
Exploring the Structural, Electronic, Magnetic, and Transport Properties of 2D Cr, Fe, and Zr Monoborides



Abstract
1. Introduction
2. Computational Approach
3. Results and Discussion
3.1. Structure and Stability
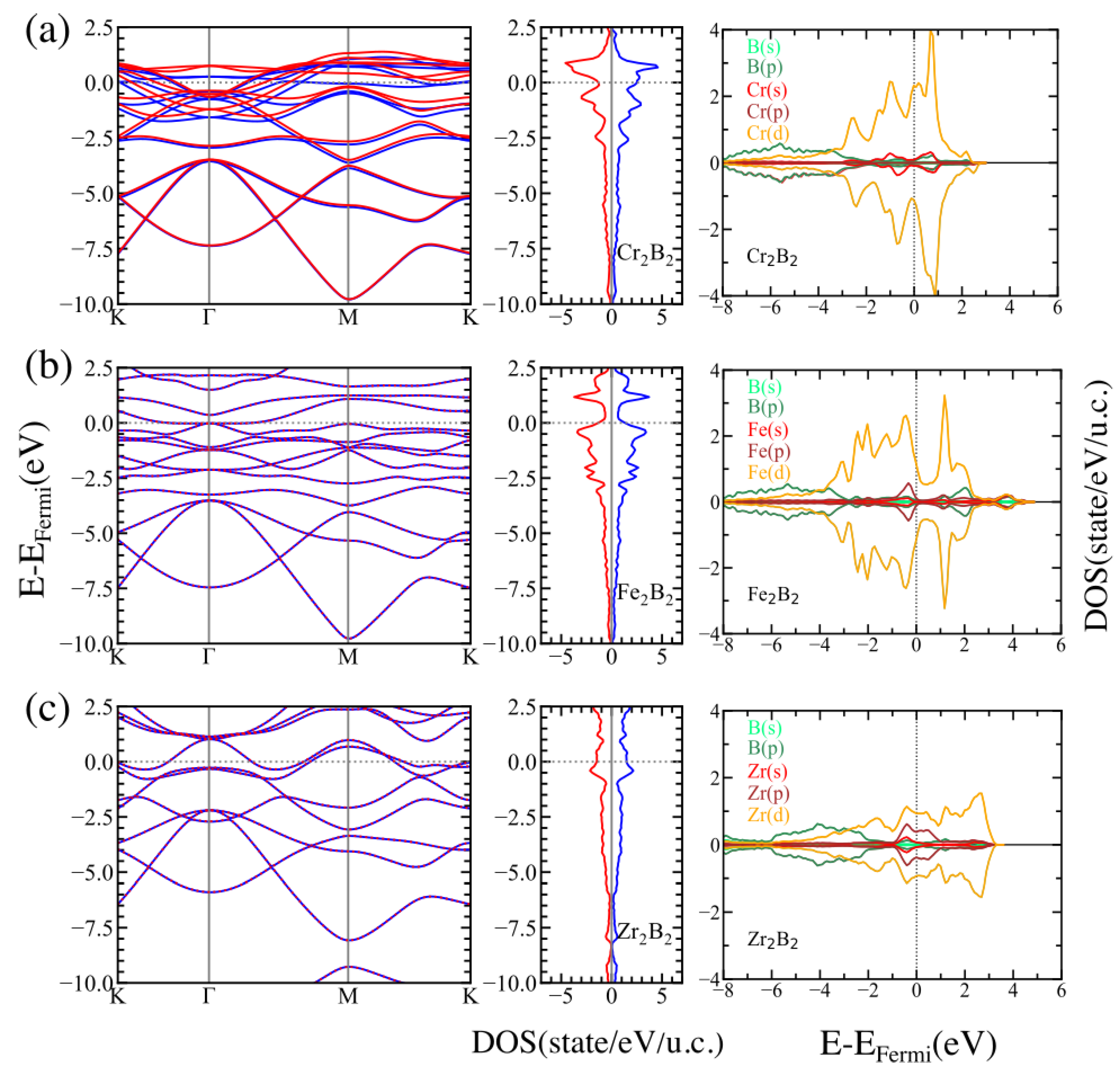
3.2. Electronic Properties
3.3. Magnetic Properties
4. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Akopov, G.; Yeung, M.T.; Kaner, R.B. Rediscovering the Crystal Chemistry of Borides. Adv. Mater. 2017, 29, 1604506. [Google Scholar] [CrossRef] [PubMed]
- Ade, M.; Hillebrecht, H. Ternary Borides Cr2AlB2, Cr3AlB4, and Cr4AlB6: The First Members of the Series (CrB2)nCrAl with n=1,2,3 and a Unifying Concept for Ternary Borides as MAB-Phases. Inorg. Chem. 2015, 54, 6122–6135. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, A.; Rosen, J.; Dahlqvist, M. Theoretical predictions of phase stability for orthorhombic and hexagonal ternary MAB phases. Phys. Chem. Chem. Phys. 2022, 24, 11249–11258. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ye, T.N.; Gong, Y.; Wu, J.; Miao, N.; Tada, T.; Hosono, H. Discovery of hexagonal ternary phase Ti2InB2 and its evolution to layered boride TiB. Nat. Commun. 2019, 10, 2284. [Google Scholar] [CrossRef]
- Alameda, L.T.; Moradifar, P.; Metzger, Z.P.; Alem, N.; Schaak, R.E. Topochemical Deintercalation of Al from MoAlB: Stepwise Etching Pathway, Layered Intergrowth Structures, and Two-Dimensional MBene. J. Am. Chem. Soc. 2018, 140, 8833–8840. [Google Scholar] [CrossRef]
- Alameda, L.T.; Lord, R.W.; Barr, J.A.; Moradifar, P.; Metzger, Z.P.; Steimle, B.C.; Holder, C.F.; Alem, N.; Sinnott, S.B.; Schaak, R.E. Multi-Step Topochemical Pathway to Metastable Mo2AlB2 and Related Two-Dimensional Nanosheet Heterostructures. J. Am. Chem. Soc. 2019, 141, 10852–10861. [Google Scholar] [CrossRef]
- Zhang, H.; Xiang, H.; zhi Dai, F.; Zhang, Z.; Zhou, Y. First demonstration of possible two-dimensional MBene CrB derived from MAB phase Cr2AlB2. J. Mater. Sci. Technol. 2018, 34, 2022–2026. [Google Scholar] [CrossRef]
- Zhang, H.; Dai, F.Z.; Xiang, H.; Wang, X.; Zhang, Z.; Zhou, Y. Phase pure and well crystalline Cr2AlB2: A key precursor for two-dimensional CrB. J. Mater. Sci. Technol. 2019, 35, 1593–1600. [Google Scholar] [CrossRef]
- Khazaei, M.; Wang, J.; Estili, M.; Ranjbar, A.; Suehara, S.; Arai, M.; Esfarjani, K.; Yunoki, S. Novel MAB phases and insights into their exfoliation into 2D MBenes. Nanoscale 2019, 11, 11305–11314. [Google Scholar] [CrossRef]
- Barinov, V.A.; Dorofeev, G.A.; Ovechkin, L.V.; Elsukov, E.P.; Ermakov, A.E. Structure and magnetic properties of the α-FeB phase obtained by mechanical working. Phys. Status Solidi (a) 1991, 123, 527–534. [Google Scholar] [CrossRef]
- Zhao, X.; Li, L.; Bao, K.; Zhu, P.; Tao, Q.; Ma, S.; Cui, T. Insight the effect of rigid boron chain substructure on mechanical, magnetic and electrical properties of β-FeB. J. Alloys Compd. 2022, 896, 162767. [Google Scholar] [CrossRef]
- Saldaña, F.I.; Defoy, E.; Janisch, D.; Rousse, G.; Autran, P.O.; Ghoridi, A.; Séné, A.; Baron, M.; Suescun, L.; Godec, Y.L.; et al. Revealing the Elusive Structure and Reactivity of Iron Boride α-FeB. Inorg. Chem. 2023, 62, 2073–2082. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Szwacki, N.; Sadrzadeh, A.; Yakobson, B.I. B80 Fullerene: An Ab Initio Prediction of Geometry, Stability, and Electronic Structure. Phys. Rev. Lett. 2007, 98, 166804. [Google Scholar] [CrossRef] [PubMed]
- Szwacki, N.G. Boron Fullerenes: A First-Principles Study. Nanoscale Res. Lett. 2007, 3, 49. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Himmetoglu, B.; Janotti, A. Transport properties of KTaO3 from first-principles. J. Phys. Condens. Matter 2016, 28, 065502. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Bo, T.; Liu, P.F.; Xu, J.; Zhang, J.; Chen, Y.; Eriksson, O.; Wang, F.; Wang, B.T. Hexagonal Ti2B2 monolayer: A promising anode material offering high rate capability for Li-ion and Na-ion batteries. Phys. Chem. Chem. Phys. 2018, 20, 22168–22178. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Li, Z.; Xiao, W.; Zhang, C.; Zhao, Y. Computational investigation of 2D 3d/4d hexagonal transition metal borides for metal-ion batteries. Electrochim. Acta 2021, 384, 138404. [Google Scholar] [CrossRef]
- Yuan, G.; Bo, T.; Qi, X.; Liu, P.F.; Huang, Z.; Wang, B.T. Monolayer Zr2B2: A promising two-dimensional anode material for Li-ion batteries. Appl. Surf. Sci. 2019, 480, 448–453. [Google Scholar] [CrossRef]
- Mir, S.H.; Yadav, V.K.; Singh, J.K. Efficient CO2 Capture and Activation on Novel Two-Dimensional Transition Metal Borides. ACS Appl. Mater. Interfaces 2022, 14, 29703–29710. [Google Scholar] [CrossRef]
- Dou, M.; Li, H.; Yao, Q.; Wang, J.; Liu, Y.; Wu, F. Room-temperature ferromagnetism in two-dimensional transition metal borides: A first-principles investigation. Phys. Chem. Chem. Phys. 2021, 23, 10615–10620. [Google Scholar] [CrossRef]
- Ozdemir, I.; Kadioglu, Y.; Yüksel, Y.; Akıncı, Ü.; Üzengi Aktürk, O.; Aktürk, E.; Ciraci, S. Columnar antiferromagnetic order of a MBene monolayer. Phys. Rev. B 2021, 103. [Google Scholar] [CrossRef]
- Qi, S.; Fan, Y.; Zhao, L.; Li, W.; Zhao, M. Two-dimensional transition metal borides as highly efficient N2 fixation catalysts. Appl. Surf. Sci. 2021, 536, 147742. [Google Scholar] [CrossRef]
- Zhang, B.; Zhou, J.; Guo, Z.; Peng, Q.; Sun, Z. Two-dimensional chromium boride MBenes with high HER catalytic activity. Appl. Surf. Sci. 2020, 500, 144248. [Google Scholar] [CrossRef]
- Shin, H.; Kang, S.; Koo, J.; Lee, H.; Kim, J.; Kwon, Y. Cohesion energetics of carbon allotropes: Quantum Monte Carlo study. J. Chem. Phys. 2014, 140, 114702. [Google Scholar] [CrossRef]
- Xu, T.; Wang, Y.; Xiong, Z.; Wang, Y.; Zhou, Y.; Li, X. A Rising 2D Star: Novel MBenes with Excellent Performance in Energy Conversion and storage. Nano-Micro Lett. 2022, 15, 6. [Google Scholar] [CrossRef]
- Şahin, H.; Cahangirov, S.; Topsakal, M.; Bekaroglu, E.; Akturk, E.; Senger, R.T.; Ciraci, S. Monolayer honeycomb structures of group-IV elements and III-V binary compounds: First-principles calculations. Phys. Rev. B 2009, 80, 155453. [Google Scholar] [CrossRef]
- Fasolino, A.; Los, J.H.; Katsnelson, M.I. Intrinsic ripples in graphene. Nat. Mater. 2007, 6, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhou, J.; Sun, Z. MBenes: Progress, challenges and future. J. Mater. Chem. A 2022, 10, 15865–15880. [Google Scholar] [CrossRef]
- Guo, Z.; Zhou, J.; Sun, Z. New two-dimensional transition metal borides for Li ion batteries and electrocatalysis. J. Mater. Chem. A 2017, 5, 23530–23535. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CrB | FeB | ZrB | ||||
|---|---|---|---|---|---|---|
| a | 2.885 | 2.919 | 2.823 | 2.913 | 3.084 | 3.159 |
| (2.860 4) (2.930 5) | (2.921 1) (2.926 2) | (2.800 4) (2.770 5) | (3.07 7) | (3.144 1) (3.160 2) (3.134 3) | ||
| b | 2.870 | 2.919 | 2.787 | 2.913 | 3.281 | 3.159 |
| (2.850 4) (2.870 5) | (2.921 1) (2.926 2) | (2.680 4) (2.820 5) | (3.27 7) | (3.144 1) (3.160 2) (3.134 3) | ||
| 1.810 | 1.685 | 1.813 | 1.682 | 1.721 | 1.824 | |
| (1.689 2) | (1.71 7) | (1.825 2) (1.891 3) | ||||
| 2.10 | 2.15 | 2.04 | 2.15 | 2.46 | 2.49 | |
| (2.11 4) | (2.05 4) | (2.43 7) | (2.49 3) | |||
| t | 2.122 | 2.662 | 2.134 | 2.680 | 2.817 | 3.382 |
| (2.651 1) (2.639 2) | (2.13 6) | (2.84 7) | (3.391 1) (3.380 2) (3.385 3) |
| CrB | FeB | ZrB | ||||
|---|---|---|---|---|---|---|
| (eV) | 6.222 | 6.201 | 6.901 | 6.830 | 8.050 | 8.087 |
| (cm) | 762.45 | 1003.50 | 772.74 | 976.01 | 928.02 | 744.12 |
| (e) | −0.76 | −0.61 | −0.37 | −0.40 | −1.17 | −0.86 |
| CrB | FeB | ZrB | ||||
|---|---|---|---|---|---|---|
| (/unit cell) | 2.56 | 0.63 | 2.69 | 0.00 | 0.00 | 0.00 |
| (/ion) | 1.03 | 0.31 | 1.26 | 2.06/−2.06 | 0.00 | 0.00 |
| (/ion) | −0.05 | −0.01 | −0.05 | 0.00 | 0.00 | 0.00 |
| MGS | FM | FM | FM | AFM | Non-magnetic | Non-magnetic |
| (meV/unit cell) | −104.38 | −0.29 | −108.12 | 46.84 | - | - |
| (K) | 403 | 1 | 418 | 181 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arias-Camacho, I.M.; Gonzalez Szwacki, N. Exploring the Structural, Electronic, Magnetic, and Transport Properties of 2D Cr, Fe, and Zr Monoborides. Materials 2023, 16, 5104. https://doi.org/10.3390/ma16145104
Arias-Camacho IM, Gonzalez Szwacki N. Exploring the Structural, Electronic, Magnetic, and Transport Properties of 2D Cr, Fe, and Zr Monoborides. Materials. 2023; 16(14):5104. https://doi.org/10.3390/ma16145104
Chicago/Turabian StyleArias-Camacho, Isabel M., and Nevill Gonzalez Szwacki. 2023. "Exploring the Structural, Electronic, Magnetic, and Transport Properties of 2D Cr, Fe, and Zr Monoborides" Materials 16, no. 14: 5104. https://doi.org/10.3390/ma16145104
APA StyleArias-Camacho, I. M., & Gonzalez Szwacki, N. (2023). Exploring the Structural, Electronic, Magnetic, and Transport Properties of 2D Cr, Fe, and Zr Monoborides. Materials, 16(14), 5104. https://doi.org/10.3390/ma16145104