

Synthesis and Characterization of Boron Carbide Nanoparticles as Potential Boron-Rich Therapeutic Carriers

 , , , , , , , , and
, , , , , , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis and Preparation of Boron Carbide Nanoparticles
2.2. Biological Activity Evaluation of the Obtained Boron Carbide Nanoparticles
2.2.1. Cell Lines
2.2.2. MTT Assay
2.2.3. Statistical Analysis
3. Results
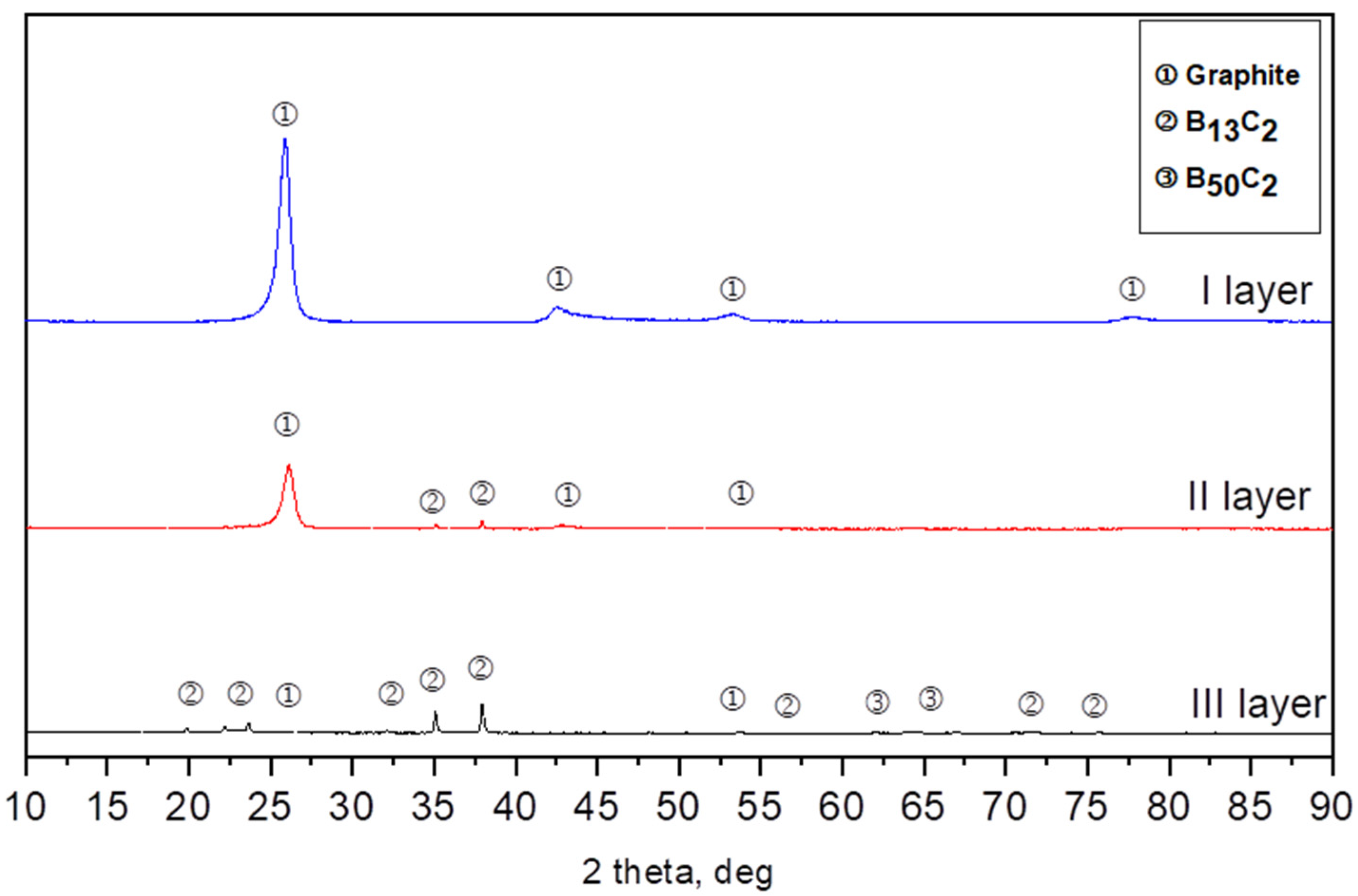
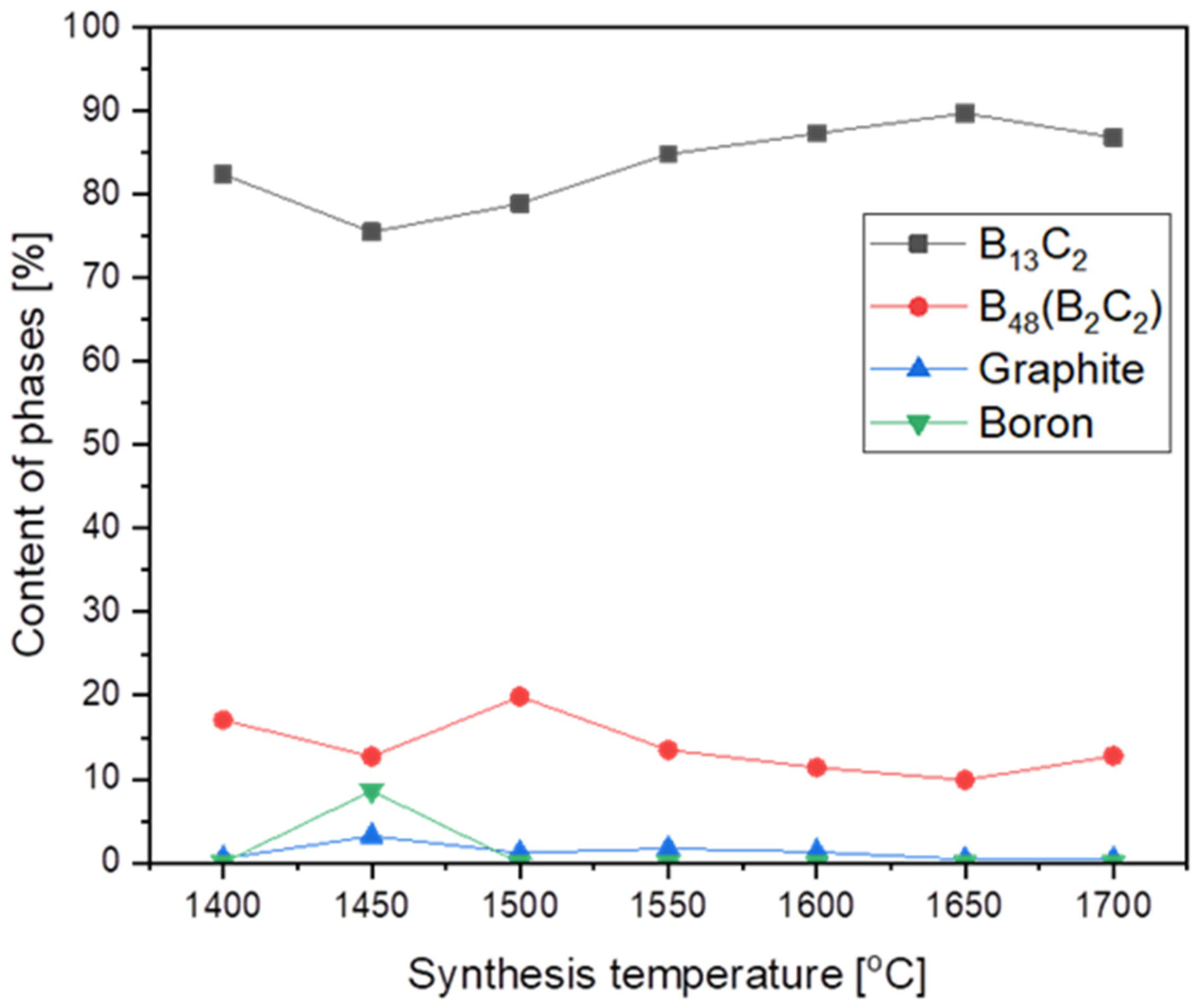
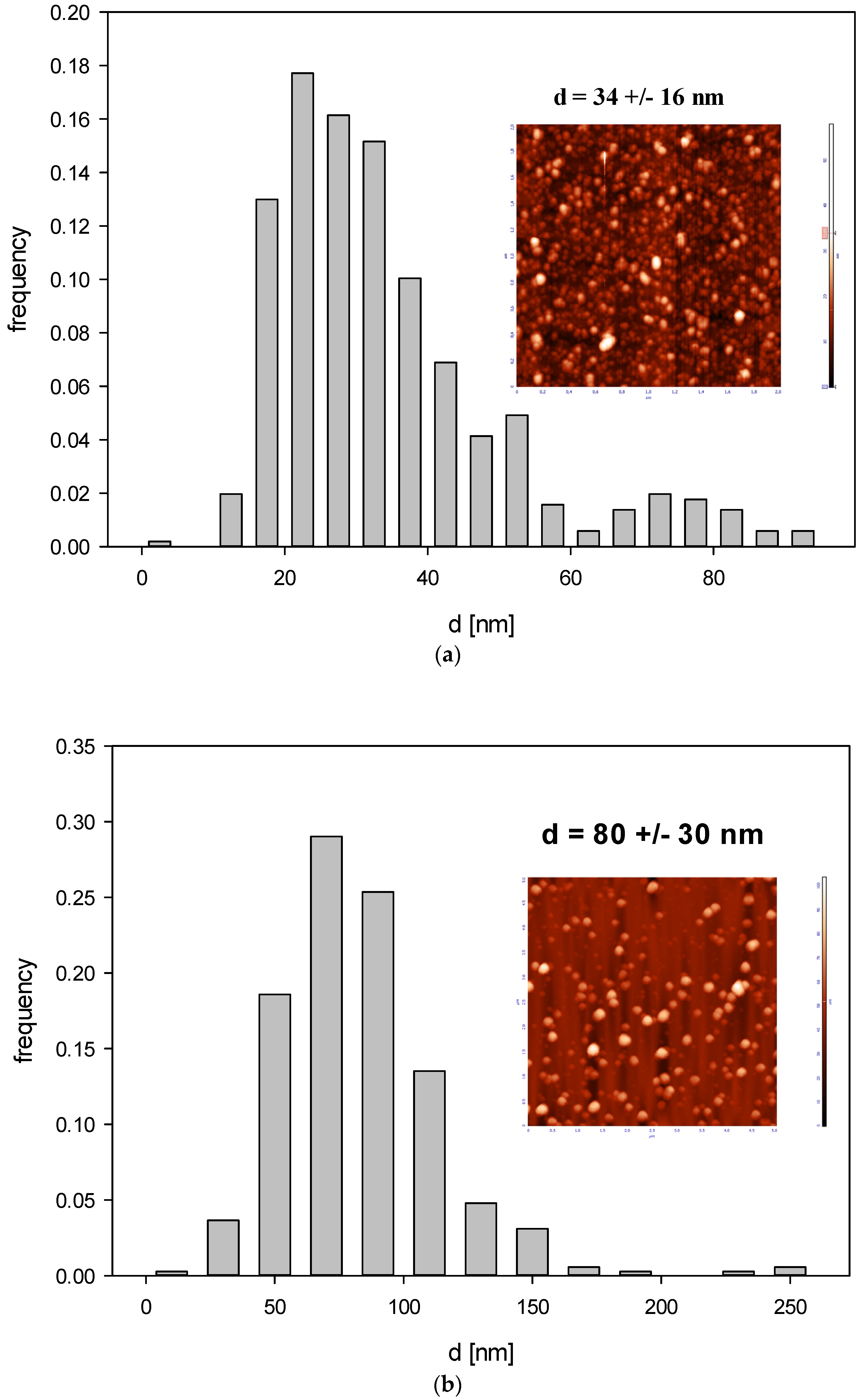
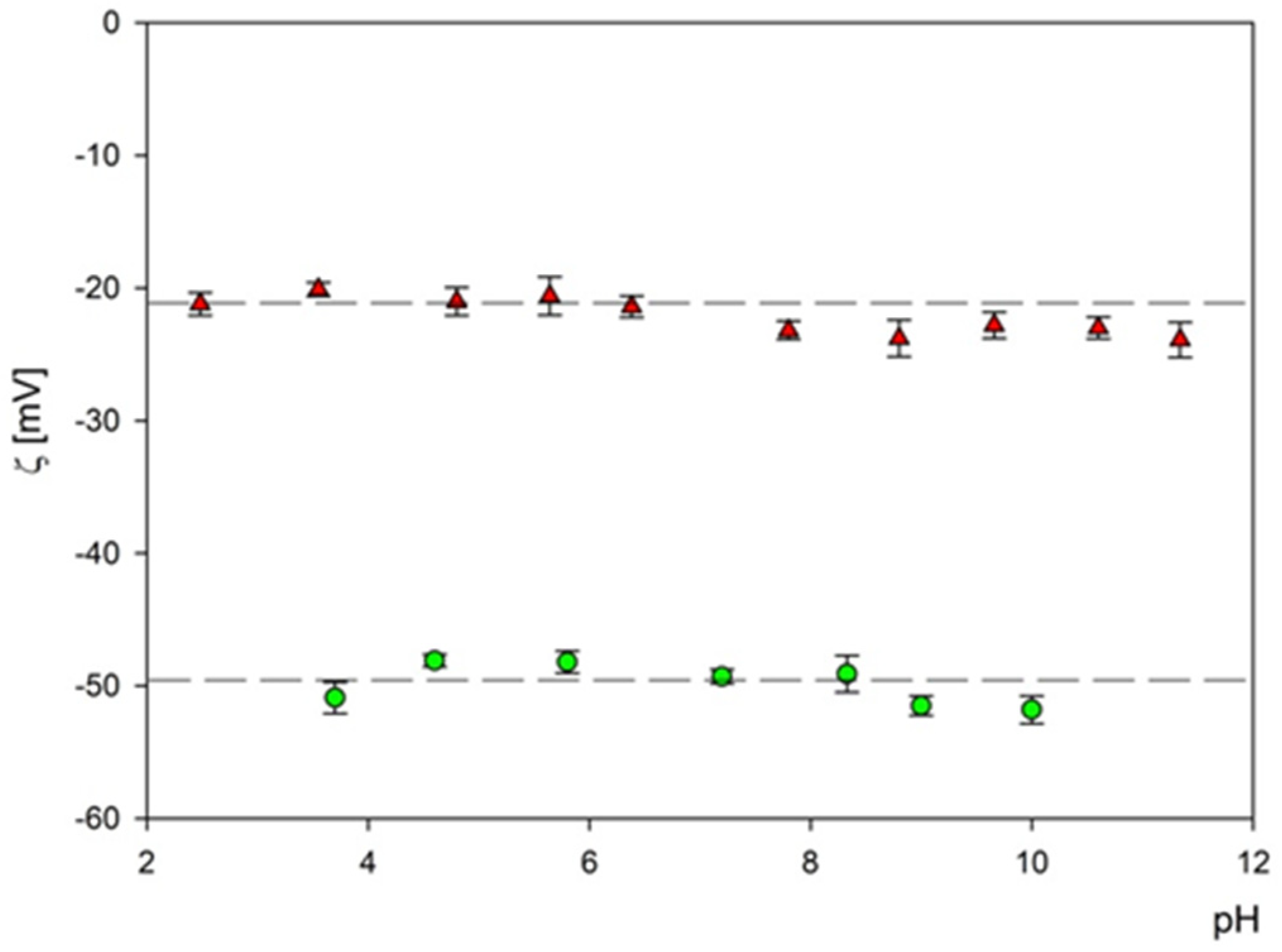
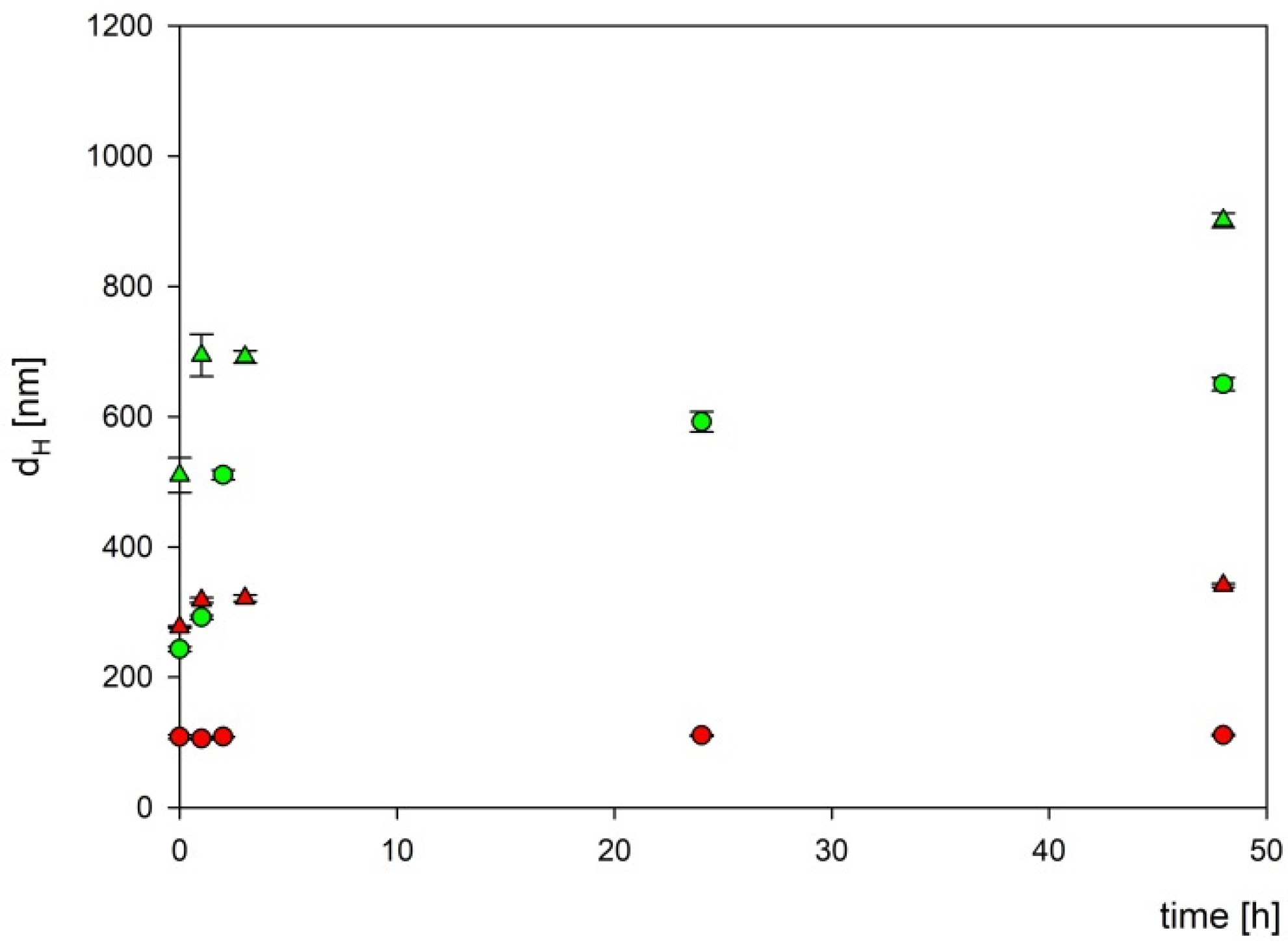
3.1. Obtaining and Analyzing the Surface of Boron Carbide Nanoparticles
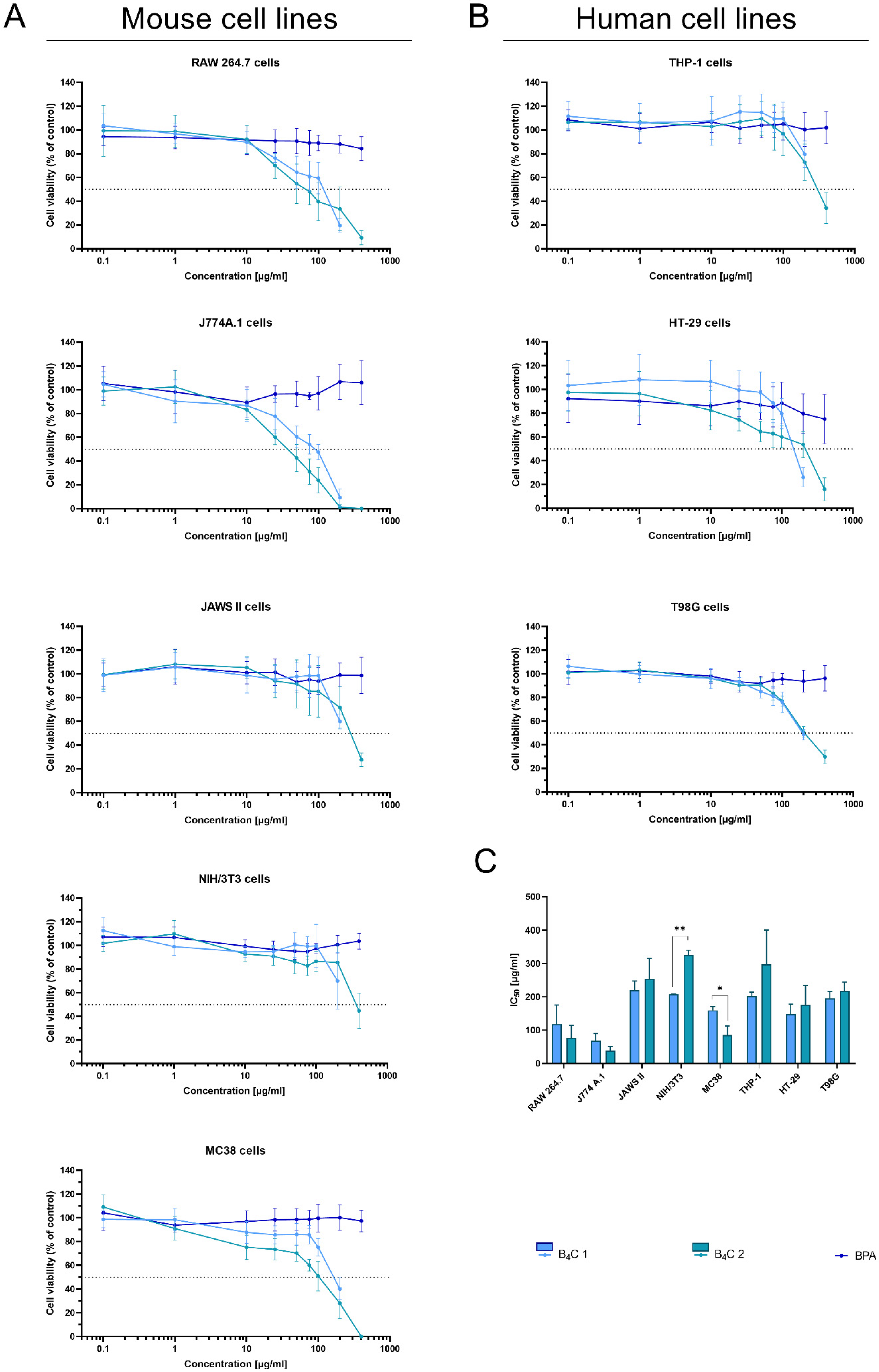
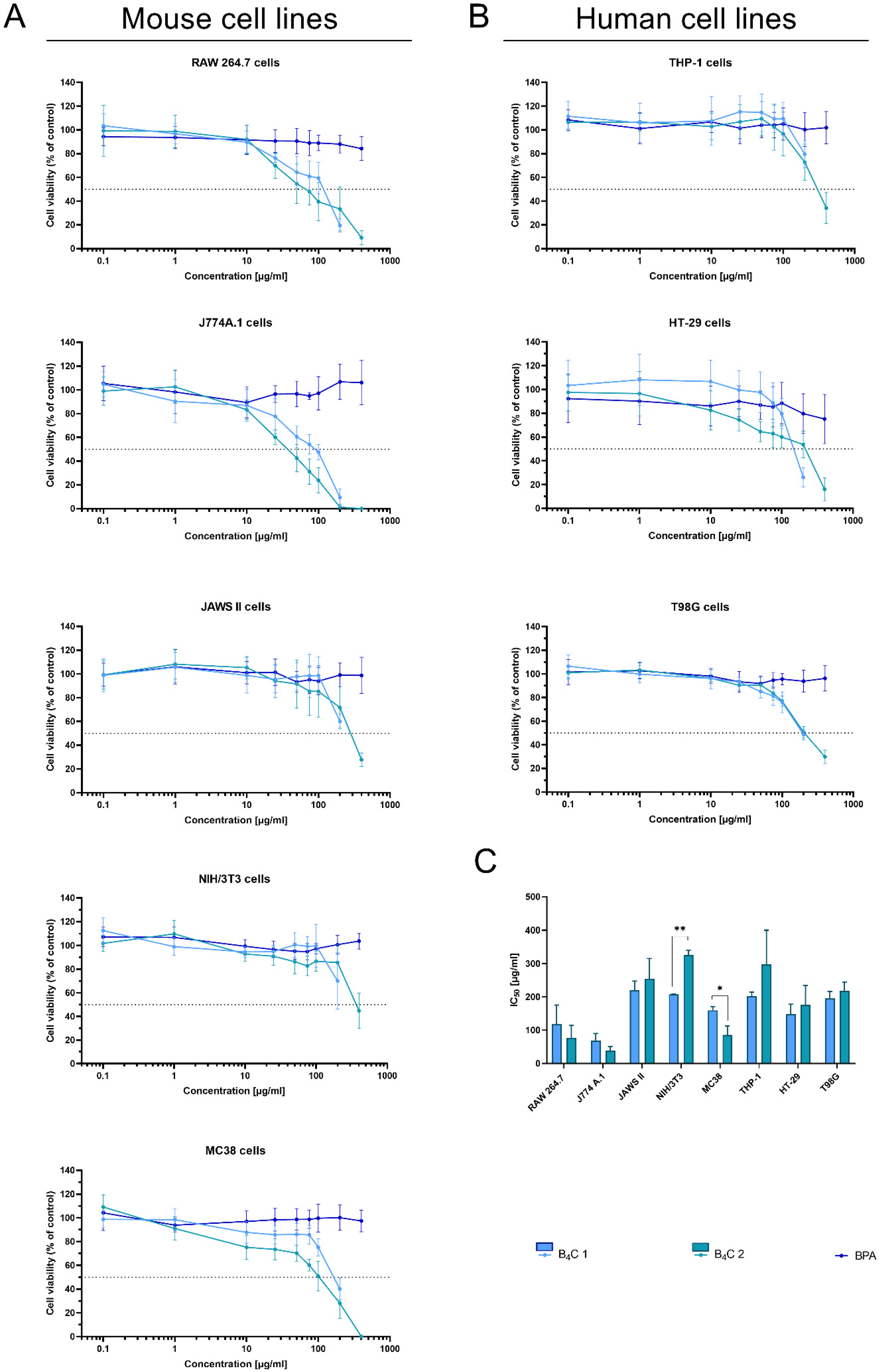
3.2. Analysis of the Cytotoxic Effect of Obtained Boron Carbide Nanoparticles
4. Conclusions
- In each obtained powder, the dominant phases of boron carbide were rhombohedral B13C2 and tetragonal B48(C2B2).
- The results suggest that the mechanism of boron carbide synthesis by the reaction between boron vapor and carbon cannot be excluded. However, this is not the only possible mechanism of boron transport.
- Boron carbides present good dispersion in water, which enhances uptake of their small-particle fraction, also by non-phagocytic cells. It can also facilitate their wide spreading in the biological environment and increase the effective delivery into the tumor site.
- The obtained boron carbide, after further surface modifications of the compound that will increase specificity for target cells, has the potential to be used as a new boron carrier in BNCT.
5. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kozień, D.; Pędzich, Z.; Jeleń, P. Synthesis and Preparation of Boron Carbide (B13C2) Nanoparticles. WIPO Patent ST 10/C PL440310, 12 September 2022. [Google Scholar]
- Thevenot, F. A Review on Boron Carbide. Key Eng. Mater. 1990, 56–57, 59–88. [Google Scholar] [CrossRef]
- Li, J.; Liu, L.; Xu, S.; Zhang, J.; Wu, Y. The Effects of Carbon Content on the Anisotropic Deformation Mechanism of Boron Carbide. Materials 2018, 11, 1861. [Google Scholar] [CrossRef] [PubMed]
- Bigdeloo, J.A.; Hadian, A.M. Synthesis of High Purity Micron Size Boron Carbide Powder from B2O3/C Precursor. Int. J. Recent Trends Eng. 2009, 1, 176–180. [Google Scholar]
- Suri, A.K.; Subramanian, C.; Sonber, J.K.; Murthy, T.C. Synthesis and Consolidation of Boron Carbide: A Review. Int. Mater. Rev. 2010, 55, 4–40. [Google Scholar] [CrossRef]
- Pierson, O.H. Handbook of Refractory Carbides and Nitrides: Properties, Characteristics, Processing and Applications; Noyes Publications: New York, NY, USA, 1996. [Google Scholar]
- Werheit, H. Boron Carbide: Consistency of Components, Lattice Parameters, Fine Structure and Chemical Composition Makes the Complex Structure Reasonable. Solid State Sci. 2016, 60, 45–54. [Google Scholar] [CrossRef]
- Xie, K.Y.; Domnich, V.; Farbaniec, L.; Chen, B.; Kuwelkar, K.; Ma, L.; McCauley, J.W.; Haber, R.A.; Ramesh, K.T.; Chen, M.; et al. Microstructural Characterization of Boron-Rich Boron Carbide. Acta Mater. 2017, 136, 202–214. [Google Scholar] [CrossRef]
- Wittig, A.; Michel, J.; Moss, R.L.; Stecher-Rasmussen, F.; Arlinghaus, H.F.; Bendel, P.; Mauri, P.L.; Altieri, S.; Hilger, R.; Salvadori, P.A.; et al. Boron Analysis and Boron Imaging in Biological Materials for Boron Neutron Capture Therapy (BNCT). Crit. Rev. Oncol. Hematol. 2008, 68, 66–90. [Google Scholar] [CrossRef]
- Moss, R.L. Critical Review, with an Optimistic Outlook, on Boron Neutron Capture Therapy (BNCT). Appl. Radiat. Isot. 2014, 88, 2–11. [Google Scholar] [CrossRef]
- Xia, K.; Ma, M.; Liu, C.; Gao, H.; Chen, Q.; He, J.; Sun, J.; Wang, H.-T.; Tian, Y.; Xing, D. Superhard and Superconducting B6C. Mater. Today Phys. 2017, 3, 76–84. [Google Scholar] [CrossRef]
- Muhlhauser, S. Z. Anorg. Chem.; 1894; Volume 5. [Google Scholar]
- Söderström, P.A.; Jaworski, G.; Valiente Dobón, J.J.; Nyberg, J.; Agramunt, J.; de Angelis, G.; Carturan, S.; Egea, J.; Erduran, M.N.; Ertürk, S.; et al. Neutron Detection and γ-Ray Suppression Using Artificial Neural Networks with the Liquid Scintillators BC-501A and BC-537. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrometers Detect. Assoc. Equip. 2019, 916, 238–245. [Google Scholar] [CrossRef]
- Ploog, K. Composition and Structure of Boron Carbides Prepared by CVD. J. Cryst. Growth 2000, 24/25, 197–204. [Google Scholar] [CrossRef]
- Will, G.; Kossobutzki, K.H. An X-Ray Diffraction Analysis of Boron Carbide, B13C2. J. Less-Common Met. 1976, 47, 43–48. [Google Scholar] [CrossRef]
- Beauvy, M. Stoichiometric Limits of Carbon-Rich Boron Carbide Phases. J. Less-Common Met. 1983, 90, 169–175. [Google Scholar] [CrossRef]
- Viala, J.C.; Bouix, J.; Gonzalez, G.; Esnouf, C. Chemical Reactivity of Aluminium with Boron Carbide. J. Mater. Sci. 1997, 32, 4559–4573. [Google Scholar] [CrossRef]
- Lartigue, S.; Male, G. Contribution to the Study of Tetragonal Compounds in the Boron Carbon System. J. Mater. Sci. Lett. 1988, 7, 153–156. [Google Scholar] [CrossRef]
- Farzaneh, F.; Golestanifard, F.; Sheikhaleslami, M.S.; Nourbakhsh, A.A. New Route for Preparing Nanosized Boron Carbide Powder via Magnesiothermic Reduction Using Mesoporous Carbon. Ceram. Int. 2015, 41, 13658–13662. [Google Scholar] [CrossRef]
- Ramos, A.S.; Taguchi, S.P.; Ramos, E.C.T.; Arantes, V.L.; Ribeiro, S. High-Energy Ball Milling of Powder B-C Mixtures. Mater. Sci. Eng. A 2006, 422, 184–188. [Google Scholar] [CrossRef]
- Sinha, A.; Mahata, T.; Sharma, B.P. Carbothermal Route for Preparation of Boron Carbide Powder from Boric Acid-Citric Acid Gel Precursor. J. Nucl. Mater. 2002, 301, 165–169. [Google Scholar] [CrossRef]
- Khanra, A.K. Production of Boron Carbide Powder by Carbothermal Synthesis of Gel Material. Bull. Mater. Sci. 2007, 30, 93–96. [Google Scholar] [CrossRef]
- Rafi-ud-din; Zahid, G.H.; Asghar, Z.; Maqbool, M.; Ahmad, E.; Azhar, T.; Subhani, T.; Shahzad, M. Ethylene Glycol Assisted Low-Temperature Synthesis of Boron Carbide Powder from Borate Citrate Precursors. J. Asian Ceram. Soc. 2014, 2, 268–274. [Google Scholar] [CrossRef]
- Kakiage, M.; Tominaga, Y.; Yanase, I.; Kobayashi, H. Synthesis of Boron Carbide Powder in Relation to Composition and Structural Homogeneity of Precursor Using Condensed Boric Acid-Polyol Product. Powder Technol. 2012, 221, 257–263. [Google Scholar] [CrossRef]
- Zakharova, K.; Mednikova, A.; Rumyantsev, V.; Genusova, T. Synthesis of Boron Carbide from Boric Acid and Carbon-Containing Precursors. Proc. Int. Conf. Nanomater. Appl. Prop. 2013, 2, 1–4. [Google Scholar]
- Rafi-ud-din; Zahid, G.H.; Ahmad, E.; Maqbool, M.; Subhani, T.; Syed, W.A.; Hussain, S.Z. Effect of Cellulose-Derived Structural Homogeneity of Precursor on the Synthesis and Morphology of Boron Carbide. J. Inorg. Organomet. Polym. Mater. 2015, 25, 995–999. [Google Scholar] [CrossRef]
- Chang, B.; Gersten, B.L.; Szewczyk, S.T.; Adams, J.W. Characterization of Boron Carbide Nanoparticles Prepared by a Solid State Thermal Reaction. Appl. Phys. A Mater. Sci. Process. 2007, 86, 83–87. [Google Scholar] [CrossRef]
- Barth, R.F.; Zhang, Z.; Liu, T. A Realistic Appraisal of Boron Neutron Capture Therapy as a Cancer Treatment Modality. Cancer Commun. 2018, 38, 36. [Google Scholar] [CrossRef]
- Kabalka, G.W.; Yao, M.L.; Marepally, S.R.; Chandra, S. Biological Evaluation of Boronated Unnatural Amino Acids as New Boron Carriers. Appl. Radiat. Isot. 2009, 67, 374–379. [Google Scholar] [CrossRef]
- Palmer, M.R.; Goorley, J.T.; Kiger, W.S.; Busse, P.M.; Riley, K.J.; Harling, O.K.; Zamenhof, R.G. Treatment Planning and Dosimetry for the Harvard-MIT Phase I Clinical Trial of Cranial Neutron Capture Therapy. Int. J. Radiat. Oncol. 2002, 53, 1361–1379. [Google Scholar] [CrossRef]
- Yoshie Ishikawa, T.I. Boron Carbide Particle as a Boron Compound for Boron Neutron Capture Therapy. J. Nucl. Med. Radiat. Ther. 2014, 5. [Google Scholar] [CrossRef]
- Mortensen, M.W.; Björkdahl, O.; Sørensen, P.G.; Hansen, T.; Jensen, M.R.; Gundersen, H.J.G.; Bjørnholm, T. Functionalization and Cellular Uptake of Boron Carbide Naraoparticles. The First Step toward T Cell-Guided Boron Neutron Capture Therapy. Bioconjug. Chem. 2006, 17, 284–290. [Google Scholar] [CrossRef]
- Türkez, H.; Arslan, M.E.; Sönmez, E.; Geyikoğlu, F.; Açıkyıldız, M.; Tatar, A. Microarray Assisted Toxicological Investigations of Boron Carbide Nanoparticles on Human Primary Alveolar Epithelial Cells. Chem. Biol. Interact. 2019, 300, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Oćwieja, M.; Adamczyk, Z.; Morga, M.; Michna, A. High Density Silver Nanoparticle Monolayers Produced by Colloid Self-Assembly on Polyelectrolyte Supporting Layers. J. Colloid Interface Sci. 2011, 364, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Pajtasz-Piasecka, E.; Szyda, A.; Rossowska, J.; Krawczenko, A.; Indrová, M.; Grabarczyk, P.; Wysocki, P.; Mackiewicz, A.; Dús, D. Loss of Tumorigenicity of Murine Colon Carcinoma MC38/0 Cell Line after Transduction with a Retroviral Vector Carrying Murine IL-12 Genes. Folia Biol. 2004, 50, 7–14. [Google Scholar]
- Morga, M.; Nattich-Rak, M.; Oćwieja, M.; Adamczyk, Z. Gold Substrates of Controlled Roughness and Electrokinetic Properties Formed by Nanoparticle Deposition. Phys. Chem. Chem. Phys. 2019, 21, 6535–6543. [Google Scholar] [CrossRef]
- Jin, G.; Xiao, F.; Liu, R. Synthesis and Biological Evaluation of a New Series of Ortho-Carboranyl Biphenyloxime Derivatives. Chem. Cent. J. 2018, 12, 76. [Google Scholar] [CrossRef]
- Mori, Y.; Suzuki, A.; Yoshino, K.; Kakihana, H. Complex Formation of P-Boronophenylalanine with Some Monosaccharides. Pigment Cell Res. 1989, 2, 273–277. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Synthesis Temperature [°C] | B13C2-dXRD [nm] | B48(B2C2)-dXRD [nm] |
|---|---|---|
| 1400 | 11.5 | 7.8 |
| 1450 | 10.7 | 8 |
| 1500 | 10.4 | 7.8 |
| 1550 | 12.4 | 10.2 |
| 1600 | 16.2 | 12.8 |
| 1650 | 13.4 | 12.6 |
| 1700 | 41 | 7.2 |
| 10−3 M NaCl | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| pH | 2.48 | 3.55 | 4.08 | 5.64 | 6.38 | 7.8 | 8.8 | 9.66 | 10.6 | 11.34 |
| µe [µm cm V−1s−1] | −1.66 | −1.58 | −1.65 | −1.61 | −1.68 | −1.81 | −1.87 | −1.77 | −1.80 | −1.87 |
| ζ [mV] | −21.2 | −20.1 | −21 | −20.6 | −21.4 | −23.2 | −23.8 | −22.8 | −23 | −23.9 |
| ζ Henry [mV] | −28.9 | −28.7 | −30.0 | −29.5 | −30.1 | −32.5 | −33.6 | −31.8 | −32.2 | −33.4 |
| dH [nm] DLS by number | 31 ± 8 | 29 ± 7 | 29 ± 7 | 28 ± 6 | 43 ± 7 | 42 ± 8 | 42 ± 9 | 42 ± 6 | 41 ± 9 | 45 ± 10 |
| Charge Density [e nm2] | −0.0281 | −0.0154 | −0.0149 | −0.0140 | −0.0142 | −0.0165 | −0.0167 | −0.0154 | −0.0157 | −0.0165 |
| Sg [nm2] ×103 | 3.02 | 2.64 | 2.64 | 2.46 | 5.81 | 5.54 | 5.54 | 5.54 | 5.28 | 6.36 |
| 10−3 M NaCl | |||||||
|---|---|---|---|---|---|---|---|
| pH | 3.7 | 4.6 | 5.8 | 7.2 | 8.33 | 9.02 | 10 |
| µe [µm cm V−1s−1] | −3.99 | −3.77 | −3.78 | −3.86 | −3.85 | −4.04 | −4.06 |
| ζ [mV] | −50.9 | −48.1 | −48.2 | −49.3 | −49.1 | −51.5 | −51.8 |
| ζ Henry [mV] | −56 | −55 | −57 | −58 | −57 | −60 | −61 |
| dH [nm] DLS by number | 66 ± 5 | 64 ± 12 | 64 ± 10 | 66 ± 8 | 70 ± 6 | 69 ± 5 | 64 ± 8 |
| Charge Density [e nm2] | −0.0335 | −0. 0302 | −0.0314 | −0.0321 | −0.0314 | −0.0337 | −0.0345 |
| Sg [nm2] ×104 | 1.37 | 1.29 | 1.29 | 1.37 | 1.54 | 1.50 | 1.29 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozień, D.; Żeliszewska, P.; Szermer-Olearnik, B.; Adamczyk, Z.; Wróblewska, A.; Szczygieł, A.; Węgierek-Ciura, K.; Mierzejewska, J.; Pajtasz-Piasecka, E.; Tokarski, T.; et al. Synthesis and Characterization of Boron Carbide Nanoparticles as Potential Boron-Rich Therapeutic Carriers. Materials 2023, 16, 6534. https://doi.org/10.3390/ma16196534
Kozień D, Żeliszewska P, Szermer-Olearnik B, Adamczyk Z, Wróblewska A, Szczygieł A, Węgierek-Ciura K, Mierzejewska J, Pajtasz-Piasecka E, Tokarski T, et al. Synthesis and Characterization of Boron Carbide Nanoparticles as Potential Boron-Rich Therapeutic Carriers. Materials. 2023; 16(19):6534. https://doi.org/10.3390/ma16196534
Chicago/Turabian StyleKozień, Dawid, Paulina Żeliszewska, Bożena Szermer-Olearnik, Zbigniew Adamczyk, Anna Wróblewska, Agnieszka Szczygieł, Katarzyna Węgierek-Ciura, Jagoda Mierzejewska, Elżbieta Pajtasz-Piasecka, Tomasz Tokarski, and et al. 2023. "Synthesis and Characterization of Boron Carbide Nanoparticles as Potential Boron-Rich Therapeutic Carriers" Materials 16, no. 19: 6534. https://doi.org/10.3390/ma16196534
APA StyleKozień, D., Żeliszewska, P., Szermer-Olearnik, B., Adamczyk, Z., Wróblewska, A., Szczygieł, A., Węgierek-Ciura, K., Mierzejewska, J., Pajtasz-Piasecka, E., Tokarski, T., Cios, G., Cudziło, S., & Pędzich, Z. (2023). Synthesis and Characterization of Boron Carbide Nanoparticles as Potential Boron-Rich Therapeutic Carriers. Materials, 16(19), 6534. https://doi.org/10.3390/ma16196534