
Chemical and Physical Modification of Lignin for Green Polymeric Composite Materials




Abstract
:1. Nomenclature and Abbreviations
2. Background
- (a)
- Fragmentation or depolymerization of lignin to yield carbon materials and chemicals rich in aromatic structures [27].
- (b)
- Chemical modification by reactions of lignin hydroxyl groups with various agents.
- (c)
- Creation of new chemically active sites.
3. Types of Modifications and Modifiers
- -
- Acyl chlorides: 10-undecenoyl chloride, oleoyl chloride [35].
- -
- -
- -
4. Chemical Modification
4.1. Esterification
4.2. Alkylation and Arylation
4.3. Epoxidation
4.4. Hydroxymethylation
4.5. Copolymerization
4.6. Amination
4.7. Silylation
4.8. Other Approaches to Chemical Modification of Lignin
5. Physical Modification
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hatakeyama, H.; Hatakeyama, T. Lignin Structure, Properties, and Applications. In Biopolymers. Lignin, Proteins, Bioactive Nanocomposites; Abe, A., Dušek, K., Kobayashi, S., Eds.; Springer Science & Business Media: Berlin, Germany, 2009; Volume 232, pp. 1–63. [Google Scholar]
- Komisarz, K.; Majka, T.M.; Pielichowski, K. Chemical Transformation of Lignosulfonates to Lignosulfonamides with Improved Thermal Characteristics. Fibers 2022, 10, 20. [Google Scholar] [CrossRef]
- Komisarz, K.; Majka, T.M.; Kurczab, M.; Pielichowski, K. Synthesis and Characterization of Thermally Stable Lignosulfonamides. Molecules 2022, 27, 7231. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Meng, X.; Pu, Y.; Ragauskas, A. Recent Advances in the Application of Functionalized Lignin in Value-Added Polymeric Materials. Polymers 2020, 12, 2277. [Google Scholar] [CrossRef] [PubMed]
- Elsheikh, A.H.; Panchal, H.; Shanmugan, S.; Muthuramalingam, T.; El-Kassas, A.M.; Ramesh, B. Recent progresses in wood-plastic composites: Pre-processing treatments, manufacturing techniques, recyclability and eco-friendly assessment. Clean. Eng. Technol. 2022, 8, 100450. [Google Scholar] [CrossRef]
- Harmsen, P.; Huijgen, W.; Bermudez, L.; Bakker, R. Literature Review of Physical and Chemical Pretreatment Processes for Lignocellulosic Biomass; Wageningen University & Research: Wageningen, The Netherlands, 2010. [Google Scholar]
- Vásquez-Garay, F.; Carrillo-Varela, I.; Vidal, C.; Reyes-Contreras, P.; Faccini, M.; Mendonça, R.T. A Review on the Lignin Biopolymer and Its Integration in the Elaboration of Sustainable Materials. Sustainability 2021, 13, 2697. [Google Scholar] [CrossRef]
- Elsheikh, A.H. Bistable Morphing Composites for Energy-Harvesting Applications. Polymers 2022, 14, 1893. [Google Scholar] [CrossRef]
- Laurichesse, S.; Avérous, L. Chemical modification of lignins: Towards biobased polymers. Prog. Polym. Sci. 2014, 39, 1266–1290. [Google Scholar] [CrossRef]
- Li, Z.; Ge, Y.; Zhang, J.; Xiao, D.; Wu, Z. Chemical Modification of Lignin and Its Environmental Application. In Sustainable Polymer Composites and Nanocomposites; Springer International Publishing: Cham, Switzerland, 2019; pp. 1345–1364. [Google Scholar]
- Figueiredo, P.; Lintinen, K.; Hirvonen, J.T.; Kostiainen, M.A.; Santos, H.A. Properties and chemical modifications of lignin: Towards lignin-based nanomaterials for biomedical applications. Prog. Mater. Sci. 2018, 93, 233–269. [Google Scholar] [CrossRef]
- Li, C.; Zhao, X.; Wang, A.; Huber, G.W.; Zhang, T. Catalytic Transformation of Lignin for the Production of Chemicals and Fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef]
- Liao, J.J.; Latif, N.H.A.; Trache, D.; Brosse, N.; Hussin, M.H. Current advancement on the isolation, characterization and application of lignin. Int. J. Biol. Macromol. 2020, 162, 985–1024. [Google Scholar] [CrossRef]
- Kleinert, T.N. Organosilv Pulping and Recovery Process. U.S. Patent No. US3585104A, 29 July 1968. [Google Scholar]
- Arapova, O.V.; Chistyakov, A.V.; Tsodikov, M.V.; Moiseev, I.I. Lignin as a Renewable Resource of Hydrocarbon Products and Energy Carriers (A Review). Pet. Chem. 2020, 60, 227–243. [Google Scholar] [CrossRef]
- Domenek, S.; Louaifi, A.; Guinault, A.; Baumberger, S. Potential of Lignins as Antioxidant Additive in Active Biodegradable Packaging Materials. J. Polym. Environ. 2013, 21, 692–701. [Google Scholar] [CrossRef] [Green Version]
- Pouteau, C.; Dole, P.; Cathala, B.; Averous, L.; Boquillon, N. Antioxidant properties of lignin in polypropylene. Polym. Degrad. Stab. 2003, 81, 9–18. [Google Scholar] [CrossRef]
- de Chirico, A.; Armanini, M.; Chini, P.; Cioccolo, G.; Provasoli, F.; Audisio, G. Flame retardants for polypropylene based on lignin. Polym. Degrad. Stab. 2003, 79, 139–145. [Google Scholar] [CrossRef]
- Henn, A.; Mattinen, M.L. Chemo-enzymatically prepared lignin nanoparticles for value-added applications. World J. Microbiol. Biotechnol. 2019, 35, 125. [Google Scholar] [CrossRef] [Green Version]
- Uzun, G.; Aydemir, D. Biocomposites from polyhydroxybutyrate and bio-fillers by solvent casting method. Bull. Mater. Sci. 2017, 40, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Kovalcik, A.; Machovsky, M.; Kozakova, Z.; Koller, M. Designing packaging materials with viscoelastic and gas barrier properties by optimized processing of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) with lignin. React. Funct. Polym. 2015, 94, 25–34. [Google Scholar] [CrossRef]
- Camargo, F.A.; Innocentini-Mei, L.H.; Lemes, A.P.; Moraes, S.G.; Durán, N. Processing and characterization of composites of poly(3-hydroxybutyrate-co- hydroxyvalerate) and lignin from sugar cane bagasse. J. Compos. Mater. 2012, 46, 417–425. [Google Scholar] [CrossRef]
- Hu, L.; Stevanovic, T.; Rodrigue, D. Compatibilization of kraft lignin-polyethylene composites using unreactive compatibilizers. J. Appl. Polym. Sci. 2014, 131, 41040. [Google Scholar] [CrossRef]
- Xu, F.; Yu, J.; Tesso, T.; Dowell, F.; Wang, D. Qualitative and quantitative analysis of lignocellulosic biomass using infrared techniques: A mini-review. Appl. Energy 2013, 104, 801–809. [Google Scholar] [CrossRef]
- Park, Y.C.; Kim, J.S. Comparison of various alkaline pretreatment methods of lignocellulosic biomass. Energy 2012, 47, 31–35. [Google Scholar] [CrossRef]
- Jawaid, M.; Sapuan, S.M.; Alotman, O.Y. Green Biocomposites Manufacturing and Properties; Springer: Berlin/Heidelberg, Germany, 2017; p. 409. [Google Scholar] [CrossRef]
- Clark, J.H.; Luque, R.; Matharu, A.S. Green chemistry, biofuels, and biorefinery. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 183–207. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.K.; Chakraborty, I.; Kar, B.B. Chemically Modified Lignin—A Potential Resource Material for Composites with Better Stability. Int. J. Sci. Environ. Technol. 2015, 4, 183–189. [Google Scholar]
- Barana, D.; Orlandi, M.; Zoia, L.; Castellani, L.; Hanel, T.; Bolck, C.; Gosselink, R. Lignin Based Functional Additives for Natural Rubber. ACS Sustain. Chem. Eng. 2018, 6, 11843–11852. [Google Scholar] [CrossRef]
- Maldhure, A.V.; Ekhe, J.D. Effect of modifications of lignin on thermal, structural, and mechanical properties of polypropylene/modified lignin blends. J. Thermoplast. Compos. Mater. 2017, 30, 625–645. [Google Scholar] [CrossRef]
- Malutan, T.; Nicu, R.; Popa, V.I. Lignin modification by epoxidation. BioResources 2008, 3, 1371–1376. [Google Scholar] [CrossRef]
- Cateto, C.A.; Barreiro, M.F.; Rodrigues, A.E.; Belgacem, M.N. Optimization study of lignin oxypropylation in view of the preparation of polyurethane rigid foams. Ind. Eng. Chem. Res. 2009, 48, 2583–2589. [Google Scholar] [CrossRef]
- Brežny, R.; Paszner, L.; Micko, M.M.; Uhrín, D. The Ion-Exchanging Lignin Derivatives Prepared by Mannich Reaction with Amino Acids. Holzforschung 1988, 42, 369–373. [Google Scholar] [CrossRef]
- Kovalenkoi, E.I.; Popova, O.V.; Aleksandrov, A.A.; Galikyan, T.G. Electrochemical Modification of Lignins. Russ. J. Electrochem. 2000, 36, 706–711. [Google Scholar] [CrossRef]
- Xing, Q.; Ruch, D.; Dubois, P.; Wu, L.; Wang, W.J. Biodegradable and High-Performance Poly(butylene adipate-co-terephthalate)-Lignin UV-Blocking Films. ACS Sustain. Chem. Eng. 2017, 5, 10342–10351. [Google Scholar] [CrossRef]
- Nevárez, L.A.M.; Casarrubias, L.B.; Celzard, A.; Fierro, V.; Muñoz, V.T.; Davila, A.C.; Lubian, J.R.T.; Sánchez, G.G. Biopolymer-based nanocomposites: Effect of lignin acetylation in cellulose triacetate films. Sci. Technol. Adv. Mater. 2011, 12, 045006. [Google Scholar] [CrossRef]
- Gordobil, O.; Delucis, R.; Egüés, I.; Labidi, J. Kraft lignin as filler in PLA to improve ductility and thermal properties. Ind. Crops Prod. 2015, 72, 46–53. [Google Scholar] [CrossRef]
- Jeong, H.; Park, J.; Kim, S.; Lee, J.; Cho, J.W. Use of acetylated softwood kraft lignin as filler in synthetic polymers. Fibers Polym. 2012, 13, 1310–1318. [Google Scholar] [CrossRef]
- Dehne, L.; Babarro, C.V.; Saake, B.; Schwarz, K.U. Influence of lignin source and esterification on properties of lignin-polyethylene blends. Ind. Crops Prod. 2016, 86, 320–328. [Google Scholar] [CrossRef]
- Yeo, J.S.; Lee, J.H.; Hwang, S.H. Effects of lignin on the volume shrinkage and mechanical properties of a styrene/unsaturated polyester/lignin ternary composite system. Compos. Part B Eng. 2017, 130, 167–173. [Google Scholar] [CrossRef]
- Thunga, M.; Chen, K.; Grewell, D.; Kessler, M.R. Bio-renewable precursor fibers from lignin/polylactide blends for conversion to carbon fibers. Carbon N. Y. 2014, 68, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Wu, H.; Kessler, M.R.K. High bio-content polyurethane composites with urethane modified lignin as filler. Polymer 2015, 69, 52–57. [Google Scholar] [CrossRef]
- Guo, J.; Chen, X.; Wang, J.; He, Y.; Xie, H.; Zheng, Q. The influence of compatibility on the structure and properties of PLA/lignin biocomposites by chemical modification. Polymers 2020, 12, 56. [Google Scholar] [CrossRef] [Green Version]
- Victor, P.A.; Gonçalves, S.B.; Machado, F. Styrene/Lignin-Based Polymeric Composites Obtained Through a Sequential Mass-Suspension Polymerization Process. J. Polym. Environ. 2018, 26, 1755–1774. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Wang, C.; Stubbs, L.P.; He, C. Carboxylated Lignin as an Effective Cohardener for Enhancing Strength and Toughness of Epoxy. Macromol. Mater. Eng. 2017, 302, 1700341. [Google Scholar] [CrossRef]
- Maldhure, A.V.; Ekhe, J.D.; Deenadayalan, E. Mechanical properties of polypropylene blended with esterified and alkylated lignin. J. Appl. Polym. Sci. 2012, 125, 1701–1712. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, S.; Fang, X.; Zhou, X.; Wang, J.; Bai, F.; Peng, S. Renewable and flexible UV-blocking film from poly(butylene succinate) and lignin. Eur. Polym. J. 2019, 116, 265–274. [Google Scholar] [CrossRef]
- Sailaja, R.R.N.; Deepthi, M.V. Mechanical and thermal properties of compatibilized composites of polyethylene and esterified lignin. Mater. Des. 2010, 31, 4369–4379. [Google Scholar] [CrossRef]
- Chirila, O.; Totolin, M.; Cazacu, G.; Dobromir, M.; Vasile, C. Lignin modification with carboxylic acids and butyrolactone under cold plasma conditions. Ind. Eng. Chem. Res. 2013, 52, 13264–13271. [Google Scholar] [CrossRef]
- Gibbons, L.; Smith, M.; Quirino, R.L. Modified lignin for composite and pellet binder applications. Int. J. Exp. Comput. Biomech. 2015, 3, 200. [Google Scholar] [CrossRef]
- Costes, L.; Laoutid, F.; Aguedo, M.; Richel, A.; Brohez, S.; Delvosalle, C.; Dubois, P. Phosphorus and nitrogen derivatization as efficient route for improvement of lignin flame retardant action in PLA. Eur. Polym. J. 2016, 84, 652–667. [Google Scholar] [CrossRef]
- Xing, W.; Yuan, H.; Zhang, P.; Yang, H.; Song, L.; Hu, Y. Functionalized lignin for halogen-free flame retardant rigid polyurethane foam: Preparation, thermal stability, fire performance and mechanical properties. J. Polym. Res. 2013, 20, 234. [Google Scholar] [CrossRef]
- Yu, Y.; Fu, S.; Song, P.; Luo, X.; Jin, Y.; Lu, F.; Wu, Q.; Ye, J. Functionalized lignin by grafting phosphorus-nitrogen improves the thermal stability and flame retardancy of polypropylene. Polym. Degrad. Stab. 2012, 97, 541–546. [Google Scholar] [CrossRef]
- Yu, Y.; Song, P.; Jin, C.; Fu, S.; Zhao, L.; Wu, Q.; Ye, J. Catalytic effects of nickel (cobalt or zinc) acetates on thermal and flammability properties of polypropylene-modified lignin composites. Ind. Eng. Chem. Res. 2012, 51, 12367–12374. [Google Scholar] [CrossRef]
- Mendis, G.P.; Weiss, S.G.; Korey, M.; Boardman, C.R.; Dietenberger, M.; Youngblood, J.P.; Howarter, J.A. Phosphorylated lignin as a halogen-free flame retardant additive for epoxy composites. Green Mater. 2016, 4, 150–159. [Google Scholar] [CrossRef]
- Wang, H.; Qiu, X.; Liu, W.; Yang, D. Facile preparation of well-combined lignin-based carbon/ZnO hybrid composite with excellent photocatalytic activity. Appl. Surf. Sci. 2017, 426, 206–216. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Y.; Fu, F.; Qian, Y.; Xiao, Y.; Yang, D.; Qiu, X. Controlled preparation of lignin/titanium dioxide hybrid composite particles with excellent UV aging resistance and its high value application. Int. J. Biol. Macromol. 2020, 150, 371–379. [Google Scholar] [CrossRef]
- Kim, S.; Park, J.; Lee, J.; Roh, H.-G.; Jeong, D.; Choi, S.; Oh, S. Potential of a bio-disintegrable polymer blend using alkyl-chain-modified lignin. Fibers Polym. 2015, 16, 744–751. [Google Scholar] [CrossRef]
- Kim, S.; Park, J.; Lee, J.; Roh, H.-G.; Jeong, D.; Choi, S.; Oh, S. Effect of alkyl-chain-modified lignin in the PLA matrix. Fibers Polym. 2014, 15, 2458–2465. [Google Scholar] [CrossRef]
- Ferdosian, F.; Yuan, Z.; Anderson, M.; Xu, C.C. Chemically modified lignin through epoxidation and its thermal properties. J-FOR 2012, 2, 11–15. [Google Scholar]
- Yang, L.; Wang, X.; Cui, Y.; Tian, Y.; Chen, H.; Wang, Z. Modification of renewable resources-lignin-by three chemical methods and its applications to polyurethane foams. Polym. Adv. Technol. 2014, 25, 1089–1098. [Google Scholar] [CrossRef]
- Popa, V.I.; Cǎpraru, A.M.; Grama, S.; Mǎluţan, T. Nanoparticles based on modified lignins with biocide properties. Cellul. Chem. Technol. 2011, 45, 221–226. [Google Scholar]
- Aini, N.M.; Othman, N.; Hussin, M.; Sahakaro, K.; Hayeemasae, N. Hydroxymethylation-Modified Lignin and Its Effectiveness as a Filler in Rubber Composites. Processes 2019, 7, 315. [Google Scholar] [CrossRef] [Green Version]
- Gilca, I.A.; Ghitescu, R.E.; Puitel, A.C.; Popa, V.I. Preparation of lignin nanoparticles by chemical modification. Iran. Polym. J. 2014, 23, 355–363. [Google Scholar] [CrossRef]
- Xiao, X.; Jiang, C.; Zhang, Y.; Cai, Z.; Yu, P. Preparation and characterization of formaldehyde-modified black liquor lignin/poly (propylene carbonate) composites. Int. J. Polym. Anal. Charact. 2018, 23, 346–353. [Google Scholar] [CrossRef]
- Jiang, C.; He, H.; Yao, X.; Yu, P.; Zhou, L.; Jia, D. The aggregation structure regulation of lignin by chemical modification and its effect on the property of lignin/styrene–butadiene rubber composites. J. Appl. Polym. Sci. 2018, 135, 45759. [Google Scholar] [CrossRef]
- Jiang, C.; Zhang, Y.; Xiao, X.; Cai, Z.; Yu, P. High-performance hydroxypropyl black liquor lignin/poly (propylene carbonate) bio-composites with enhanced natural degradability. Polym. Test. 2018, 72, 348–356. [Google Scholar] [CrossRef]
- Zhou, H.; Shi, X.; Wu, W.; An, X.; Tian, Y.; Qiao, Y. Facile preparation of lignosulfonate/N-methylaniline composite and its application in efficient removal of Cr(VI) from aqueous solutions. Int. J. Biol. Macromol. 2020, 154, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Kai, D.; Zhang, K.; Jiang, L.; Wong, H.Z.; Li, Z.; Zhang, Z.; Loh, X.J. Sustainable and Antioxidant Lignin-Polyester Copolymers and Nanofibers for Potential Healthcare Applications. ACS Sustain. Chem. Eng. 2017, 5, 6016–6025. [Google Scholar] [CrossRef]
- Kai, D.; Chong, H.M.; Chow, L.P.; Jiang, L.; Lin, Q.; Zhang, K.; Zhang, H.; Zhang, Z.; Loh, X.J. Strong and biocompatible lignin /poly (3-hydroxybutyrate) composite nanofibers. Compos. Sci. Technol. 2018, 158, 26–33. [Google Scholar] [CrossRef]
- Chung, Y.-L.; Olsson, J.V.; Li, R.J.; Frank, C.W.; Waymouth, R.M.; Billington, S.L.; Sattely, E.S. A renewable lignin-lactide copolymer and application in biobased composites. ACS Sustain. Chem. Eng. 2013, 1, 1231–1238. [Google Scholar] [CrossRef]
- Kai, D.; Zhang, K.; Liow, S.S.; Loh, X.J. New Dual Functional PHB-Grafted Lignin Copolymer: Synthesis, Mechanical Properties, and Biocompatibility Studies. ACS Appl. Bio Mater. 2019, 2, 127–134. [Google Scholar] [CrossRef]
- Du, X.; Li, J.; Lindström, M.E. Modification of industrial softwood kraft lignin using Mannich reaction with and without phenolation pretreatment. Ind. Crops Prod. 2014, 52, 729–735. [Google Scholar] [CrossRef]
- Ge, Y.; Song, Q.; Li, Z. A Mannich base biosorbent derived from alkaline lignin for lead removal from aqueous solution. J. Ind. Eng. Chem. 2015, 23, 228–234. [Google Scholar] [CrossRef]
- Qin, L.; Ge, Y.; Deng, B.; Li, Z. Poly (ethylene imine) anchored lignin composite for heavy metals capturing in water. J. Taiwan Inst. Chem. Eng. 2017, 71, 84–90. [Google Scholar] [CrossRef]
- Teng, X.; Xu, H.; Song, W.; Shi, J.; Xin, J.; Hiscox, W.C.; Zhang, J. Preparation and Properties of Hydrogels Based on PEGylated Lignosulfonate Amine. ACS Omega 2017, 2, 251–259. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, F.; Zhang, H.; Wang, J. Preparation of a Polyhydric Aminated Lignin and Its Use in the Preparation of Polyurethane Film. J. Wood Chem. Technol. 2017, 37, 323–333. [Google Scholar] [CrossRef]
- Liu, J.; Liu, H.F.; Deng, L.; Liao, B.; Guo, Q.X. Improving aging resistance and mechanical properties of waterborne polyurethanes modified by lignin amines. J. Appl. Polym. Sci. 2013, 130, 1736–1742. [Google Scholar] [CrossRef]
- Nikafshar, S.; Fang, Z.; Nejad, M. Development of a Novel Curing Accelerator-Blowing Agent for Formulating Epoxy Rigid Foam Containing Aminated-Lignin. Ind. Eng. Chem. Res. 2020, 59, 15146–15154. [Google Scholar] [CrossRef]
- Zhang, R.; Xiao, X.; Tai, Q.; Huang, H.; Hu, Y. Modification of lignin and its application as char agent in intumescent flame-retardant poly(lactic acid). Polym. Eng. Sci. 2012, 52, 2620–2626. [Google Scholar] [CrossRef]
- Musilová, L.; Mráček, A.; Kovalcik, A.; Smolka, P.; Minařík, A.; Humpolíček, P.; Vícha, R.; Ponížil, P. Hyaluronan hydrogels modified by glycinated Kraft lignin: Morphology, swelling, viscoelastic properties and biocompatibility. Carbohydr. Polym. 2017, 181, 394–403. [Google Scholar] [CrossRef]
- Bula, K.; Klapiszewski, Ł.; Jesionowski, T. A novel functional silica/lignin hybrid material as a potential bio-based polypropylene filler. Polym. Compos. 2015, 36, 913–922. [Google Scholar] [CrossRef]
- Klapiszewski, Ł.; Pawlak, F.; Tomaszewska, J.; Jesionowski, T. Preparation and characterization of novel pvc/silica-lignin composites. Polymers 2015, 7, 1767–1788. [Google Scholar] [CrossRef] [Green Version]
- Borysiak, S.; Klapiszewski, Ł.; Bula, K.; Jesionowski, T. Nucleation ability of advanced functional silica/lignin hybrid fillers in polypropylene composites. J. Therm. Anal. Calorim. 2016, 126, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Yeo, J.S.; Seong, D.W.; Hwang, S.H. Chemical surface modification of lignin particle and its application as filler in the polypropylene composites. J. Ind. Eng. Chem. 2015, 31, 80–85. [Google Scholar] [CrossRef]
- Buono, P.; Duval, A.; Verge, P.; Averous, L.; Habibi, Y. New Insights on the Chemical Modification of Lignin: Acetylation versus Silylation. ACS Sustain. Chem. Eng. 2016, 4, 5212–5222. [Google Scholar] [CrossRef]
- Song, Y.; Zong, X.; Wang, N.; Yan, N.; Shan, X.; Li, J. Preparation of γ -divinyl-3-aminopropyltriethoxysilane modified lignin and its application in flame retardant poly(lactic acid). Materials 2018, 11, 1505. [Google Scholar] [CrossRef] [PubMed]
- Ferry, L.; Dorez, G.; Taguet, A.; Otazaghine, B.; Lopez-Cuesta, J.M. Chemical modification of lignin by phosphorus molecules to improve the fire behavior of polybutylene succinate. Polym. Degrad. Stab. 2015, 113, 135–143. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Y.; Yu, J.; Li, S.; Wang, J.; Wang, C.; Chu, F. Integration of lignin and acrylic monomers towards grafted copolymers by free radical polymerization. Int. J. Biol. Macromol. 2014, 67, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Zong, E.; Liu, X.; Liu, L.; Wang, J.; Song, P.; Ma, Z.; Ding, J.; Fu, S. Graft Polymerization of Acrylic Monomers onto Lignin with CaCl2-H2O2 as Initiator: Preparation, Mechanism, Characterization, and Application in Poly(lactic acid). ACS Sustain. Chem. Eng. 2018, 6, 337–348. [Google Scholar] [CrossRef]
- Liu, L.; Qian, M.; Song, P.; Huang, G.; Yu, Y.; Fu, S. Fabrication of Green Lignin-based Flame Retardants for Enhancing the Thermal and Fire Retardancy Properties of Polypropylene/Wood Composites. ACS Sustain. Chem. Eng. 2016, 4, 2422–2431. [Google Scholar] [CrossRef]
- Liu, L.; Huang, G.; Song, P.; Yu, Y.; Fu, S. Converting industrial alkali lignin to biobased functional additives for improving fire behavior and smoke suppression of polybutylene succinate. ACS Sustain. Chem. Eng. 2016, 4, 4732–4742. [Google Scholar] [CrossRef]
- Hoffmann, A.; Nong, J.P.; Porzel, A.; Bremer, M.; Fischer, S. Modification of Lignoboost Kraft Lignin from softwoods with dihydroxybenzenes. React. Funct. Polym. 2019, 142, 112–118. [Google Scholar] [CrossRef]
- Yu, S.; Wu, S.; Liu, Y.; Li, L.; Ge, X. Highly effective esterification of lignin to produce a pharmaceutical intermediate using novel silica mesoporous molecular sieves as catalysts. J. Taiwan Inst. Chem. Eng. 2020, 109, 26–34. [Google Scholar] [CrossRef]
- Thielemans, W.; Wool, R.P. Lignin esters for use in unsaturated thermosets: Lignin modification and solubility modeling. Biomacromolecules 2005, 6, 1895–1905. [Google Scholar] [CrossRef]
- Liu, Y.; Cheng, G.; Baráth, E.; Shi, H.; Lercher, J.A. Alkylation of lignin-derived aromatic oxygenates with cyclic alcohols on acidic zeolites. Appl. Catal. B Environ. 2021, 281, 119424. [Google Scholar] [CrossRef]
- Khalil, H.P.S.A.; Marliana, M.M.; Issam, A.M.; Bakare, I.O. Exploring isolated lignin material from oil palm biomass waste in green composites. Mater. Des. 2011, 32, 2604–2610. [Google Scholar] [CrossRef]
- Dong, C.; Meng, X.; Leu, S.-Y.; Xu, L.; Wu, Z.; Cravotto, G.; Fang, Z. Enhancing α-etherification of lignin in Eucalyptus diol pretreatment to improve lignin monomer production. Ind. Crops Prod. 2022, 185, 115130. [Google Scholar] [CrossRef]
- Nair, V.; Panigrahy, A.; Vinu, R. Development of novel chitosan-lignin composites for adsorption of dyes and metal ions from wastewater. Chem. Eng. J. 2014, 254, 491–502. [Google Scholar] [CrossRef]
- Panesar, S.S.; Jacob, S.; Misra, M.; Mohanty, A.K. Functionalization of lignin: Fundamental studies on aqueous graft copolymerization with vinyl acetate. Ind. Crops Prod. 2013, 46, 191–196. [Google Scholar] [CrossRef]
- Matsushita, Y.; Yasuda, S. Reactivity of a condensed-type lignin model compound in the Mannich reaction and preparation of cationic surfactant from sulfuric acid lignin. J. Wood Sci. 2003, 49, 166–171. [Google Scholar] [CrossRef]
- Yue, X.; Chen, F.; Zhou, X. Improved Interfacial Bonding of Pvc/Wood-Flour Composites By Lignin Amine Modification. BioResources 2011, 6, 2022–2034. [Google Scholar] [CrossRef]
- Matsushita, Y.; Imai, M.; Tamura, T.; Fukushima, K. Preparation and evaluation of a dispersant for gypsum paste from acid hydrolysis lignin. J. Appl. Polym. Sci. 2005, 98, 2508–2513. [Google Scholar] [CrossRef]
- Gřundělová, L.; Gregorova, A.; Mráček, A.; Vícha, R.; Smolka, P.; Minařík, A. Viscoelastic and mechanical properties of hyaluronan films and hydrogels modified by carbodiimide. Carbohydr. Polym. 2015, 119, 142–148. [Google Scholar] [CrossRef]
- Dizhbite, T.; Zakis, G.; Kizima, A.; Lazareva, E.; Rossinskaya, G.; Jurkjane, V.; Telysheva, G.; Viesturs, U. Lignin a useful bioresource for the production of sorp-tion-active materials. Bioresour. Technol. 1999, 67, 221–228. [Google Scholar] [CrossRef]
- Telysheva, G.; Rossinskaya, G. Characterisation of lignin from acid hydroly-sis of wood. In Proceedings of the Third International Forum on Sulfur-Free Lignin, Fribourg, Switzerland, 29 February–1 March 1996; pp. 25–30. [Google Scholar]
- Glasser, W.G.; Gratzl, J.S.; Collins, J.J.; Forss, K.; McCarthy, J.L. Preparation and Characterization of Acetyl Lignin Sulfonate Methyl Esters. Macromolecules 1975, 8, 565–573. [Google Scholar] [CrossRef]
- Oiivares, M.; Guzmfin, J.A.; Natho, A.; Saavedra, A. Kraft lignin utilization in adhesives. Wood Sci. Technol. 1988, 22, 157–165. [Google Scholar] [CrossRef]
- Dolenko, A.J.; Clarke, M.R. Resin binders from kraft lignin. For. Prod. J. 1978, 28, 41–46. [Google Scholar]
- Rao, N.R.; Rao, T.V.; Reddy, S.V.S.R.; Rao, B.S. The effect of gamma irradiation on physical, thermal and antioxidant properties of kraft lignin. J. Radiat. Res. Appl. Sci. 2015, 8, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Liu, W.; Shahabadi, S.I.S.; Xu, J.; Lu, X. Sheet-Like Lignin Particles as Multifunctional Fillers in Polypropylene. ACS Sustain. Chem. Eng. 2016, 4, 4997–5004. [Google Scholar] [CrossRef]
- El-Zawawy, W.K.; Ibrahim, M.M.; Belgacem, M.N.; Dufresneb, A. Characterization of the effects of lignin and lignin complex particles as filler on a polystyrene film. Mater. Chem. Phys. 2011, 131, 348–357. [Google Scholar] [CrossRef]
- Souza, J.R.; Araujo, J.R.; Archanjo, B.S.; Simão, R.A. Cross-linked lignin coatings produced by UV light and SF6 plasma treatments. Prog. Org. Coat. 2019, 128, 82–89. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Explanation |
|---|---|
| CHEMICALS | |
| H2SO4 | sulfuric acid |
| THF | tetrahydrofuran |
| NaOH | sodium hydroxide |
| DMF | dimethylformamide |
| TEC | triethyl citrate |
| ATBC | acetyl tributyl citrate |
| TCP | tricresyl phosphate |
| BMA | n-butyl methacrylate |
| DVB | divinylbenzene |
| HCl | hydrochloric acid |
| NH4OH | ammonia aqueous solution |
| KOH | potassium hydroxide |
| HFP | 1,1,1,3,3,3,-hexafluoro-2-propanol |
| LBL | LignoBoost Lignin |
| HMAP | 4-hydroxy-3-methoxyacetophenone |
| DETA | diethylenetriamine |
| APP | ammonium polyphosphate |
| EDC | N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride |
| TDMSCl | tert-butyldimethylsilyl chloride |
| DVAPTS | γ-divinyl-3-aminopropyltriethoxysilane |
| PEI | poly(ethylene imine) |
| DEP | Diethyl phosphite |
| WP | wood powder |
| AAL | acetic acid lignin |
| BBL | biobutanol lignin |
| P2O5 | phosphorus pentoxide |
| V | vanilin |
| POLYMERS | |
| LDPE | low-density polyethylene |
| HDPE | high-density polyethylene |
| PP | polypropylene |
| PS | polystyrene |
| PET | poly(ethylene terephthalate) |
| PU | polyurethanes |
| NR | natural rubber |
| BR | butadiene rubber |
| PBAT | poly(butylene adipate-co-terephthalate) |
| PBS | poly(butylene succinate) |
| PHA | polyhydroxyalkanoates |
| PLA | polylactide |
| PAN | polyacrylonitrile |
| PPC | poly(propylene carbonate) |
| PCLLA | poly(ε-caprolactone-co-lactide) |
| PCL | polycaprolactone |
| PLLA | poly(L-lactic acid) |
| PHB | poly(3-hydroxybutyrate) |
| WPU | waterborne polyurethanes |
| PVC | poly(vinyl chloride) |
| PMMA | poly(methyl methacrylate) |
| PBZMA | poly(benzyl methacrylate) |
| PEMA | poly(ethyl methacrylate) |
| MATERIAL SAMPLES | |
| CTA | cellulose triacetate |
| MALig | maleic anhydride-modified kraft lignin |
| CELig | dichloroethane-modified kraft lignin |
| CMLig | dichloromethane-modified kraft lignin |
| CBLig | chlorobenzene-modified kraft lignin |
| LCC | lignin-carbohydrate complex |
| PUFs | polyurethane foams |
| LPB | lignin-PHB copolymer |
| LPHC | random lignin-PHB-PCL copolymer |
| LPH+C | block copolymer of lignin with PHB and PCL |
| LPC+H | block copolymer of lignin with PCL and PHB |
| P-LBL | phenolated LignoBoost Lignin |
| PLCD | poly(ethylene imine)-anchored lignin composite |
| UM-Lig | urea-modified lignin |
| CLig | lignin modified with DVAPTS |
| PNZn-lignin | product of reaction of lignin with PEI, DEP, and zinc acetate |
| AAL-g-PBMA | acetic acid lignin grafted with side chains of poly(butyl methacrylate) |
| AAL-g-PMMA | acetic acid lignin grafted with side chains of poly(methyl methacrylate) |
| BBL-g-PBZMA | biobutanol lignin grafted with side chains of poly(benzyl methacrylate) |
| BBL-g-PEMA | biobutanol lignin grafted with side chains of poly(ethyl methacrylate) |
| LBL_ct | LignoBoost lignin modified with catechol |
| LBL_rs | LignoBoost lignin modified with resorcinol |
| LBL_hq | LignoBoost lignin modified with hydroquinone |
| CHARACTERIZATION METHODS | |
| SEM | Scanning electron microscopy |
| XPS | X-ray photoelectron spectroscopy |
| FTIR | Fourier-transform infrared spectroscopy |
| 1H-NMR | Proton nuclear magnetic resonance |
| DSC | Differential scanning calorimetry |
| TGA | Thermogravimetric analysis |
| PROPERTIES | |
| LOI | Limiting oxygen index |
| PHRR | Peak heat release rate |
| THR | Total heat release |
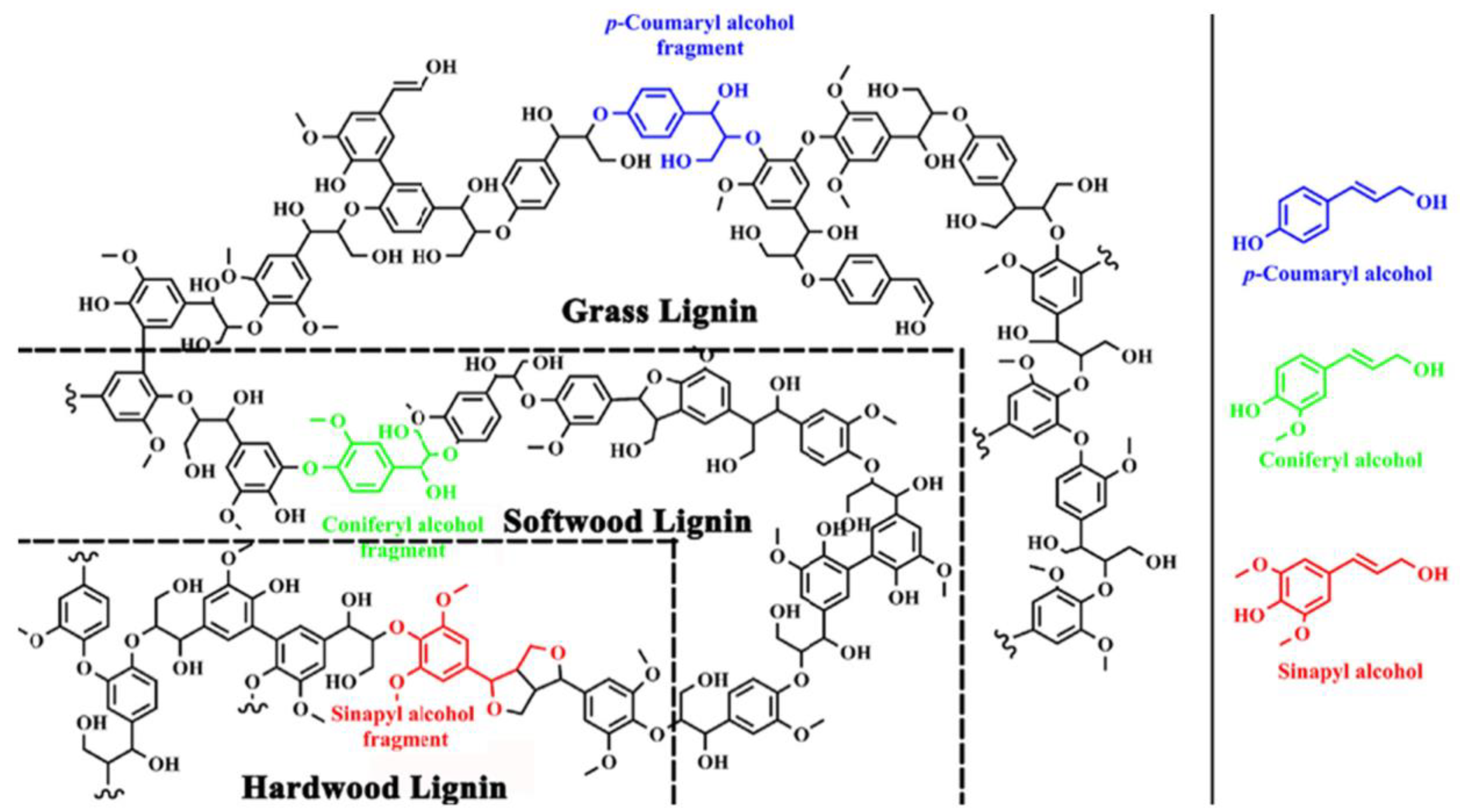
| Monolignol | Conifer Wood | Broadleaf Wood | Grass |
|---|---|---|---|
| Sinapyl alcohol | 0–1% | 50–75% | 25–50% |
| Coniferyl alcohol | 90–95% | 25–50% | 25–50% |
| p-Coumaryl alcohol | 0.5–3.4% | trace amounts | 10–25% |
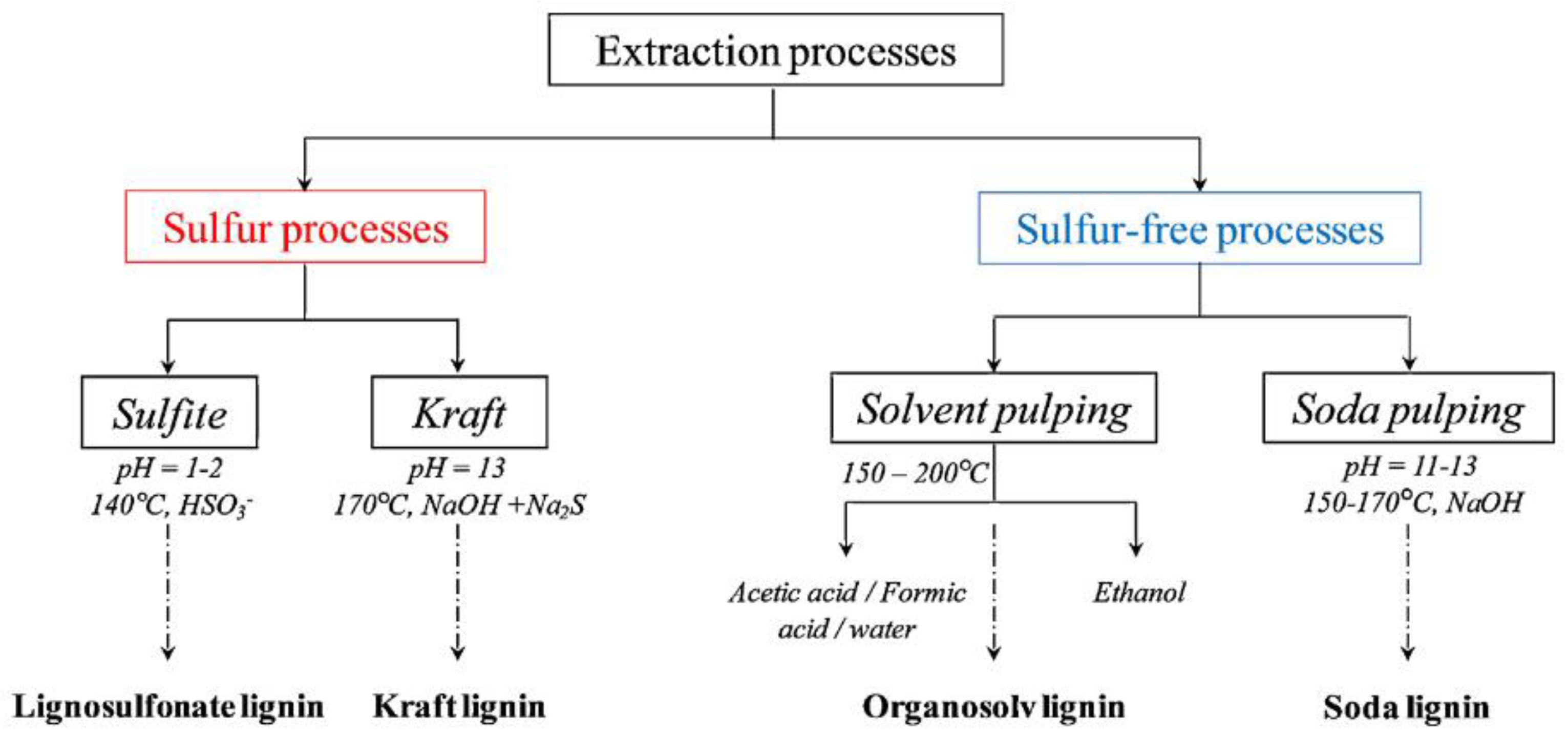
| Lignin Properties | Sulfur Lignins | Sulfur-Free Lignins | ||
|---|---|---|---|---|
| Kraft | Lignosulfonate | Soda | Organosolv | |
| Raw material | Softwood Hardwood | Softwood Hardwood | Annual plants | Softwood Hardwood Annual plants |
| Solubility | Alkali Organic solvents | Water | Alkali | Wide range of organic solvents |
| Number-average molar mass (Mn g·mol−1) | 1000–3000 | 15,000–50,000 | 800–3000 | 500–5000 |
| Dispersity | 2.5–3.5 | 6–8 | 2.5–3.5 | 1.5–2.5 |
| Tg (°C) | 140–150 | 130 | 140 | 90–110 |
| Type of Modification | Chemical Reaction | Chemical Agent |
|---|---|---|
| Chemical modification | Esterification | Acyl chlorides |
| Carboxylic anhydrides | ||
| Carboxylic acids | ||
| Lactones | ||
| Alkylation and arylation | Chlorinated hydrocarbons | |
| Heterocyclic hydrocarbons | ||
| Carboxylic acids | ||
| Epoxidation | Epichlorohydrin derivatives | |
| Hydroxymethylation | Aldehydes | |
| Cyclic ethers | ||
| Copolymerization | Anilines | |
| Lactides | ||
| Lactones | ||
| Amination | Amine compounds | |
| Silylation | Silica-containing agents | |
| Methylolation | Aldehydes | |
| Demethylation | Dichromate salt with aldehydes or organic acids | |
| Sulfonation | Aqueous solution of sulfur dioxide | |
| Alkoxylation | Cyclic ethers | |
| Hydrolysis | Hydrochloric acids, chlorosulfonic acid | |
| Other | Phosphorous compounds | |
| Acrylic compounds | ||
| Ketones | ||
| Benzenediols | ||
| Physical modification | - | Freeze-drying |
| UV irradiation | ||
| Gamma irradiation | ||
| Plasma treatment | ||
| Ultrasonic homogenization and sorption of compounds onto the surface |
| Type of Lignin | Modifying Agents | Conditions | Properties after Modification |
|---|---|---|---|
| Soda lignin | 10-undecenoyl chloride, oleoyl chloride | 65 °C, 46 h, no additional solvents or catalysts |
|
| Kraft lignin | Acetic anhydride, propionic anhydride | 85 °C |
|
| Soda lignin | Butyric anhydride | 50 °C, overnight reaction, catalyst: 1-methylimidazole |
|
| Kraft lignin | H2SO4, γ-valerolactone, acetyl ketene, butyric anhydride | γ-valerolactone: solution of lignin in γ-valerolactone stirred at 140 °C for 5 h, after addition of acetyl ketene stirred at 90 °C for 1 h butyrated lignin: stirred at 120 °C for 24 h, catalyst: 1-methylimidazole |
|
| Organosolv lignin | Oleic acid, lactic acid, butyric acid, butyrolactone | Lignin powder impregnated with 5 wt.% reagent solution, followed by cold plasma modification at 500 Hz, 50 W for 60 min |
|
| Kraft lignin | Succinic anhydride | Dissolution of lignin in pyridine under sonication, followed by addition of succinic anhydride and stirring overnight at 70 °C |
|
| Kraft lignin | Maleic anhydride, dichloroethane | Maleic anhydride: lignin was added in small portions at 100 °C to molten maleic anhydride, then the reaction mixture was placed in a microwave oven (2.45 GHz) for 20 min Dichloroethane: lignin was mixed with an excess of dichloroethane, catalyst: anhydrous aluminum chloride, and refluxed for 20 min in a modified microwave oven |
|
| Alkali (kraft) lignin | Maleic anhydride | After dissolution of lignin in 20 wt.% aqueous NaOH solution and 1 h of stirring, maleic anhydride was added and the reaction continued for 4 h at 70 °C |
|
| Alkali (kraft) lignin, dealkali lignin | Tung oil, acrylic acid, n-butyl methacrylate | Tung oil: 50–60 °C, 2 h, mechanical stirring, alkaline conditions Acrylic acid: reagents refluxed for 24 h, acidic conditions n-butyl methacrylate: 50 °C, 2 h, stirring, alkaline conditions |
|
| Kraft lignin | Phthalic anhydride | 120 °C, 3 h, continuous stirring, in presence of pyridine |
|
| Type of Lignin | Modifying Agents | Conditions | Properties after Modification |
|---|---|---|---|
| Kraft lignin | Dichloromethane, chlorobenzene | Dichloromethane: lignin and dichloromethane refluxed for 1 h Chlorobenzene: lignin mixed with chlorobenzene, 0.5 h in room temperature followed by reflux for 2 h at boiling point Both reaction used anhydrous aluminum chloride as a catalyst |
|
| Kraft lignin | γ-butyrolactone, tetrahydrofuran | γ-butyrolactone: 200 °C, 1 h, 1 wt.% of H2SO4 as a catalyst, followed by another 1 h at 250 °C THF: 30 min at 50 °C, then 30 min at 100 °C and 1 h at 150 °C, 1 wt.% of H2SO4 as a catalyst |
|
| Kraft lignin | Lactic acid, tetrahydrofuran | Lactic acid: low molecular weight PLA obtained by condensation was combined with lignin and 2 wt.% of lactic acid (catalyst) and stirred for 2 h at 180 °C THF: 30 min at 100 °C, then 2 h at 150 °C, 1 wt.% of H2SO4 as a catalyst |
|
| Type of Lignin | Modifying Agents | Conditions | Properties after Modification |
|---|---|---|---|
| Organosolv lignin | Epichlorohydrin | Depolymerized lignin was reacted with epichlorohydrin under alkaline conditions for 3 or 5 h, at 50, 70, and 90 °C, with varying epichlorohydrin/lignin molar ratios: 1, 2, 4, and 10 |
|
| Alkali (kraft) lignin, hydroxy-methylated kraft lignin | Epichlorohydrin | Epoxidation: 50, 70, or 90 °C, and 3, 5, or 7 h; lignin:NaOH (w/w) ratio, 1:3 or 1:6, lignin:epichlorohydrin (w/w) ratio, 1:10 | |
| Alkali (kraft) lignin | Epichlorohydrin | After dissolution of lignin in the mixture of 5 wt.% NaOH and 37% formaldehyde solution, the solution was stirred at 80 °C for 2 h, followed by the addition of epoxy chloropropane, and then stirred for 4 h at 80 °C |
| Type of Lignin | Modifying Agents | Conditions | Properties after Modification |
|---|---|---|---|
| Alkali (kraft) lignin | Formaldehyde | After dissolution of lignin in NaOH solution, formaldehyde (2.5:1 molar ratio of formaldehyde:lignin) was added, followed by heating to 50 °C and adjusting pH with HCl |
|
| Alkali (kraft) lignin | Formaldehyde | After dissolution of lignin in the mixture of distilled water, 5 wt.% of NaOH, and a 37% formaldehyde solution, the solution was stirred at 80 °C for 4 h, followed by the pH adjustment with HCl | |
| Kraft lignin | Formaldehyde | Mixture of lignin with distilled water stirred for 2 h at room temperature, followed by the addition of NaOH and NH4OH as a catalyst and shaking of the whole mixture for 2 h. After the introduction of 37 wt.% of formaldehyde solution, the reaction was carried out for 4 h at 85 °C and then ended by pH adjustment with HCl |
|
| Kraft lignin | Formaldehyde | Mixture of lignin with distilled water stirred for 2 h at room temperature, followed by heating the mixture to a set temperature (50, 72.5, 95 °C) and addition of NaOH and NH4OH as a catalyst, and stirring the mixture for 2 h. After the introduction of the 37 wt.% formaldehyde solution (lignin:aldehyde ratio between 1:2 and 2:1), the reaction was carried out for another 2 h and ended by pH adjustment with HCl |
|
| Kraft lignin | Formaldehyde | Lignin dispersion in deionized water with pH adjusted to 12 by the addition of NaOH was heated to 90 °C, followed by the dropwise addition of formaldehyde, and reacted for 3 h | |
| Kraft lignin | Propylene oxide | Lignin was dispersed in deionized water and the pH was adjusted to 12 by the addition of NaOH. Then, propylene oxide was added dropwise and the reaction continued at 30 °C for 10 h |
|
| Kraft lignin, soda lignin, Organosolv lignin | Propylene oxide | Set amounts of lignin/propylene oxide (w/v)—10/90, 20/80, 30/70, and 40/60—and KOH as a catalyst were placed in a reactor and heated under stirring to 160 °C |
| Type of Lignin | Modifying Agents | Conditions | Properties after Modification |
|---|---|---|---|
| Alkali (kraft) lignin | ε-caprolactone, L-lactide | Lignin, ε-caprolactone, and L-lactide with set mass ratios (2:2.4:5.6, 2:4:4, 2:5.6:2.4) and 0.5 wt.% (of monomer) of tin(II) 2-ethylhexanoate as a catalyst were placed in a flask and stirred at 130 °C for 24 h, nitrogen atmosphere |
|
| Kraft lignin | L-lactide | Lignin and L-lactide in set mass ratios (10/90, 20/80, 30/70, 40/60, 50/50) were placed in a reactor and reacted for 3.5 h at 130 °C in the presence of triazabicyclodecene as a catalyst. Reaction ended by the addition of acetic acid solution in dichloromethane |
|
| Alkali (kraft) lignin | β-butyrolactone, ε-caprolactone | LPB: lignin and β-butyrolactone (weight ratio 2:8) and 5 wt.% (of monomer) of tin(II) 2-ethylhexanoate as a catalyst were placed in a flask and stirred at 350 rpm, at 130 °C for 4 h, nitrogen atmosphere LPHC: lignin reacted with both β-butyrolactone and ε-caprolactone (weight ratio 2:4:4) under analogous conditions for 8 h LPH+C/LPC+H: lignin reacted first with β-butyrolactone or ε-caprolactone (weight ratio 2:4:4) for 4 h, followed by the addition of the latter monomer and continuation for another 4 h |
|
| Alkali (kraft) lignin | β-butyrolactone | Lignin and β-butyrolactone (weight ratio 2:8) as well as tin(II) 2-ethylhexanoate (catalyst) were reacted at 130 °C for 24 h, under nitrogen atmosphere |
|
| Type of Lignin | Modifying Agents | Conditions | Properties after Modification |
|---|---|---|---|
| Kraft lignin, alkali lignin | DETA, formaldehyde | Lignin and DETA were dissolved in distilled water, and after the adjustment of pH to alkaline, formaldehyde was added at room temperature while stirring and reaction continued after heating to 50 °C for 4 h |
|
| Alkaline lignin | Urea, formaldehyde | Lignin dissolved in NaOH aqueous solution was mixed with urea and formaldehyde, then refluxed at 70 °C for 10 h under mechanical stirring |
|
| Kraft lignin | Glycine, sodium acetate trihydrate, formaldehyde | Lignin was mixed with glycine and sodium acetate trihydrate solution in acetic acid, then formaldehyde was added at 50 °C |
|
| Type of Lignin | Modifying Agents | Conditions | Properties after Modification |
|---|---|---|---|
| Kraft lignin | Silica | Reagents were combined via grinding in a planetary ball mill for 12 h with intervals of 15 min, after which the direction of rotation was changed. Every 2 h, the mill was switched off for 5 min to avoid overheating |
|
| Kraft lignin | Silica | Reagents were combined using a grinder mortar for 2 h, with a 2 min break after each 30 min of grinding to avoid overheating |
|
| Soda lignin | Tert-butyldimethylsilyl chloride | TDMSCl and imidazole were added to lignin dissolved in DMF and the reaction was carried out at room temperature for up to 18 h |
|
| Soda lignin | γ-divinyl-3-aminopropyl-triethoxysilane | After dissolution of DVAPTS in a 5:95 (v/v) mixture of water and ethanol, it was combined with soda lignin and stirred |
|
| Type of Lignin | Conditions | Properties after Modification |
|---|---|---|
| Alkali lignin | Colloidal solutions of lignin (50 and 100 mg/mL) in deionized water were dispersed in an ultrasonic bath, frozen with liquid nitrogen, and freeze-dried |
|
| Alkali lignin, vanilin | Both alkali lignin and vanillin underwent sorption of metal cations—Fe3+, Ni2+, and Co2+—to form metal surface complexes |
|
| Soda lignin | Using the spin-coating method, thin films from dispersions of soda lignin in an acetone:water mixture (9:1) were prepared. Substrates were divided into: untreated control group, SF6-plasma treated group (15 and 30 min of exposure), and UV-light irradiated group (15 and 30 min of exposure) |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komisarz, K.; Majka, T.M.; Pielichowski, K. Chemical and Physical Modification of Lignin for Green Polymeric Composite Materials. Materials 2023, 16, 16. https://doi.org/10.3390/ma16010016
Komisarz K, Majka TM, Pielichowski K. Chemical and Physical Modification of Lignin for Green Polymeric Composite Materials. Materials. 2023; 16(1):16. https://doi.org/10.3390/ma16010016
Chicago/Turabian StyleKomisarz, Karolina, Tomasz M. Majka, and Krzysztof Pielichowski. 2023. "Chemical and Physical Modification of Lignin for Green Polymeric Composite Materials" Materials 16, no. 1: 16. https://doi.org/10.3390/ma16010016
APA StyleKomisarz, K., Majka, T. M., & Pielichowski, K. (2023). Chemical and Physical Modification of Lignin for Green Polymeric Composite Materials. Materials, 16(1), 16. https://doi.org/10.3390/ma16010016