate magnesium complexes and their application in the stereoselective polymerization of rac-lactide and rac-β-butyrolactone. Dalton Trans. 2016, 45, 10942–10953. [Google Scholar] [CrossRef] [PubMed]
- Poirier, V.; Roisnel, T.; Carpentier, J.-F.; Sarazin, Y. Versatile catalytic systems based on complexes of zinc, magnesium and calcium supported by a bulky bis(morpholinomethyl)phenoxy ligand for the large-scale immortal ring-opening polymerisation of cyclic esters. Dalton Trans. 2009, 44, 9820–9827. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ma, H. Highly Active Magnesium Initiators for Ring-Opening Polymerization of rac-Lactide. Macromolecules 2010, 43, 6535–6537. [Google Scholar] [CrossRef]
- Song, S.; Ma, H.; Yang, Y. Magnesium complexes supported by salan-like ligands: Synthesis, characterization and their application in the ring-opening polymerization of rac-lactide. Dalton Trans. 2013, 42, 14200–14211. [Google Scholar] [CrossRef]
- Rosen, T.; Goldberg, I.; Venditto, V.; Kol, M. Tailor-Made Stereoblock Copolymers of Poly(lactic acid) by a Truly Living Polymerization Catalyst. J. Am. Chem. Soc. 2016, 138, 12041–12044. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Kan, C.; Wang, H.; Ma, H. Highly Active Chiral Oxazolinyl Aminophenolate Magnesium Initiators for Isoselective Ring-Opening Polymerization of rac-Lactide: Dinuclearity Induced Enantiomorphic Site Control. Macromolecules 2018, 51, 5304–5312. [Google Scholar] [CrossRef]
- Dutta, S.; Hung, W.-C.; Huang, B.-H.; Lin, C.-C. Recent Developments in Metal-Catalyzed Ring-Opening Polymerization of Lactides and Glycolides: Preparation of Polylactides, Polyglycolide, and Poly(lactide-co-glycolide). In Synthetic Biodegradable Polymers; Rieger, B., Künkel, A., Coates, G.W., Reichardt, R., Dinjus, E., Zevaco, T.A., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 219–283. [Google Scholar]
- Wheaton, C.A.; Hayes, P.G.; Ireland, B.J. Complexes of Mg, Ca and Zn as homogeneous catalysts for lactide polymerization. Dalton Trans. 2009, 25, 4832–4846. [Google Scholar] [CrossRef] [PubMed]
- Platel, R.H.; Hodgson, L.M.; Williams, C.K. Biocompatible Initiators for Lactide Polymerization. Polym. Rev. 2008, 48, 11–63. [Google Scholar] [CrossRef]
- dos Santos Vieira, I.; Herres-Pawlis, S. Lactide Polymerisation with Complexes of Neutral N-Donors – New Strategies for Robust Catalysts. Eur. J. Inorg. Chem. 2012, 2012, 765–774. [Google Scholar] [CrossRef]
- Bouyhayi, M.; Sarazin, Y.; Casagrande Jr, O.L.; Carpentier, J.-F. Aluminum, calcium and zinc complexes supported by potentially tridentate iminophenolate ligands: Synthesis and use in the ring-opening polymerization of lactide. Appl. Organomet. Chem. 2012, 26, 681–688. [Google Scholar] [CrossRef]
- Dai, Z.-R.; Yin, C.-F.; Wang, C.; Wu, J.-C. Zinc bis-Schiff base complexes: Synthesis, structure, and application in ring-opening polymerization of rac-lactide. Chin. Chem. Lett. 2016, 27, 1649–1654. [Google Scholar] [CrossRef]
- Huang, M.; Pan, C.; Ma, H. Ring-opening polymerization of rac-lactide and α-methyltrimethylene carbonate catalyzed by magnesium and zinc complexes derived from binaphthyl-based iminophenolate ligands. Dalton Trans. 2015, 44, 12420–12431. [Google Scholar] [CrossRef]
- Duan, R.; Gao, B.; Li, X.; Pang, X.; Wang, X.; Shao, H.; Chen, X. Zinc complexes bearing tridentate O,N,O-type half-Salen ligands for ring-opening polymerization of lactide. Polymer 2015, 71, 1–7. [Google Scholar] [CrossRef]
- Hollingsworth, T.S.; Hollingsworth, R.L.; Rosen, T.; Groysman, S. Zinc bimetallics supported by a xanthene-bridged dinucleating ligand: Synthesis, characterization, and lactide polymerization studies. RSC Adv. 2017, 7, 41819–41829. [Google Scholar] [CrossRef]
- Bakewell, C.; Fateh-Iravani, G.; Beh, D.W.; Myers, D.; Tabthong, S.; Hormnirun, P.; White, A.J.P.; Long, N.; Williams, C.K. Comparing a series of 8-quinolinolato complexes of aluminium, titanium and zinc as initiators for the ring-opening polymerization of rac-lactide. Dalton Trans. 2015, 44, 12326–12337. [Google Scholar] [CrossRef]
- Platel, R.H.; White, A.J.P.; Williams, C.K. Stereocontrolled lactide polymerisation determined by the nuclearity of the yttrium initiator. Chem. Commun. 2009, 27, 4115–4117. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, M.H.; Choojun, K.; Chow, A.S.; Fraenkel, G. Molecular Dynamics and Ligand Exchange in Magnesium Complexes: Evidence for both Dissociative and Associative Ligand Exchange. Angew. Chem. Int. Ed. 2013, 52, 3264–3266. [Google Scholar] [CrossRef] [PubMed]
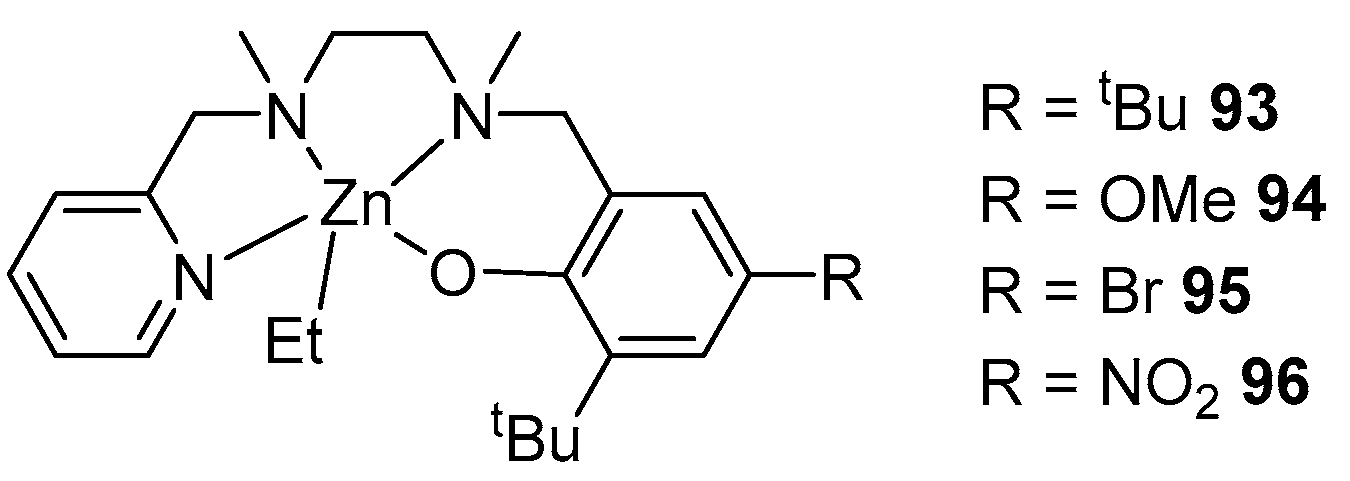
- Kuchuk, E.A.; Mankaev, B.N.; Zaitsev, K.V.; Zabalov, M.V.; Zakharova, E.A.; Oprunenko, Y.F.; Churakov, A.V.; Lermontova, E.K.; Zaitseva, G.S.; Karlov, S.S. Toward the Synthesis of Heteroleptic Zinc ROP Initiators Based on Pyridine-Containing Monoalcohols by Tuning Ligand Substituents. Organometallics 2023, 42, 2549–2557. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, W.; Liu, D.; Li, S.; Liu, X.; Cui, D.; Chen, X. Magnesium and Zinc Complexes Supported by N,O-Bidentate Pyridyl Functionalized Alkoxy Ligands: Synthesis and Immortal ROP of ε-CL and l-LA. Organometallics 2012, 31, 4182–4190. [Google Scholar] [CrossRef]
- Knight, P.D.; White, A.J.P.; Williams, C.K. Dinuclear Zinc Complexes Using Pentadentate Phenolate Ligands. Inorg. Chem. 2008, 47, 11711–11719. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, H.; Ma, H. Stereoselective Polymerization of rac-Lactide Catalyzed by Zinc Complexes with Tetradentate Aminophenolate Ligands in Different Coordination Patterns: Kinetics and Mechanism. Inorg. Chem. 2015, 54, 5839–5854. [Google Scholar] [CrossRef]
- Huang, M.; Ma, H. Magnesium and Zinc Complexes Supported by N,N,O Tridentate Ligands: Synthesis and Catalysis in the Ring-Opening Polymerization of rac-Lactide and α-Methyltrimethylene Carbonate. Eur. J. Inorg. Chem. 2016, 2016, 3791–3803. [Google Scholar] [CrossRef]
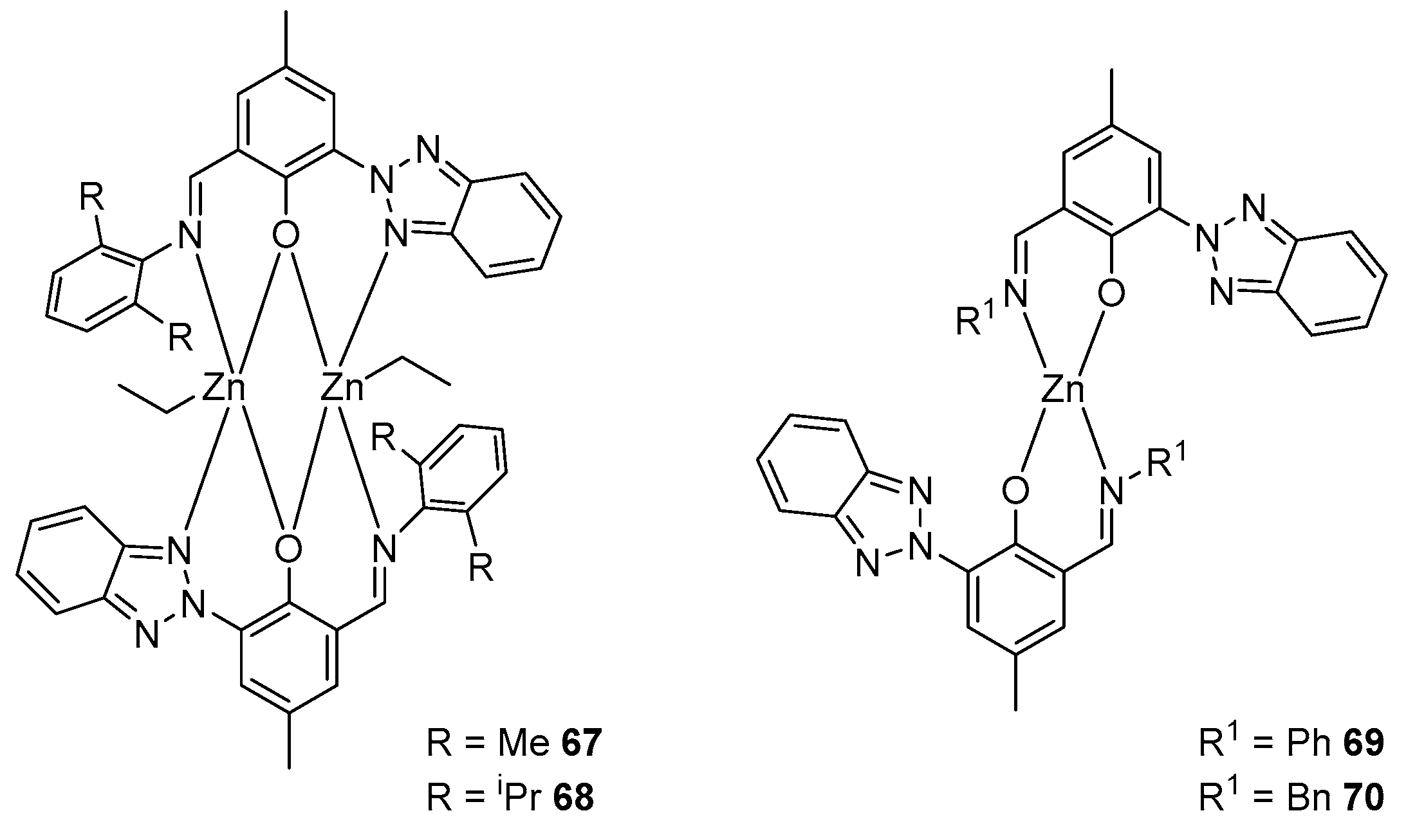
- Chang, C.-H.; Chuang, H.-J.; Chen, T.-Y.; Li, C.-Y.; Lin, C.-H.; Lee, T.-Y.; Ko, B.-T.; Huang, H.-Y. Di-nuclear zinc complexes containing tridentate imino-benzotriazole phenolate derivatives as efficient catalysts for ring-opening polymerization of cyclic esters and copolymerization of phthalic anhydride with cyclohexene oxide. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 714–725. [Google Scholar] [CrossRef]
- Wang, L.; Ma, H. Zinc complexes supported by multidentate aminophenolate ligands: Synthesis, structure and catalysis in ring-opening polymerization of rac-lactide. Dalton Trans. 2010, 39, 7897–7910. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, Y.; Ma, H. Exploring Steric Effects in Diastereoselective Synthesis of Chiral Aminophenolate Zinc Complexes and Stereoselective Ring-Opening Polymerization of rac-Lactide. Inorg. Chem. 2016, 55, 7356–7372. [Google Scholar] [CrossRef] [PubMed]
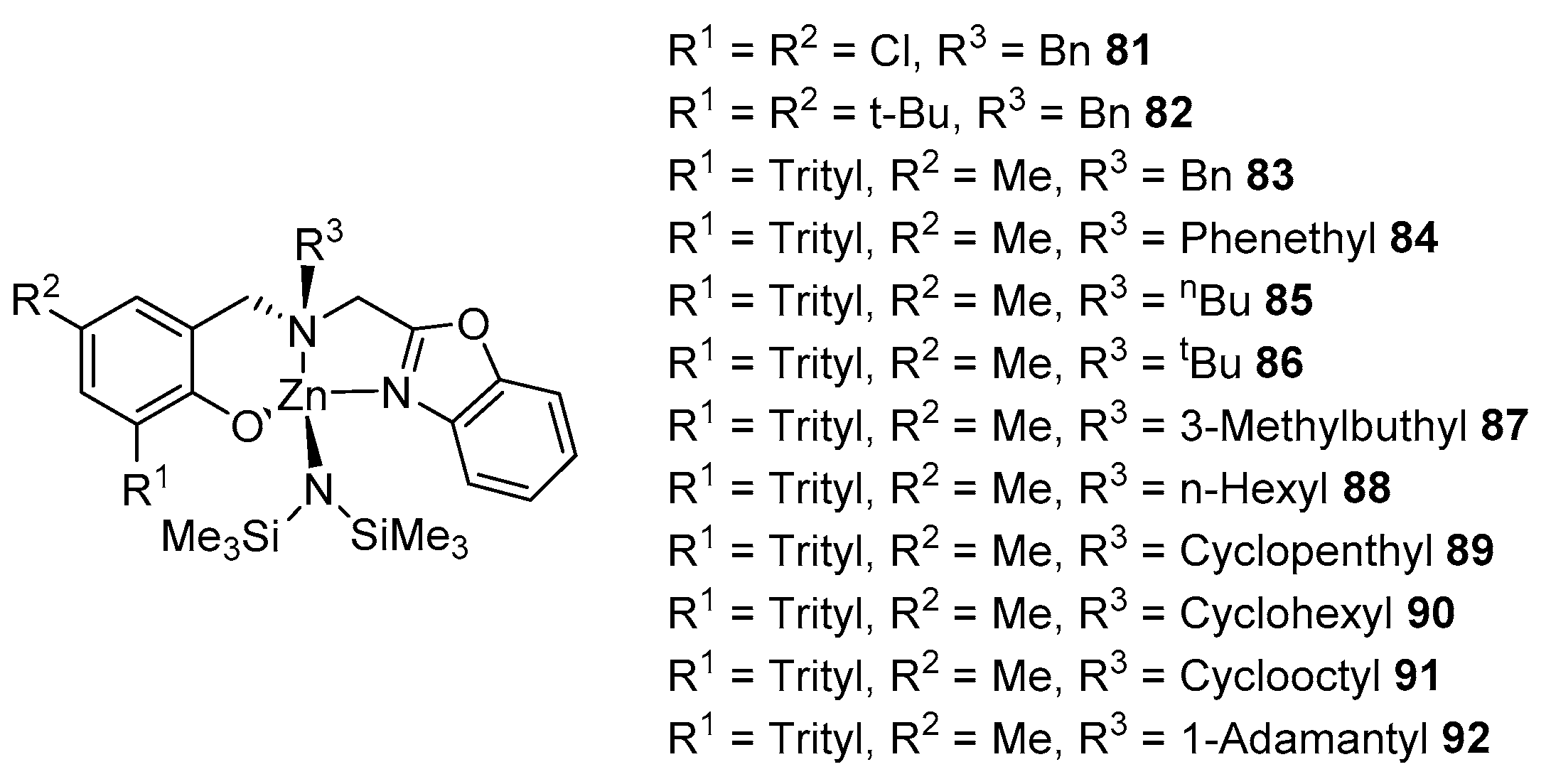
- Hu, J.; Kan, C.; Ma, H. Exploring Steric Effects of Zinc Complexes Bearing Achiral Benzoxazolyl Aminophenolate Ligands in Isoselective Polymerization of rac-Lactide. Inorg. Chem. 2018, 57, 11240–11251. [Google Scholar] [CrossRef] [PubMed]
- Stasiw, D.E.; Luke, A.M.; Rosen, T.; League, A.B.; Mandal, M.; Neisen, B.D.; Cramer, C.J.; Kol, M.; Tolman, W.B. Mechanism of the Polymerization of rac-Lactide by Fast Zinc Alkoxide Catalysts. Inorg. Chem. 2017, 56, 14366–14372. [Google Scholar] [CrossRef] [PubMed]
- McKeown, P.; Brown-Humes, J.; Davidson, M.G.; Mahon, M.F.; Woodman, T.J.; Jones, M.D. Ligands and complexes based on piperidine and their exploitation of the ring opening polymerisation of rac-lactide. Dalton Trans. 2017, 46, 5048–5057. [Google Scholar] [CrossRef] [PubMed]
- Dumitrescu, A.; Martin-Vaca, B.; Gornitzka, H.; Cazaux, J.-B.; Bourissou, D.; Bertrand, G. Zinc(II), Samarium(III) and Tin(II) Complexes Featuring a Tridentate Nitrogen Donor for the Ring-Opening Copolymerization of (D,L)-Lactide and Glycolide. Eur. J. Inorg. Chem. 2002, 2002, 1948–1951. [Google Scholar] [CrossRef]
- Kuchuk, E.A.; Mankaev, B.N.; Serova, V.A.; Zaitsev, K.V.; Churakov, A.V.; Oprunenko, Y.F.; Zaitseva, G.S.; Karlov, S.S. New dialkylenetriamine zinc complexes as highly efficient ROP catalysts. Mendeleev Commun. 2020, 30, 596–598. [Google Scholar] [CrossRef]
- Sauer, A.; Kapelski, A.; Fliedel, C.; Dagorne, S.; Kol, M.; Okuda, J. Structurally well-defined group 4 metal complexes as initiators for the ring-opening polymerization of lactide monomers. Dalton Trans. 2013, 42, 9007–9023. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Lu, W.-Y.; Chen, Y.-J.; Hsu, S.C.N.; Ou, S.-W.; Peng, W.-T.; Jheng, N.-Y.; Lai, Y.-C.; Wu, B.-S.; Chung, H.; et al. Synthesis, characterization, and catalytic activity of titanium iminophenoxide complexes in relation to the ring-opening polymerization of L-lactide and ε-caprolactone. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 327–333. [Google Scholar] [CrossRef]
- Kumar Saha, T.; Rajashekhar, B.; Chakraborty, D. Alkoxides of group 4 metals containing the bis(imino)phenoxide ligand: Synthesis, structural characterization and polymerization studies. RSC Adv. 2012, 2, 307–318. [Google Scholar] [CrossRef]
- Saha, T.K.; Mandal, M.; Chakraborty, D.; Ramkumar, V. Imino phenoxide complexes of group 4 metals: Synthesis, structural characterization and polymerization studies. New J. Chem. 2013, 37, 949–960. [Google Scholar] [CrossRef]
- Li, C.-Y.; Yu, C.-J.; Ko, B.-T. Facile Synthesis of Well-Defined Titanium Alkoxides Based on Benzotriazole Phenoxide Ligands: Efficient Catalysts for Ring-Opening Polymerization of Cyclic Esters. Organometallics 2013, 32, 172–180. [Google Scholar] [CrossRef]
- Stjerndahl, A.; Wistrand, A.F.; Albertsson, A.-C. Industrial Utilization of Tin-Initiated Resorbable Polymers: Synthesis on a Large Scale with a Low Amount of Initiator Residue. Biomacromolecules 2007, 8, 937–940. [Google Scholar] [CrossRef] [PubMed]
- Dorgan, J.R.; Janzen, J.; Clayton, M.P.; Hait, S.B.; Knauss, D.M. Melt rheology of variable L-content poly(lactic acid). J. Rheol. 2005, 49, 607–619. [Google Scholar] [CrossRef]
- Gendler, S.; Segal, S.; Goldberg, I.; Goldschmidt, Z.; Kol, M. Titanium and Zirconium Complexes of Dianionic and Trianionic Amine−Phenolate-Type Ligands in Catalysis of Lactide Polymerization. Inorg. Chem. 2006, 45, 4783–4790. [Google Scholar] [CrossRef] [PubMed]
- Chmura, A.J.; Davidson, M.G.; Frankis, C.J.; Jones, M.D.; Lunn, M.D. Highly active and stereoselective zirconium and hafnium alkoxide initiators for solvent-free ring-opening polymerization of rac-lactide. Chem. Commun. 2008, 11, 1293–1295. [Google Scholar] [CrossRef] [PubMed]
- Kuchuk, E.A.; Zaitsev, K.V.; Mamedova, F.A.; Churakov, A.V.; Zaitseva, G.S.; Lemenovsky, D.A.; Karlov, S.S. Synthesis, structure, and catalytic activity of new aluminum and titanium complexes based on aminobisphenolate ligands containing bulky substituents. Russ. Chem. Bull. 2016, 65, 1743–1749. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, S.; Zhou, S. Aluminum alkyl complexes: Synthesis, structure, and application in ROP of cyclic esters. Dalton Trans. 2016, 45, 4471–4485. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, Y.; Sun, W.-H.; Wang, L.; Redshaw, C. Dimethylaluminium aldiminophenolates: Synthesis, characterization and ring-opening polymerization behavior towards lactides. Dalton Trans. 2012, 41, 11587–11596. [Google Scholar] [CrossRef]
- Pappalardo, D.; Annunziata, L.; Pellecchia, C. Living Ring-Opening Homo- and Copolymerization of ε-Caprolactone and l- and d,l-Lactides by Dimethyl(salicylaldiminato)aluminum Compounds. Macromolecules 2009, 42, 6056–6062. [Google Scholar] [CrossRef]
- Meduri, A.; Fuoco, T.; Lamberti, M.; Pellecchia, C.; Pappalardo, D. Versatile Copolymerization of Glycolide and rac-Lactide by Dimethyl(salicylaldiminato)aluminum Compounds. Macromolecules 2014, 47, 534–543. [Google Scholar] [CrossRef]
- Chang, M.-C.; Lu, W.-Y.; Chang, H.-Y.; Lai, Y.-C.; Chiang, M.Y.; Chen, H.-Y.; Chen, H.-Y. Comparative Study of Aluminum Complexes Bearing N,O- and N,S-Schiff Base in Ring-Opening Polymerization of ε-Caprolactone and l-Lactide. Inorg. Chem. 2015, 54, 11292–11298. [Google Scholar] [CrossRef] [PubMed]
- Hancock, S.L.; Mahon, M.F.; Jones, M.D. Salen complexes based on 1,4-diaminocyclohexane and their exploitation for the polymerisation of rac-lactide. New J. Chem. 2013, 37, 1996–2001. [Google Scholar] [CrossRef]
- Qian, F.; Liu, K.; Ma, H. Amidinate aluminium complexes: Synthesis, characterization and ring-opening polymerization of rac-lactide. Dalton Trans. 2010, 39, 8071–8083. [Google Scholar] [CrossRef] [PubMed]
- Tabthong, S.; Nanok, T.; Kongsaeree, P.; Prabpai, S.; Hormnirun, P. Monomethylaluminum and dimethylaluminum pyrrolylaldiminates for the ring-opening polymerization of rac-lactide: Effects of ligand structure and coordination geometry. Dalton Trans. 2014, 43, 1348–1359. [Google Scholar] [CrossRef] [PubMed]
- Kamavichanurat, S.; Jampakaew, K.; Hormnirun, P. Controlled and effective ring-opening (co)polymerization of rac-lactide, ε-caprolactone and ε-decalactone by β-pyrimidyl enolate aluminum complexes. Polym. Chem. 2023, 14, 1752–1772. [Google Scholar] [CrossRef]
- Di Iulio, C.; Jones, M.D.; Mahon, M.F. Synthesis of Al(III) silsesquioxane complexes and their exploitation for the ring opening polymerisation of rac-lactide. J. Organomet. Chem. 2012, 718, 96–100. [Google Scholar] [CrossRef]
- Isnard, F.; Lamberti, M.; Lettieri, L.; D’Auria, I.; Press, K.; Troiano, R.; Mazzeo, M. Bimetallic salen aluminum complexes: Cooperation between reactive centers in the ring-opening polymerization of lactides and epoxides. Dalton Trans. 2016, 45, 16001–16010. [Google Scholar] [CrossRef]
- Matsubara, K.; Terata, C.; Sekine, H.; Yamatani, K.; Harada, T.; Eda, K.; Dan, M.; Koga, Y.; Yasuniwa, M. Stereoselective ring-opening polymerization of D,L-lactide, initiated by aluminum isopropoxides bearing tridentate nonchiral schiff-base ligands. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 957–966. [Google Scholar] [CrossRef]
- Kireenko, M.M.; Kuchuk, E.A.; Zaitsev, K.V.; Tafeenko, V.A.; Oprunenko, Y.F.; Churakov, A.V.; Lermontova, E.K.; Zaitseva, G.S.; Karlov, S.S. Aluminum complexes based on pyridine substituted alcohols: Synthesis, structure, and catalytic application in ROP. Dalton Trans. 2015, 44, 11963–11976. [Google Scholar] [CrossRef]
- Cross, E.D.; Allan, L.E.N.; Decken, A.; Shaver, M.P. Aluminum salen and salan complexes in the ring-opening polymerization of cyclic esters: Controlled immortal and copolymerization of rac-β-butyrolactone and rac-lactide. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 1137–1146. [Google Scholar] [CrossRef]
- Luo, W.; Shi, T.; Liu, S.; Zuo, W.; Li, Z. Well-Designed Unsymmetrical Salphen-Al Complexes: Synthesis, Characterization, and Ring-Opening Polymerization Catalysis. Organometallics 2017, 36, 1736–1742. [Google Scholar] [CrossRef]
- Emig, N.; Nguyen, H.; Krautscheid, H.; Réau, R.; Cazaux, J.-B.; Bertrand, G. Neutral and Cationic Tetracoordinated Aluminum Complexes Featuring Tridentate Nitrogen Donors: Synthesis, Structure, and Catalytic Activity for the Ring-Opening Polymerization of Propylene Oxide and (d,l)-Lactide. Organometallics 1998, 17, 3599–3608. [Google Scholar] [CrossRef]
- Schwarz, A.D.; Chu, Z.; Mountford, P. Sulfonamide-Supported Aluminum Catalysts for the Ring-Opening Polymerization of rac-Lactide. Organometallics 2010, 29, 1246–1260. [Google Scholar] [CrossRef]
- Kuchuk, E.A.; Kireenko, M.M.; Mankaev, B.N.; Zaitsev, K.V.; Churakov, A.V.; Lermontova, E.K.; Kuz’mina, L.G.; Oprunenko, Y.F.; Zhirnov, A.E.; Zaitseva, G.S.; et al. Diamidoamine Aluminum Complexes: Synthesis, Structure, L–Lactide and ϵ-Caprolactone Polymerization. ChemistrySelect 2021, 6, 10243–10249. [Google Scholar] [CrossRef]
- Blake, M.P.; Schwarz, A.D.; Mountford, P. Sulfonamide, Phenolate, and Directing Ligand-Free Indium Initiators for the Ring-Opening Polymerization of rac-Lactide. Organometallics 2011, 30, 1202–1214. [Google Scholar] [CrossRef]
- Douglas, A.F.; Patrick, B.O.; Mehrkhodavandi, P. A Highly Active Chiral Indium Catalyst for Living Lactide Polymerization. Angew. Chem. Int. Ed. 2008, 47, 2290–2293. [Google Scholar] [CrossRef]
- Osten, K.M.; Yu, I.; Duffy, I.R.; Lagaditis, P.O.; Yu, J.C.C.; Wallis, C.J.; Mehrkhodavandi, P. Effects of ligand tuning on dinuclear indium catalysts for lactide polymerization. Dalton Trans. 2012, 41, 8123–8134. [Google Scholar] [CrossRef]
- Yu, I.; Acosta-Ramírez, A.; Mehrkhodavandi, P. Mechanism of Living Lactide Polymerization by Dinuclear Indium Catalysts and Its Impact on Isoselectivity. J. Am. Chem. Soc. 2012, 134, 12758–12773. [Google Scholar] [CrossRef]
- Aluthge, D.C.; Patrick, B.O.; Mehrkhodavandi, P. A highly active and site selective indium catalyst for lactide polymerization. Chem. Commun. 2013, 49, 4295–4297. [Google Scholar] [CrossRef]
- Peckermann, I.; Kapelski, A.; Spaniol, T.P.; Okuda, J. Indium Complexes Supported by 1,ω-Dithiaalkanediyl-Bridged Bis(phenolato) Ligands: Synthesis, Structure, and Controlled Ring-Opening Polymerization of l-Lactide. Inorg. Chem. 2009, 48, 5526–5534. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Wang, M.; Zhang, P.; Wang, L.; Zhu, F.; Sun, L. Preparation and structure of an enantiomeric water-bridged dinuclear indium complex containing two homochiral N atoms and its performance as an initiator in polymerization of rac-lactide. Inorg. Chem. Commun. 2010, 13, 968–971. [Google Scholar] [CrossRef]
- Buffet, J.-C.; Okuda, J.; Arnold, P.L. Chiral Indium Alkoxide Complexes as Initiators for the Stereoselective Ring-Opening Polymerization of rac-Lactide. Inorg. Chem. 2010, 49, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A.; Hillmyer, M.A.; Tolman, W.B. Stereoselective and controlled polymerization of d,l-lactide using indium(iii) trichloride. Chem. Commun. 2009, 19, 2736–2737. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A.; Knight, S.C.; Gupta, A.K.; Yao, L.J.; Hillmyer, M.A.; Tolman, W.B. Mechanistic Study of the Stereoselective Polymerization of d,l-Lactide Using Indium(III) Halides. J. Am. Chem. Soc. 2010, 132, 11649–11657. [Google Scholar] [CrossRef] [PubMed]
- Normand, M.; Kirillov, E.; Roisnel, T.; Carpentier, J.-F. Indium Complexes of Fluorinated Dialkoxy-Diimino Salen-like Ligands for Ring-Opening Polymerization of rac-Lactide: How Does Indium Compare to Aluminum? Organometallics 2012, 31, 1448–1457. [Google Scholar] [CrossRef]
- Hsieh, I.P.; Huang, C.-H.; Lee, H.M.; Kuo, P.-C.; Huang, J.-H.; Lee, H.-I.; Cheng, J.-T.; Lee, G.-H. Indium complexes incorporating bidentate substituted pyrrole ligand: Synthesis, characterization, and ring-opening polymerization of ε-caprolactone. Inorg. Chim. Acta 2006, 359, 497–504. [Google Scholar] [CrossRef]
- Xu, C.; Yu, I.; Mehrkhodavandi, P. Highly controlled immortal polymerization of β-butyrolactone by a dinuclear indium catalyst. Chem. Commun. 2012, 48, 6806–6808. [Google Scholar] [CrossRef]
- Osten, K.M.; Mehrkhodavandi, P. Indium Catalysts for Ring Opening Polymerization: Exploring the Importance of Catalyst Aggregation. Acc. Chem. Res. 2017, 50, 2861–2869. [Google Scholar] [CrossRef]
- Quan, S.M.; Diaconescu, P.L. High activity of an indium alkoxide complex toward ring opening polymerization of cyclic esters. Chem. Commun. 2015, 51, 9643–9646. [Google Scholar] [CrossRef]
- Kremer, A.B.; Osten, K.M.; Yu, I.; Ebrahimi, T.; Aluthge, D.C.; Mehrkhodavandi, P. Dinucleating Ligand Platforms Supporting Indium and Zinc Catalysts for Cyclic Ester Polymerization. Inorg. Chem. 2016, 55, 5365–5374. [Google Scholar] [CrossRef]
- Aluthge, D.C.; Yan, E.X.; Ahn, J.M.; Mehrkhodavandi, P. Role of Aggregation in the Synthesis and Polymerization Activity of SalBinap Indium Alkoxide Complexes. Inorg. Chem. 2014, 53, 6828–6836. [Google Scholar] [CrossRef] [PubMed]
- Aluthge, D.C.; Ahn, J.M.; Mehrkhodavandi, P. Overcoming aggregation in indium salen catalysts for isoselective lactide polymerization. Chem. Sci. 2015, 6, 5284–5292. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Gowda, R.R.; Jagan, R.; Chakraborty, D. Gallium and indium complexes containing the bis(imino)phenoxide ligand: Synthesis, structural characterization and polymerization studies. Dalton Trans. 2015, 44, 10410–10422. [Google Scholar] [CrossRef] [PubMed]
- Osten, K.M.; Aluthge, D.C.; Mehrkhodavandi, P. The effect of steric changes on the isoselectivity of dinuclear indium catalysts for lactide polymerization. Dalton Trans. 2015, 44, 6126–6139. [Google Scholar] [CrossRef] [PubMed]
- Diaz, C.; Fu, J.; Soobrattee, S.; Cao, L.; Nyamayaro, K.; Goonesinghe, C.; Patrick, B.O.; Mehrkhodavandi, P. Comparison of Imine- and Phosphinimine-Supported Indium Complexes: Tuning the Reactivity for the Sequential and Simultaneous Copolymerization of Lactide and ε-Caprolactone. Inorg. Chem. 2022, 61, 3763–3773. [Google Scholar] [CrossRef] [PubMed]
- Fußstetter, H. Book Review: The Chemistry of the Semiconductor Industry. By S. J. Moss and A. Ledwith. Angew Chem. Int. 1988, 27, 1739–1740. [Google Scholar] [CrossRef]
- Hart, M.M.; Smith, C.F.; Yancey, S.T.; Adamson, R.H. Toxicity and Antitumor Activity of Gallium Nitrate and Periodically Related Metal Salts22. JNCI J. Natl. Cancer Inst. 1971, 47, 1121–1128. [Google Scholar]
- Bernstein, L.R. Mechanisms of Therapeutic Activity for Gallium. Pharmacol. Rev. 1998, 50, 665. [Google Scholar]
- Nordberg, G.F.; Fowler, B.A.; Nordberg, M. Handbook on the Toxicology of Metals; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- MacDonald, J.; Shaver, M. Green Polymer Chemistry: Biobased Materials and Biocatalysis; Cheng, H.N., Gross, R.A., Smith, P.B., Eds.; American Chemical Society: Washington, DC, USA, 2015. [Google Scholar]
- Specklin, D.; Fliedel, C.; Hild, F.; Mameri, S.; Karmazin, L.; Bailly, C.; Dagorne, S. Mononuclear salen-gallium complexes for iso-selective ring-opening polymerization (ROP) of rac-lactide. Dalton Trans. 2017, 46, 12824–12834. [Google Scholar] [CrossRef]
- Atwood, D.A.; Harvey, M.J. Group 13 Compounds Incorporating Salen Ligands. Chem. Rev. 2001, 101, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Horeglad, P.; Kruk, P.; Pécaut, J. Heteroselective Polymerization of rac-Lactide in the Presence of Dialkylgallium Alkoxides: The Effect of Lewis Base on Polymerization Stereoselectivity. Organometallics 2010, 29, 3729–3734. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Gallucci, J.C.; Zhen, H.; Huffman, J.C. Three-Coordinate Zinc Amide and Phenoxide Complexes Supported by a Bulky Schiff Base Ligand. Inorg. Chem. 2001, 40, 5051–5054. [Google Scholar] [CrossRef] [PubMed]
- Horeglad, P.; Litwińska, A.; Żukowska, G.Z.; Kubicki, D.; Szczepaniak, G.; Dranka, M.; Zachara, J. The influence of organosuperbases on the structure and activity of dialkylgallium alkoxides in the polymerization of rac-lactide: The road to stereo diblock PLA copolymers. Appl. Organomet. Chem. 2013, 27, 328–336. [Google Scholar] [CrossRef]
- Horeglad, P.; Szczepaniak, G.; Dranka, M.; Zachara, J. The first facile stereoselectivity switch in the polymerization of rac-lactide—From heteroselective to isoselective dialkylgallium alkoxides with the help of N-heterocyclic carbenes. Chem. Commun. 2012, 48, 1171–1173. [Google Scholar] [CrossRef] [PubMed]
- Hild, F.; Neehaul, N.; Bier, F.; Wirsum, M.; Gourlaouen, C.; Dagorne, S. Synthesis and Structural Characterization of Various N,O,N-Chelated Aluminum and Gallium Complexes for the Efficient ROP of Cyclic Esters and Carbonates: How Do Aluminum and Gallium Derivatives Compare ? Organometallics 2013, 32, 587–598. [Google Scholar] [CrossRef]
- Hild, F.; Dagorne, S. A Discrete N,O,N-Supported Gallium Amido Complex for the Intermolecular Hydroamination of Terminal Alkynes. Organometallics 2012, 31, 1189–1194. [Google Scholar] [CrossRef]
- Bakewell, C.; White, A.J.P.; Long, N.J.; Williams, C.K. 8-Quinolinolato Gallium Complexes: Iso-selective Initiators for rac-Lactide Polymerization. Inorg. Chem. 2013, 52, 12561–12567. [Google Scholar] [CrossRef]
- Bakewell, C.; Platel, R.H.; Cary, S.K.; Hubbard, S.M.; Roaf, J.M.; Levine, A.C.; White, A.J.P.; Long, N.J.; Haaf, M.; Williams, C.K. Bis(8-quinolinolato)aluminum ethyl complexes: Iso-Selective Initiators for rac-Lactide Polymerization. Organometallics 2012, 31, 4729–4736. [Google Scholar] [CrossRef]
- Kremer, A.B.; Andrews, R.J.; Milner, M.J.; Zhang, X.R.; Ebrahimi, T.; Patrick, B.O.; Diaconescu, P.L.; Mehrkhodavandi, P. A Comparison of Gallium and Indium Alkoxide Complexes as Catalysts for Ring-Opening Polymerization of Lactide. Inorg. Chem. 2017, 56, 1375–1385. [Google Scholar] [CrossRef]
- Osten, K.M.; Aluthge, D.C.; Patrick, B.O.; Mehrkhodavandi, P. Probing the Role of Secondary versus Tertiary Amine Donor Ligands for Indium Catalysts in Lactide Polymerization. Inorg. Chem. 2014, 53, 9897–9906. [Google Scholar] [CrossRef] [PubMed]
- Mankaev, B.N.; Hasanova, L.F.; Churakov, A.V.; Egorov, M.P.; Karlov, S.S. Gallium (III) Complexes Based on Aminobisphenolate Ligands: Extremely High Active ROP-Initiators from Well-Known and Easily Accessible Compounds. Int. J. Mol. Sci. 2022, 23, 15649. [Google Scholar] [CrossRef] [PubMed]
- Zabalov, M.V.; Mankaev, B.N.; Egorov, M.P.; Karlov, S.S. The Novel Gallium Aminobisphenolate Initiator of the Ring-Opening Copolymerization of L-Lactide and ε-Caprolactone: A Computational Study. Int. J. Mol. Sci. 2022, 23, 15523. [Google Scholar] [CrossRef] [PubMed]
- Bawa, K.K.; Oh, J.K. Stimulus-Responsive Degradable Polylactide-Based Block Copolymer Nanoassemblies for Controlled/Enhanced Drug Delivery. Mol. Pharm. 2017, 14, 2460–2474. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yang, H.; Wang, R.; Cheng, R.; Meng, F.; Wei, W.; Zhong, Z. Versatile Synthesis of Functional Biodegradable Polymers by Combining Ring-Opening Polymerization and Postpolymerization Modification via Michael-Type Addition Reaction. Macromolecules 2010, 43, 201–207. [Google Scholar] [CrossRef]
- Koning, C.; Van Duin, M.; Pagnoulle, C.; Jerome, R. Strategies for compatibilization of polymer blends. Prog. Polym. Sci. 1998, 23, 707–757. [Google Scholar] [CrossRef]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgotten polymer—Polycaprolactone in the 21st century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef]
- Tabata, Y.; Abe, H. Effects of composition and sequential structure on thermal properties for copolymer of 3-hydroxybutyrate and lactate units. Polym. Degrad. Stab. 2013, 98, 1796–1803. [Google Scholar] [CrossRef]
- Jeffery, B.J.; Whitelaw, E.L.; Garcia-Vivo, D.; Stewart, J.A.; Mahon, M.F.; Davidson, M.G.; Jones, M.D. Group 4 initiators for the stereoselective ROP of rac-β-butyrolactone and its copolymerization with rac-lactide. Chem. Commun. 2011, 47, 12328–12330. [Google Scholar] [CrossRef]
- Aluthge, D.C.; Xu, C.; Othman, N.; Noroozi, N.; Hatzikiriakos, S.G.; Mehrkhodavandi, P. PLA–PHB–PLA Triblock Copolymers: Synthesis by Sequential Addition and Investigation of Mechanical and Rheological Properties. Macromolecules 2013, 46, 3965–3974. [Google Scholar] [CrossRef]
- Yu, I.; Ebrahimi, T.; Hatzikiriakos, S.G.; Mehrkhodavandi, P. Star-shaped PHB–PLA block copolymers: Immortal polymerization with dinuclear indium catalysts. Dalton Trans. 2015, 44, 14248–14254. [Google Scholar] [CrossRef] [PubMed]
- Fagerland, J.; Finne-Wistrand, A.; Pappalardo, D. Modulating the thermal properties of poly(hydroxybutyrate) by the copolymerization of rac-β-butyrolactone with lactide. New J. Chem. 2016, 40, 7671–7679. [Google Scholar] [CrossRef]
- Hori, Y.; Takahashi, Y.; Yamaguchi, A.; Nishishita, T. Ring-opening copolymerization of optically active. beta.-butyrolactone with several lactones catalyzed by distannoxane complexes: Synthesis of new biodegradable polyesters. Macromolecules 1993, 26, 4388–4390. [Google Scholar] [CrossRef]
- Luciano, E.; Buonerba, A.; Grassi, A.; Milione, S.; Capacchione, C. Thioetherphenolate group 4 metal complexes in the ring opening polymerization of rac-β-Butyrolactone. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 3132–3139. [Google Scholar] [CrossRef]
- Feng, J.; Zhuo, R.-X.; Zhang, X.-Z. Construction of functional aliphatic polycarbonates for biomedical applications. Prog. Polym. Sci. 2012, 37, 211–236. [Google Scholar] [CrossRef]
- Cameron, D.J.A.; Shaver, M.P. Aliphatic polyester polymer stars: Synthesis, properties and applications in biomedicine and nanotechnology. Chem. Soc. Rev. 2011, 40, 1761–1776. [Google Scholar] [CrossRef] [PubMed]
- Darensbourg, D.J.; Choi, W.; Karroonnirun, O.; Bhuvanesh, N. Ring-Opening Polymerization of Cyclic Monomers by Complexes Derived from Biocompatible Metals. Production of Poly(lactide), Poly(trimethylene carbonate), and Their Copolymers. Macromolecules 2008, 41, 3493–3502. [Google Scholar] [CrossRef]
- Pospiech, D.; Komber, H.; Jehnichen, D.; Häussler, L.; Eckstein, K.; Scheibner, H.; Janke, A.; Kricheldorf, H.R.; Petermann, O. Multiblock Copolymers of l-Lactide and Trimethylene Carbonate. Biomacromolecules 2005, 6, 439–446. [Google Scholar] [CrossRef]
- Jie, C.; Zhu, K.J. Preparation, Characterization and Biodegradable Characteristics of Poly(D,L-lactide-co-1,3-trimethylene carbonate). Polym. Int. 1997, 42, 373–379. [Google Scholar] [CrossRef]
- Hu, G.; Wang, Y.; Wang, Y.; Zhu, X.; Yuan, D.; Yao, Y. Random copolymerization of trimethylene carbonate with l-lactide initiated by amine-bridged bis(phenolate) neodymium alkoxides. Chem. Commun. 2023, 59, 6921–6924. [Google Scholar] [CrossRef]
- Hua, X.; Liu, X.; Cui, D. Sequence controlled copolymerization of lactide and a functional cyclic carbonate using stereoselective aluminum catalysts. Polym. Chem. 2019, 10, 4042–4048. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, Y.; Liu, X.; Chen, X.; Cui, D.; Chen, E.Y.X. Protic compound mediated living cross-chain-transfer polymerization of rac-lactide: Synthesis of isotactic (crystalline)–heterotactic (amorphous) stereomultiblock polylactide. Chem. Commun. 2012, 48, 6375–6377. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, Q.; Cui, Y.; He, J.; Zhang, Y. Living/controlled ring-opening (co)polymerization of lactones by Al-based catalysts with different sidearms. Dalton Trans. 2019, 48, 7167–7178. [Google Scholar] [CrossRef] [PubMed]
- Nair, K.L.; Jagadeeshan, S.; Nair, S.A.; Kumar, G.S.V. Evaluation of triblock copolymeric micelles of δ- valerolactone and poly (ethylene glycol) as a competent vector for doxorubicin delivery against cancer. J. Nanobiotechnol. 2011, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Gamma-valerolactone, a sustainable platform molecule derived from lignocellulosic biomass. Green Chem. 2013, 15, 584–595. [Google Scholar] [CrossRef]
- Hu, Q.; Jie, S.-Y.; Braunstein, P.; Li, B.-G. Ring-opening Copolymerization of ε-Caprolactone and δ-Valerolactone Catalyzed by a 2,6-Bis(amino)phenol Zinc Complex. Chin. J. Polym. Sci. 2020, 38, 240–247. [Google Scholar] [CrossRef]
- García-Valle, F.M.; Cuenca, T.; Mosquera, M.E.G.; Milione, S.; Cano, J. Ring-Opening Polymerization (ROP) of cyclic esters by a versatile aluminum Diphenoxyimine Complex: From polylactide to random copolymers. Eur. Polym. J. 2020, 125, 109527. [Google Scholar] [CrossRef]
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
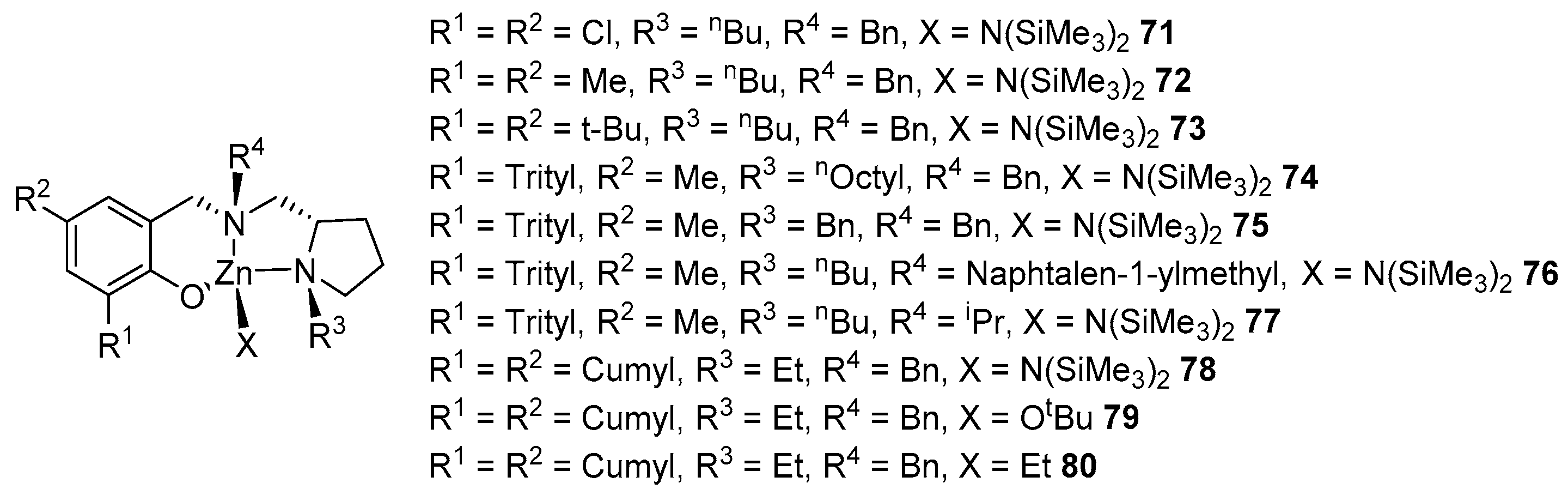{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
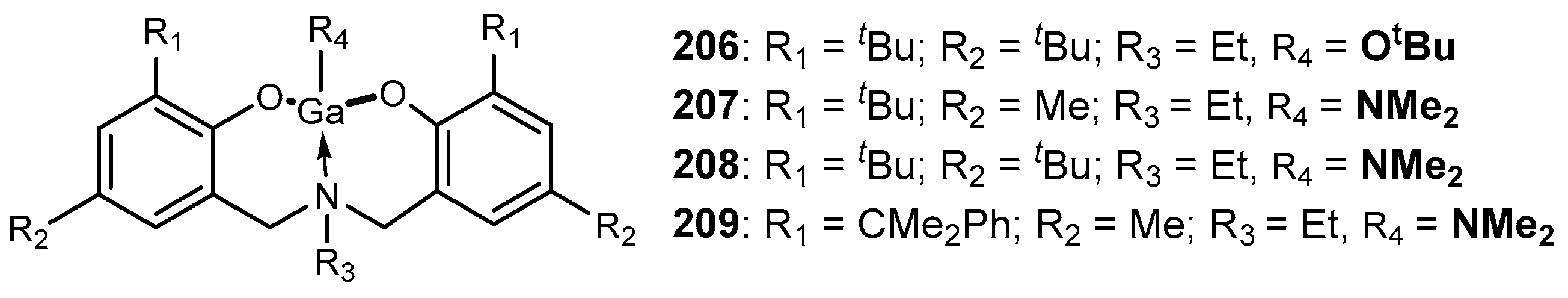{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






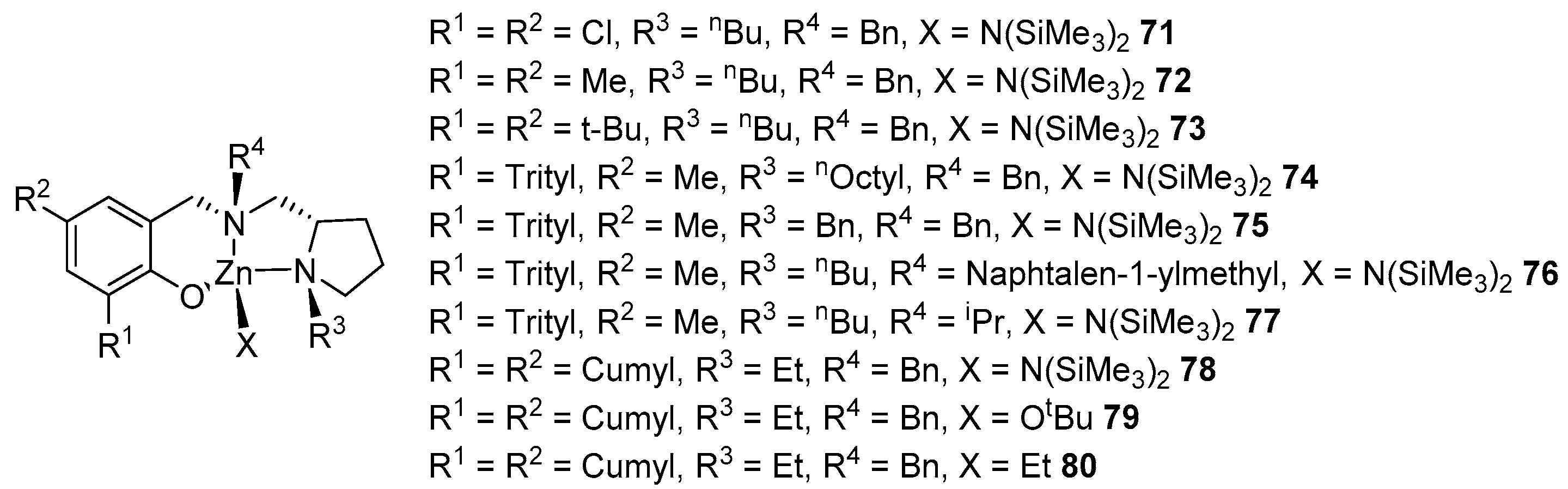









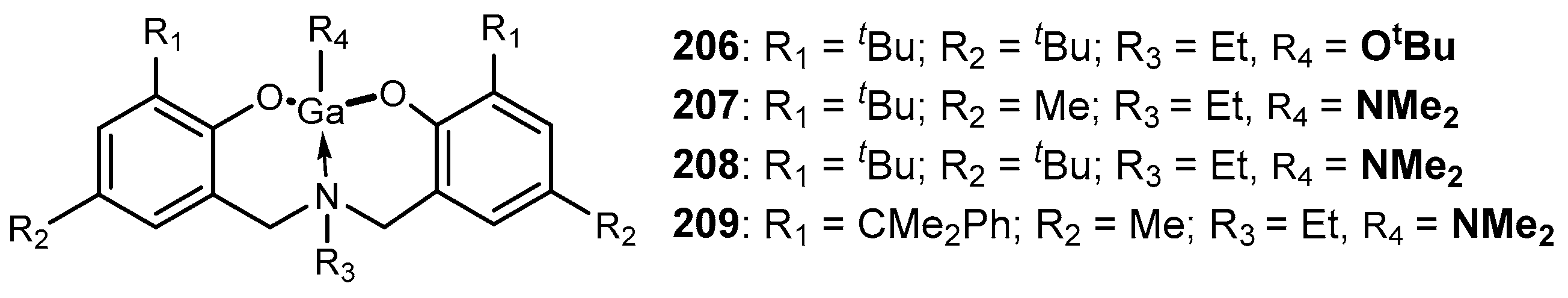
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mankaev, B.N.; Karlov, S.S. Metal Complexes in the Synthesis of Biodegradable Polymers: Achievements and Prospects. Materials 2023, 16, 6682. https://doi.org/10.3390/ma16206682
Mankaev BN, Karlov SS. Metal Complexes in the Synthesis of Biodegradable Polymers: Achievements and Prospects. Materials. 2023; 16(20):6682. https://doi.org/10.3390/ma16206682
Chicago/Turabian StyleMankaev, Badma N., and Sergey S. Karlov. 2023. "Metal Complexes in the Synthesis of Biodegradable Polymers: Achievements and Prospects" Materials 16, no. 20: 6682. https://doi.org/10.3390/ma16206682
APA StyleMankaev, B. N., & Karlov, S. S. (2023). Metal Complexes in the Synthesis of Biodegradable Polymers: Achievements and Prospects. Materials, 16(20), 6682. https://doi.org/10.3390/ma16206682