
GIPAW Pseudopotentials of d Elements for Solid-State NMR



Abstract
:1. Introduction
2. Theoretical Background
3. Method and Computational Details
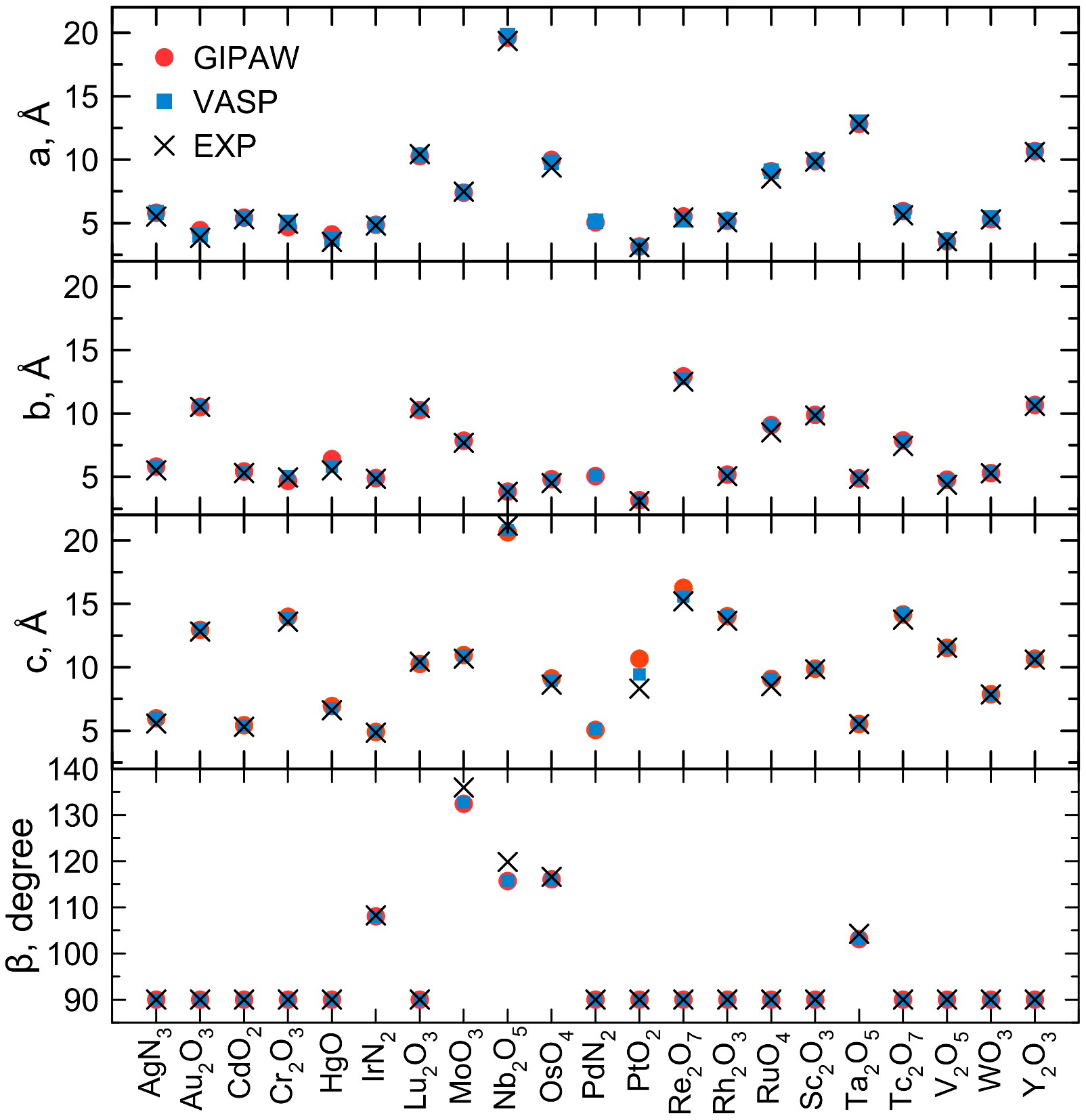
4. Results and Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ditchfield, R. Molecular Orbital Theory of Magnetic Shielding and Magnetic Susceptibility. J. Chem. Phys. 1972, 56, 5688–5691. [Google Scholar] [CrossRef]
- Pickard, C.J.; Mauri, F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B 2001, 63, 245101. [Google Scholar] [CrossRef] [Green Version]
- Joyce, S.A.; Yates, J.R.; Pickard, C.J.; Mauri, F. A first principles theory of nuclear magnetic resonance J-coupling in solid-state systems. J. Chem. Phys. 2007, 127, 204107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charpentier, T. The PAW/GIPAW approach for computing NMR parameters: A new dimension added to NMR study of solids. Solid State Nucl. Magn. Reson. 2011, 40, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.R. DFT-GIPAW 27Al NMR Simulations for Intermetallics: Accuracy Issues and Magnetic Screening Mechanisms. J. Phys. Chem. C 2019, 123, 9371–9381. [Google Scholar] [CrossRef]
- Moudrakovski, I.; Lang, S.; Patchkovskii, S.; Ripmeester, J. High Field 33S Solid State NMR and First-Principles Calculations in Potassium Sulfates. J. Phys. Chem. A 2010, 114, 309–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, E.M.J.; Yartsev, A.; Rensmo, H.; Sundström, V. Photocurrent Spectra and Fast Kinetic Studies of P3HT/PCBM Mixed with a Dye for Photoconversion in the Near-IR Region. J. Phys. Chem. C 2009, 113, 3014–3020. [Google Scholar] [CrossRef]
- Widdifield, C.M.; Bryce, D.L. Solid-State 79/81Br NMR and Gauge-Including Projector-Augmented Wave Study of Structure, Symmetry, and Hydration State in Alkaline Earth Metal Bromides. J. Phys. Chem. A 2010, 114, 2102–2116. [Google Scholar] [CrossRef]
- Lister, S.E.; Soleilhavoup, A.; Withers, R.L.; Hodgkinson, P.; Evans, J.S.O. Structures and Phase Transitions in (MoO2)2P2O7. Inorg. Chem. 2010, 49, 2290–2301. [Google Scholar] [CrossRef]
- Sutrisno, A.; Terskikh, V.V.; Huang, Y. A natural abundance 33S solid-state NMR study of layered transition metaldisulfides at ultrahigh magnetic field. Chem. Commun. 2009, 2, 186–188. [Google Scholar] [CrossRef] [Green Version]
- Griffin, J.M.; Wimperis, S.; Berry, A.J.; Pickard, C.J.; Ashbrook, S.E. Solid-State 17O NMR Spectroscopy of Hydrous Magnesium Silicates: Evidence for Proton Dynamics. J. Phys. Chem. C 2009, 113, 465–471. [Google Scholar] [CrossRef]
- Choi, M.; Matsunaga, K.; Oba, F.; Tanaka, I. 27Al NMR Chemical Shifts in Oxide Crystals: A First-Principles Study. J. Phys. Chem. C 2009, 113, 3869–3873. [Google Scholar] [CrossRef]
- Dumez, J.N.; Pickard, C.J. Calculation of NMR chemical shifts in organic solids: Accounting for motional effects. J. Chem. Phys. 2009, 130, 104701. [Google Scholar] [CrossRef] [PubMed]
- Hung, I.; Uldry, A.C.; Becker-Baldus, J.; Webber, A.L.; Wong, A.; Smith, M.E.; Joyce, S.A.; Yates, J.R.; Pickard, C.J.; Dupree, R.; et al. Probing Heteronuclear 15N-17O and 13C-17O Connectivities and Proximities by Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2009, 131, 1820–1834. [Google Scholar] [CrossRef]
- O’Dell, L.A.; Schurko, R.W. Static solid-state 14N NMR and computational studies of nitrogen EFG tensors in some crystalline amino acids. Phys. Chem. Chem. Phys. 2009, 11, 7069–7077. [Google Scholar] [CrossRef] [PubMed]
- Bachelet, G.B.; Hamann, D.R.; Schlüter, M. Pseudopotentials that work: From H to Pu. Phys. Rev. B 1982, 26, 4199–4228. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baseggio, O.; Bonfà, P.; Brunato, D.; Car, R.; Carnimeo, I.; Cavazzoni, C.; de Gironcoli, S.; Delugas, P.; Ferrari Ruffino, F.; et al. Quantum ESPRESSO toward the exascale. J. Chem. Phys. 2020, 152, 154105. [Google Scholar] [CrossRef] [Green Version]
- Koelling, D.D.; Harmon, B.N. A technique for relativistic spin-polarised calculations. J. Phys. Solid State Phys. 1977, 10, 3107–3114. [Google Scholar] [CrossRef]
- Rappe, A.M.; Rabe, K.M.; Kaxiras, E.; Joannopoulos, J.D. Optimized pseudopotentials. Phys. Rev. B 1990, 41, 1227–1230. [Google Scholar] [CrossRef]
- Louie, S.G.; Froyen, S.; Cohen, M.L. Nonlinear ionic pseudopotentials in spin-density-functional calculations. Phys. Rev. B 1982, 26, 1738–1742. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef] [Green Version]
- Broyden, C.G. The Convergence of a Class of Double-Rank Minimization Algorithms 1. General Considerations. IMA J. Appl. Math. 1970, 6, 76–90. [Google Scholar] [CrossRef]
- Goldfarb, D. A Family of Variable-Metric Methods Derived by Variational Means. Math. Comp. 1970, 24, 23–26. [Google Scholar] [CrossRef]
- Shanno, D.F. Conditioning of Quasi-Newton Methods for Function Minimization. Math. Comp. 1970, 24, 647–656. [Google Scholar] [CrossRef]
- Steihaug, T. Practical Methods of Optimization Volume 1: Unconstrained Optimization; Wiley: New York, NY, USA, 1980. [Google Scholar]
- Ho, K.M.; Fu, C.L.; Harmon, B.N.; Weber, W.; Hamann, D.R. Vibrational Frequencies and Structural Properties of Transition Metals via Total-Energy Calculations. Phys. Rev. Lett. 1982, 49, 673–676. [Google Scholar] [CrossRef] [Green Version]
- Hedin, L. New Method for Calculating the One-Particle Green’s Function with Application to the Electron-Gas Problem. Phys. Rev. A 1965, 139, 795. [Google Scholar] [CrossRef]
- Martin, R.M.; Reining, L.; Ceperley, D. Interacting Electrons; Cambridge University Press: Cambridge, UK, 2016. [Google Scholar]
- Yates, J.R.; Dobbins, S.E.; Pickard, C.J.; Mauri, F.; Ghi, P.Y.; Harris, R.K. A combined first principles computational and solid-state NMR study of a molecular crystal: Flurbiprofen. Phys. Chem. Chem. Phys. 2005, 7, 1402–1407. [Google Scholar] [CrossRef]
- Cococcioni, M.; de Gironcoli, S. Linear response approach to the calculation of the effective interaction parameters in the LDA+U method. Phys. Rev. B 2005, 71, 035105. [Google Scholar] [CrossRef] [Green Version]
- Trinité, V.; Vast, N.; Hayoun, M. Effects of localization of the semicore density on the physical properties of transition and noble metals. J. Phys. Condens. Matter 2008, 20, 235239. [Google Scholar] [CrossRef]
- Cramer, C.J.; Truhlar, D.G. Density functional theory for transition metals and transition metal chemistry. Phys. Chem. Chem. Phys. 2009, 11, 10757–10816. [Google Scholar] [CrossRef]
- Hansen, M.R.; Madsen, G.K.H.; Jakobsen, H.J.; Skibsted, J. Evaluation of 27Al and 51V Electric Field Gradients and the Crystal Structure for Aluminum Orthovanadate (AlVO4) by Density Functional Theory Calculations. J. Phys. Chem. B 2006, 110, 5975–5983. [Google Scholar] [CrossRef]
- Yan, Z.; Chen, B.; Huang, Y. A solid-state NMR study of the formation of molecular sieve SAPO-34. Solid State Nucl. Magn. Reson. 2009, 35, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 1993, 48, 13115–13118. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J.; Hafner, J. Theory of the crystal structures of selenium and tellurium: The effect of generalized-gradient corrections to the local-density approximation. Phys. Rev. B 1994, 50, 13181–13185. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J.; Hafner, J. Ab initio Force Constant Approach to Phonon Dispersion Relations of Diamond and Graphite. Europhys. Lett. (EPL) 1995, 32, 729–734. [Google Scholar] [CrossRef]
- Bak, M.; Rasmussen, J.T.; Nielsen, N.C. SIMPSON: A General Simulation Program for Solid-State NMR Spectroscopy. J. Magn. Reson. 2000, 147, 296–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paruzzo, F.M.; Hofstetter, A.; Musil, F.; De, S.; Ceriotti, M.; Emsley, L. Chemical shifts in molecular solids by machine learning. Nat. Commun. 2018, 9, 4501. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| System | System | ||||
|---|---|---|---|---|---|
| AgN | 2542.54 | 0.04 | PtO | −7557.22 | 1.20 |
| AuO | 1115.14 | 2.45 | ReO | −1064.07 | 3.44 |
| CdO | 3322.38 | 0.09 | RhO | −10,829.68 | 0.05 |
| CrO | −6933.31 | −0.07 | RuO | −2968.18 | 0.02 |
| HgO | 6700.46 | −15.05 | ScO | 677.59 | −0.25 |
| IrN | −4132.37 | −4.95 | TaO | 2529.32 | 2.76 |
| LuO | 5317.24 | −1.03 | TcO | −2249.87 | 0.43 |
| MoO | −1196.63 | −2.30 | VO | −1447.62 | −0.35 |
| NbO | 102.85 | −1.06 | WO | 752.99 | 5.18 |
| OsO | −2337.18 | −0.06 | YO | 1928.05 | −0.41 |
| PdN | −3407.89 | −0.37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tantardini, C.; Kvashnin, A.G.; Ceresoli, D. GIPAW Pseudopotentials of d Elements for Solid-State NMR. Materials 2022, 15, 3347. https://doi.org/10.3390/ma15093347
Tantardini C, Kvashnin AG, Ceresoli D. GIPAW Pseudopotentials of d Elements for Solid-State NMR. Materials. 2022; 15(9):3347. https://doi.org/10.3390/ma15093347
Chicago/Turabian StyleTantardini, Christian, Alexander G. Kvashnin, and Davide Ceresoli. 2022. "GIPAW Pseudopotentials of d Elements for Solid-State NMR" Materials 15, no. 9: 3347. https://doi.org/10.3390/ma15093347
APA StyleTantardini, C., Kvashnin, A. G., & Ceresoli, D. (2022). GIPAW Pseudopotentials of d Elements for Solid-State NMR. Materials, 15(9), 3347. https://doi.org/10.3390/ma15093347