
Hydrophobic, Thermal Shock-and-Corrosion-Resistant XSBR Latex-Modified Lightweight Class G Cement Composites in Geothermal Well Energy Storage Systems

Abstract
:1. Introduction
2. Materials and Methods
2.1. Starting Materials
2.2. Cement Formulas and Sample Preparation
2.3. Measurements
3. Results and Discussion
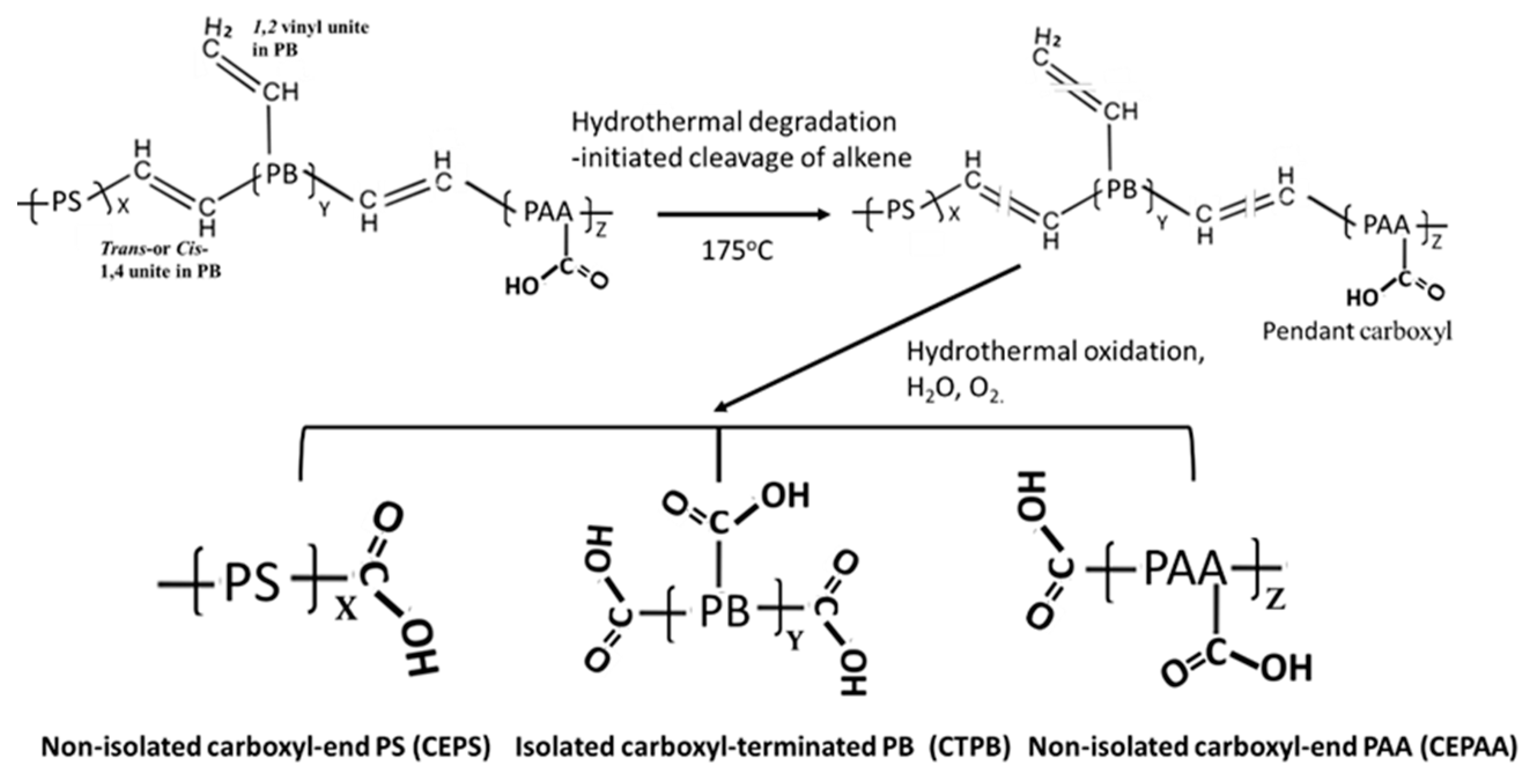
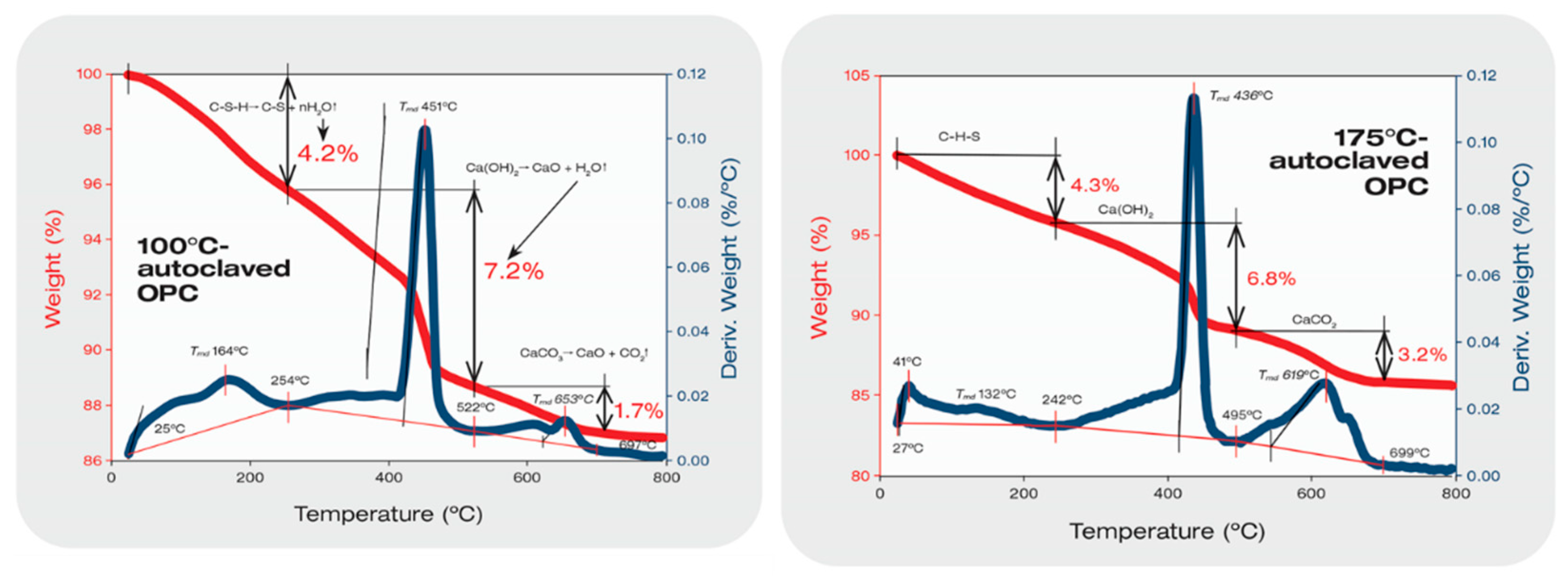
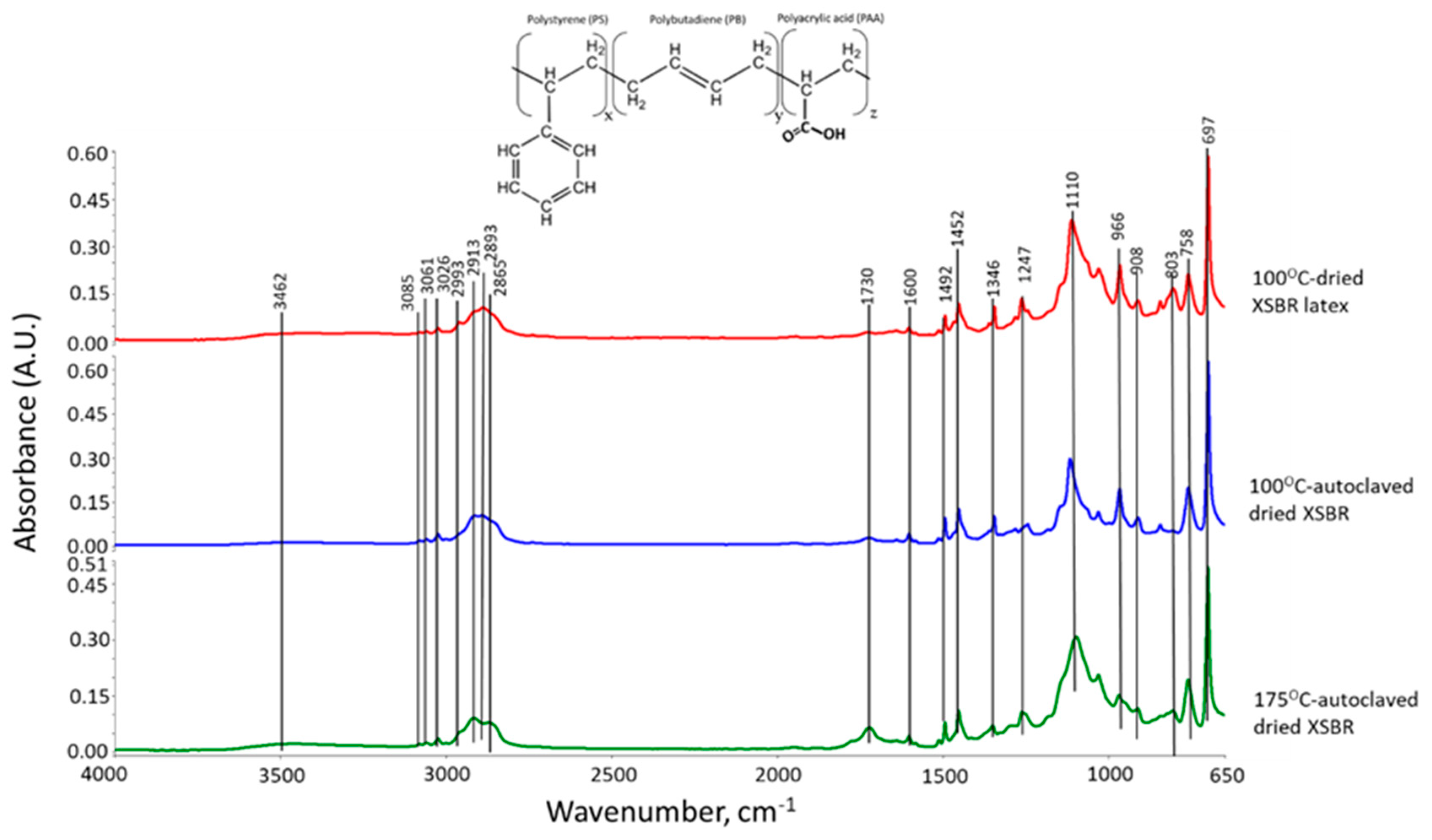
3.1. Hydrothermal Stability of XSBR
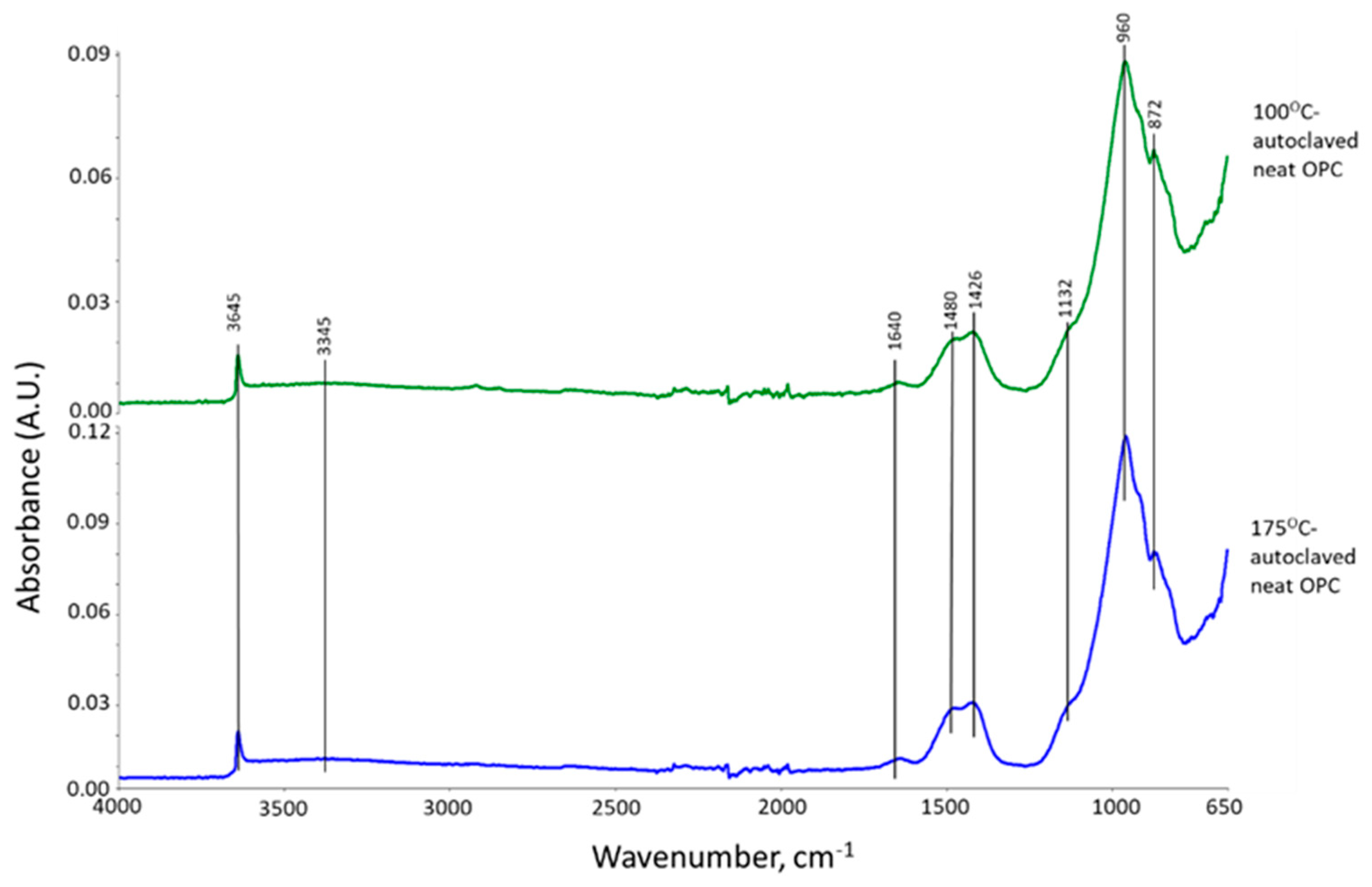
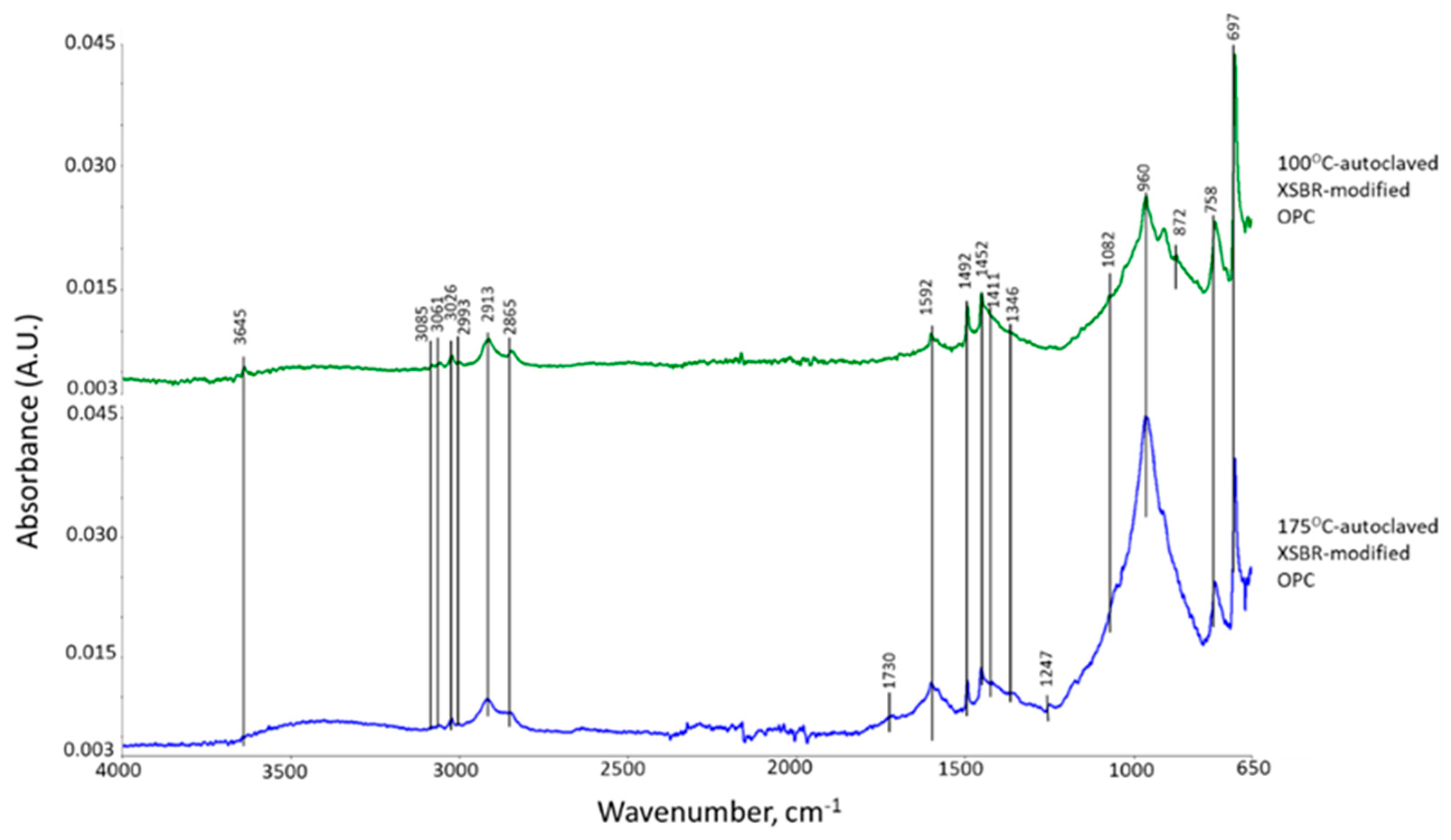
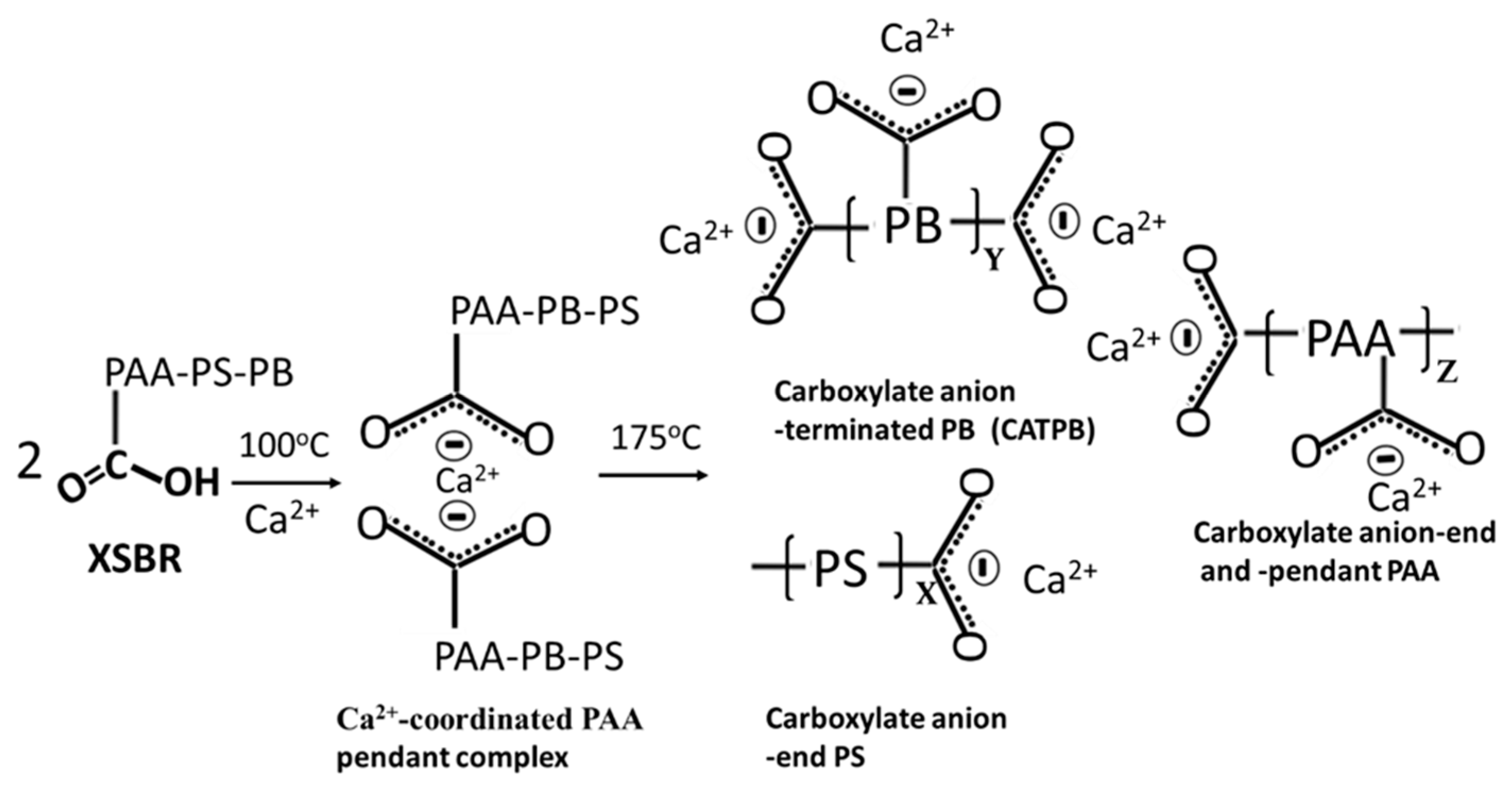
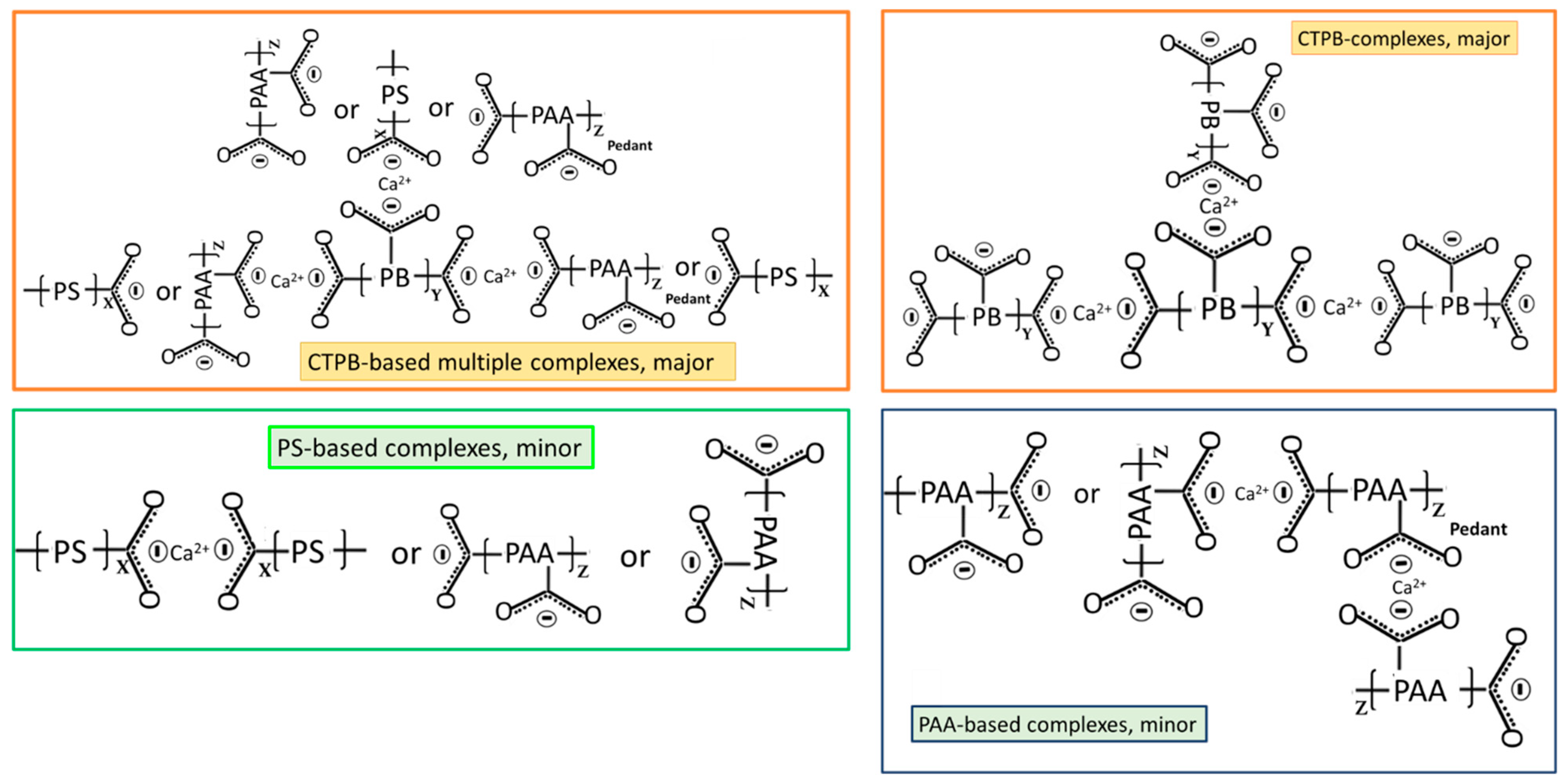
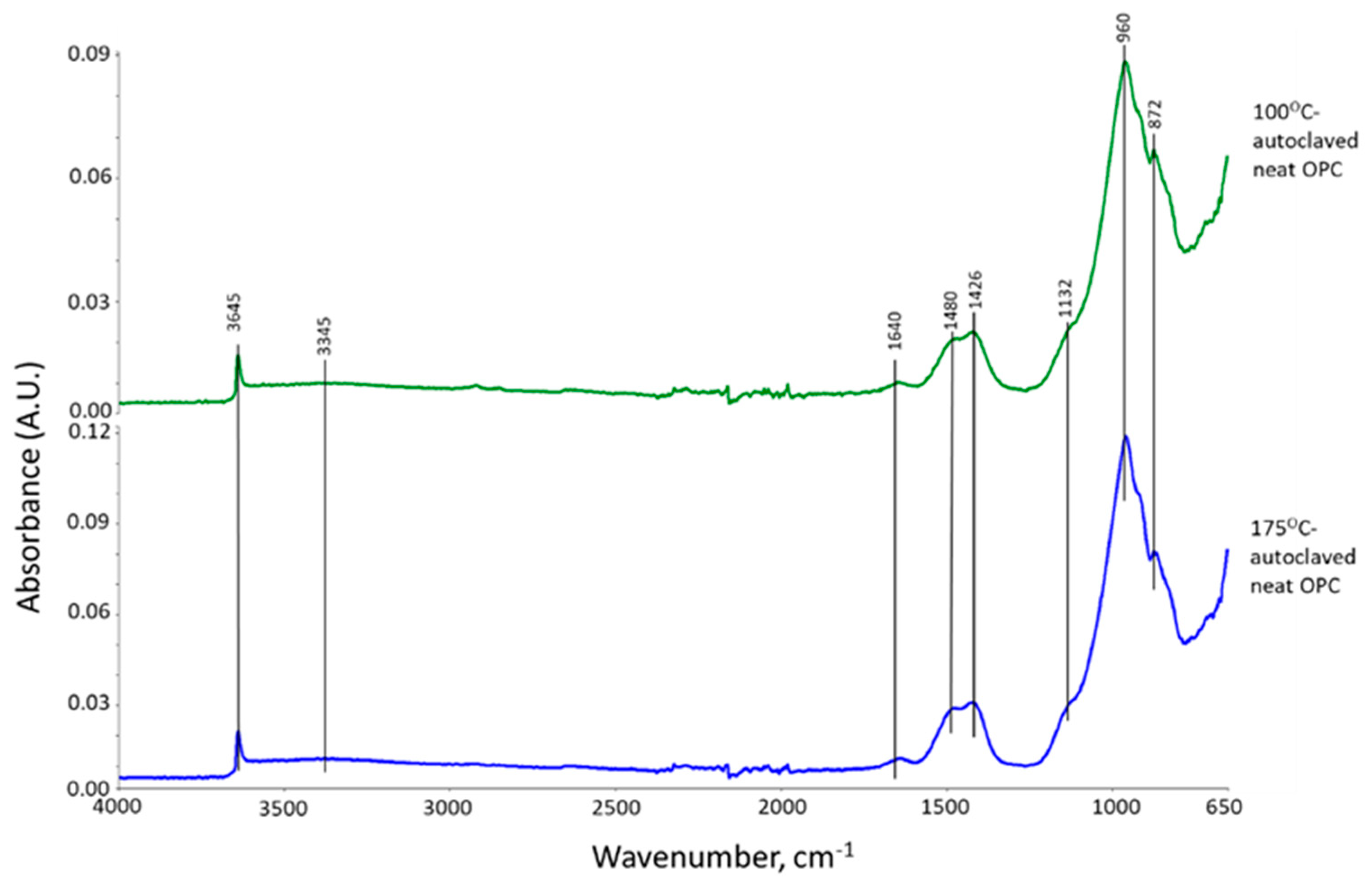
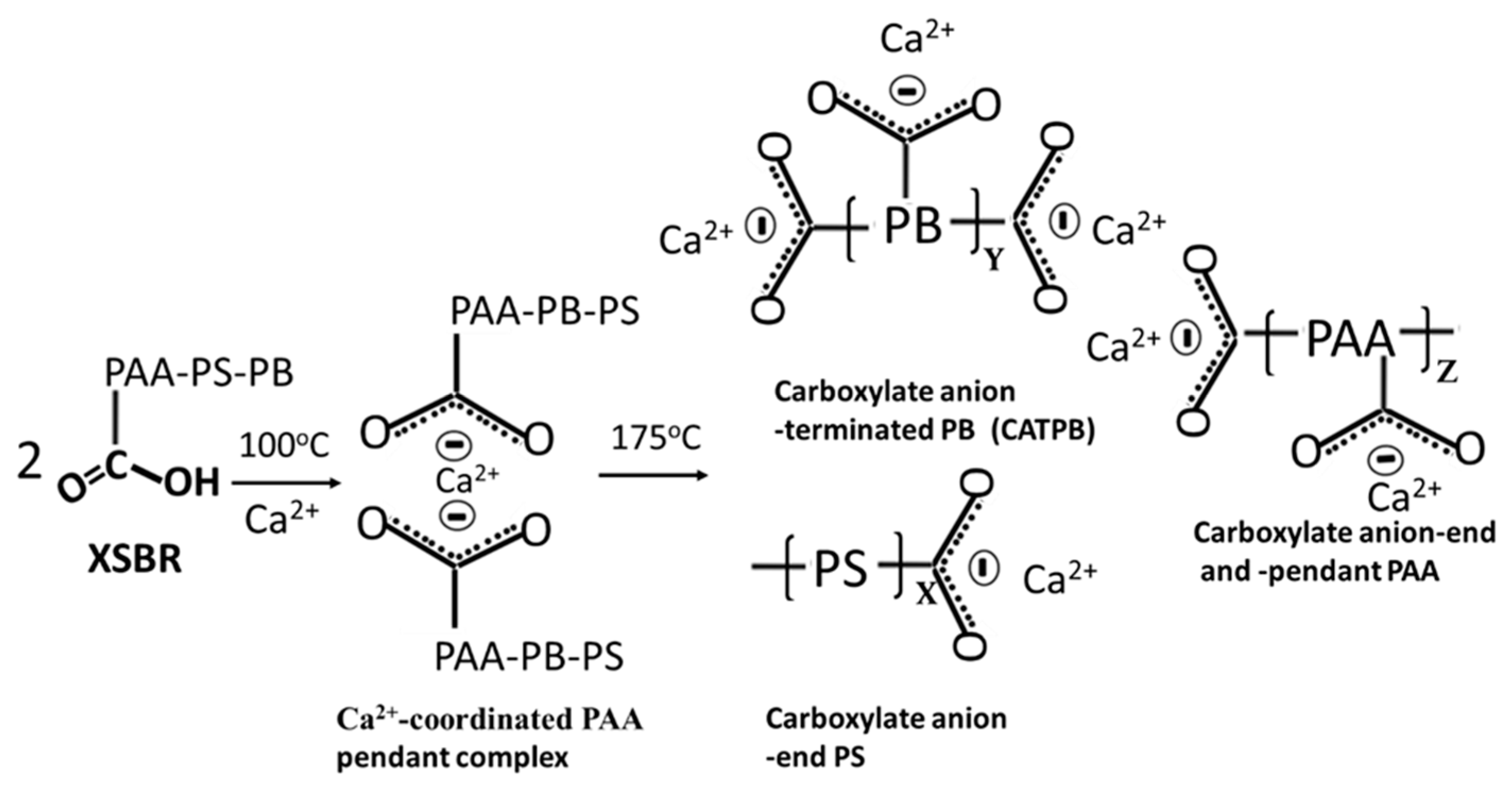
3.2. Products of Hydrothermal Reaction between XSBR and OPC
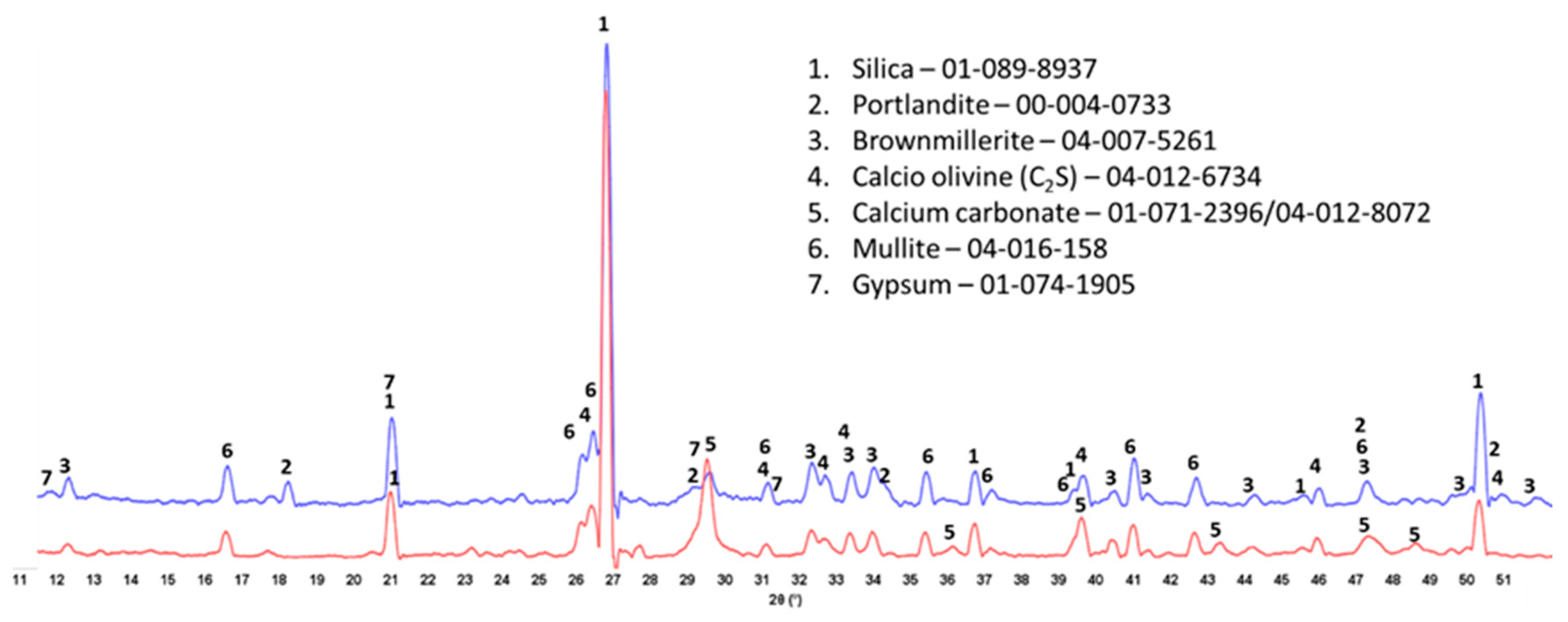
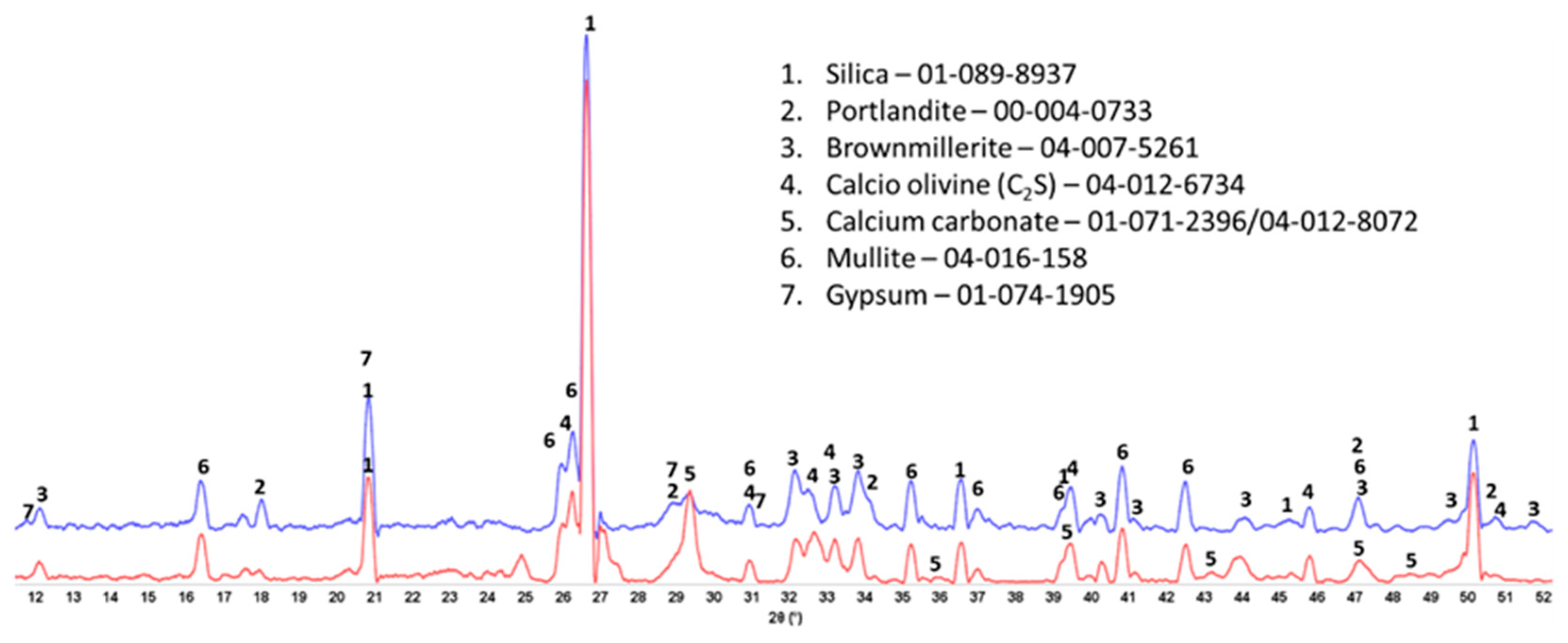
3.3. Crystalline Phase Composition
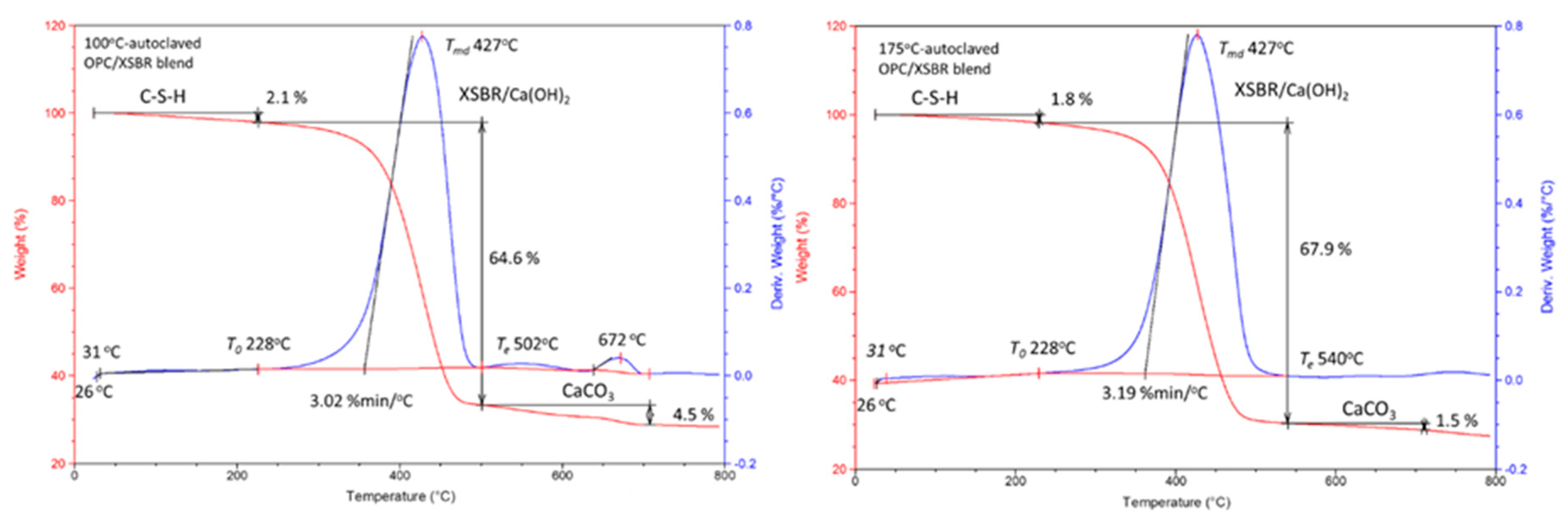
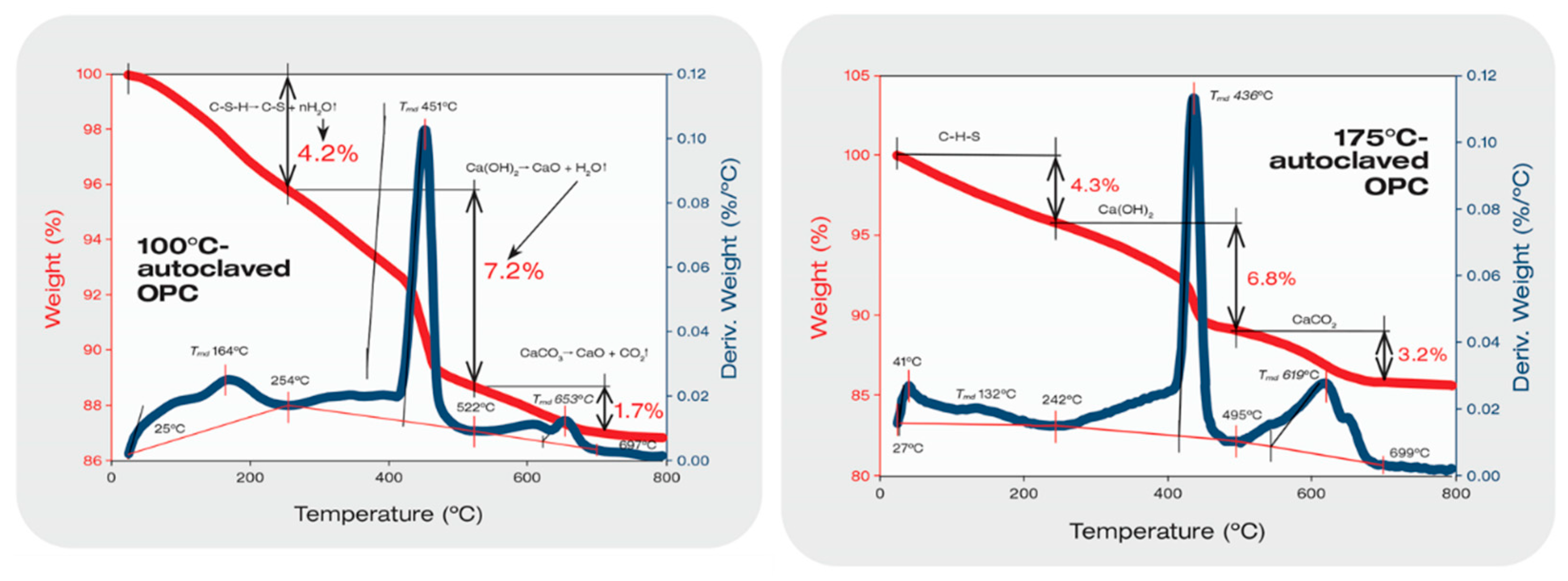
3.4. Thermal Stability of XSBR-OPC Reaction Products
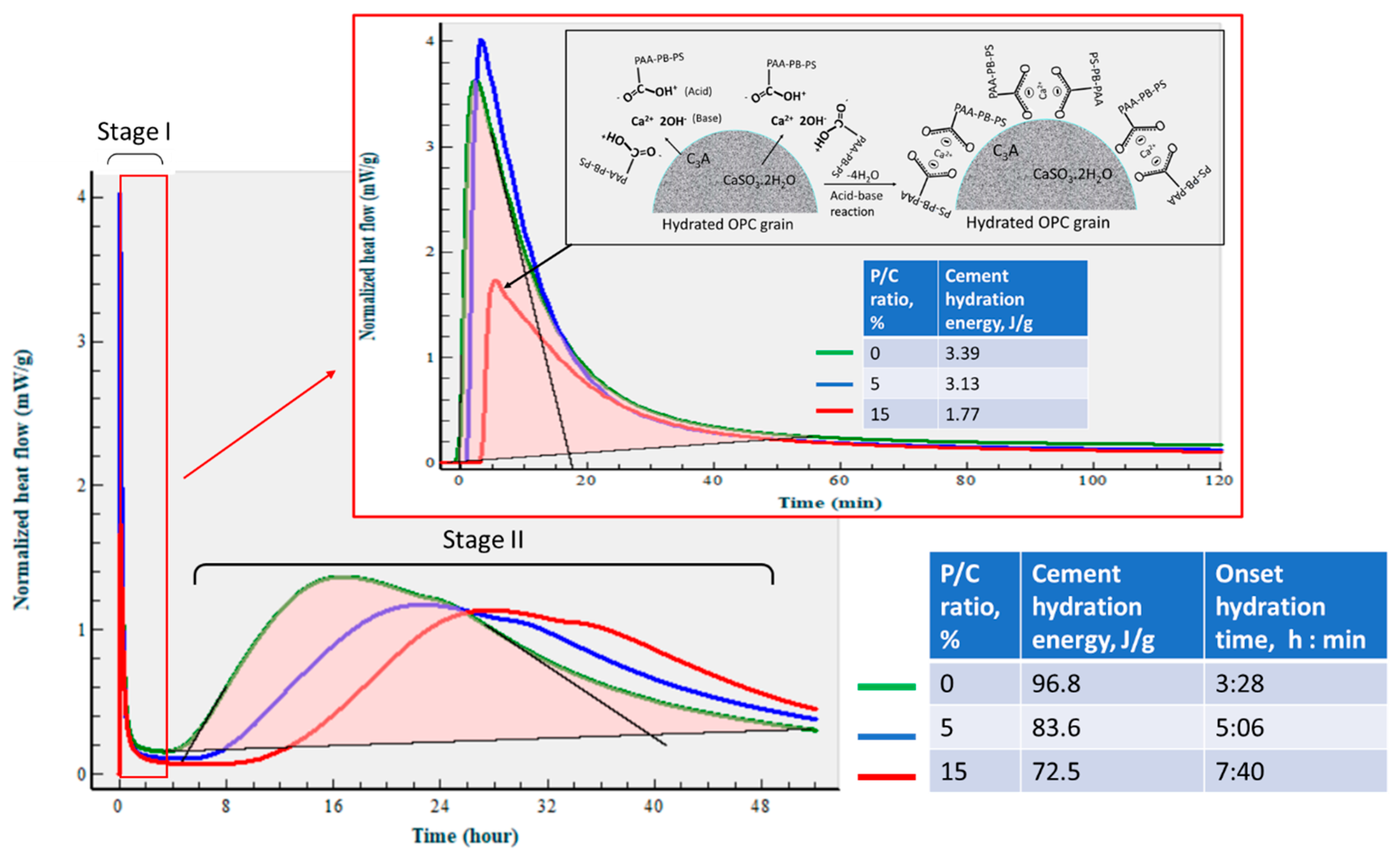
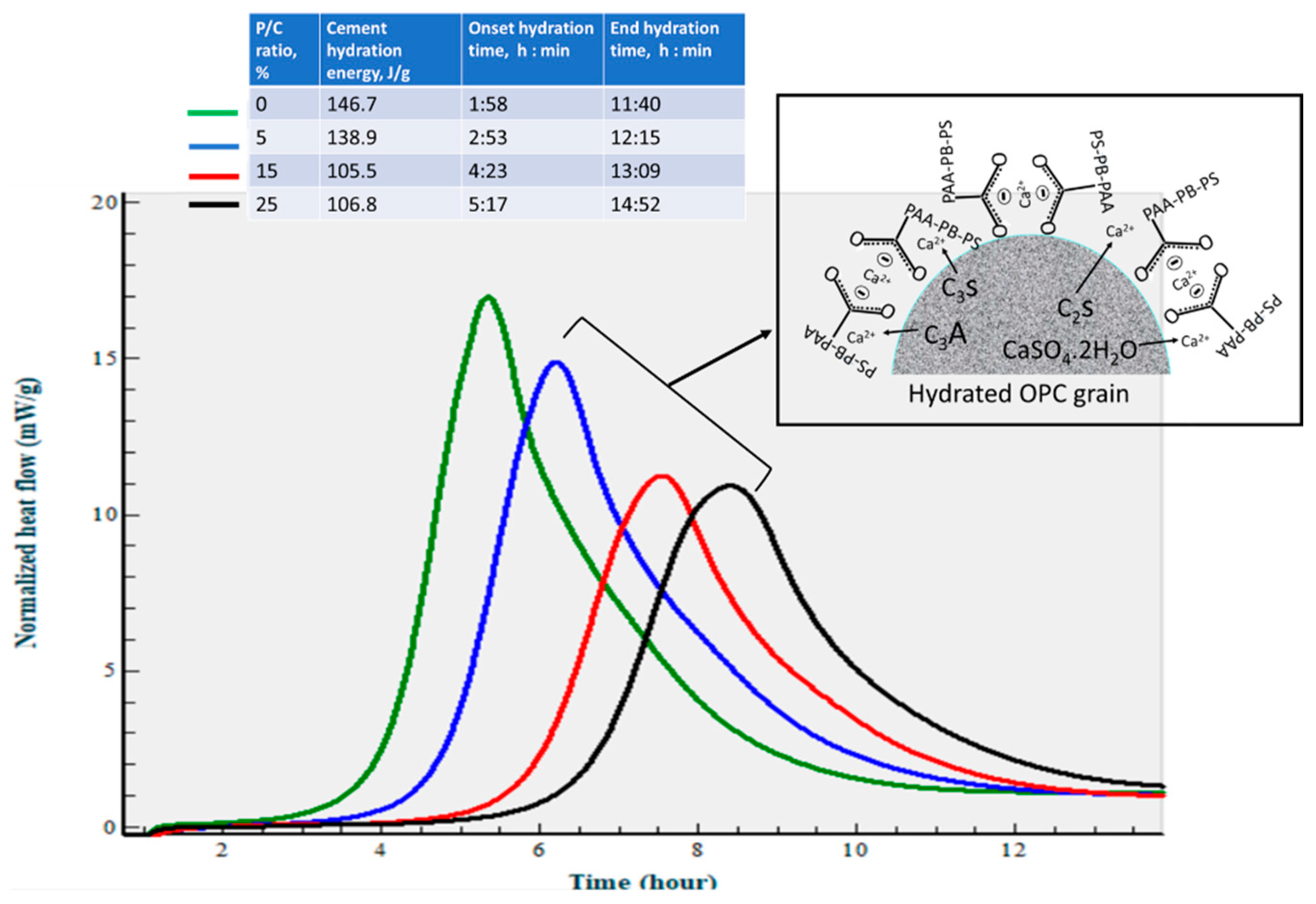
3.5. Changes in Hydration Behavior of OPC by XSBR
3.6. Slurry Properties
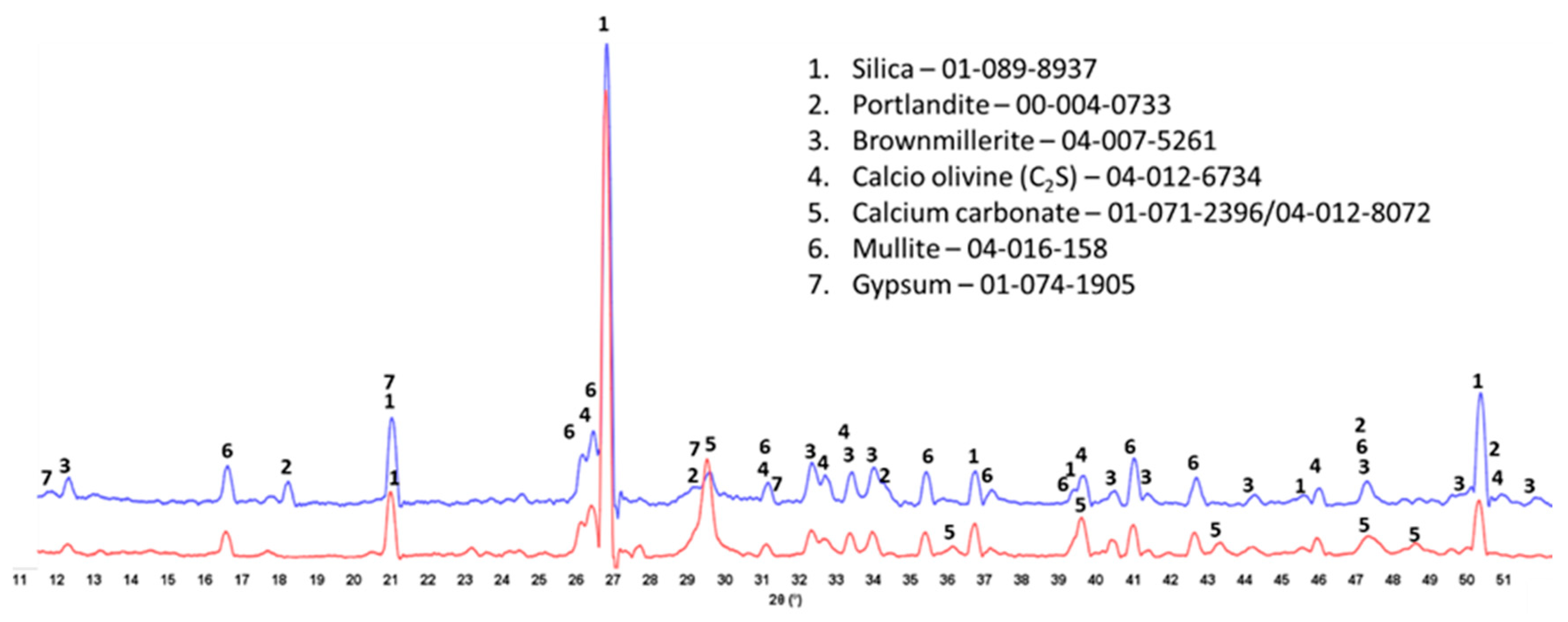
3.7. Crystalline Phase Compositions
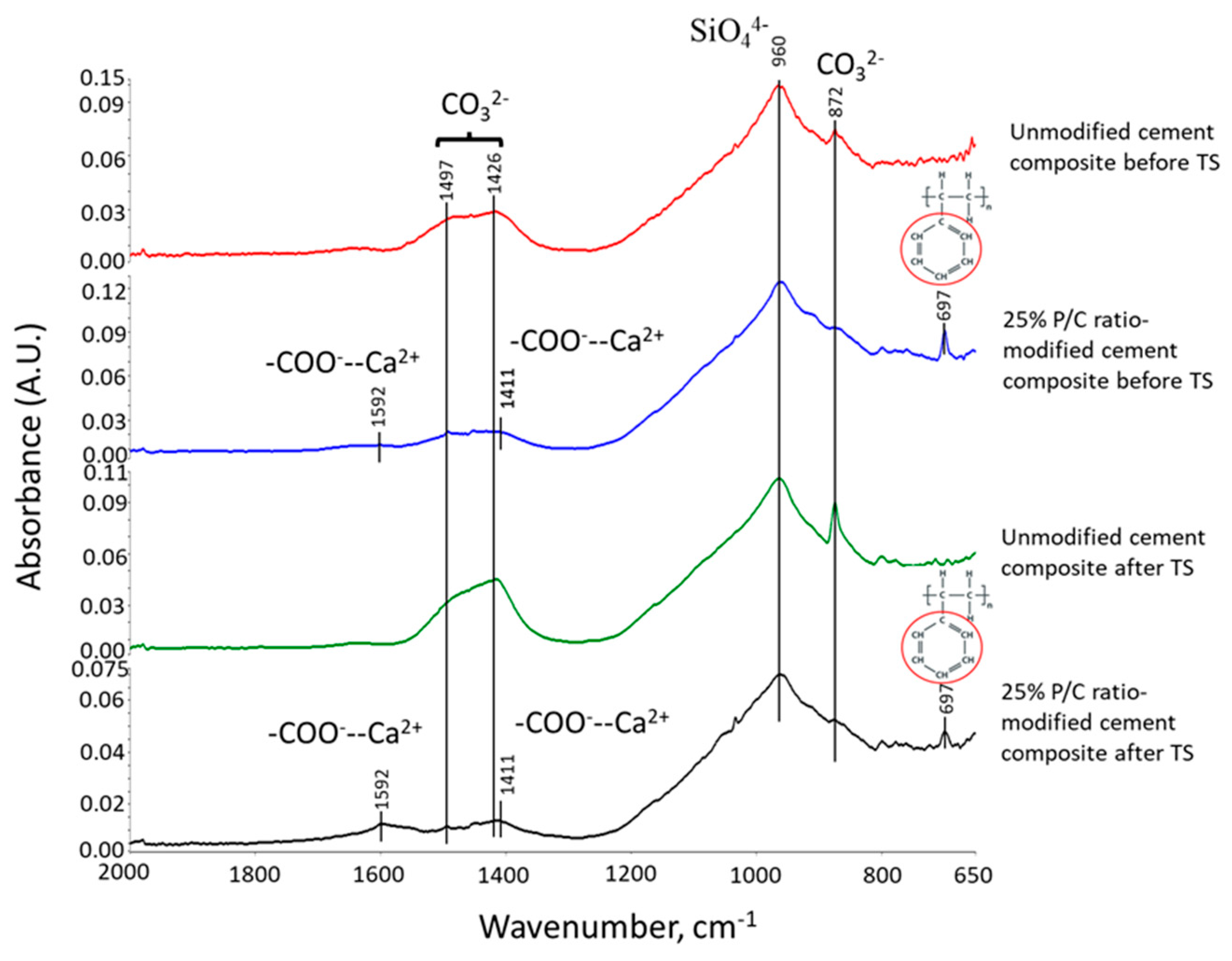
3.8. Molecular Alterations of XSBR in OPC Composites after Thermal Shock (TS) Tests
3.9. Water Repellency of Composite Surfaces
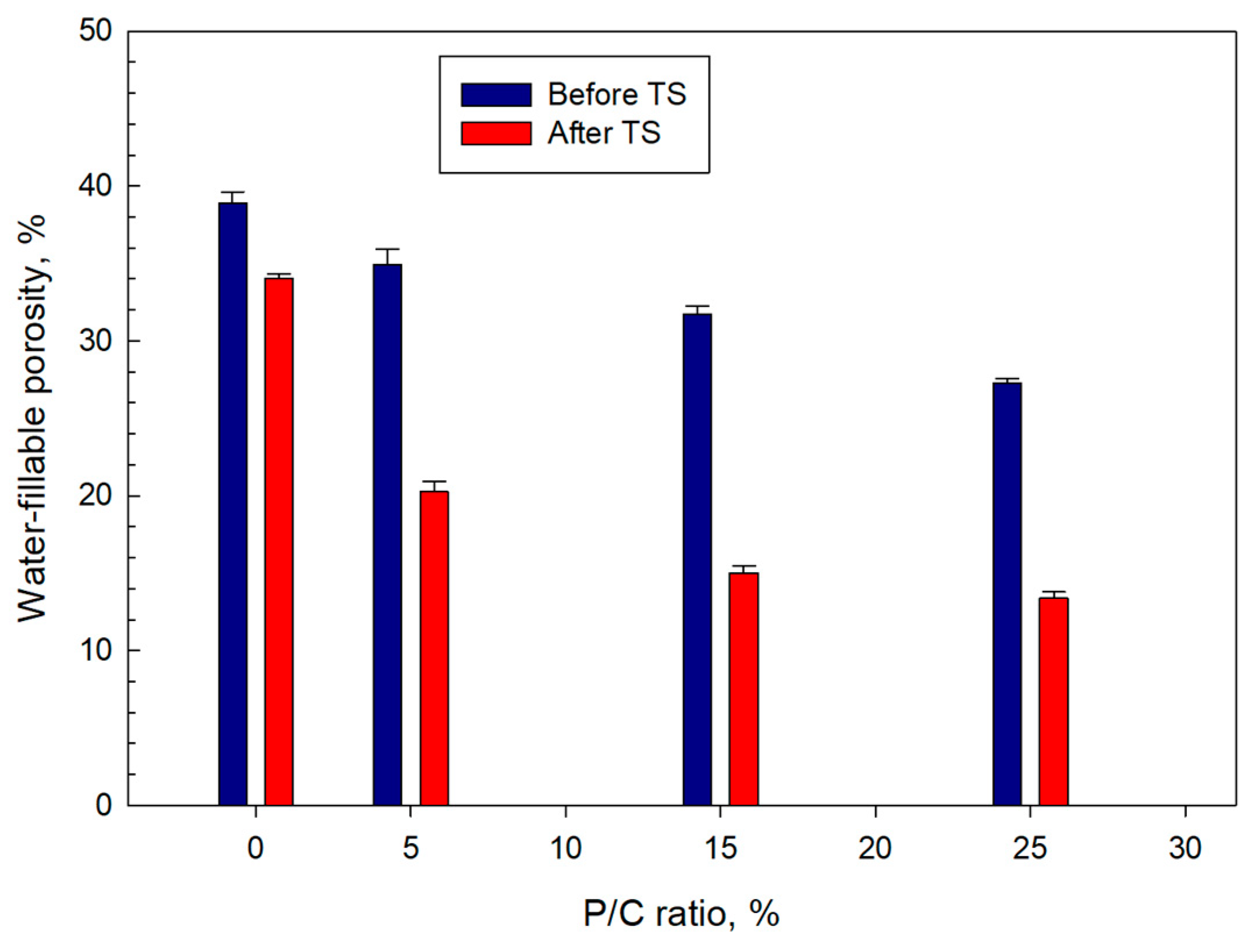
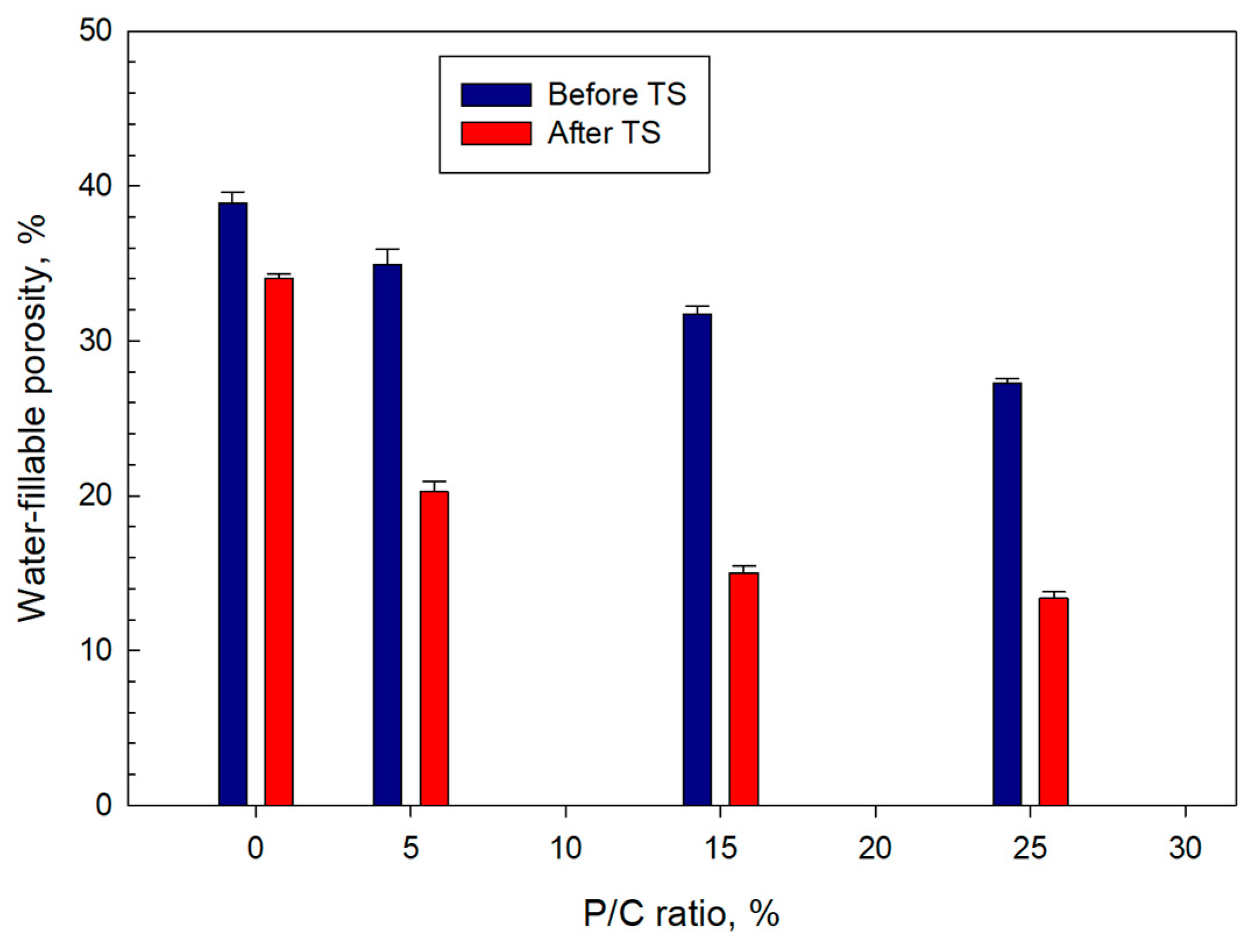
3.10. Water-Fillable Porosity of Composites
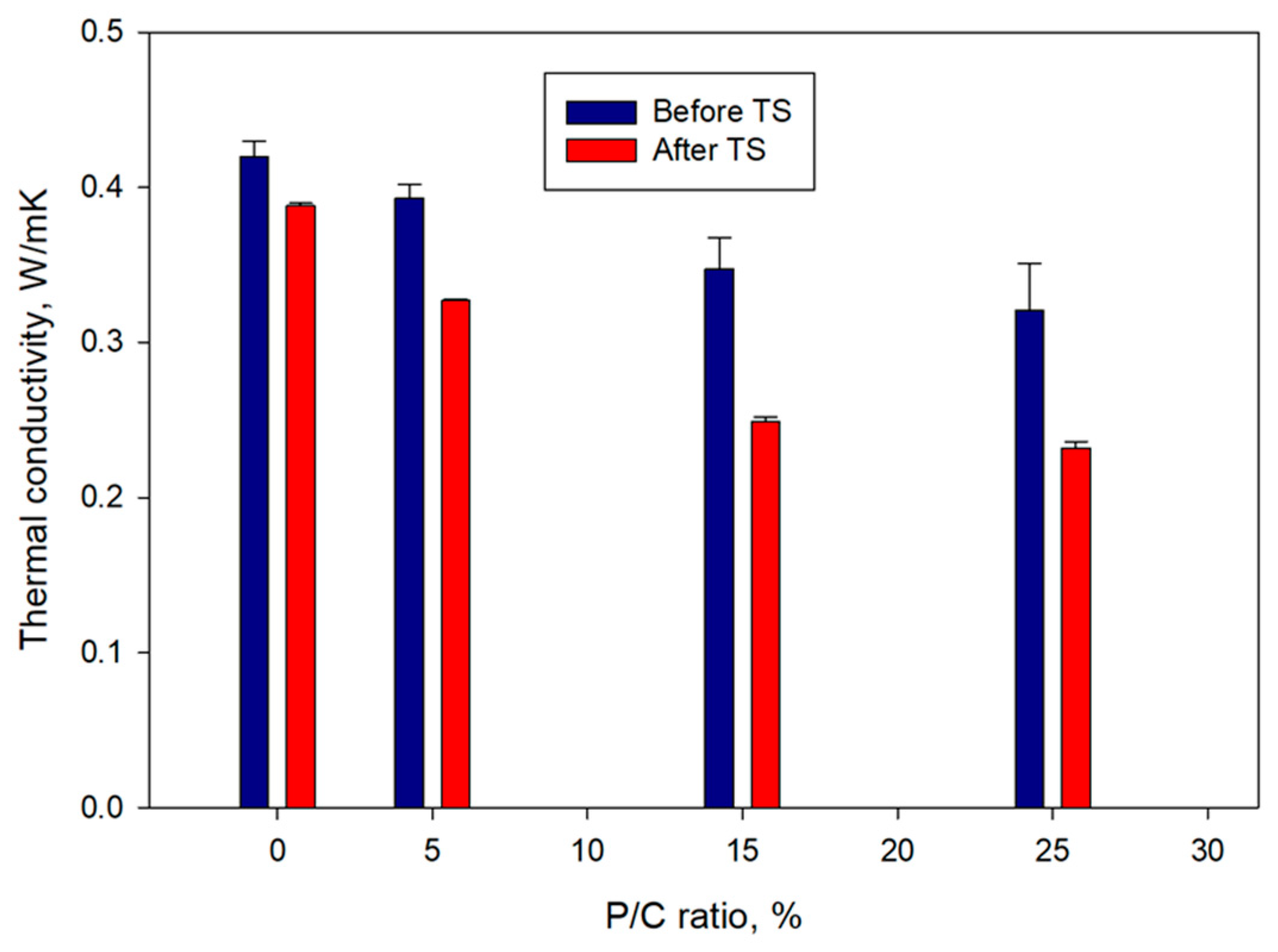
3.11. Thermal Conductivity
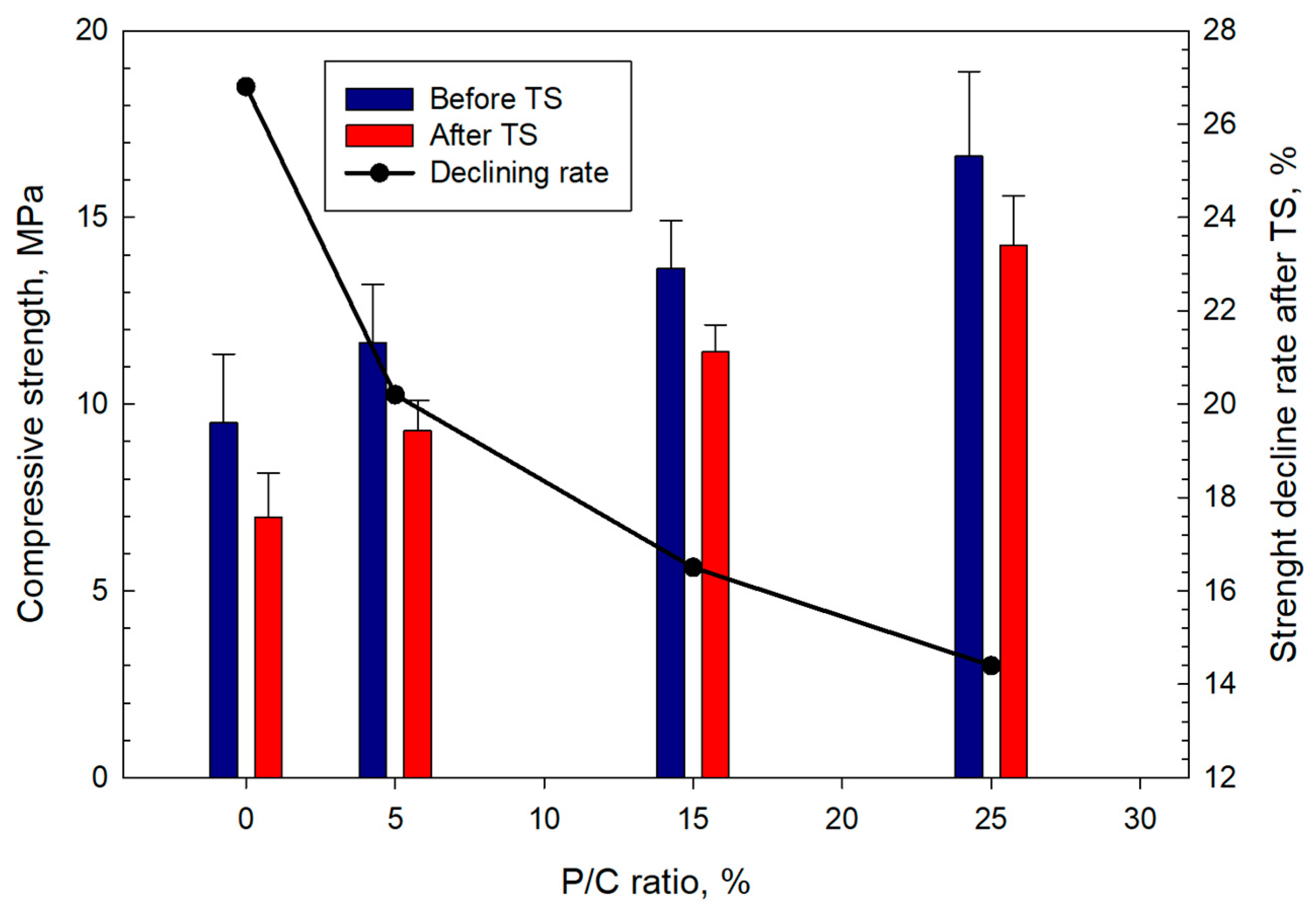
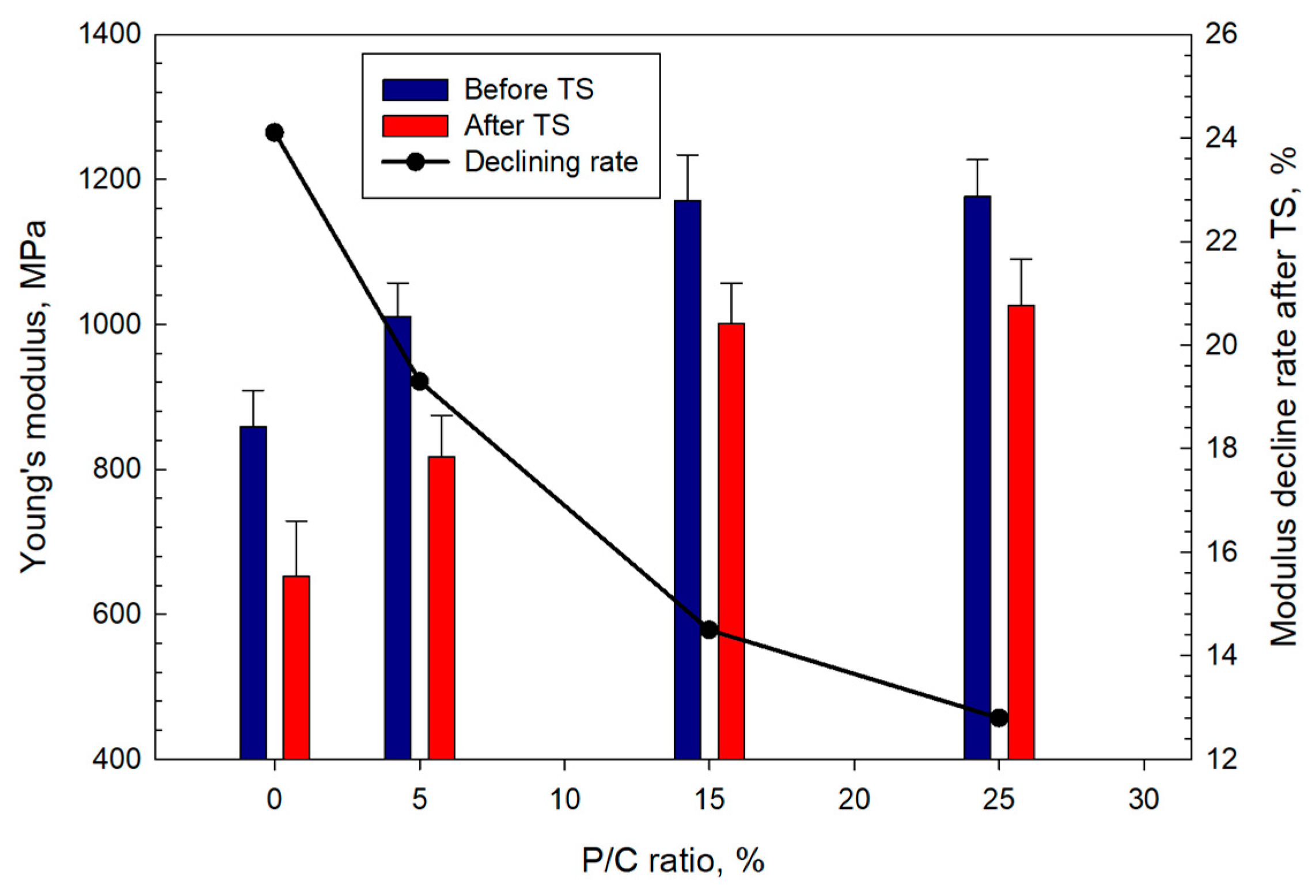
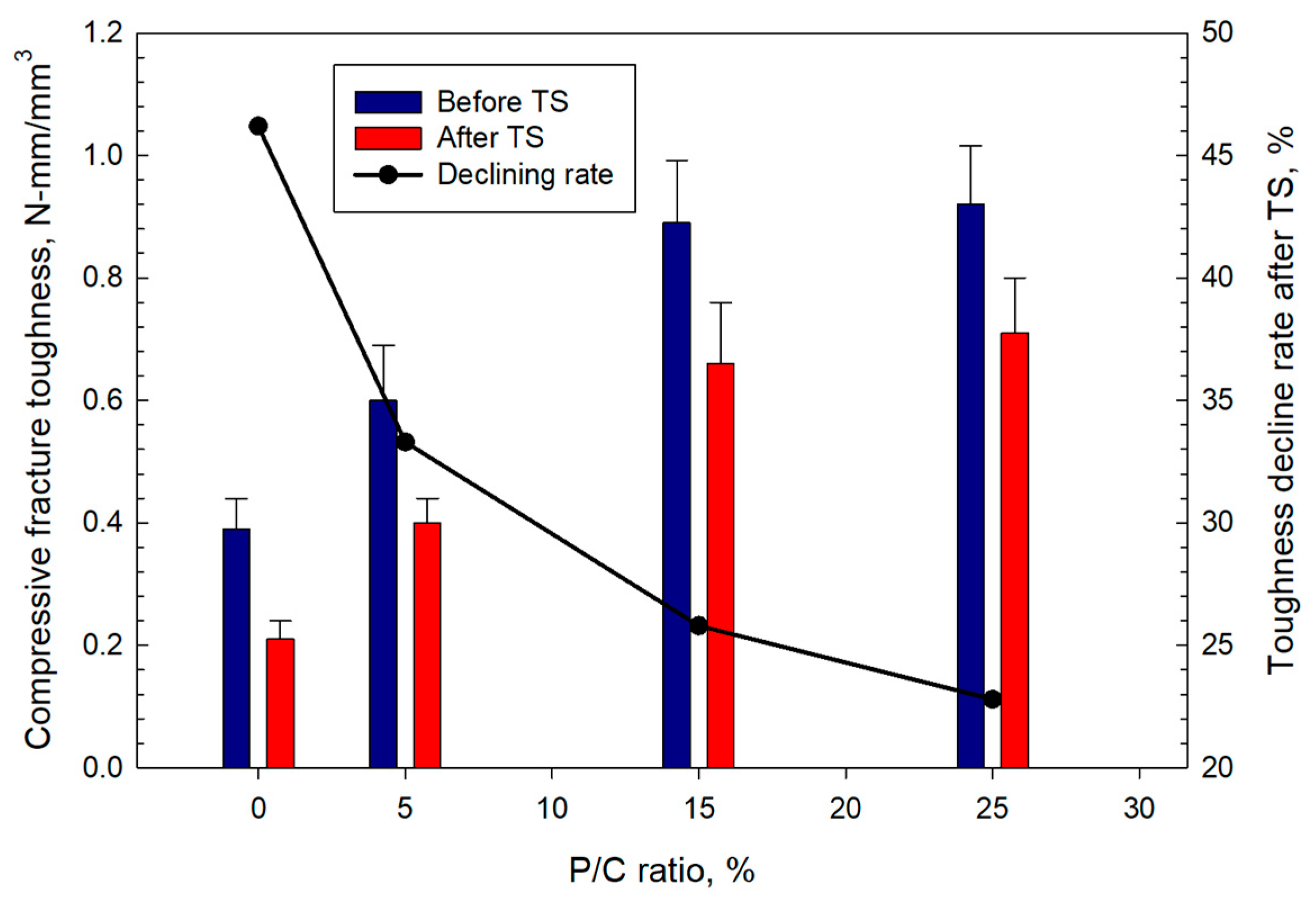
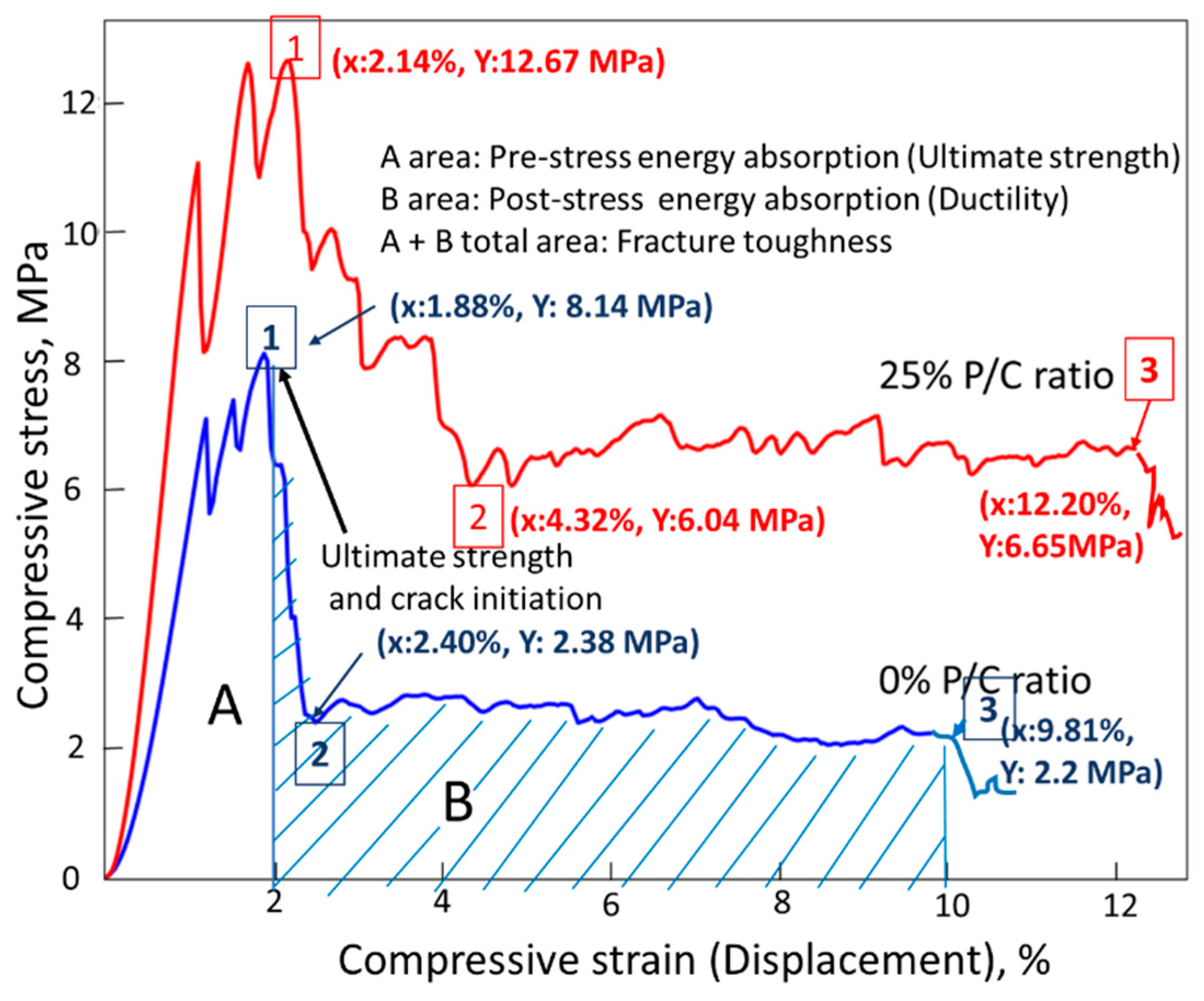
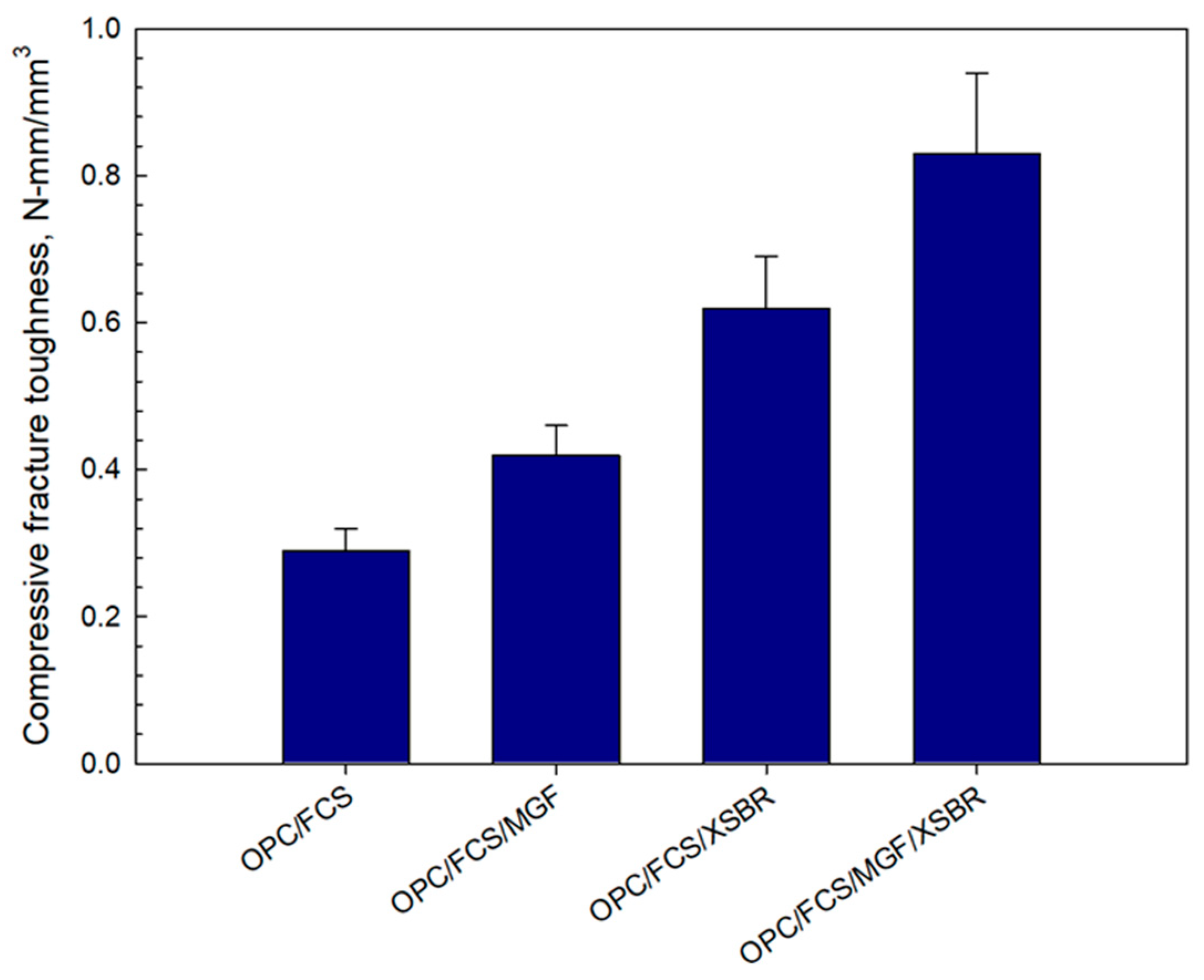
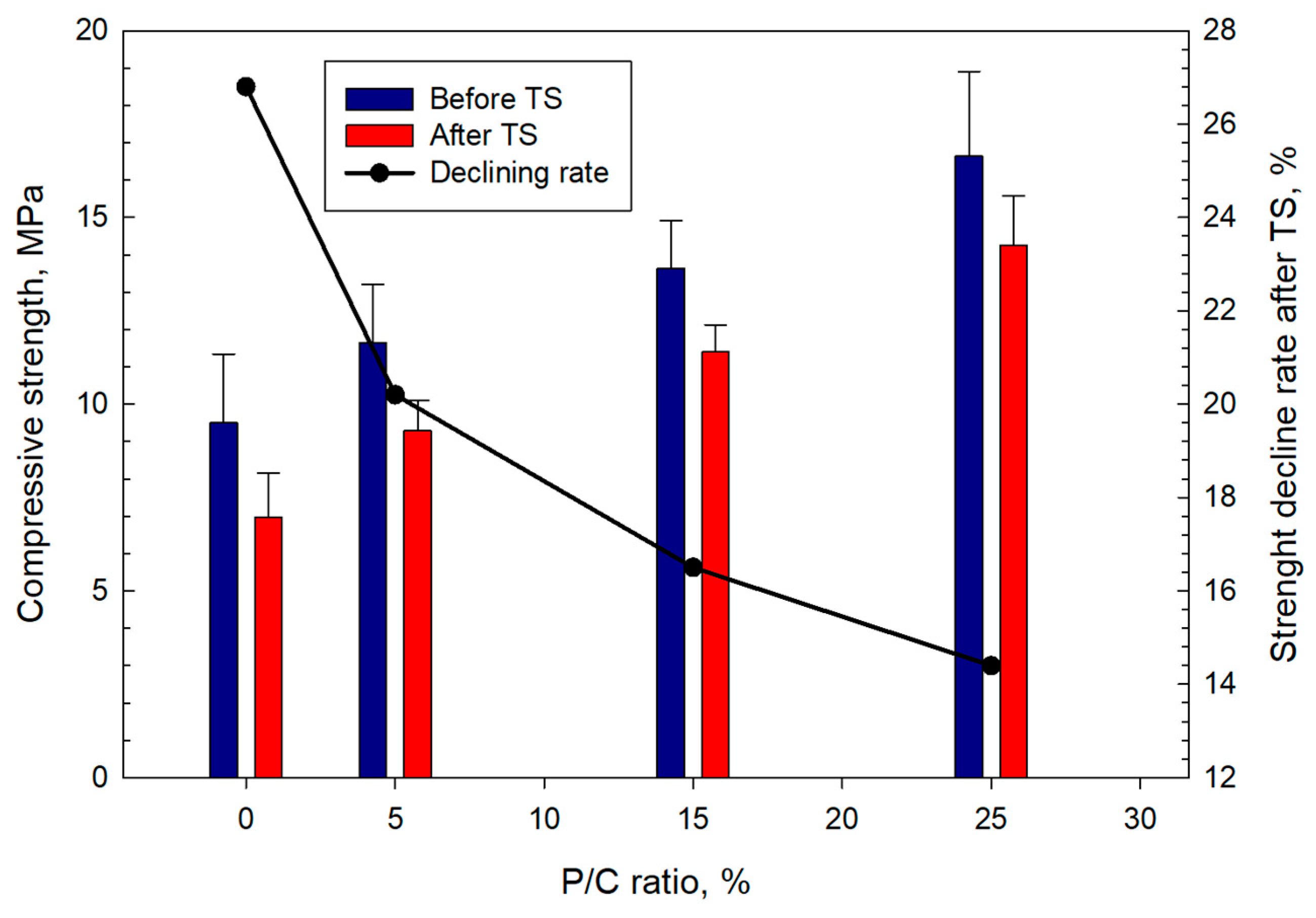
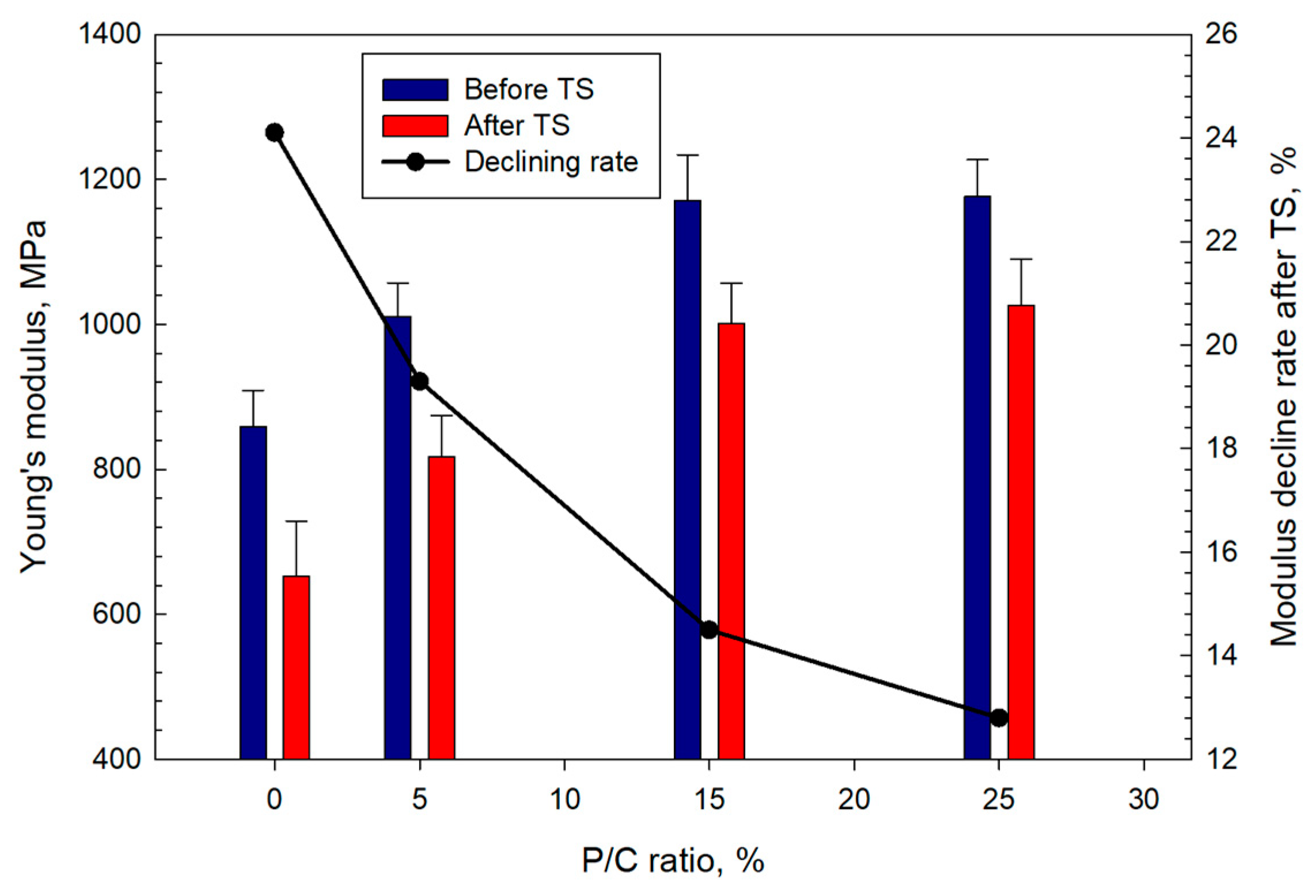
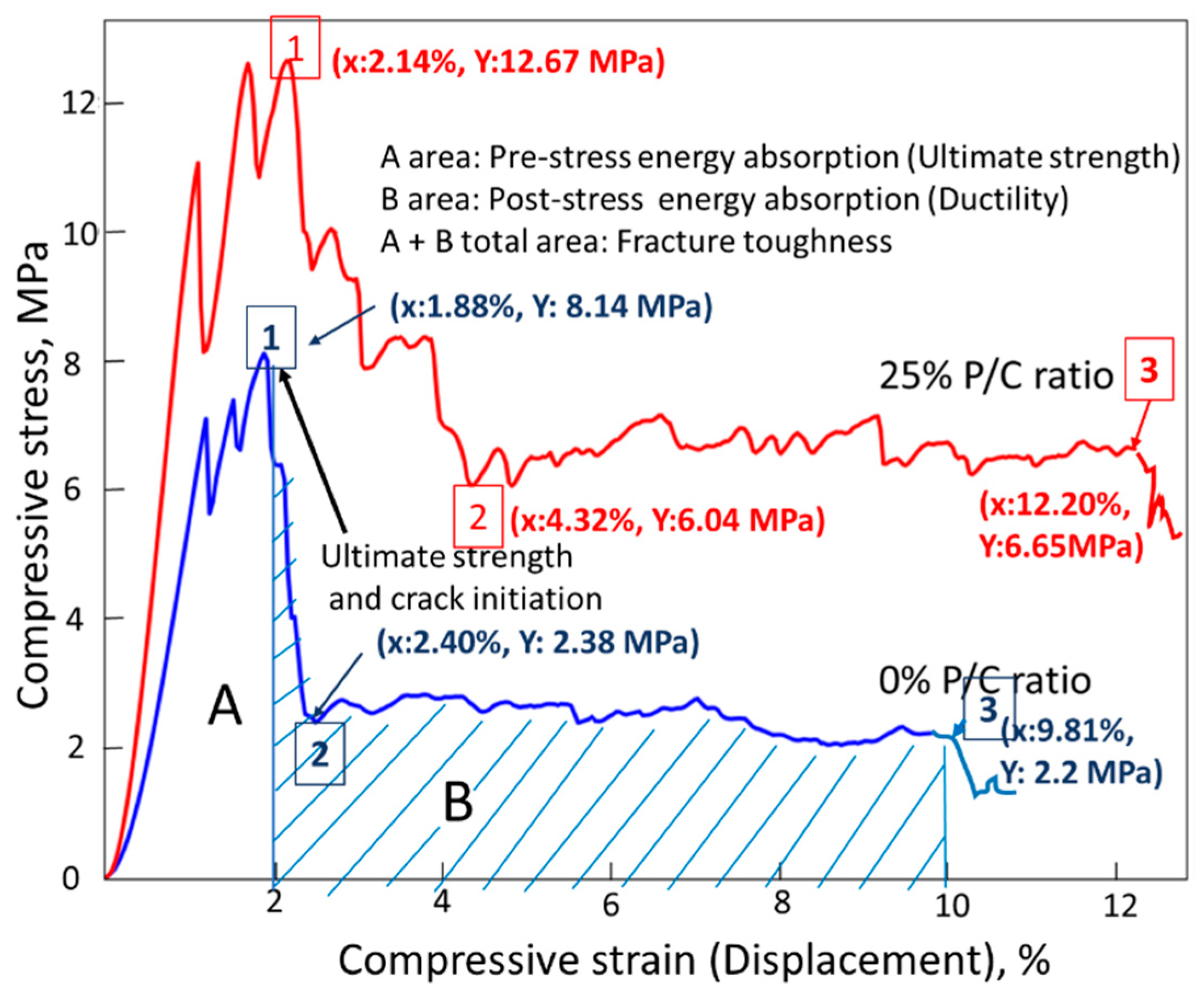
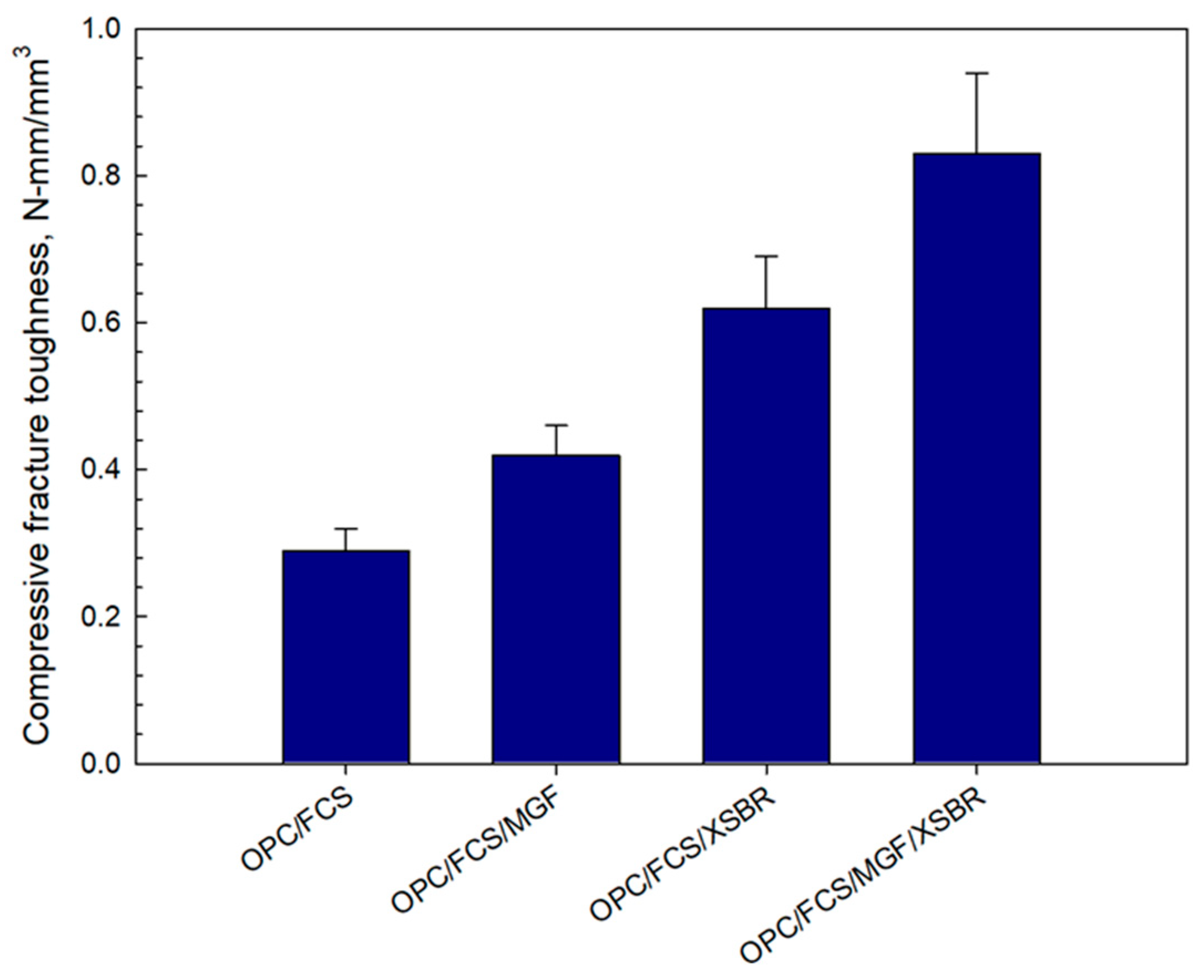
3.12. Mechanical Properties
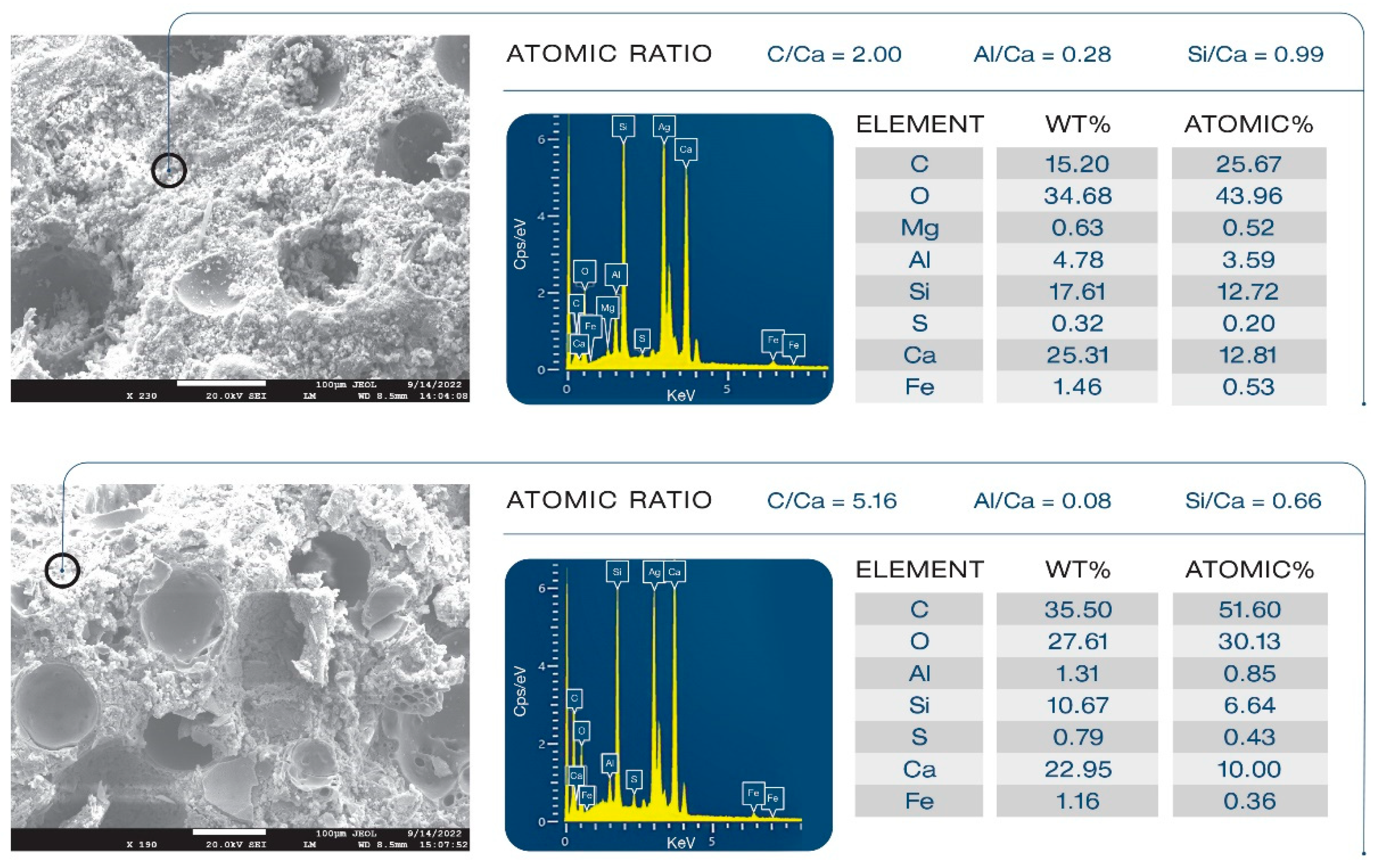
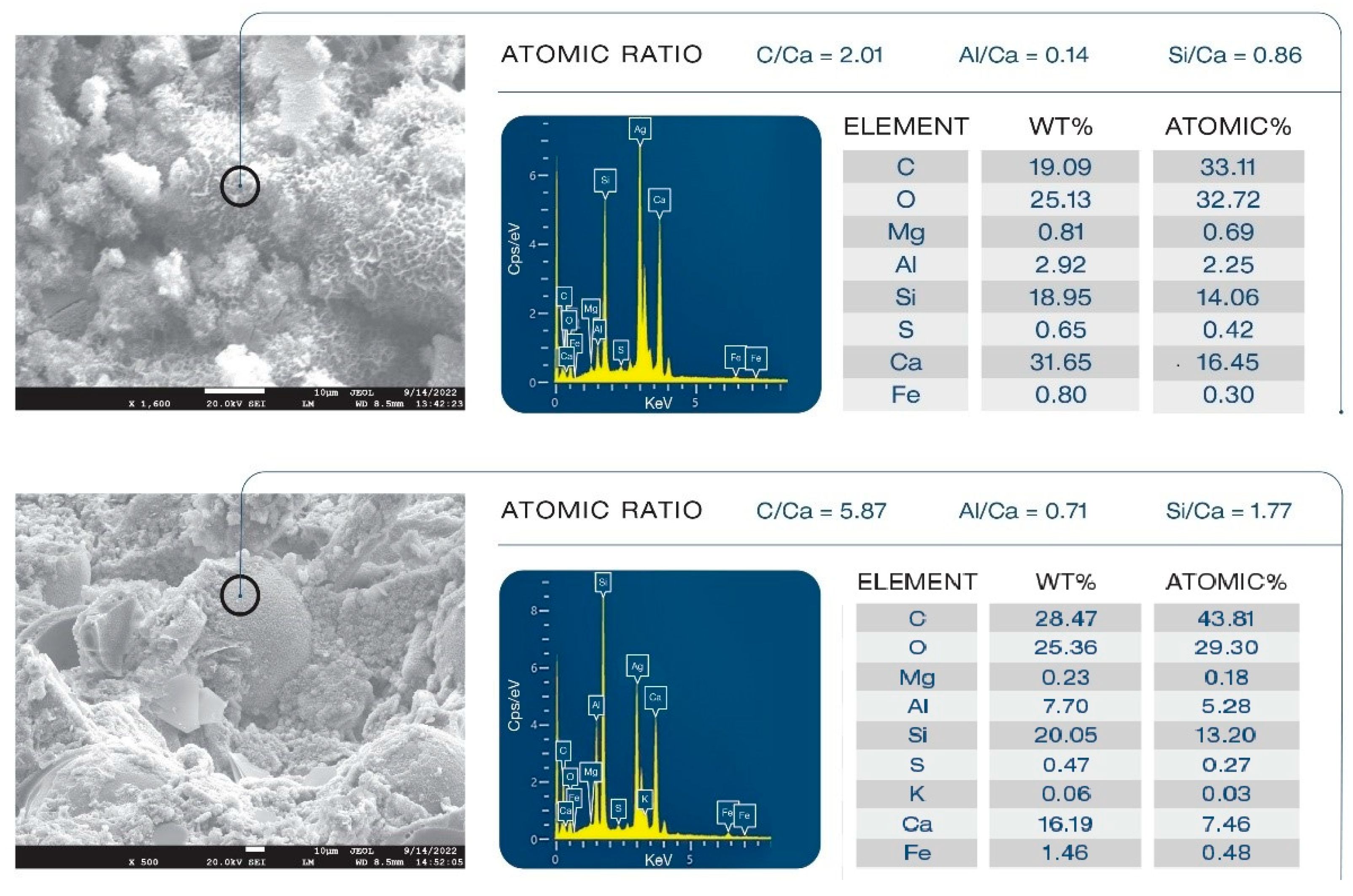
3.13. Morphological Alterations
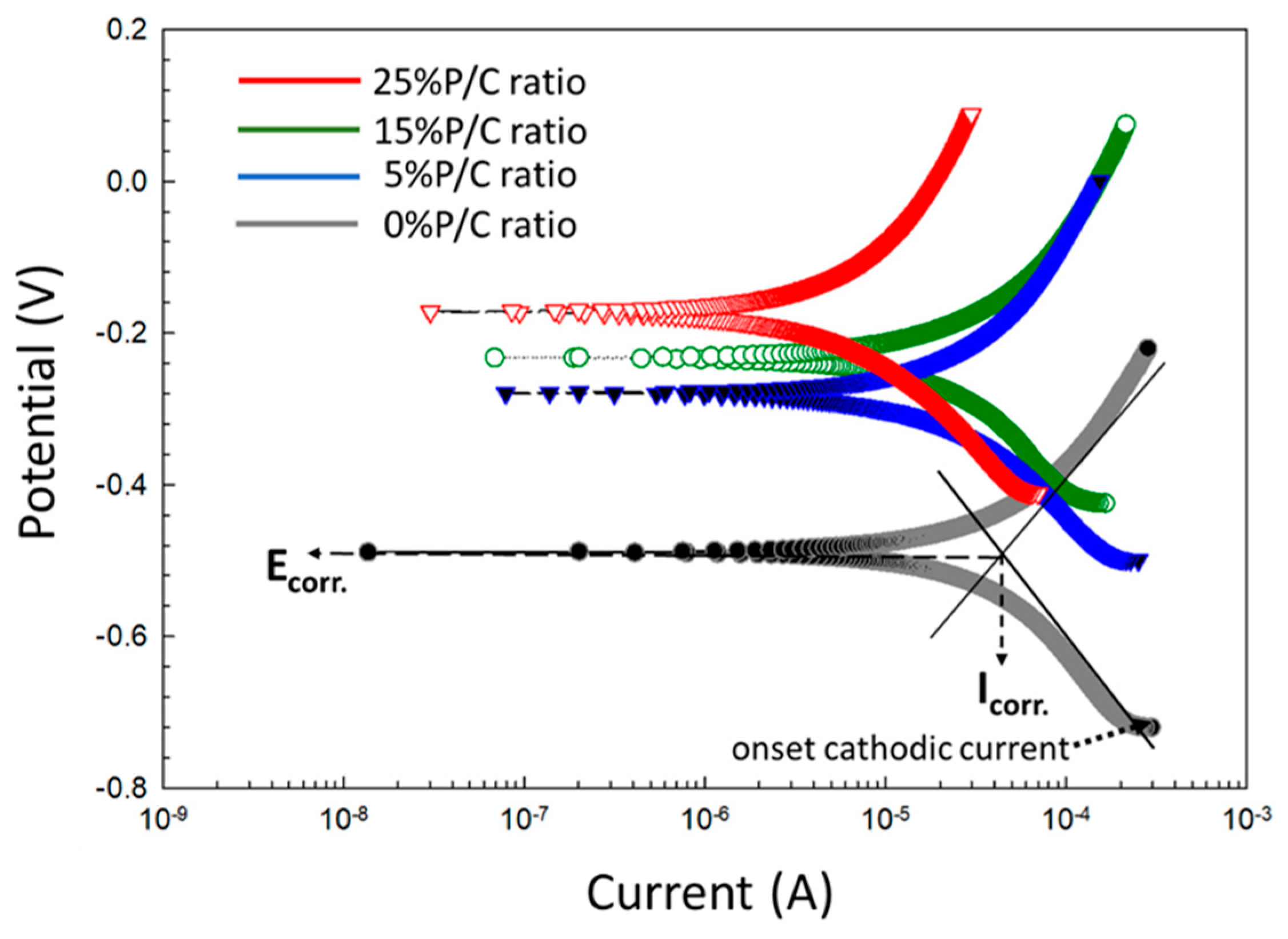
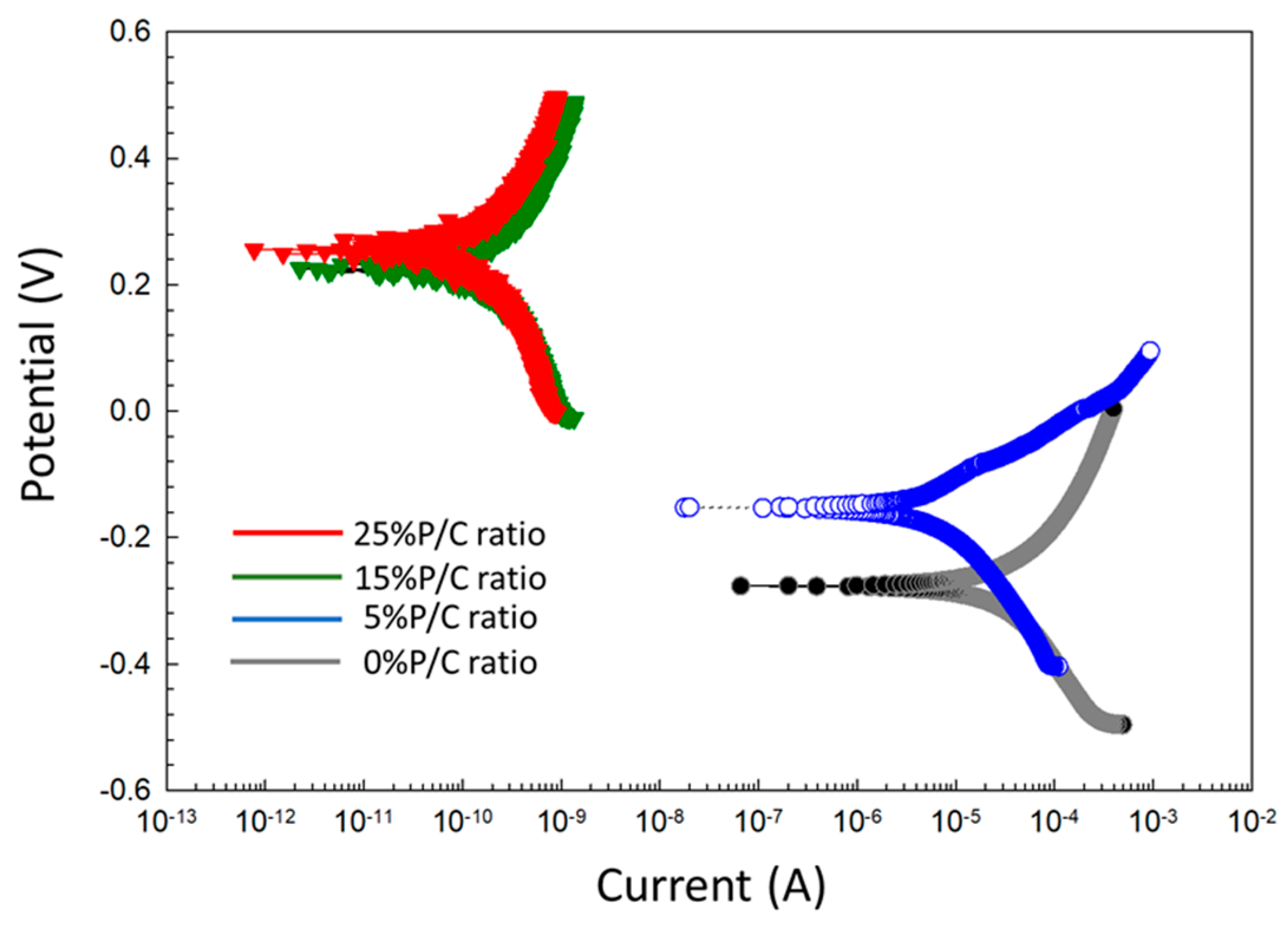
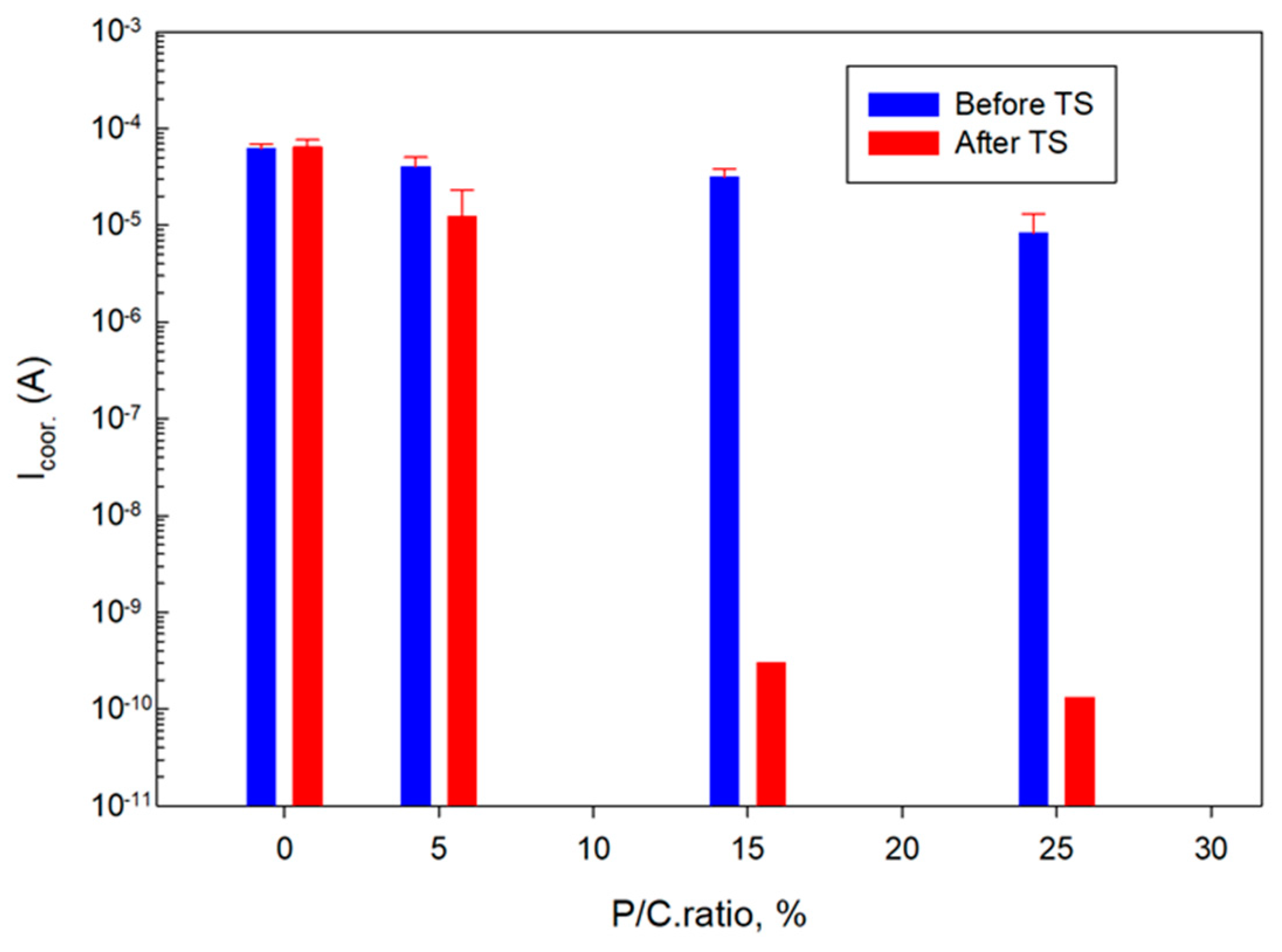
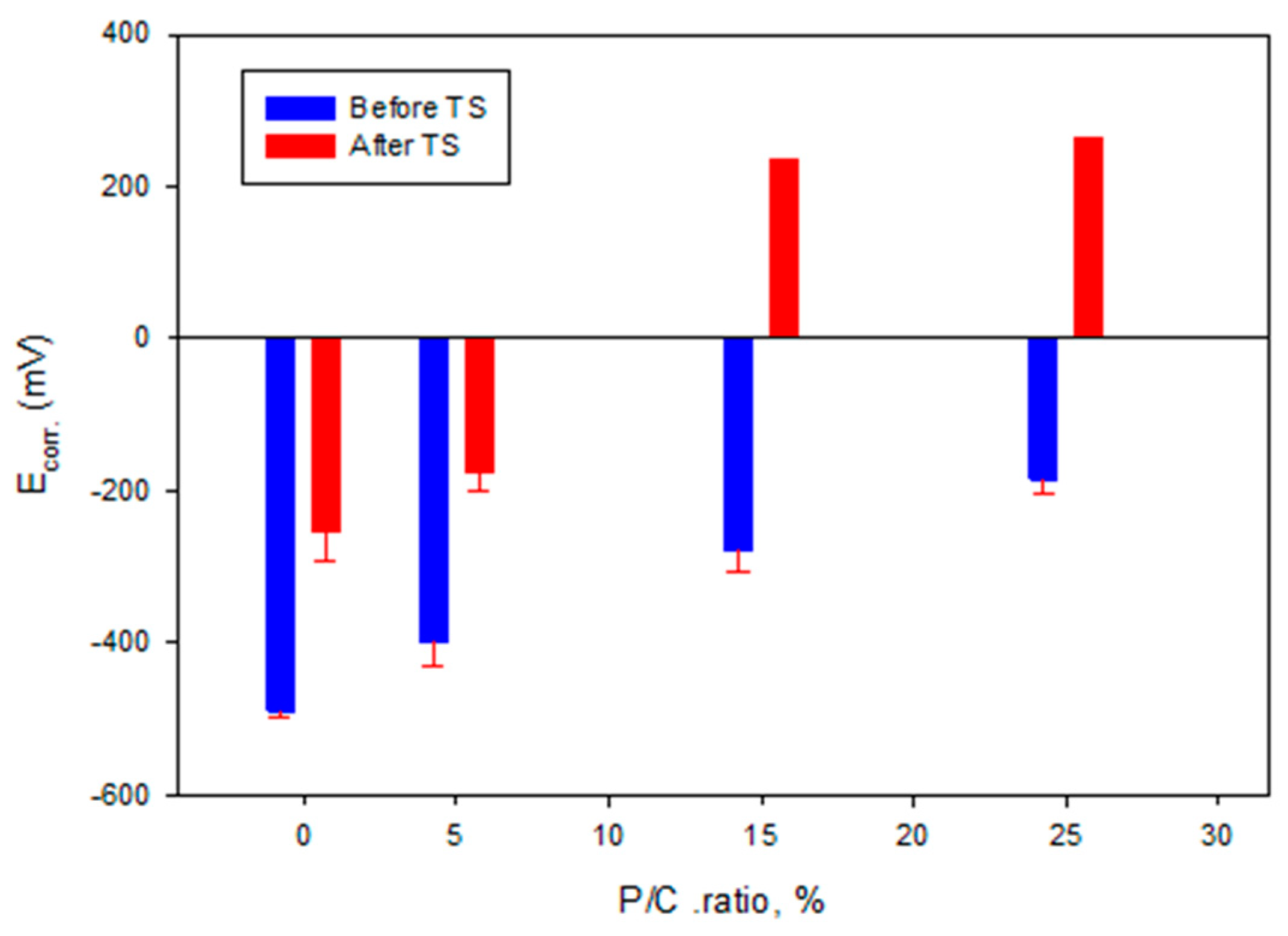
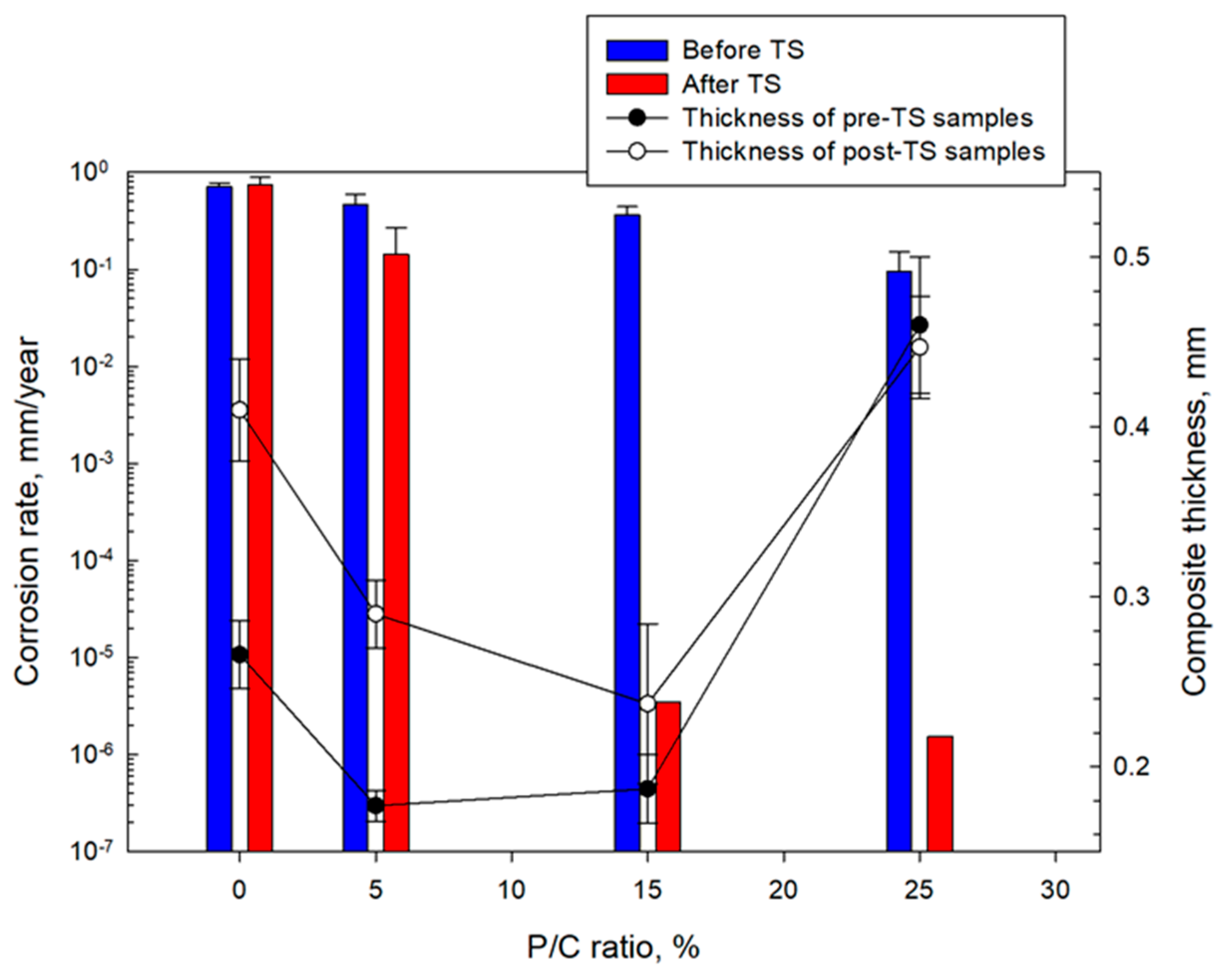
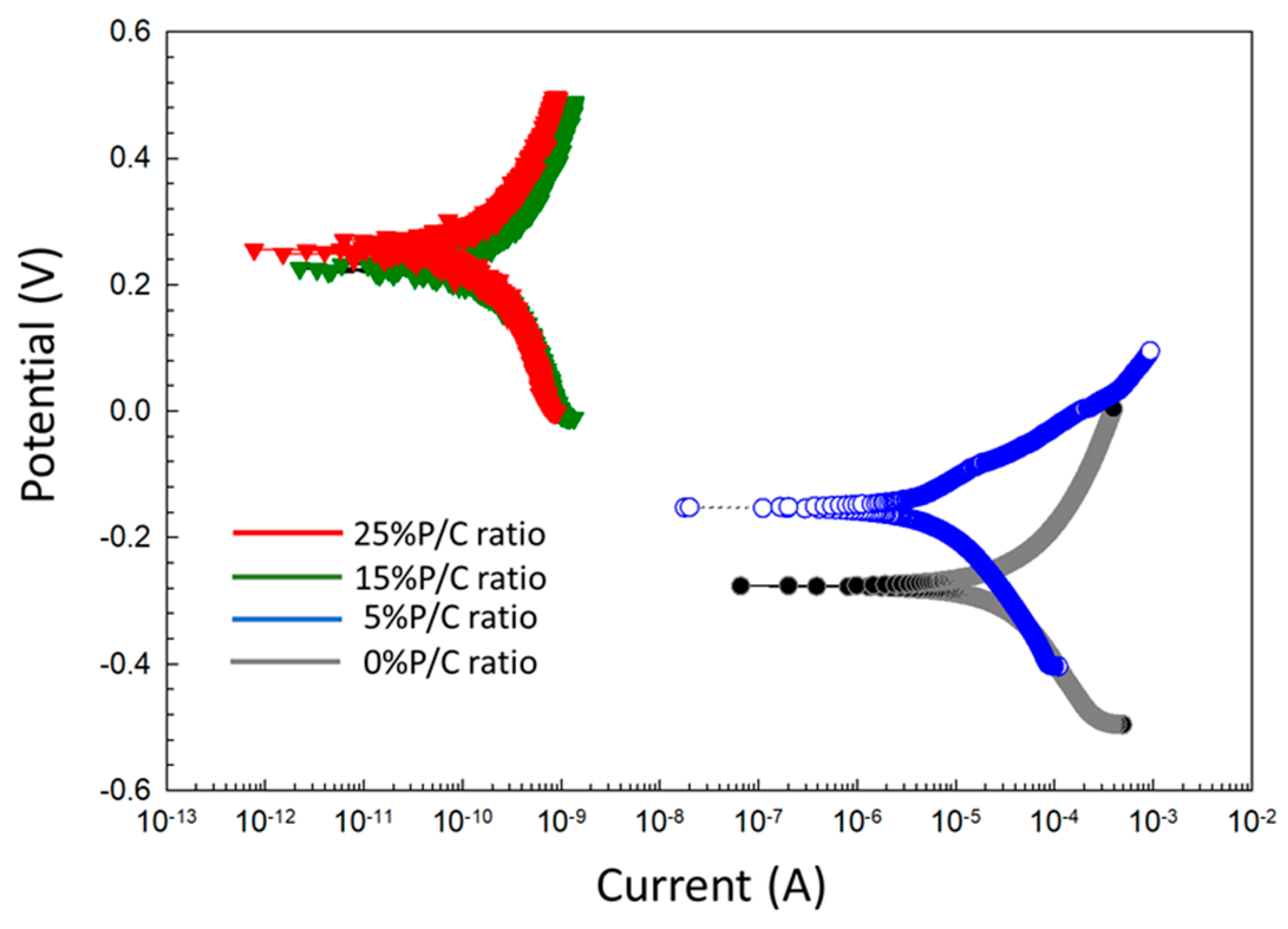
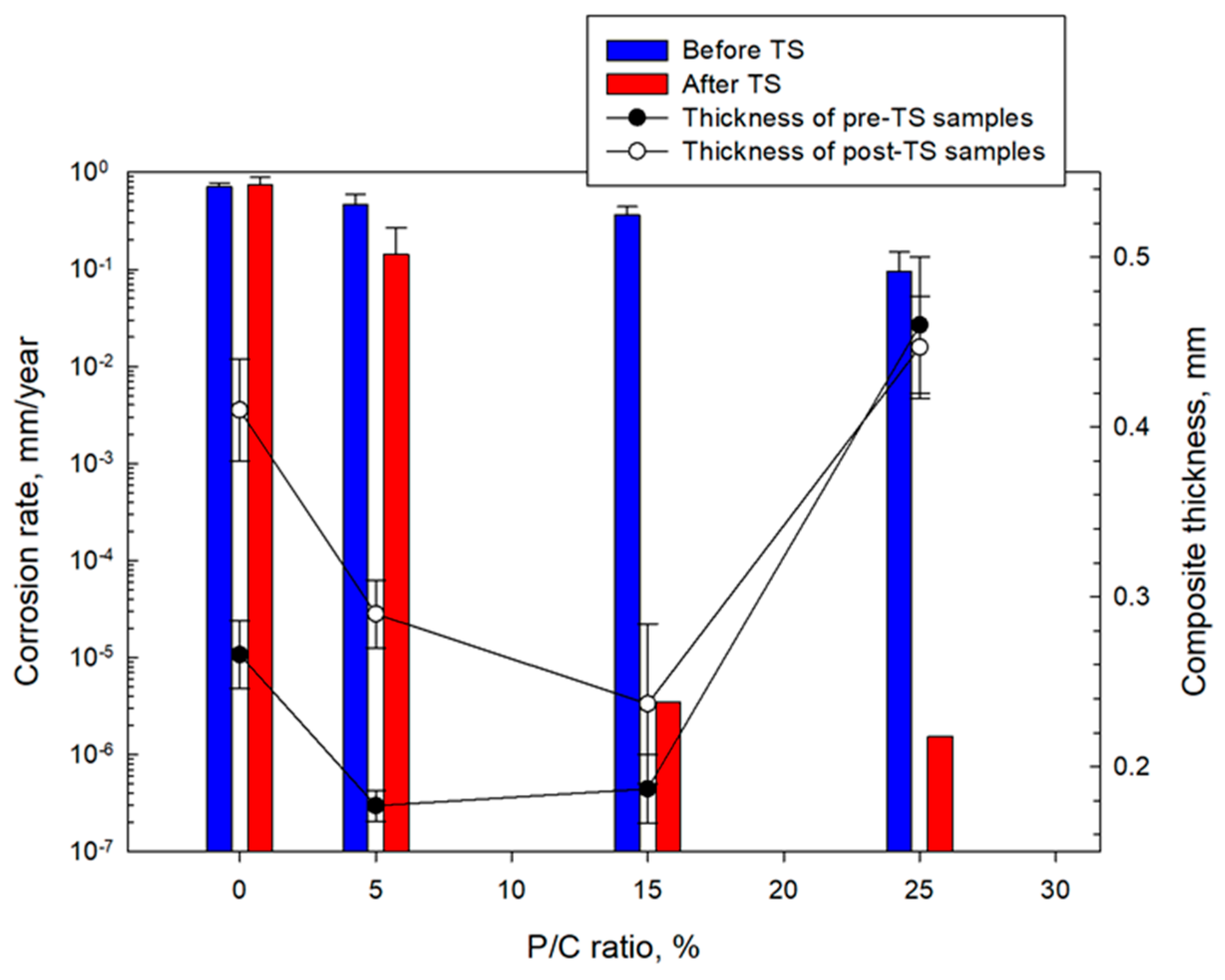
3.14. Corrosion Mitigation of CS
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sugama, T.; Pyatina, T. Hydrophobic Lightweight Cement with Thermal Shock Resistance and Thermal Insulating Properties for Energy-Storage Geothermal Well Systems. Materials 2021, 14, 6679. [Google Scholar] [CrossRef] [PubMed]
- Sugama, T.; Pyatina, T. Thermally Insulating, Thermal Shock Resistant Calcium Aluminate Phosphate Cement Composites for Reservoir Thermal Energy Storage. Materials 2022, 15, 6328. [Google Scholar] [CrossRef] [PubMed]
- Sugama, T.; Pyatina, T. Functional Acrylic Polymer as Corrosion Inhibitor of Carbon Steel in Autoclaved Air Foamed Sodium Silicate-Activated Calcium Aluminate/Class F Fly Ash Cement. Engineering 2013, 5, 887–901. [Google Scholar] [CrossRef]
- Xu, J.; Hong, B.; Lu, G.; Li, T.; Wang, S.; Wang, C.; Wang, D. Carboxylated Styrene-Butadiene Latex (XSB) in Asphalt Modification towards Cleaner Production and Enhanced Performance of Pavement in Cold Regions. J. Clean. Prod. 2022, 372, 133653. [Google Scholar] [CrossRef]
- Baueregger, S.; Perello, M.; Plank, J. Influence of Carboxylated Styrene-Butadiene Latex Copolymer on Portland Cement Hydration. Cem. Concr. Compos. 2015, 63, 42–50. [Google Scholar] [CrossRef]
- de Campos Vitorino, F.; Dweck, J.; Ferrara, L.; Filho, R.D.T. Effect of Plain and Carboxylated Styrene-Butadiene Rubber on the Rheological Behavior of Silica Fume-Class G Portland Cement Slurries. J. Mater. Res. Technol. 2020, 9, 5364–5377. [Google Scholar] [CrossRef]
- Alimardani, M.; Abbassi-Sourki, F. New and Emerging Applications of Carboxylated Styrene Butadiene Rubber Latex in Polymer Composites and Blends: Review from Structure to Future Prospective. J. Compos. Mater. 2015, 49, 1267–1282. [Google Scholar] [CrossRef]
- Zhao, J.; Hu, M.; Liu, W.; Feng, J.; Zhang, H.; Liu, M.; Guo, J. Toughening Effects of Well-Dispersed Carboxylated Styrene-Butadiene Latex Powders on the Properties of Oil Well Cement. Constr. Build. Mater. 2022, 340, 127768. [Google Scholar] [CrossRef]
- Xing, Y.; Hu, M.; Cao, C.; Yu, J.; Zhao, J.; Zheng, H.; Guo, J. Dispersion Mechanism of Styrene–Butadiene Rubber Powder Modified by Itaconic Acid and Its Toughening Effect on Oil Well Cement. Materials 2022, 15, 8345. [Google Scholar] [CrossRef]
- Omosebi, O.; Maheshwari, H.; Ahmed, R.; Shah, S.; Osisanya, S.; Hassani, S.; DeBruijn, G.; Cornell, W.; Simon, D. Degradation of Well Cement in HPHT Acidic Environment: Effects of CO2 Concentration and Pressure. Cem. Concr. Compos. 2016, 74, 54–70. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, M.; Song, J.; Wang, C.; Wang, X.; Hamad, B.A. Study on the Corrosion Change Law and Prediction Model of Cement Stone in Oil Wells with CO2 Corrosion in Ultra-High-Temperature Acid Gas Wells. Constr. Build. Mater. 2022, 323, 125879. [Google Scholar] [CrossRef]
- Xu, B.; Yuan, B.; Wang, Y.; Zeng, S.; Yang, Y. Nanosilica-Latex Reduction Carbonation-Induced Degradation in Cement of CO2 Geological Storage Wells. J. Nat. Gas Sci. Eng. 2019, 65, 237–247. [Google Scholar] [CrossRef]
- Abid, K.; Gholami, R.; Elochukwu, H.; Mostofi, M.; Bing, C.H.; Muktadir, G. A Methodology to Improve Nanosilica Based Cements Used in CO2 Sequestration Sites. Petroleum 2018, 4, 198–208. [Google Scholar] [CrossRef]
- Mahmoud, A.A.; Elkatatny, S. Improving Class G Cement Carbonation Resistance for Applications of Geologic Carbon Sequestration Using Synthetic Polypropylene Fiber. J. Nat. Gas Sci. Eng. 2020, 76, 103184. [Google Scholar] [CrossRef]
- Mahmoud, A.A.; Elkatatny, S. Mitigating CO2 Reaction with Hydrated Oil Well Cement under Geologic Carbon Sequestration Using Nanoclay Particles. J. Nat. Gas Sci. Eng. 2019, 68, 102902. [Google Scholar] [CrossRef]
- Ramalho, R.V.A.; Alves, S.M.; de Oliveira Freitas, J.C.; de Sena Costa, B.L.; Bezerra, U.T. Evaluation of Mechanical Properties of Cement Slurries Containing SBR Latex Subjected to High Temperatures. J. Pet. Sci. Eng. 2019, 178, 787–794. [Google Scholar] [CrossRef]
- Martínez-Barrera, G.; López, H.; Castaño, V.M.; Rodríguez, R. Studies on the Rubber Phase Stability in Gamma Irradiated Polystyrene-SBR Blends by Using FT-IR and Raman Spectroscopy. Radiat. Phys. Chem. 2004, 69, 155–162. [Google Scholar] [CrossRef]
- Smith, B.C. The Infrared Spectroscopy of Alkenes. Spectroscopy 2016, 31, 28–34. [Google Scholar]
- Smith, B.C. Group Wavenumbers and an Introduction to the Spectroscopy of Benzene Rings. Spectroscopy 2015, 30, 18–23. [Google Scholar]
- Bruno, C.; Luna, B.; Araú, M.; Siqueira, D.D.; Diniz, D.; Morais, S.; Antô, E.; Filho, S.; Vinícius, M.; Fook, L. Incorporation of a Recycled Rubber Compound from the Shoe Industry in Polystyrene: Effect of SBS Compatibilizer Content. J. Elastomers Plast. 2020, 52, 3–28. [Google Scholar] [CrossRef]
- Smith, B.C. More Theory and Practice: The Thorny Problem of Mixtures and More on Straight Chain Alkanes. Spectroscopy 2015, 30, 26–31. [Google Scholar]
- Wang, F.; Chang, L.; Hu, Y.; Wu, G.; Liu, H. Synthesis and Properties of In-Situ Bulk High Impact Polystyrene Toughened by High Cis-1,4 Polybutadiene. Polymers 2019, 11, 791. [Google Scholar] [CrossRef]
- Masson, J.-F.; Pelletier, L.; Collins, P. Rapid FTIR Method for Quantification of Styrene-Butadiene Type Copolymers in Bitumen. J. Appl. Polym. Sci. 2000, 79, 1034–1041. [Google Scholar] [CrossRef]
- Smith, B.C. C=O Bond, Part III: Carboxylic Acids. Spectroscopy 2018, 33, 14–20. [Google Scholar]
- De Sarkar, M.; De, P.P.; Bhowmick, A.K. Diimide Reduction of Carboxylated Styrene-Butadiene Rubber in Latex Stage. Polymer 2000, 41, 907–915. [Google Scholar] [CrossRef]
- Gómez-Fernández, J.C.; Villalaín, J. The Use of FT-IR for Quantitative Studies of the Apparent PK(a) of Lipid Carboxyl Groups and the Dehydration Degree of the Phosphate Group of Phospholipids. Chem. Phys. Lipids 1998, 96, 41–52. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Sahu, P.; Bhajiwala, H.; Mohanty, S.; Gupta, V.; Bhowmick, A.K. Synthesis, Characterization and Properties of Self-Healable Ionomeric Carboxylated Styrene-Butadiene Polymer. J. Mater. Sci. 2019, 54, 14986–14999. [Google Scholar] [CrossRef]
- Reed, S.F. Telechelic Diene Prepolymers. Carboxyl-Terminated Polydienes. J. Polym. Sci. Part A-1 1971, 9, 2147–2153. [Google Scholar] [CrossRef]
- Dai, L.; Wang, X.; Bu, Z.; Li, B.-G.; Jie, S. Facile Access to Carboxyl-Terminated Polybutadiene and Polyethylene from Cis-Polybutadiene Rubber. J. Appl. Polym. Sci. 2018, 136, 46934. [Google Scholar] [CrossRef]
- Liu, X. Oxidation Reactions of Alkenes. In Organic Chemistry I; Kwantlen Polytechnic University: Surrey, BC, Canada, 2021. [Google Scholar]
- Yousuf, M.; Mollah, A.; Hess, T.R.; Tsai, Y.N.; Cocke, D.L. An FTIR and XPS Investigations of the Effects of Carbonation on the Solidification/Stabilization of Cement Based Systems-Portland Type V with Zinc. Cem. Concr. Res. 1993, 23, 773–784. [Google Scholar] [CrossRef]
- Ylmen, R.; Jaglid, U. Carbonation of Portland Cement Studied by Diffuse Reflection Fourier Transform Infrared Spectroscopy. Int. J. Concr. Struct. Mater. 2013, 7, 119–125. [Google Scholar] [CrossRef]
- Efimov, A.; Pogareva, V.; Shashkin, A. Water-Related Bands in the IR Absorption Spectra of Silicate Glasses. J. Non-Cryst. Solids 2003, 332, 93114. [Google Scholar] [CrossRef]
- Xyla, A.G.; Koutsoukos, P.G. Quantitative Analysis of Calcium Carbonate Polymorphs by Infrared Spectroscopy. J. Chem. Soc. Furuday Trans. I 1989, 85, 3165–3172. [Google Scholar] [CrossRef]
- Gunasekaran, S.; Anbalagan, G.; Pandi, S. Raman and Infrared Spectra of Carbonate of Calcite Structure. J. Raman Spectrosc. 2006, 37, 892–899. [Google Scholar] [CrossRef]
- Ylmén, R.; Jäglid, U.; Steenari, B.M.; Panas, I. Early Hydration and Setting of Portland Cement Monitored by IR, SEM and Vicat Techniques. Cem. Concr. Res. 2009, 39, 433–439. [Google Scholar] [CrossRef]
- Thompson, M.; Palmer, R. In Situ Fourier Transform Infrared Diffuse Reflectance and Photoacoustic Spectroscopy Characterization of Sulfur-Oxygen Species Resulting from the Reaction of SO2 with CaCO3. Appl. Spectrosc. 1988, 42, 945–951. [Google Scholar] [CrossRef]
- Taddei, P.; Modena, E.; Tinti, A.; Siboni, F.; Prati, C.; Gandolfi, M.G. Effect of the Fluoride Content on the Bioactivity of Calcium Silicate-Based Endodontic Cements. Ceram. Int. 2014, 40, 4095–4107. [Google Scholar] [CrossRef]
- Mollah, M.Y.A.; Yu, W.; Schennach, R.; Cocke, D.L. Fourier Transform Infrared Spectroscopic Investigation of the Early Hydration of Portland Cement and the Influence of Sodium Lignosulfonate. Cem. Concr. Res. 2000, 30, 267–273. [Google Scholar] [CrossRef]
- Smith, B.C. The Carbonyl Group, Part V: Carboxylates-Coming Clean. Spectroscopy 2018, 33, 20–23. [Google Scholar]
- Xu, C.; Nie, J.; Wu, W.; Fu, L.; Lin, B. Design of Self-Healable Supramolecular Hybrid Network Based on Carboxylated Styrene Butadiene Rubber and Nano-Chitosan. Carbohydr. Polym. 2019, 205, 410–419. [Google Scholar] [CrossRef]
- Encarnación Ruiz-Agudo, E.; Kudłacz, K.; Putnis, C.V.; Putnis, A.; Rodriguez-Navarro, C. Dissolution and Carbonation of Portlandite [Ca(OH)2] Single Crystals. Environ. Sci. Technol. 2013, 47, 11342–11349. [Google Scholar] [CrossRef] [PubMed]
- Galan, I.; Glasser, F.P.; Baza, D.; Andrade, C. Assessment of the Protective Effect of Carbonation on Portlandite Crystals. Cem. Concr. Res. 2015, 74, 68–77. [Google Scholar] [CrossRef]
- Boualleg, S.; Bencheikh, M.; Belagraa, L.; Daoudi, A.; Chikouche, M.A. The Combined Effect of the Initial Cure and the Type of Cement on the Natural Carbonation, the Portlandite Content, and Nonevaporable Water in Blended Cement. Adv. Mater. Sci. Eng. 2017, 2017, 5634713. [Google Scholar] [CrossRef]
- Fan, J.; Guo, J.; Chen, D.; Hu, M.; Cao, L.; Xu, Y.; Wang, M. Effects of Submicron Core-Shell Latexes with Different Functional Groups on the Adsorption and Cement Hydration. Constr. Build. Mater. 2018, 183, 127–138. [Google Scholar] [CrossRef]
- Garbev, K.; Bornefeld, M.; Beuchle, G.; Stemmermann, P. Cell Dimensions and Composition of Nanocrystalline Calcium Silicate Hydrate Solid Solutions. Part 2: X-Ray and Thermogravimetry Study. J. Am. Ceram. Soc. 2008, 91, 3015–3023. [Google Scholar] [CrossRef]
- L’Hôpital, E.; Lothenbach, B.; Kulik, D.A.; Scrivener, K. Influence of Calcium to Silica Ratio on Aluminium Uptake in Calcium Silicate Hydrate. Cem. Concr. Res. 2016, 85, 111–121. [Google Scholar] [CrossRef]
- Steiner, S.; Lothenbach, B.; Proske, T.; Borgschulte, A.; Winnefeld, F. Effect of Relative Humidity on the Carbonation Rate of Portlandite, Calcium Silicate Hydrates and Ettringite. Cem. Concr. Res. 2020, 135, 106116. [Google Scholar] [CrossRef]
- Gu, P.; Beaudoin, J.J. A Conduction Calorimetric Study of Early Hydration of Ordinary Portland Cement/High Alumina Cement Pastes. J. Mater. Sci. 1997, 32, 3875–3881. [Google Scholar] [CrossRef]
- Dittrich, S.; Neubauer, J.; Goetz-Neunhoeffer, F. The Influence of Fly Ash on the Hydration of OPC within the First 44 h—A Quantitative in Situ XRD and Heat Flow Calorimetry Study. Cem. Concr. Res. 2014, 56, 129–138. [Google Scholar] [CrossRef]
- Nguyen, H.; Carvelli, V.; Kunther, W.; Illikainen, M.; Kinnunen, P. Phase Evolution and Mechanical Performance of an Ettringite-Based Binder during Hydrothermal Aging. Cem. Concr. Res. 2021, 143, 106403. [Google Scholar] [CrossRef]
- Flores-Vivian, I.; Hejazi, V.; Kozhukhova, M.I.; Nosonovsky, M.; Sobolev, K. Self-Assembling Particle-Siloxane Coatings for Superhydrophobic Concrete. ACS Appl. Mater. Interfaces 2013, 5, 13284–13294. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Oxide Composition, wt.% | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Al2O3 | CaO | SiO2 | Fe2O3 | Na2O | K2O | TiO2 | MgO | SO3 | |
| Class G cement | 3.0 | 67.6 | 18.4 | 3.9 | 0.3 | 1.3 | - | - | 5.5 |
| Silica flour | - | - | 100 | - | - | - | - | - | - |
| FCSs | 35.0 | 2.7 | 50.1 | 7.1 | 0.30 | 3.1 | 1.6 | - | - |
| E-type MGFs | 11.4 | 28.6 | 55.0 | 0.9 | 0.6 | - | 0.7 | 2.8 | - |
| ΔA1730 cm−1 | ΔA1600 cm−1 | ΔA1730 cm−1/ΔA1600 cm−1 | ΔA966 cm−1 | ΔA908 cm−1 | |
|---|---|---|---|---|---|
| 100 °C-dried XSBR | 0.008 | 0.017 | 0.471 | 0.125 | 0.034 |
| 100 °C-autoclaved dried XSBR | 0.015 | 0.031 | 0.484 | 0.125 | 0.038 |
| 175 °C-autoclaved dried XSBR | 0.042 | 0.02 | 2.1 | 0.03 | 0.017 |
| Property | P/C Ratio, % | |||
|---|---|---|---|---|
| 0 | 5 | 15 | 25 | |
| Water-to-composite ratio (W/C) | 0.51 | 0.45 | 0.37 | 0.31 |
| Density, d/cm3 | 1.26 | 1.11 | 1.06 | 1.04 |
| Slump size, mm | 60 | 64 | 72 | 75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugama, T.; Pyatina, T. Hydrophobic, Thermal Shock-and-Corrosion-Resistant XSBR Latex-Modified Lightweight Class G Cement Composites in Geothermal Well Energy Storage Systems. Materials 2023, 16, 5792. https://doi.org/10.3390/ma16175792
Sugama T, Pyatina T. Hydrophobic, Thermal Shock-and-Corrosion-Resistant XSBR Latex-Modified Lightweight Class G Cement Composites in Geothermal Well Energy Storage Systems. Materials. 2023; 16(17):5792. https://doi.org/10.3390/ma16175792
Chicago/Turabian StyleSugama, Toshifumi, and Tatiana Pyatina. 2023. "Hydrophobic, Thermal Shock-and-Corrosion-Resistant XSBR Latex-Modified Lightweight Class G Cement Composites in Geothermal Well Energy Storage Systems" Materials 16, no. 17: 5792. https://doi.org/10.3390/ma16175792
APA StyleSugama, T., & Pyatina, T. (2023). Hydrophobic, Thermal Shock-and-Corrosion-Resistant XSBR Latex-Modified Lightweight Class G Cement Composites in Geothermal Well Energy Storage Systems. Materials, 16(17), 5792. https://doi.org/10.3390/ma16175792