
Cu2O/SnO2 Heterostructures: Role of the Synthesis Procedure on PEC CO2 Conversion




Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
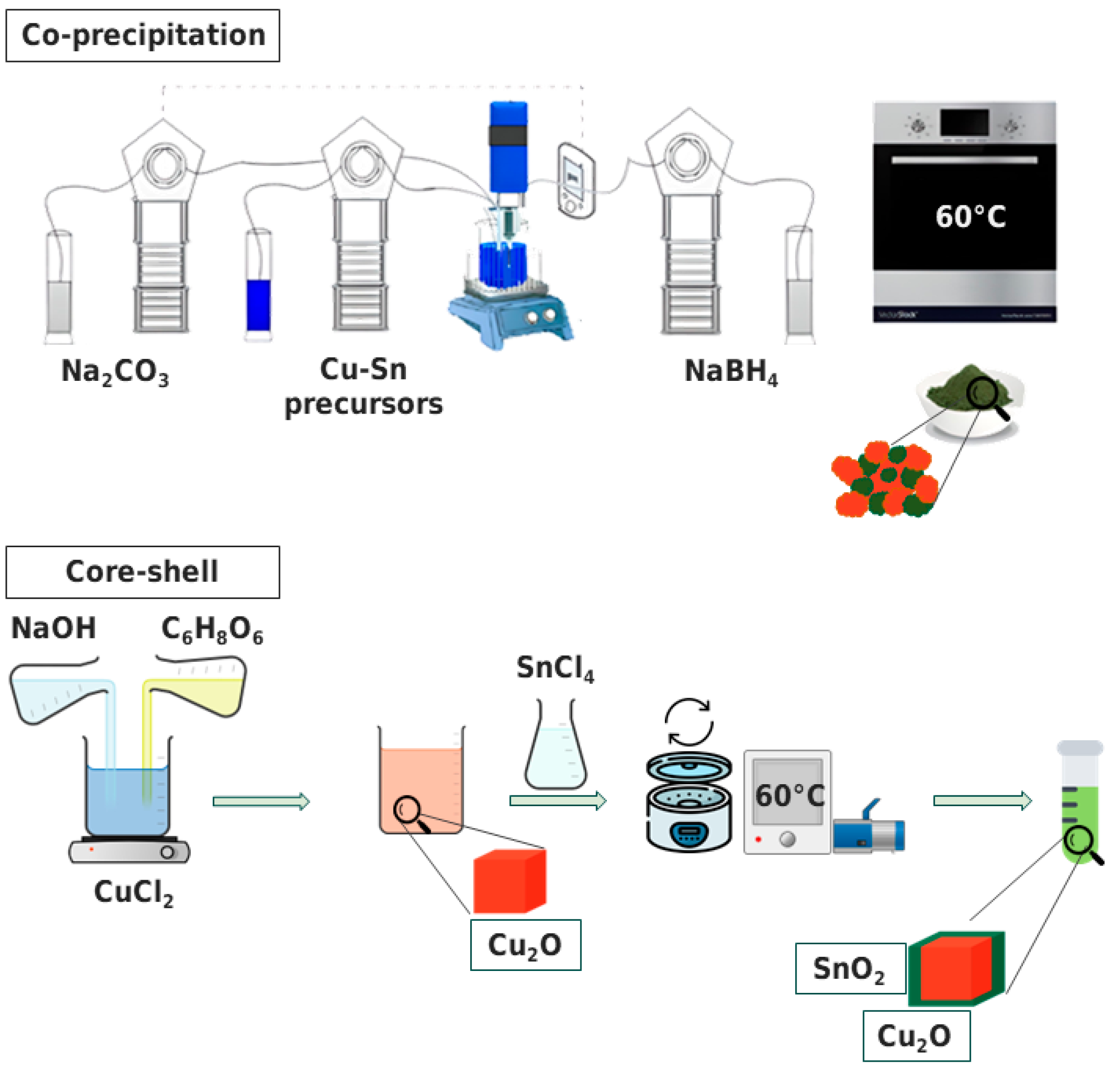
2.1.1. Co-Precipitation Method
2.1.2. Core–Shell Method
2.2. Synthesis Procedure
2.2.1. Co-Precipitation Method
2.2.2. Core–Shell Method
2.3. Characterization
2.4. Photo-Electrode Preparation
2.5. PEC Set-Up and Product Analysis
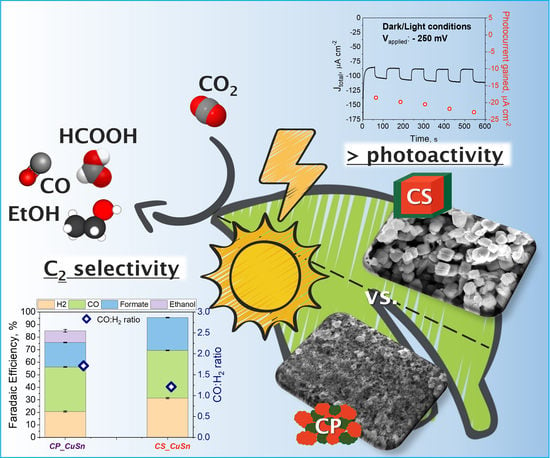
3. Results
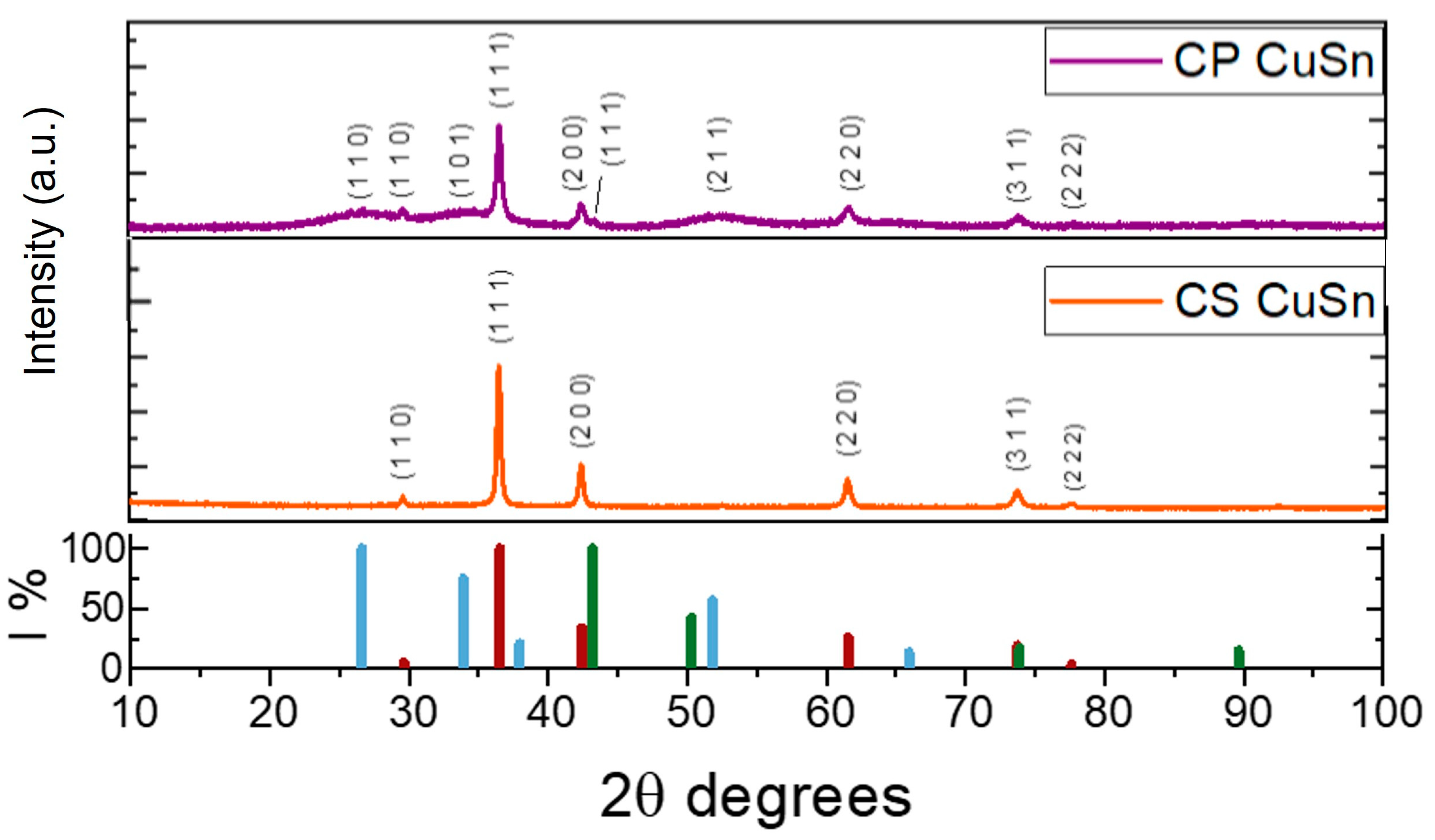
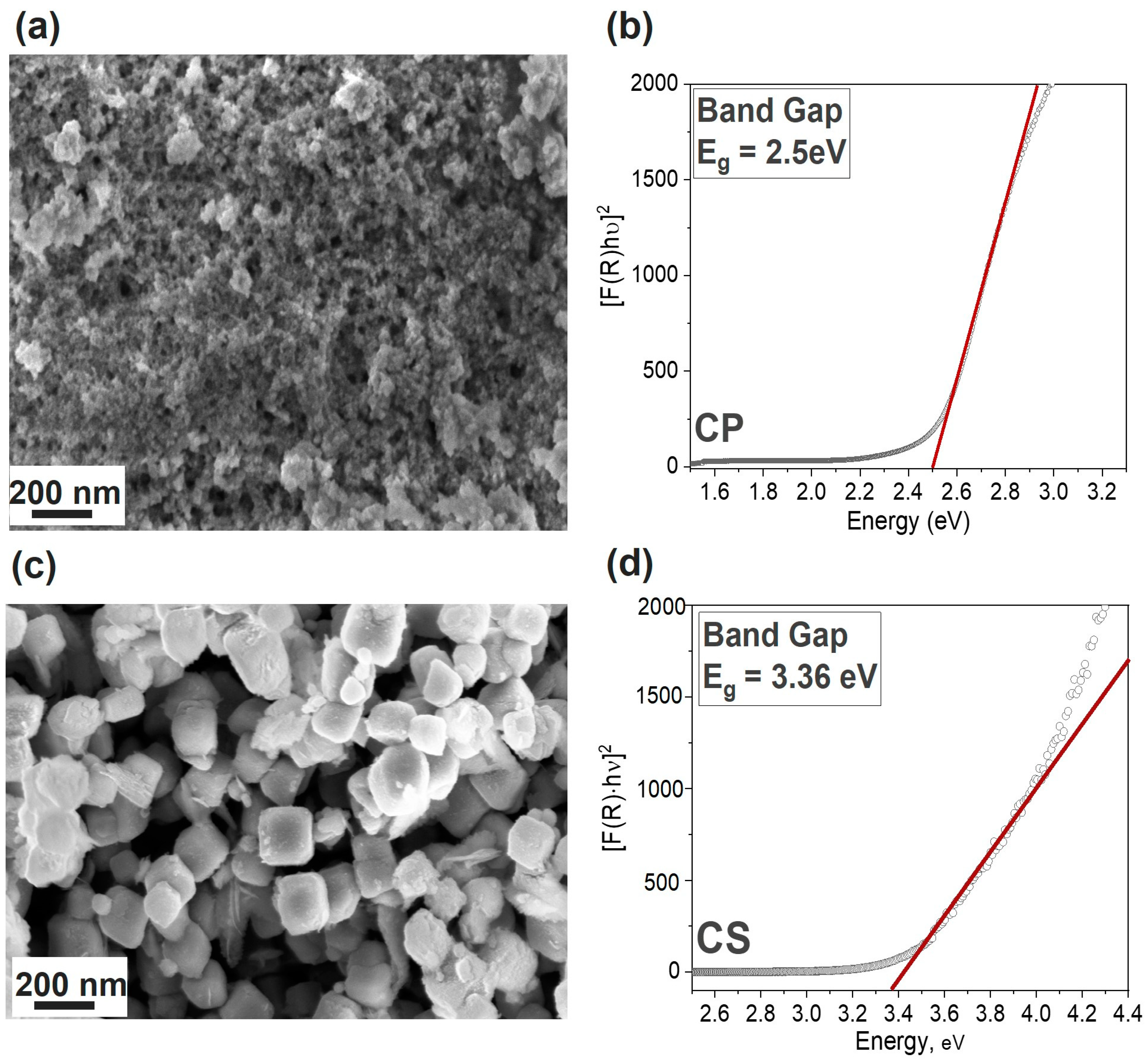
3.1. Physico-Chemical Comparison
3.2. PEC Measurements
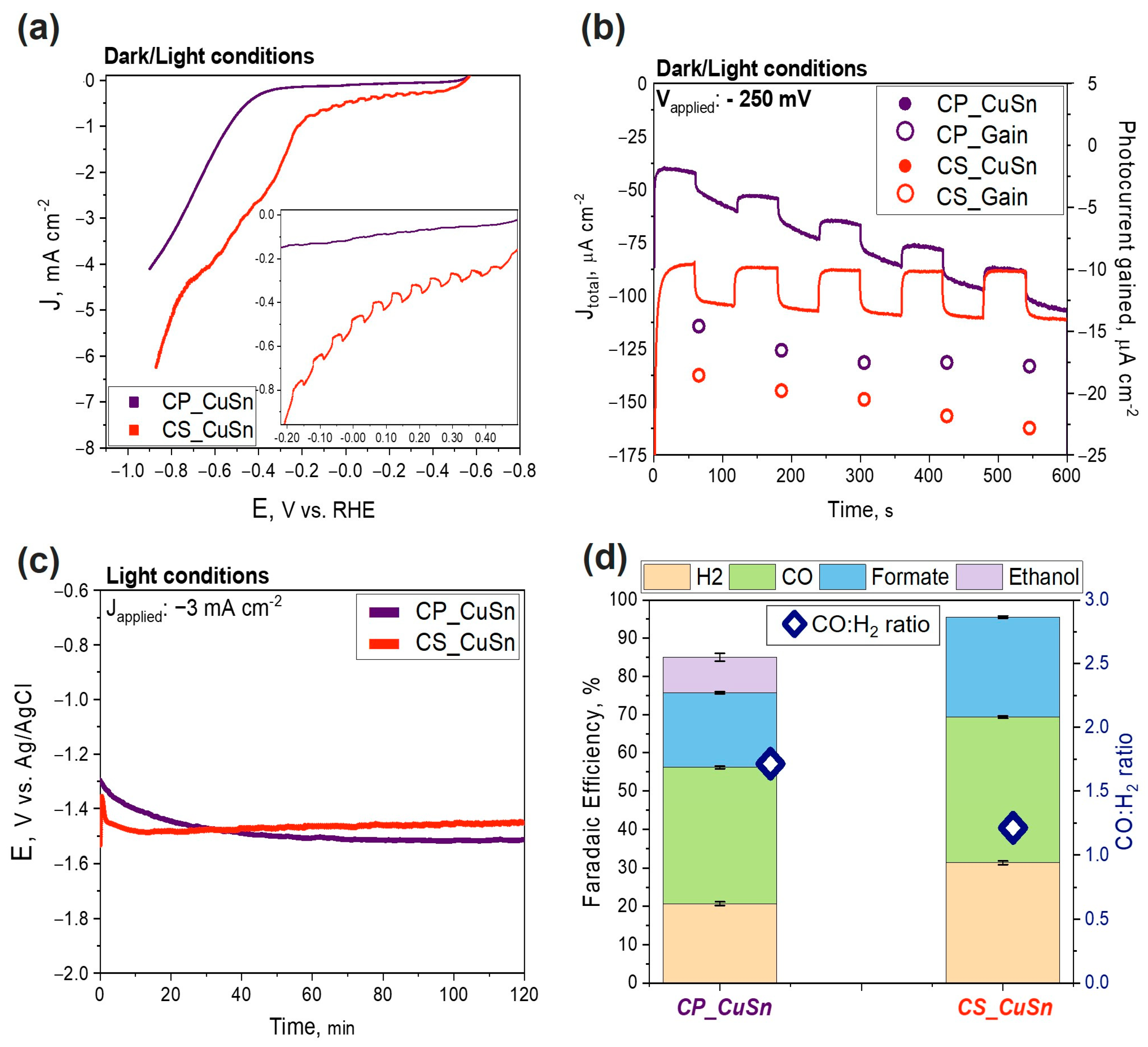
3.2.1. PEC CO2 Reduction Test
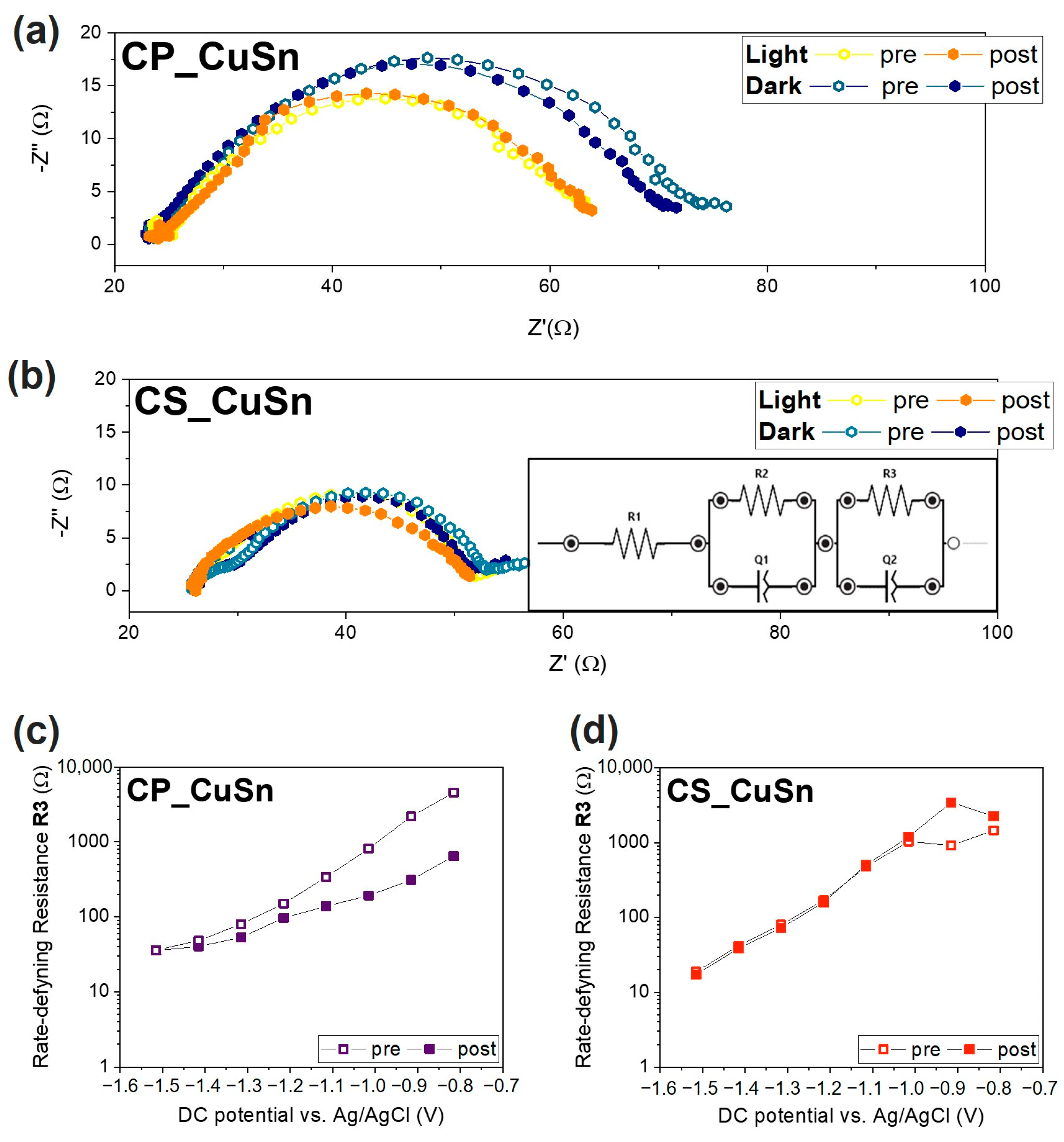
3.2.2. Electrochemical Impedance Spectroscopy Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- IPCC. Summary for Policymakers: Climate Change 2022 Impacts, Adaptation and Vulnerability_Working Group II Contribution to the Sixth Assessment Report of the Intergovernamental Panel on Climate Change; IPCC: Geneva, Switzerland, 2022; ISBN 9781009325844. [Google Scholar]
- Korner, C. Biosphere Responses to CO2 Enrichment. Ecol. Appl. 2000, 10, 1590. [Google Scholar] [CrossRef]
- Arndt, D.S.; Blunden, J.; Dunn, R.J.H. State of the Climate in 2019. Bull. Am. Meteorol. Soc. 2020, 101, S1–S8. [Google Scholar] [CrossRef]
- Guzmán, H.; Salomone, F.; Batuecas, E.; Tommasi, T.; Russo, N.; Bensaid, S.; Hernández, S. How to Make Sustainable CO2 Conversion to Methanol: Thermocatalytic versus Electrocatalytic Technology. Chem. Eng. J. 2021, 417, 127973. [Google Scholar] [CrossRef]
- Zhang, N.; Long, R.; Gao, C.; Xiong, Y. Recent Progress on Advanced Design for Photoelectrochemical Reduction of CO2 to Fuels. Sci. China Mater. 2018, 61, 771–805. [Google Scholar] [CrossRef]
- Hu, J.; Zhai, C.; Zhu, M. Photo-Responsive Metal/Semiconductor Hybrid Nanostructure: A Promising Electrocatalyst for Solar Light Enhanced Fuel Cell Reaction. Chinese Chem. Lett. 2021, 32, 1348–1358. [Google Scholar] [CrossRef]
- Ribbing, C.G.; Roos, A. Copper Oxides (Cu2O, CuO). Handb. Opt. Constants Solids 1997, 2, 875–882. [Google Scholar] [CrossRef]
- Li, S.; Mo, Q.L.; Xiao, Y.; Xiao, F.X. Maneuvering Cuprous Oxide-Based Photocathodes for Solar-to-Fuel Conversion. Coord. Chem. Rev. 2023, 477, 214948. [Google Scholar] [CrossRef]
- Mohan, S.; Honnappa, B.; Augustin, A.; Shanmugam, M.; Chuaicham, C.; Sasaki, K.; Ramasamy, B.; Sekar, K. A Critical Study of Cu2O: Synthesis and Its Application in CO2 Reduction by Photochemical and Electrochemical Approaches. Catalysts 2022, 12, 445. [Google Scholar] [CrossRef]
- Baran, T.; Visibile, A.; Busch, M.; He, X.; Wojtyla, S.; Rondinini, S.; Minguzzi, A.; Vertova, A. Copper Oxide-Based Photocatalysts and Photocathodes: Fundamentals and Recent Advances. Molecules 2021, 26, 7271. [Google Scholar] [CrossRef] [PubMed]
- Paracchino, A.; Laporte, V.; Sivula, K.; Grätzel, M.; Thimsen, E. Highly Active Oxide Photocathode for Photoelectrochemical Water Reduction. Nat. Mater. 2011, 10, 456–461. [Google Scholar] [CrossRef]
- Wu, L.; Tsui, L.K.; Swami, N.; Zangari, G. Photoelectrochemical Stability of Electrodeposited Cu2O Films. J. Phys. Chem. C 2010, 114, 11551–11556. [Google Scholar] [CrossRef]
- Chen, S.; Wang, L.W. Thermodynamic Oxidation and Reduction Potentials of Photocatalytic Semiconductors in Aqueous Solution. Chem. Mater. 2012, 24, 3659–3666. [Google Scholar] [CrossRef]
- Liu, G.; Zheng, F.; Li, J.; Zeng, G.; Ye, Y.; Larson, D.M.; Yano, J.; Crumlin, E.J.; Ager, J.W.; Wang, L.W.; et al. Investigation and Mitigation of Degradation Mechanisms in Cu2O Photoelectrodes for CO2 Reduction to Ethylene. Nat. Energy 2021, 6, 1124–1132. [Google Scholar] [CrossRef]
- Batzill, M.; Diebold, U. The Surface and Materials Science of Tin Oxide. Prog. Surf. Sci. 2005, 79, 47–154. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, C.; Wallace, G.G. Tin Nanoparticles Decorated Copper Oxide Nanowires for Selective Electrochemical Reduction of Aqueous CO2 to CO. J. Mater. Chem. A 2016, 4, 10710–10718. [Google Scholar] [CrossRef]
- Sarfraz, S.; Garcia-Esparza, A.T.; Jedidi, A.; Cavallo, L.; Takanabe, K. Cu-Sn Bimetallic Catalyst for Selective Aqueous Electroreduction of CO2 to CO. ACS Catal. 2016, 6, 2842–2851. [Google Scholar] [CrossRef]
- Li, Y.; Cui, F.; Ross, M.B.; Kim, D.; Sun, Y.; Yang, P. Structure-Sensitive CO2 Electroreduction to Hydrocarbons on Ultrathin 5-Fold Twinned Copper Nanowires. Nano Lett. 2017, 17, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.J.; Uddin, M.J.; Alaboina, P. Review of Nanotechnology for Cathode Materials in Batteries; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 9780323429962. [Google Scholar]
- Rane, A.V.; Kanny, K.; Abitha, V.K.; Thomas, S.; Thomas, S. Methods for Synthesis of Nanoparticles and Fabrication of Nanocomposites; Elsevier: Amsterdam, The Netherlands, 2018; ISBN 9780081019757. [Google Scholar]
- Guzmán, H.; Roldán, D.; Russo, N.; Hernández, S. Ultrasound-Assisted Synthesis of Copper-Based Catalysts for the Electrocatalytic CO2 Reduction: Effect of Ultrasound Irradiation, Precursor Concentration and Calcination Temperature. Sustain. Mater. Technol. 2023, 35, e00557. [Google Scholar] [CrossRef]
- Allahyari, S.; Haghighi, M.; Ebadi, A.; Hosseinzadeh, S. Ultrasound Assisted Co-Precipitation of Nanostructured CuO-ZnO-Al2O3 over HZSM-5: Effect of Precursor and Irradiation Power on Nanocatalyst Properties and Catalytic Performance for Direct Syngas to DME. Ultrason. Sonochem. 2014, 21, 663–673. [Google Scholar] [CrossRef]
- Pinjari, D.V.; Prasad, K.; Gogate, P.R.; Mhaske, S.T.; Pandit, A.B. Synthesis of Titanium Dioxide by Ultrasound Assisted Sol-Gel Technique: Effect of Calcination and Sonication Time. Ultrason. Sonochem. 2015, 23, 185–191. [Google Scholar] [CrossRef]
- Wang, H.; Chen, L.; Feng, Y.; Chen, H. Exploiting Core-Shell Synergy for Nanosynthesis and Mechanistic Investigation. Acc. Chem. Res. 2013, 46, 1636–1646. [Google Scholar] [CrossRef]
- Wang, Z.; Luan, D.; Boey, F.Y.C.; Lou, X.W. Fast Formation of SnO2 Nanoboxes with Enhanced Lithium Storage Capability. J. Am. Chem. Soc. 2011, 133, 4738–4741. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.N.; Li, M.; Hua, B.; Duan, N.; Ding, S.; Bergens, S.; Shankar, K.; Luo, J.L. A Rational Design of Cu2O−SnO2 Core-Shell Catalyst for Highly Selective CO2-to-CO Conversion. ChemCatChem 2019, 11, 4147–4153. [Google Scholar] [CrossRef]
- Zoli, M.; Roldán, D.; Guzmán, H.; Castellino, M.; Chiodoni, A.; Bejtka, K.; Russo, N.; Hernández, S. Facile and Scalable Synthesis of Cu2O-SnO2 Catalyst for the Photoelectrochemical CO2 Conversion. Catal. Today 2023, 413–415, 113985. [Google Scholar] [CrossRef]
- Kumar, B.; Llorente, M.; Froehlich, J.; Dang, T.; Sathrum, A.; Kubiak, C.P. Photochemical and Photoelectrochemical Reduction of CO2. Annu. Rev. Phys. Chem. 2012, 63, 541–569. [Google Scholar] [CrossRef]
- Guzmán, H.; Zammillo, F.; Roldán, D.; Galletti, C.; Russo, N.; Hernández, S. Investigation of Gas Diffusion Electrode Systems for the Electrochemical CO2 Conversion. Catalysts 2021, 11, 482. [Google Scholar] [CrossRef]
- Sacco, A. Electrochemical Impedance Spectroscopy: Fundamentals and Application in Dye-Sensitized Solar Cells. Renew. Sustain. Energy Rev. 2017, 79, 814–829. [Google Scholar] [CrossRef]
- Reddy, K.M.; Manorama, S.V.; Reddy, A.R. Bandgap Studies on Anatase Titanium Dioxide Nanoparticles. Mater. Chem. Phys. 2003, 78, 239–245. [Google Scholar] [CrossRef]
- Liu, L.; Sun, W.; Yang, W.; Li, Q.; Shang, J.K. Post-Illumination Activity of SnO2 Nanoparticle-Decorated Cu2O Nanocubes by H2O2 Production in Dark from Photocatalytic “Memory”. Sci. Rep. 2016, 6, 20878. [Google Scholar] [CrossRef]
- Pagare, P.K.; Torane, A.P. Band Gap Varied Cuprous Oxide (Cu2O) Thin Films as a Tool for Glucose Sensing. Microchim. Acta 2016, 183, 2983–2989. [Google Scholar] [CrossRef]
- Kamarulzaman, N.; Abdul Aziz, N.D.; Kasim, M.F.; Chayed, N.F.; Yahaya Subban, R.H.; Badar, N. Anomalies in Wide Band Gap SnO2 Nanostructures. J. Solid State Chem. 2019, 277, 271–280. [Google Scholar] [CrossRef]
- Schreier, M.; Luo, J.; Gao, P.; Moehl, T.; Mayer, M.T.; Grätzel, M. Covalent Immobilization of a Molecular Catalyst on Cu2O Photocathodes for CO2 Reduction. J. Am. Chem. Soc. 2016, 138, 1938–1946. [Google Scholar] [CrossRef]
- Won, D.H.; Choi, C.H.; Chung, J.; Woo, S.I. Photoelectrochemical Production of Formic Acid and Methanol from Carbon Dioxide on Metal-Decorated CuO/Cu2O-Layered Thin Films under Visible Light Irradiation. Appl. Catal. B Environ. 2014, 158–159, 217–223. [Google Scholar] [CrossRef]
- de Brito, J.F.; Araujo, A.R.; Rajeshwar, K.; Zanoni, M.V.B. Photoelectrochemical Reduction of CO2 on Cu/Cu2O Films: Product Distribution and PH Effects. Chem. Eng. J. 2015, 264, 302–309. [Google Scholar] [CrossRef]
- Kortlever, R.; Tan, K.H.; Kwon, Y.; Koper, M.T.M. Electrochemical Carbon Dioxide and Bicarbonate Reduction on Copper in Weakly Alkaline Media. J. Solid State Electrochem. 2013, 17, 1843–1849. [Google Scholar] [CrossRef]
- Zhang, Y.; Pan, D.; Tao, Y.; Shang, H.; Zhang, D.; Li, G.; Li, H. Photoelectrocatalytic Reduction of CO2 to Syngas via SnOx-Enhanced Cu2O Nanowires Photocathodes. Adv. Funct. Mater. 2022, 32, 2109600. [Google Scholar] [CrossRef]
- Chen, P.; Zhang, Y.; Zhou, Y.; Dong, F. Photoelectrocatalytic Carbon Dioxide Reduction: Fundamental, Advances and Challenges. Nano Mater. Sci. 2021, 3, 344–367. [Google Scholar] [CrossRef]
- Hendy, C.M.; Smith, G.C.; Xu, Z.; Lian, T.; Jui, N.T. Radical Chain Reduction via Carbon Dioxide Radical Anion (CO2•−). J. Am. Chem. Soc. 2021, 143, 8987–8992. [Google Scholar] [CrossRef]
- Kohlpaintner, C.W.; Fischer, R.W.; Cornils, B. Aqueous Biphasic Catalysis: Ruhrchemie/Rhône-Poulenc Oxo Process. Appl. Catal. A Gen. 2001, 221, 219–225. [Google Scholar] [CrossRef]
- Xu, H.; Rebollar, D.; He, H.; Chong, L.; Liu, Y.; Liu, C.; Sun, C.J.; Li, T.; Muntean, J.V.; Winans, R.E.; et al. Highly Selective Electrocatalytic CO2 Reduction to Ethanol by Metallic Clusters Dynamically Formed from Atomically Dispersed Copper. Nat. Energy 2020, 5, 623–632. [Google Scholar] [CrossRef]
- Pardo Pérez, L.C.; Arndt, A.; Stojkovikj, S.; Ahmet, I.Y.; Arens, J.T.; Dattila, F.; Wendt, R.; Guilherme Buzanich, A.; Radtke, M.; Davies, V.; et al. Determining Structure-Activity Relationships in Oxide Derived Cu-Sn Catalysts During CO2 Electroreduction Using X-ray Spectroscopy. Adv. Energy Mater. 2022, 12, 2103328. [Google Scholar] [CrossRef]
- Zeng, J.; Bejtka, K.; Ju, W.; Castellino, M.; Chiodoni, A.; Sacco, A.; Farkhondehfal, M.A.; Hernández, S.; Rentsch, D.; Battaglia, C.; et al. Advanced Cu-Sn Foam for Selectively Converting CO2 to CO in Aqueous Solution. Appl. Catal. B Environ. 2018, 236, 475–482. [Google Scholar] [CrossRef]
- Dunwell, M.; Lu, Q.; Heyes, J.M.; Rosen, J.; Chen, J.G.; Yan, Y.; Jiao, F.; Xu, B. The Central Role of Bicarbonate in the Electrochemical Reduction of Carbon Dioxide on Gold. J. Am. Chem. Soc. 2017, 139, 3774–3783. [Google Scholar] [CrossRef]
- Sawicka-Chudy, P.; Sibiński, M.; Wisz, G.; Rybak-Wilusz, E.; Cholewa, M. Numerical Analysis and Optimization of Cu2O/TiO2, CuO/TiO2, Heterojunction Solar Cells Using SCAPS. J. Phys. Conf. Ser. 2018, 1033, 012002. [Google Scholar] [CrossRef]
- Ravichandran, A.T.; Dhanabalan, K.; Vasuhi, A.; Chandramohan, R.; Mantha, S. Morphology, Bandgap, and Grain Size Tailoring in Cu2O Thin Film by SILAR Method. IEEE Trans. Nanotechnol. 2015, 14, 108–112. [Google Scholar] [CrossRef]
- Huang, M.H. Semiconductor Nanocrystals Possessing Broadly Size- and Facet-Dependent Optical Properties. J. Chin. Chem. Soc. 2021, 68, 45–50. [Google Scholar] [CrossRef]
- Nursanto, E.B.; Jeon, H.S.; Kim, C.; Jee, M.S.; Koh, J.H.; Hwang, Y.J.; Min, B.K. Gold Catalyst Reactivity for CO2 Electro-Reduction: From Nano Particle to Layer. Catal. Today 2016, 260, 107–111. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Kaneko, K. Determination of Pore Size and Pore Size Distribution. 1. Adsorbents and Catalysts. J. Memb. Sci. 1994, 96, 59–89. [Google Scholar] [CrossRef]
- Roy, A.; Jadhav, H.S.; Gil Seo, J. Cu2O/CuO Electrocatalyst for Electrochemical Reduction of Carbon Dioxide to Methanol. Electroanalysis 2021, 33, 705–712. [Google Scholar] [CrossRef]
- Cox, P.A. The Electronic Structure and Chemistry of Solids; Oxford University: Oxford, UK, 1987; ISBN 9780198552048. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | ICP-OES | N2 Physisorption | |||
|---|---|---|---|---|---|
| Sn at.% | Cu:Sn |
BET Surface Area (m2 g−1) |
Pore Volume (cm3 g−1) |
Pore Size (nm) | |
| CP_CuSn | 32.3 | 2:1 | 142 | 0.12 | 5.9 |
| CS_CuSn | 23.4 | 3:1 | 64 | 0.08 | 5.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zoli, M.; Guzmán, H.; Sacco, A.; Russo, N.; Hernández, S. Cu2O/SnO2 Heterostructures: Role of the Synthesis Procedure on PEC CO2 Conversion. Materials 2023, 16, 4497. https://doi.org/10.3390/ma16134497
Zoli M, Guzmán H, Sacco A, Russo N, Hernández S. Cu2O/SnO2 Heterostructures: Role of the Synthesis Procedure on PEC CO2 Conversion. Materials. 2023; 16(13):4497. https://doi.org/10.3390/ma16134497
Chicago/Turabian StyleZoli, Maddalena, Hilmar Guzmán, Adriano Sacco, Nunzio Russo, and Simelys Hernández. 2023. "Cu2O/SnO2 Heterostructures: Role of the Synthesis Procedure on PEC CO2 Conversion" Materials 16, no. 13: 4497. https://doi.org/10.3390/ma16134497
APA StyleZoli, M., Guzmán, H., Sacco, A., Russo, N., & Hernández, S. (2023). Cu2O/SnO2 Heterostructures: Role of the Synthesis Procedure on PEC CO2 Conversion. Materials, 16(13), 4497. https://doi.org/10.3390/ma16134497