
The Enhancement of CO Oxidation Performance and Stability in SO2 and H2S Environment on Pd-Au/FeOX/Al2O3 Catalysts
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Methods
2.1.1. FeOx/Al2O3 Preparation
2.1.2. Preparation of Au/FeOx/Al2O3 Catalysts
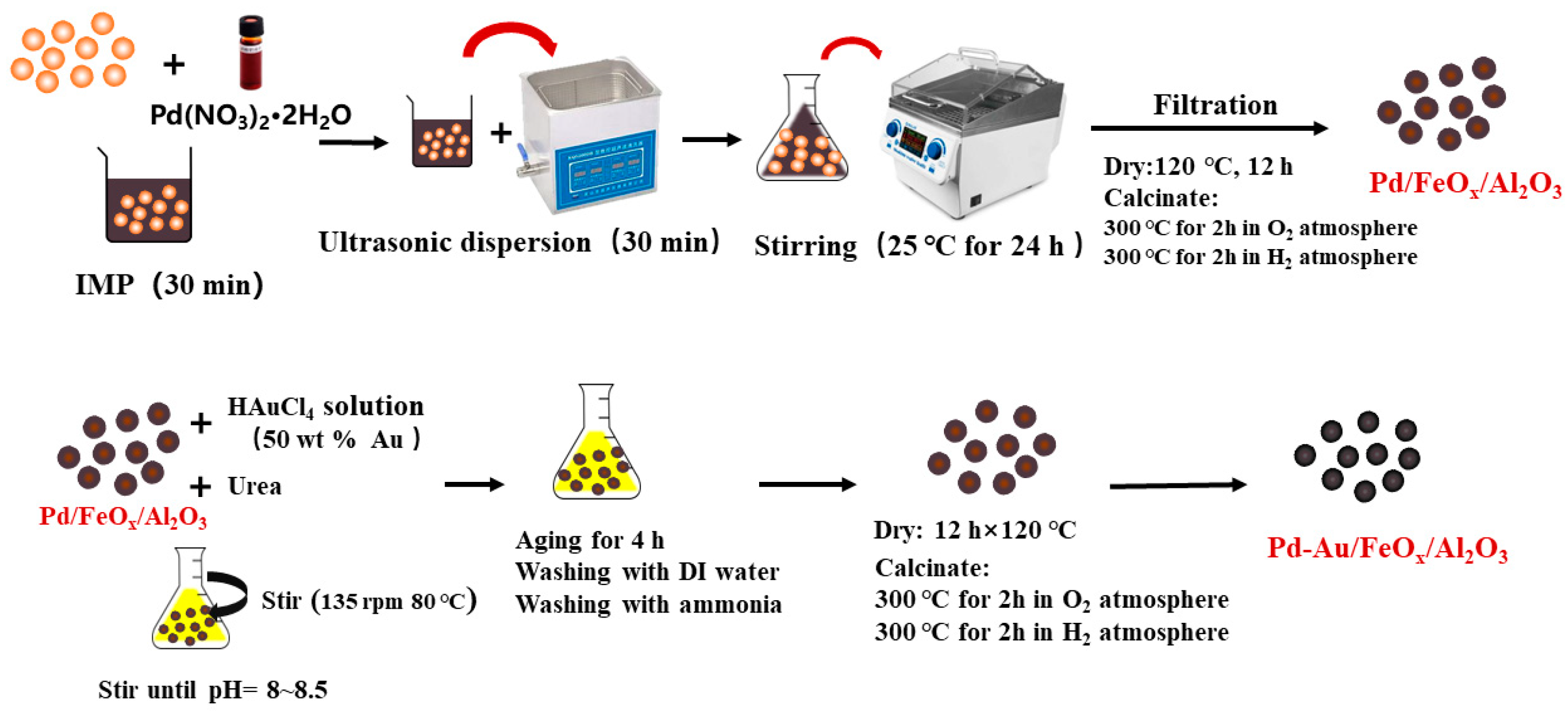
2.1.3. Preparation of Pd-Au/FeOx/Al2O3 Catalysts
- (1)
- Preparation of Pd/FeOx/Al2O3 catalysts
- (2)
- Preparation of Pd-Au/FeOx/Al2O3 Catalysts
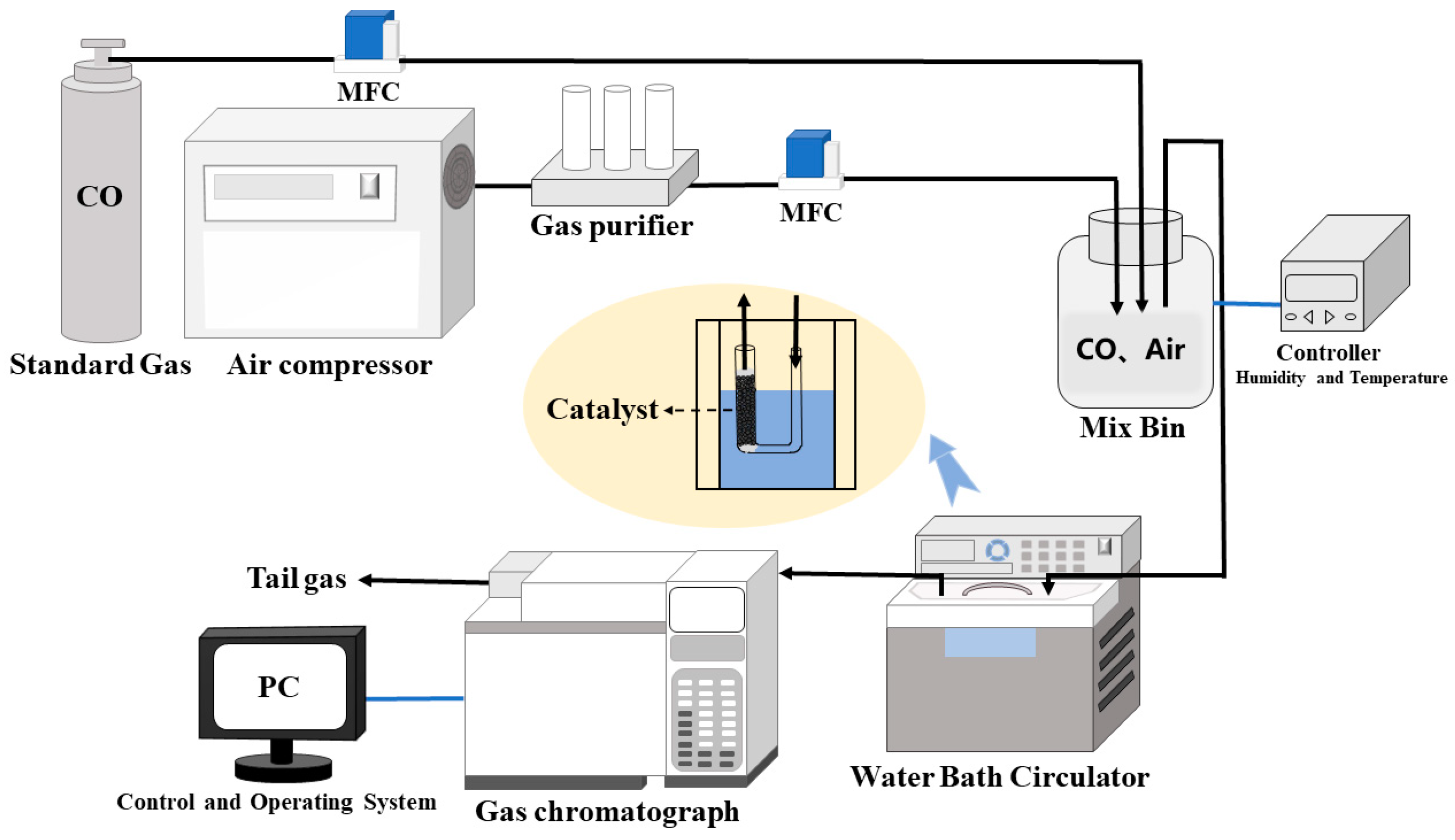
2.2. Test Methods
2.3. SO2 and H2S Pretreatment
2.4. Evaluation of Catalytic Activity
2.5. Evaluation of the Stability of the Catalysts
2.6. DFT Calculations
3. Results and Discussion
3.1. Physical and Chemical Characterization of Catalysts
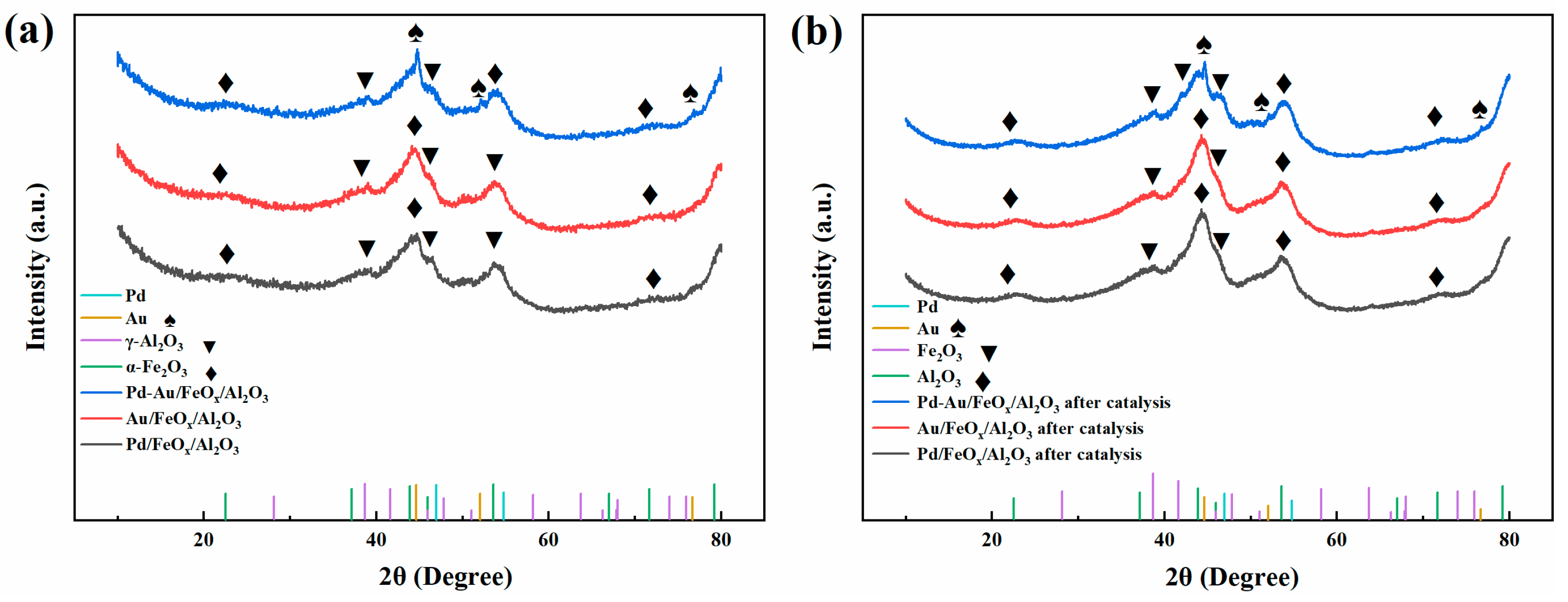
3.1.1. XRD Analysis
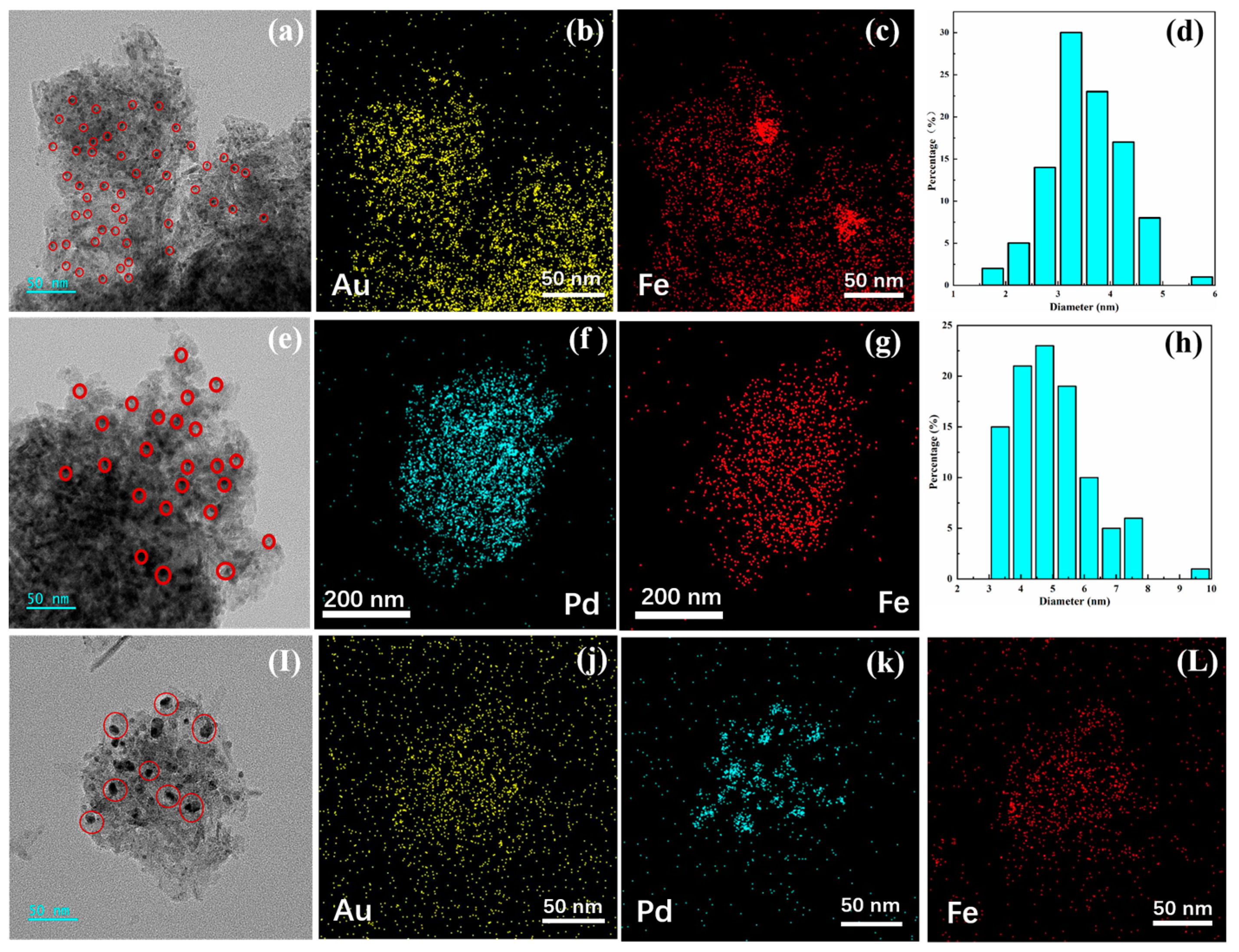
3.1.2. HRTEM Analysis
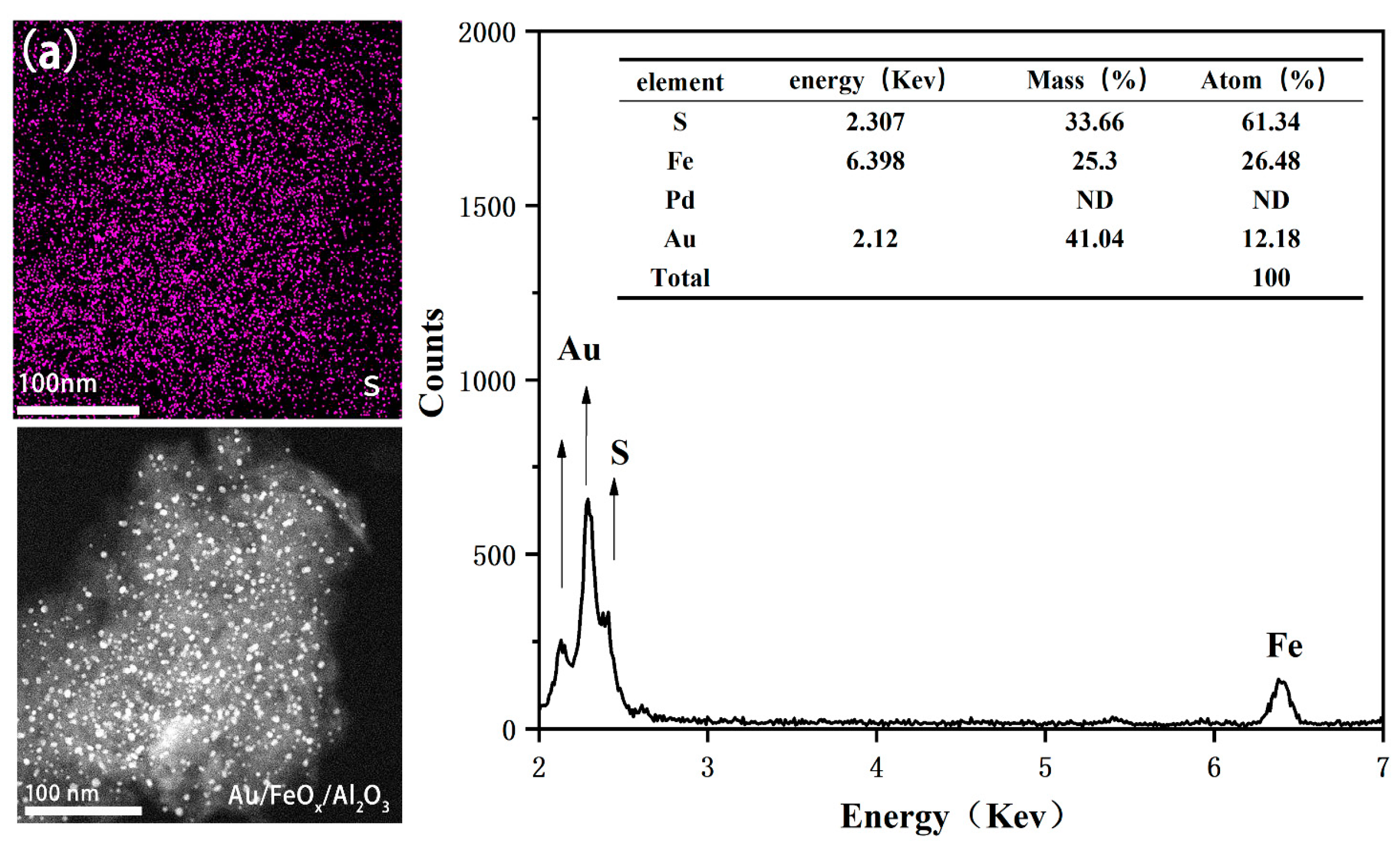
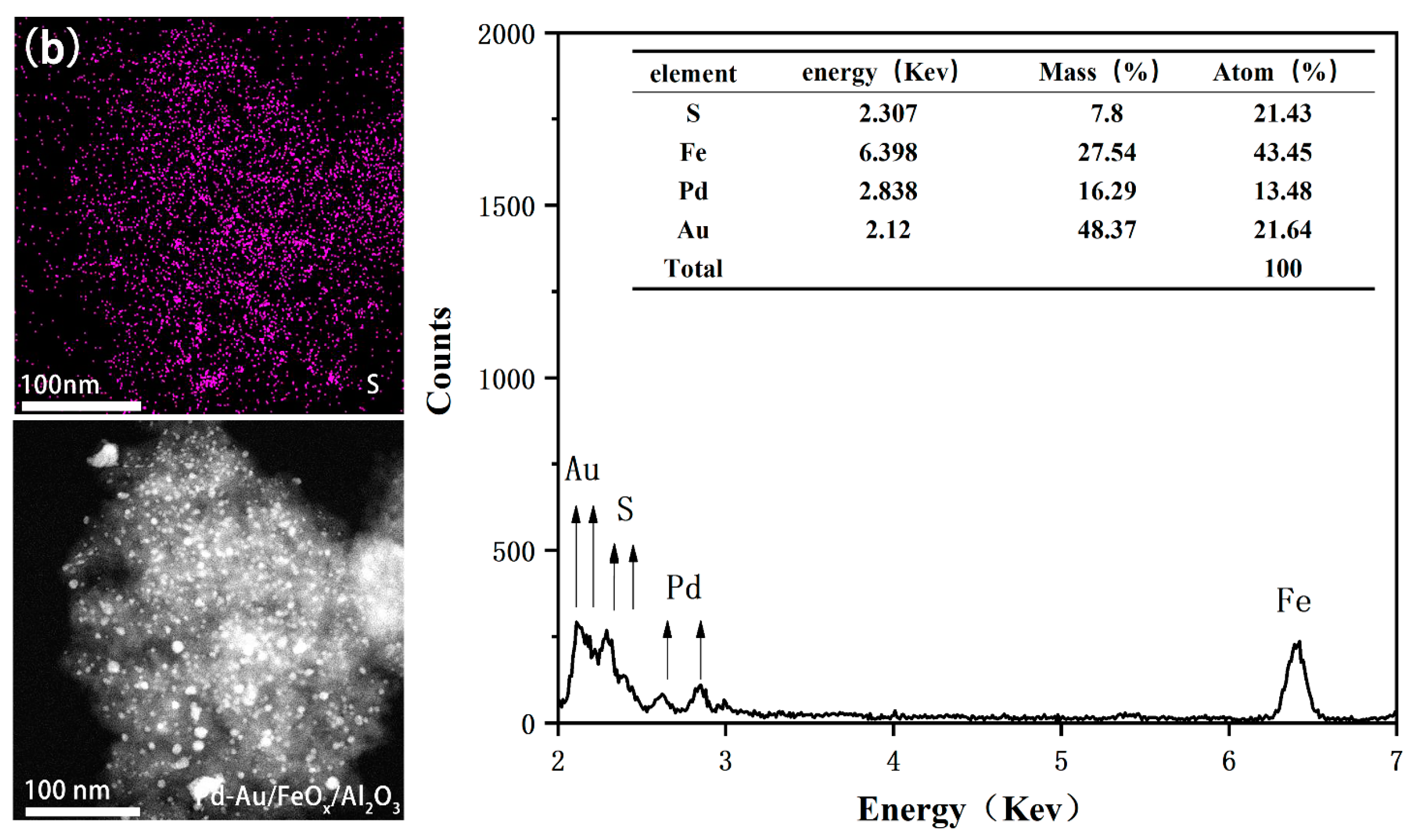
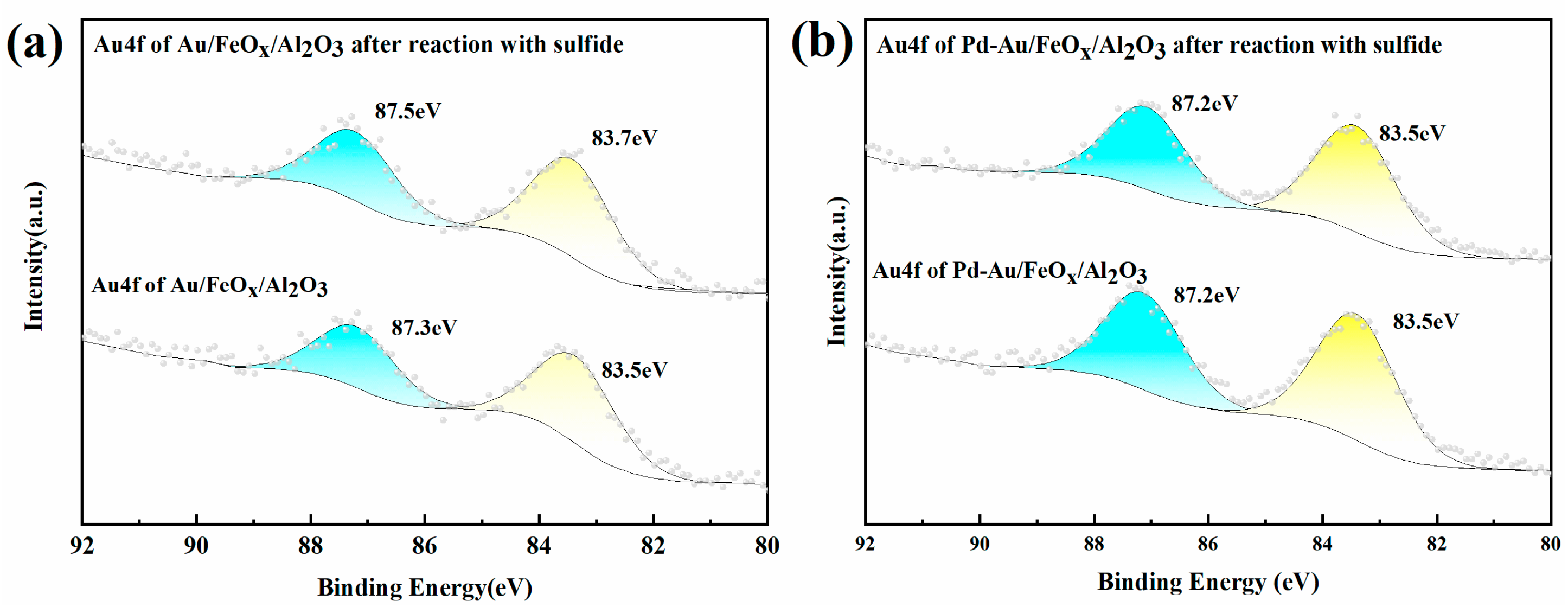
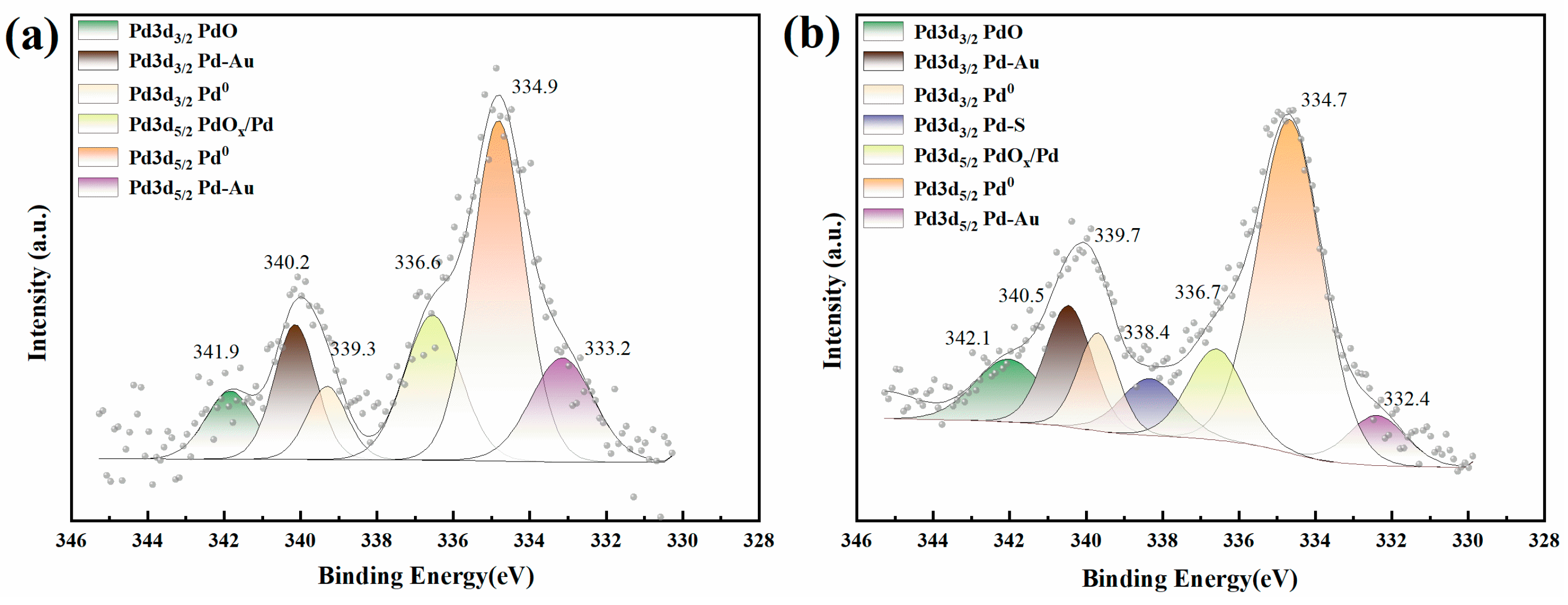
3.1.3. XPS Analysis
3.2. CO Oxidation Catalyst Performance
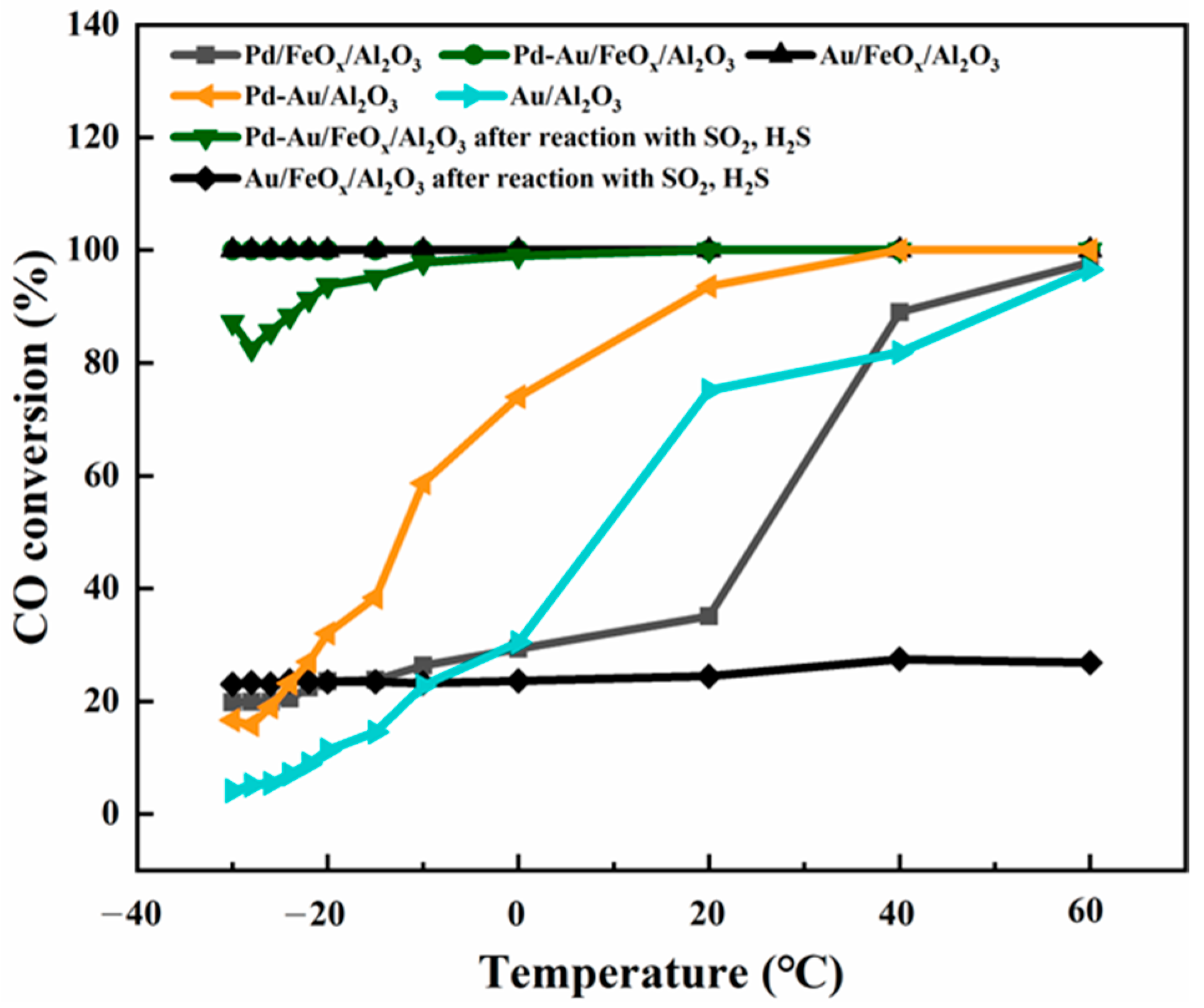
3.2.1. Evaluating Catalytic Activity
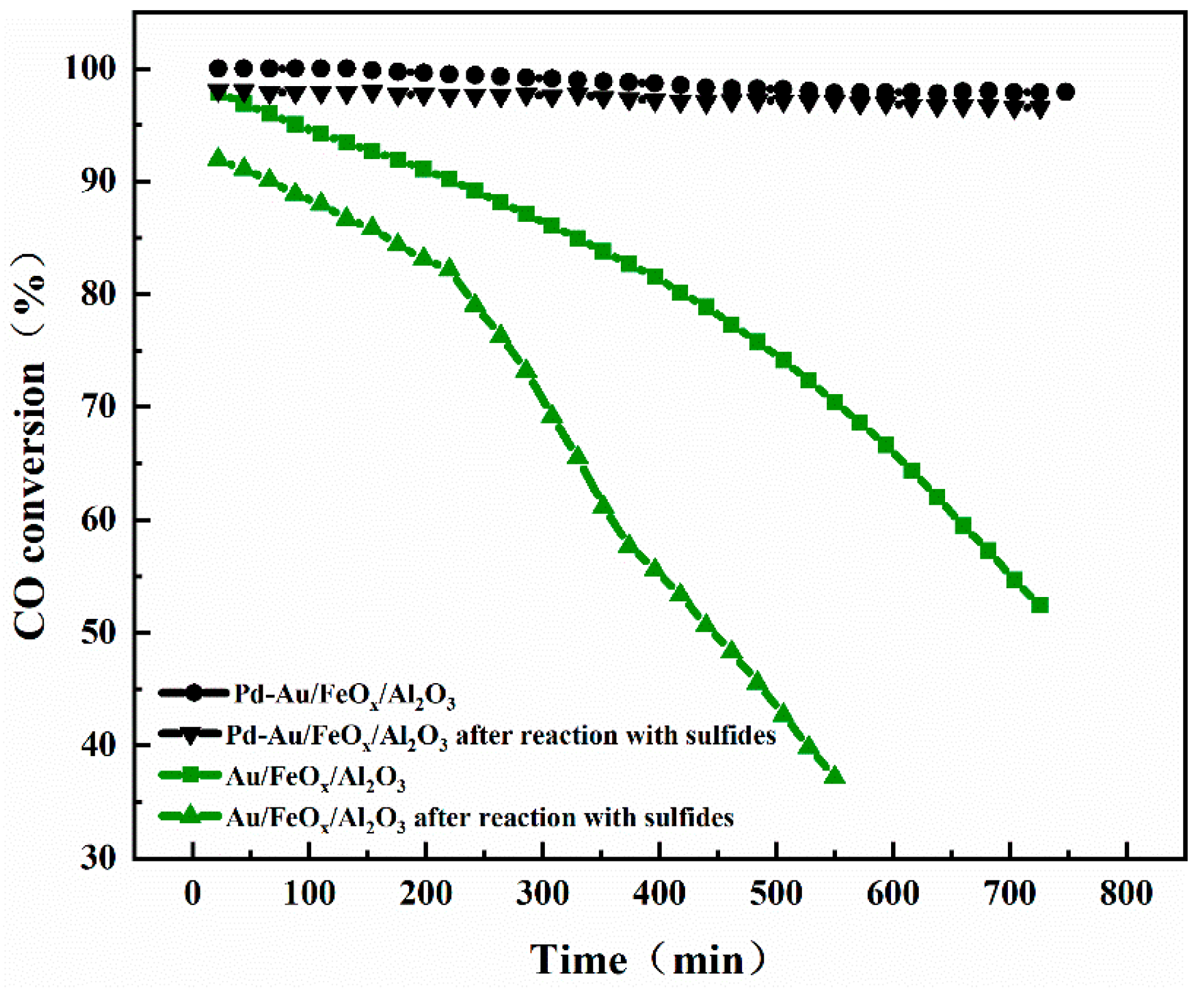
3.2.2. Evaluating Catalyst Stability
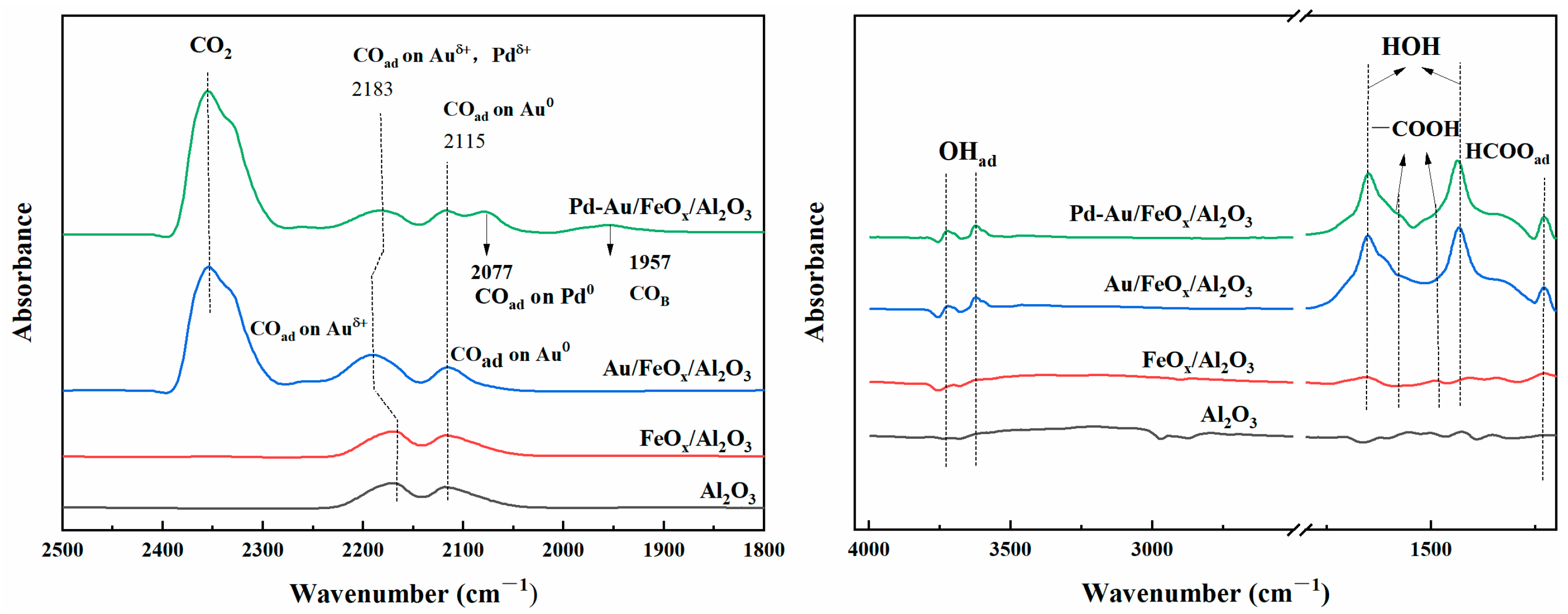
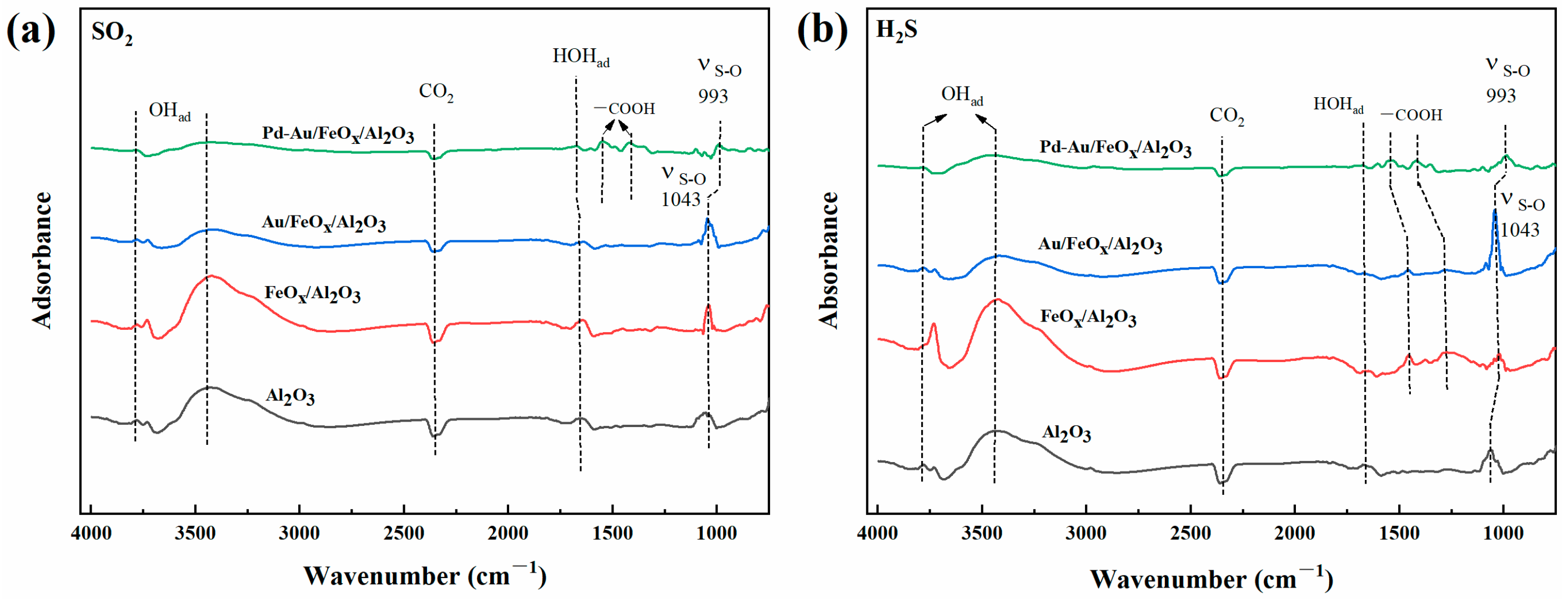
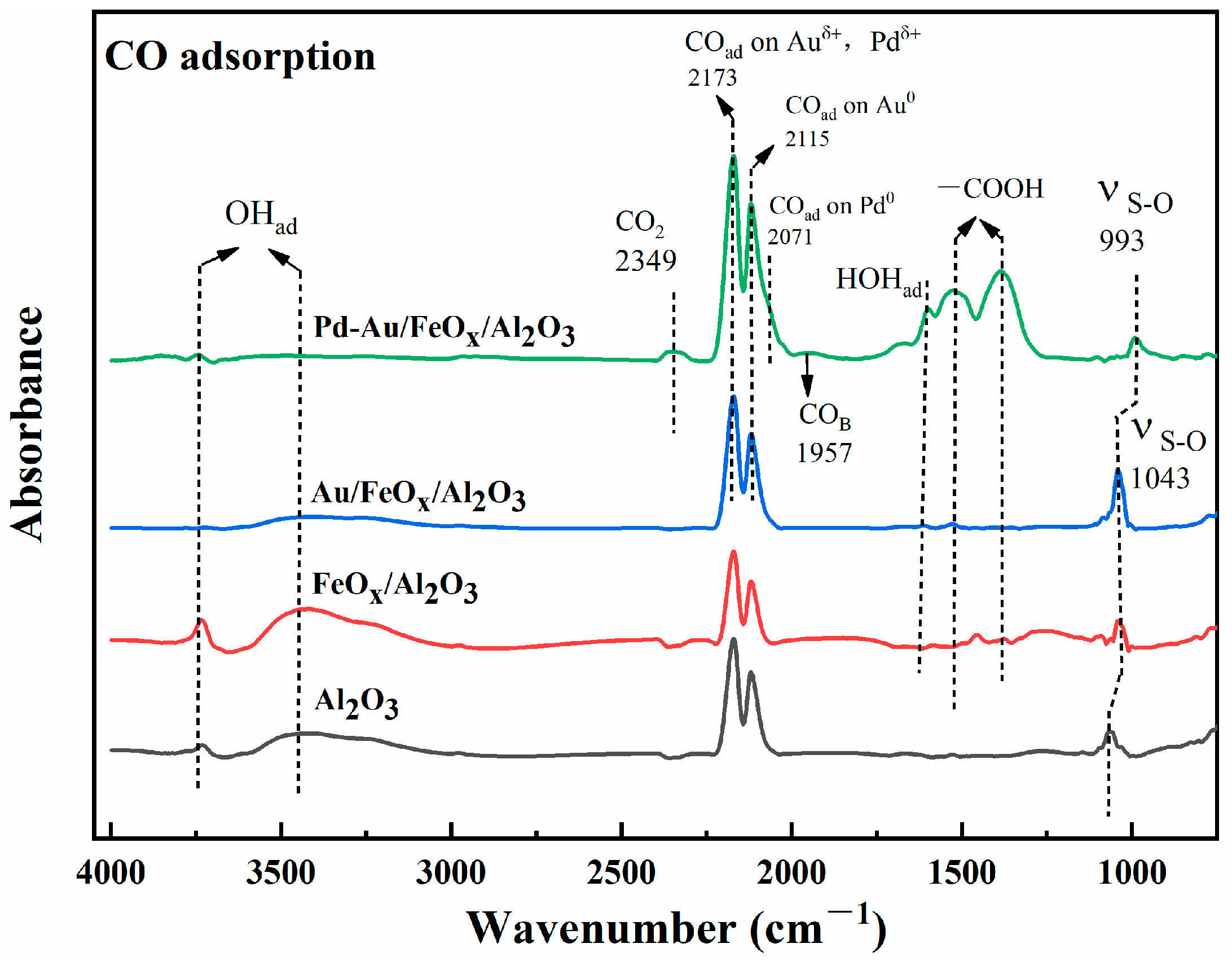
3.2.3. Mechanistic Analysis of CO Oxidation
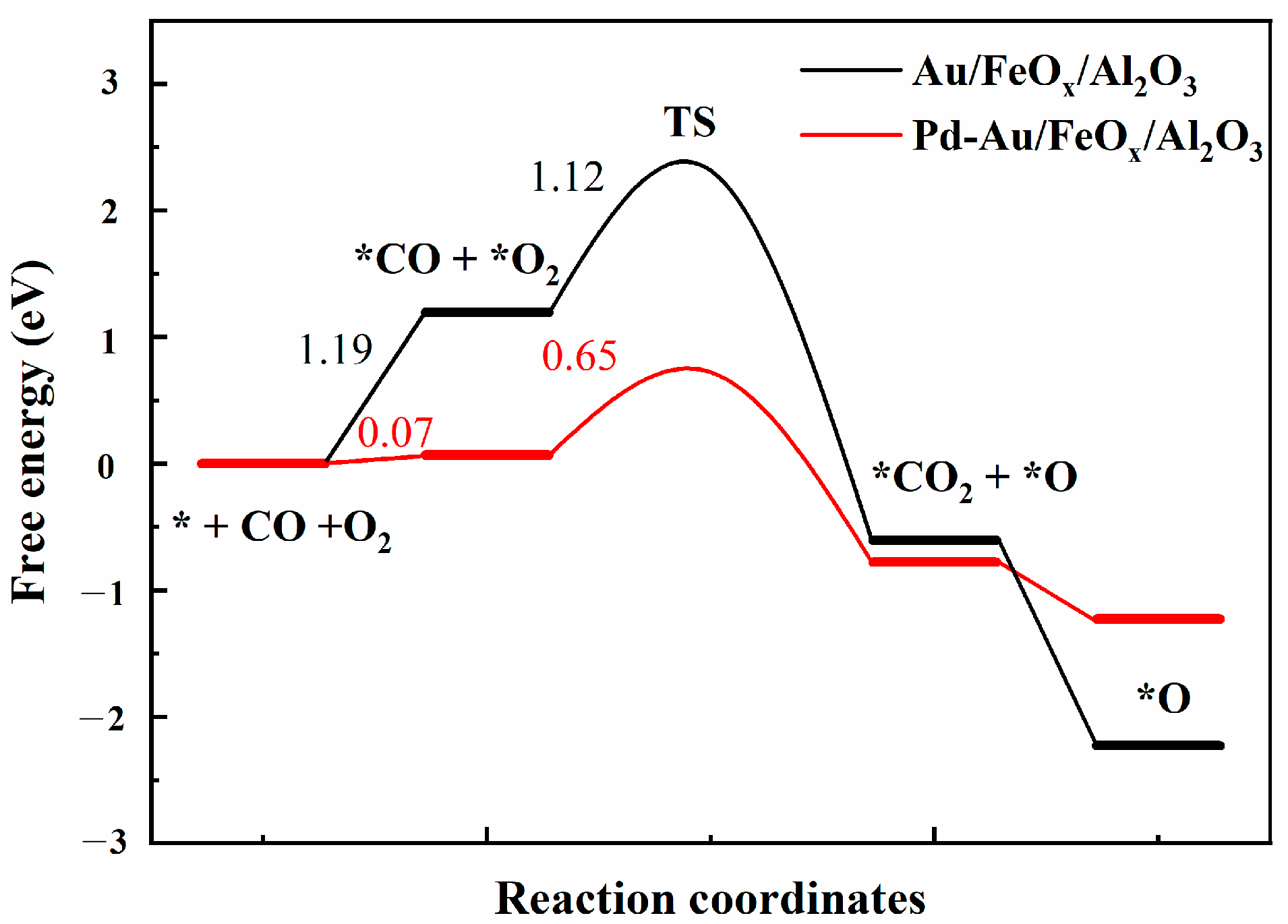
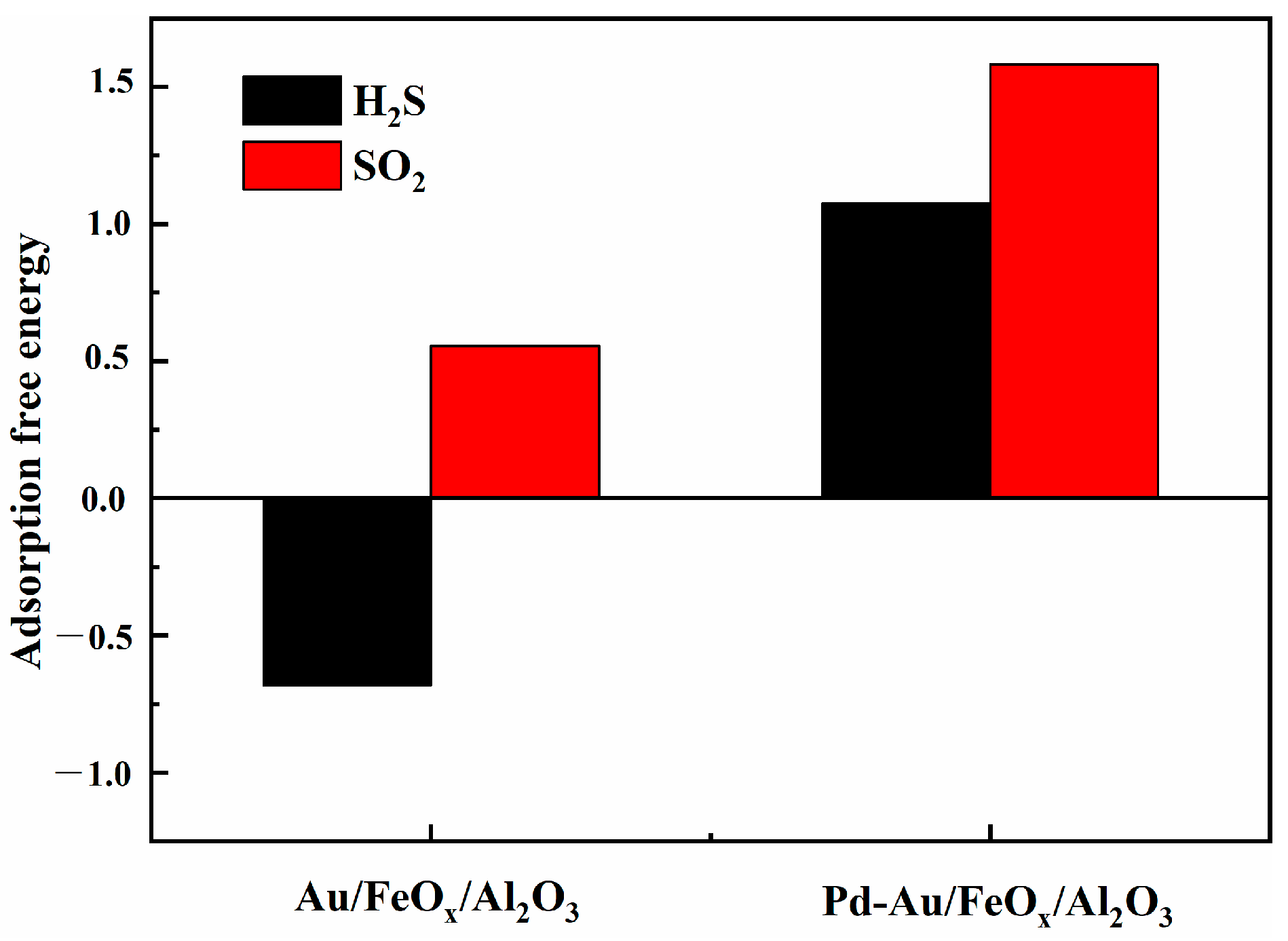
3.2.4. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rose, J.J.; Wang, L.; Xu, Q.Z.; McTiernan, C.F.; Shiva, S.; Tejero, J.; Gladwin, M.T. Carbon Monoxide Poisoning: Pathogenesis, Management, and Future Directions of Therapy. Am. J. Respir. Crit. Care Med. 2017, 195, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Guan, J. Applications of single-atom catalysts. Microchem. J. 2022, 15, 38–70. [Google Scholar] [CrossRef]
- Xie, X.; Zhang, X.; Xie, M.; Xiong, L.; Sun, H.; Lu, Y.; Mu, Q.; Rummeli, M.H.; Xu, J.; Li, S.; et al. Au-activated N motifs in non-coherent cupric porphyrin metal organic frameworks for promoting and stabilizing ethylene production. Nat. Commun. 2022, 13, 63. [Google Scholar] [CrossRef]
- Qin, L.; Wang, Z.; Fu, Y.; Lai, C.; Liu, X.; Li, B.; Liu, S.; Yi, H.; Li, L.; Zhang, M.; et al. Gold nanoparticles-modified MnFe2O4 with synergistic catalysis for photo-Fenton degradation of tetracycline under neutral pH. J. Hazard. Mater. 2021, 414, 125448. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Cheng, J.; He, W.; Li, Y.; Mao, J.; Zheng, X.; Chen, C.; Cui, C.; Hao, Q. Interfacial electronic modulation of Ni3S2 nanosheet arrays decorated with Au nanoparticles boosts overall water splitting. Appl. Catal. B Environ. 2022, 304, 120935. [Google Scholar] [CrossRef]
- Jiang, X.; Huang, J.; Bi, Z.; Ni, W.; Gurzadyan, G.; Zhu, Y.; Zhang, Z. Plasmonic Active “Hot Spots”-Confined Photocatalytic CO2 Reduction with High Selectivity for CH4 Production. Adv. Mater. 2022, 34, 2109330. [Google Scholar] [CrossRef]
- Jiang, W.; Low, J.; Mao, K.; Duan, D.; Chen, S.; Liu, W.; Pao, C.-W.; Ma, J.; Sang, S.; Shu, C.; et al. Pd-Modified ZnO-Au Enabling Alkoxy Intermediates Formation and Dehydrogenation for Photocatalytic Conversion of Methane to Ethylene. J. Am. Chem. Soc. 2021, 143, 269–278. [Google Scholar] [CrossRef]
- Feng, C.; Liu, X.; Zhu, T.; Tian, M. Catalytic oxidation of CO on noble metal-based catalysts. Environ. Sci. Pollut. Res. 2021, 28, 24847–24871. [Google Scholar] [CrossRef]
- Wu, F.; He, L.; Li, W.C.; Lu, R.; Wang, Y.; Lu, A.H. Highly dispersed boron-nitride/CuOx-supported Au nanoparticles for catalytic CO oxidation at low temperatures. Chin. J. Catal. 2021, 42, 388–395. [Google Scholar] [CrossRef]
- Amrute, A.P.; De Bellis, J.; Felderhoff, M.; Schuth, F. Mechanochemical Synthesis of Catalytic Materials. Chem.-A Eur. J. 2021, 27, 6819–6847. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, Y.; Du, W.; Xu, J.; Wang, Q.; Song, N.; Qian, G.; Duan, X.; Zhou, X. Engineering gold impregnated uncalcined TS-1 to boost catalytic formation of propylene oxide. Appl. Catal. B Environ. 2022, 319, 121837. [Google Scholar] [CrossRef]
- Nishio, H.; Miura, H.; Kamata, K.; Shishido, T. Deposition of highly dispersed gold nanoparticles onto metal phosphates by deposition-precipitation with aqueous ammonia. Catal. Sci. Technol. 2021, 11, 7141–7150. [Google Scholar] [CrossRef]
- Zhu, D.; Xu, Y.; Shi, J.; Zou, X.; Zhang, W.; Huang, X.; Li, Z. Selective enrichment and electrochemical determination of Cu in mushroom using L-Cysteine functionalized Fe3O4@Au nanoparticles. Microchem. J. 2021, 165, 106148. [Google Scholar] [CrossRef]
- Nakahara, R.; Sakai, M.; Kimura, T.; Yamamoto, M.; Syouji, A.; Hara, K.; Kouno, T. Lasing action of ZnO nanowires grown by mist chemical vapor deposition using thin Au layer on c-plane sapphire substrate. Jpn. J. Appl. Phys. 2021, 60, 058002. [Google Scholar] [CrossRef]
- Li, Z.; Wu, L.; Guo, J.; Shao, Y.; Song, Y.; Ding, Y.; Zhu, L.; Yao, X. Light-Promoted Minisci Coupling Reaction of Ethers and Aza Aromatics Catalyzed by Au/TiO2 Heterogeneous Photocatalyst. ChemCatChem 2021, 13, 3671–3678. [Google Scholar] [CrossRef]
- Dobrosz-Gomez, I.; Gomez-Garcia, M.A.; Rynkowski, J.M. The Origin of Au/Ce1-xZrxO2 Catalyst’s Active Sites in Low-Temperature CO Oxidation. Catalysts 2020, 10, 1312. [Google Scholar] [CrossRef]
- Yang, S.S.; Huang, Z.Y.; Wu, P.X.; Li, Y.H.; Dong, X.B.; Li, C.Q.; Zhu, N.Y.; Duan, X.D.; Dionysiou, D.D. Rapid removal of tetrabromobisphenol A by alpha-Fe2O3-x@Graphene@Montmorillonite catalyst with oxygen vacancies through peroxymonosulfate activation: Role of halogen and alpha-hydroxyalkyl radicals. Appl. Catal. B Environ. 2020, 260, 118129. [Google Scholar] [CrossRef]
- Zhang, S.C.; Liu, Z.F.; Chen, D.; Yan, W.G. An efficient hole transfer pathway on hematite integrated by ultrathin Al2O3 interlayer and novel CuCoOx cocatalyst for efficient photoelectrochemical water oxidation. Appl. Catal. B Environ. 2020, 277, 119197. [Google Scholar] [CrossRef]
- Yang, J.; Hu, S.Y.; Fang, Y.R.; Hoang, S.; Li, L.; Yang, W.W.; Liang, Z.F.; Wu, J.; Hu, J.P.; Xiao, W.; et al. Oxygen Vacancy Promoted O2 Activation over Perovskite Oxide for Low-Temperature CO Oxidation. ACS Catal. 2019, 9, 9751–9763. [Google Scholar] [CrossRef]
- Ma, G.Y.; Wang, L.; Wang, X.R.; Li, L.; Ma, H.F. CO Oxidation over Alumina-Supported Copper Catalysts. Catalysts 2022, 12, 1030. [Google Scholar] [CrossRef]
- Wang, C.L.; Yao, Q.; Cao, L.N.; Li, J.J.; Chen, S.; Lu, J.L. Precise Tailoring of Ir-FeOx Interfaces for Improved Catalytic Performance in Preferential Oxidation of Carbon Monoxide in Hydrogen. J. Phys. Chem. C 2019, 123, 29262–29270. [Google Scholar] [CrossRef]
- Rio, E.D.; Blanco, G.; Collins, S.; Haro, M.L.; Chen, X.W.; Delgado, J.J.; Calvino, J.J.; Bernal, S. CO Oxidation Activity of a Au/Ceria-Zirconia Catalyst Prepared by Deposition-Precipitation with Urea. Top. Catal. 2011, 54, 931–940. [Google Scholar]
- Chrouda, A.; Ahmed, S.M.A.; Elamin, M.B. Preparation of Nanocatalysts Using Deposition Precipitation with Urea: Mechanism, Advantages and Results. ChemBioEng Rev. 2022, 9, 248–264. [Google Scholar] [CrossRef]
- Valechha, D.; Megarajan, S.K.; Fakeeha, A.H.; Al-Fatesh, A.S.; Labhasetwar, N.K. Effect of SO2 on Catalytic CO Oxidation Over Nano-Structured, Mesoporous Au/Ce1-xZrxO2 Catalysts. Catal. Lett. 2017, 147, 2893–2900. [Google Scholar] [CrossRef]
- Chen, D.C.; Tang, J.; Zhang, X.X.; Cui, H.; Li, Y. Sulfur dioxide adsorbed on pristine and Au dimer decorated gamma-graphyne: A density functional theory study. Appl. Surf. Sci. 2018, 458, 781–789. [Google Scholar] [CrossRef]
- Spezzati, G.; Benavidez, A.D.; DeLaRiva, A.T.; Su, Y.Q.; Hofmann, J.P.; Asahina, S.; Olivier, E.J.; Neethling, J.H.; Miller, J.T.; Datye, A.K.; et al. CO oxidation by Pd supported on CeO2(100) and CeO2(111) facets. Appl. Catal. B Environ. 2019, 243, 36–46. [Google Scholar] [CrossRef]
- Bi, F.K.; Zhang, X.D.; Xiang, S.; Wang, Y.Y. Effect of Pd loading on ZrO2 support resulting from pyrolysis of UiO-66: Application to CO oxidation. J. Colloid Interface Sci. 2020, 573, 11–20. [Google Scholar] [CrossRef]
- Peterson, E.J.; Delariva, A.T.; Lin, S.; Johnson, R.S.; Guo, H.; Miller, J.T.; Kwak, J.H.; Peden, C.H.F.; Kiefer, B.; Allard, L.F.; et al. Low-temperature carbon monoxide oxidation catalysed by regenerable atomically dispersed palladium on alumina. Nat. Commun. 2014, 5, 4885. [Google Scholar] [CrossRef]
- Sharma, A.K.; Mehara, P.; Das, P. Recent Advances in Supported Bimetallic Pd-Au Catalysts:Development and Applications in Organic Synthesis with Focused Catalytic Action Study. ACS Catal. 2022, 12, 6672–6701. [Google Scholar] [CrossRef]
- He, Y.; Luan, C.; Fang, Y.; Feng, X.; Peng, X.; Yang, G.; Tsubaki, N. Low-temperature direct conversion of methane to methanol over carbon materials supported Pd-Au nanoparticles. Catal. Today 2020, 339, 48–53. [Google Scholar] [CrossRef]
- Zhou, Q.; Luo, S.; Zhang, M.; Liao, N. Selective and efficient hydrogen separation of Pd-Au-Ag ternary alloy membrane. Int. J. Hydrogen Energy 2022, 47, 13054–13061. [Google Scholar] [CrossRef]
- Bathla, A.; Pal, B. Superior co-catalytic activity of Pd(core)@Au(shell) nanocatalyst imparted to TiO2 for the selective hydrogenation under solar radiations. Sol. Energy 2020, 205, 292–301. [Google Scholar] [CrossRef]
- Zhang, J.; Alexandrova, A.N. The Golden Crown: A Single Au Atom that Boosts the CO Oxidation Catalyzed by a Palladium Cluster on Titania Surfaces. J. Phys. Chem. Lett. 2013, 4, 2250–2255. [Google Scholar] [CrossRef]
- Wilburn, M.S.; Epling, W.S. SO2 adsorption and desorption characteristics of bimetallic Pd-Pt catalysts: Pd: Pt ratio dependency. Catal. Today 2019, 320, 11–19. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient Iterative Schemes for ab initio Total-energy Calculations Using a Plane-wave Basis Set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Norskov, J.K. Improved Adsorption Energetics within Density-functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals. Phys. Rev. B 1999, 59, 7413. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type Density Functional Constructed with a Long-range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.H.; Tao, T.; Zhao, S.Z.; Yu, Q.J.; Gao, F.Y.; Zhou, Y.S.; Tang, X.L. Promoted adsorption of methyl mercaptan by gamma-Al2O3 catalyst loaded with Cu/Mn. Environ. Technol. Innov. 2021, 21, 101349. [Google Scholar] [CrossRef]
- Lassoued, A.; Dkhil, B.; Gadri, A.; Ammar, S. Control of the shape and size of iron oxide (alpha-Fe2O3) nanoparticles synthesized through the chemical precipitation method. Results Phys. 2017, 7, 3007–3015. [Google Scholar] [CrossRef]
- Jayaseelan, C.; Ramkumar, R.; Rahuman, A.A.; Perumal, P. Green synthesis of gold nanoparticles using seed aqueous extract of Abelmoschus esculentus and its antifungal activity. Ind. Crops Prod. 2013, 45, 423–429. [Google Scholar] [CrossRef]
- Bukhtiyarov, A.V.; Prosvirin, I.P.; Panafidin, M.A.; Fedorov, A.Y.; Klyushin, A.Y.; Knop-Gericke, A.; Zubavichus, Y.V.; Bukhtiyarov, V.I. Near-Ambient Pressure XPS and MS Study of CO Oxidation over Model Pd-Au/HOPG Catalysts: The Effect of the Metal Ratio. Nanomaterials 2021, 11, 3292. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.; Luo, L.F.; Jiang, Z.Q.; Huang, W.X. Alloying Au surface with Pd reduces the intrinsic activity in catalyzing CO oxidation. Catal. Today 2017, 280, 253–258. [Google Scholar] [CrossRef]
- Bukhtiyarov, A.V.; Prosvirin, I.P.; Saraev, A.A.; Klyushin, A.Y.; Knop-Gericke, A.; Bukhtiyarov, V.I. In situ formation of the active sites in Pd-Au bimetallic nanocatalysts for CO oxidation: NAP (near ambient pressure) XPS and MS study. Faraday Discuss. 2018, 208, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Modelska, M.; Binczarski, M.J.; Kaminski, Z.; Karski, S.; Kolesinska, B.; Mierczynski, P.; Severino, C.J.; Stanishevsky, A.; Witonska, I.A. Bimetallic Pd-Au/SiO2 Catalysts for Reduction of Furfural in Water. Catalysts 2020, 10, 444. [Google Scholar] [CrossRef]
- Huang, X.Y.; Akdim, O.; Douthwaite, M.; Wang, K.; Zhao, L.; Lewis, R.J.; Pattisson, S.; Daniel, I.T.; Miedziak, P.J.; Shaw, G.; et al. Au-Pd separation enhances bimetallic catalysis of alcohol oxidation. Nature 2022, 603, 271–275. [Google Scholar] [CrossRef]
- Bukhtiyarov, A.V.; Prosvirin, I.P.; Bukhtiyarov, V.I. XPS/STM study of model bimetallic Pd-Au/HOPG catalysts. Appl. Surf. Sci. 2016, 367, 214–221. [Google Scholar] [CrossRef]
- Chenakin, S.P.; Melaet, G.; Szukiewicz, R.; Kruse, N. XPS study of the surface chemical state of a Pd/(SiO2+TiO2) catalyst after methane oxidation and SO2 treatment. J. Catal. 2014, 312, 1–11. [Google Scholar] [CrossRef]
- Venezia, A.M.; Murania, R.; Pantaleo, G.; Deganello, G. Pd and PdAu on mesoporous silica for methane oxidation: Effect of SO2. J. Catal. 2007, 251, 94–102. [Google Scholar] [CrossRef]
- Liotta, L.F.; Di Carlo, G.; Pantaleo, G.; Venezia, A.M.; Deganello, G.; Borla, E.M.; Pidria, M.F. Pd/Co3O4 catalyst for CH4 emissions abatement: Study of SO2 poisoning effect. Top. Catal. 2007, 42–43, 425–428. [Google Scholar] [CrossRef]
- Spezzati, G.; Su, Y.Q.; Hofmann, J.P.; Benavidez, A.D.; DeLaRiva, A.T.; McCabe, J.; Datye, A.K.; Hensen, E.J.M. Atomically Dispersed Pd-O Species on CeO(2)(111) as Highly Active Sites for Low-Temperature CO Oxidation. ACS Catal. 2017, 7, 6887–6891. [Google Scholar] [CrossRef] [PubMed]
- Zorn, K.; Giorgio, S.; Halwax, E.; Henry, C.R.; Gronbeck, H.; Rupprechter, G. CO Oxidation on Technological Pd-Al2O3 Catalysts: Oxidation State and Activity. J. Phys. Chem. C 2011, 115, 1103–1111. [Google Scholar] [CrossRef]
- Celorrio, V.; Quaino, P.M.; Santos, E.; Florez-Montano, J.; Humphrey, J.J.L.; Guillen-Villafuerte, O.; Plana, D.; Lazaro, M.J.; Pastor, E.; Fermin, D.J. Strain Effects on the Oxidation of CO and HCOOH on Au-Pd Core-Shell Nanoparticles. ACS Catal. 2017, 7, 1673–1680. [Google Scholar] [CrossRef]
- Tran-Thuy, T.M.; Yu, T.L.; Lin, S.D. How H2O may influence ambient CO oxidation over Au/BN. Appl. Catal. B Environ. 2022, 314, 121492. [Google Scholar] [CrossRef]
- Li, S.; Hasan, N.; Ma, H.; Li, O.L.; Lee, B.; Jia, Y.F.; Liu, C. Significantly enhanced photocatalytic activity by surface acid corrosion treatment and Au nanoparticles decoration on the surface of SnFe2O4 nano-octahedron. Sep. Purif. Technol. 2022, 299, 121650. [Google Scholar] [CrossRef]
- Li, Q.Z.; Wu, C.J.; Wang, K.; Wang, X.X.; Chen, X.; Dai, W.X.; Fu, X.Z. Comparison of the catalytic performance of Au/TiO2 prepared by in situ photodeposition and deposition precipitation methods for CO oxidation at room temperature under visible light irradiation. Catal. Sci. Technol. 2022, 12, 237–249. [Google Scholar] [CrossRef]
- Martin, N.M.; Skoglundh, M.; Smedler, G.; Raj, A.; Thompsett, D.; Velin, P.; Martinez-Casado, F.J.; Matej, Z.; Balmes, O.; Carlsson, P.A. CO Oxidation and Site Speciation for Alloyed Palladium-Platinum Model Catalysts Studied by in Situ FTIR Spectroscopy. J. Phys. Chem. C 2017, 121, 26321–26329. [Google Scholar] [CrossRef]
- Jia, S.H.; Pu, G.; Xiong, W.C.; Wang, P.C.; Gao, J.; Yuan, C. Investigation on Simultaneous Removal of SO2 and NO over a Cu-Fe/TiO2 Catalyst Using Vaporized H2O2: An Analysis on SO2 Effect. Ind. Eng. Chem. Res. 2021, 60, 13474–13484. [Google Scholar] [CrossRef]
- Shigenobu, S.; Sugiyama, T.; Hojo, H.; Einaga, H. Enhanced Benzene Oxidation of Sintered Pd/gamma-Al2O3 Catalysts by SO2 Treatment. Catal. Lett. 2022, 42, 33–37. [Google Scholar]
- Ye, Y.L.; Xie, J.L.; De, F.; Wang, X.H.; He, F.; Jin, Q.Q. Effect of acid treatment on surfaces of activated carbon supported catalysts for NO and SO2 removal. Fuller. Nanotub. Carbon Nanostruct. 2022, 30, 297–305. [Google Scholar] [CrossRef]
- Maumau, T.R.; Modibedi, R.M.; Mathe, M.K. Electro-oxidation of alcohols using carbon supported gold, palladium catalysts in alkaline media. In Proceedings of the 1st Africa Energy Materials Conference (AEMC), Pretoria, South Africa, 28–31 March 2017. [Google Scholar]
- Han, Y.F.; Wang, J.H.; Kumar, D.; Yan, Z.; Goodman, D.W. A kinetic study of vinyl acetate synthesis over Pd-based catalysts: Kinetics of vinyl acetate synthesis over Pd-Au/SiO2 and Pd/SiO2 catalysts. J. Catal. 2005, 232, 467–475. [Google Scholar] [CrossRef]
- Gao, F.; Wang, Y.L.; Goodman, D.W. Reaction Kinetics and Polarization-Modulation Infrared Reflection Absorption Spectroscopy (PM-IRAS) Investigation of CO Oxidation over Supported Pd-Au Alloy Catalysts. Phys. Chem. Chem. Phys. 2010, 114, 4036–4043. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pd 3d 3/2 | Pd 3d 5/2 | ||||||
|---|---|---|---|---|---|---|---|
| PdO | Pd-Au | Pd0 | Pd-S | PdOx/Pd | Pd0 | Pd-Au | |
| Pd-Au/FeOx/Al2O3 (Atom, %) | 9.84 | 14.95 | 13.76 | \ | 14.81 | 20.36 | 26.29 |
| Pd-Au/FeOx/Al2O3 after reacting with sulphides (Atom, %) | 9.02 | 12.01 | 8.26 | 7.07 | 11.18 | 46.11 | 6.36 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Q.; Wang, X.; Liu, Y.; Kong, W.; Ren, S.; Liang, Y.; Tang, M.; Zhou, S.; Dong, Y. The Enhancement of CO Oxidation Performance and Stability in SO2 and H2S Environment on Pd-Au/FeOX/Al2O3 Catalysts. Materials 2023, 16, 3755. https://doi.org/10.3390/ma16103755
He Q, Wang X, Liu Y, Kong W, Ren S, Liang Y, Tang M, Zhou S, Dong Y. The Enhancement of CO Oxidation Performance and Stability in SO2 and H2S Environment on Pd-Au/FeOX/Al2O3 Catalysts. Materials. 2023; 16(10):3755. https://doi.org/10.3390/ma16103755
Chicago/Turabian StyleHe, Qingrong, Xuwei Wang, Yimeng Liu, Weimin Kong, Shanshan Ren, Yun Liang, Min Tang, Shuyuan Zhou, and Yanchun Dong. 2023. "The Enhancement of CO Oxidation Performance and Stability in SO2 and H2S Environment on Pd-Au/FeOX/Al2O3 Catalysts" Materials 16, no. 10: 3755. https://doi.org/10.3390/ma16103755
APA StyleHe, Q., Wang, X., Liu, Y., Kong, W., Ren, S., Liang, Y., Tang, M., Zhou, S., & Dong, Y. (2023). The Enhancement of CO Oxidation Performance and Stability in SO2 and H2S Environment on Pd-Au/FeOX/Al2O3 Catalysts. Materials, 16(10), 3755. https://doi.org/10.3390/ma16103755