1. Introduction
PUs are materials used in a wide range of industrial sectors, such as in appliances, furniture and the automotive and construction industries [
1,
2,
3]. According to their specific chemistry, they can be commercially available as solid thermoplastics (TPU) or as foams [
4]. PU foams can be further classified as flexible foams (used for applications such as mattresses and automotive seats) or rigid foams (used for applications such as thermal insulation and structural materials) [
1,
2,
3,
4]. PUs are typically prepared by
mixing two reactants, namely a polyether polyol with a poly-isocyanate (methylene diphenylene or toluene diisocyanate), forming the solid polymer [
5,
6,
7].
The chemistry involved in the synthesis of rigid PUs is complex because several simultaneous reactions are involved. The simultaneity of the reactions is due to the extreme reactivity of the isocyanate group (−N=C=O) towards the hydrogen active compounds, such as those containing the −OH and −NH functional groups [
6]. However, among the reactions taking place during the foaming process, two reactions are the main processes leading to the formation of a PU foam. On one hand, the
polymerization reaction (also known as gelling) between isocyanate and polyol leads to the formation of the urethane group [
1,
2,
3,
4,
5,
6]. Concurrent to the gelling reaction, the isocyanate reacts with water (included in the polyol) to give an unstable carbamic acid, which spontaneously decomposes into CO
2 (as a chemical blowing agent, CBA) and the corresponding amine in the so-called blowing reaction. Therefore, the generated gas provides the polymer expansion [
1,
2,
3,
4,
5,
6]. The amine may further react with another isocyanate molecule to give urea. In addition to CBAs, several physical blowing agents (PBAs) may be used as co-blowing agents to further promote the polymer expansion. Typically, PBAs are liquids with a low boiling temperature (e.g., pentane) that undergo evaporation due to the exothermicity of the reacting medium [
1,
2,
3,
4,
5,
6].
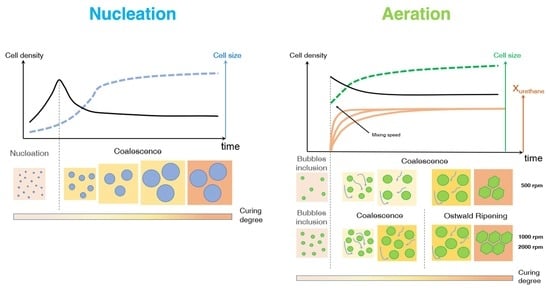
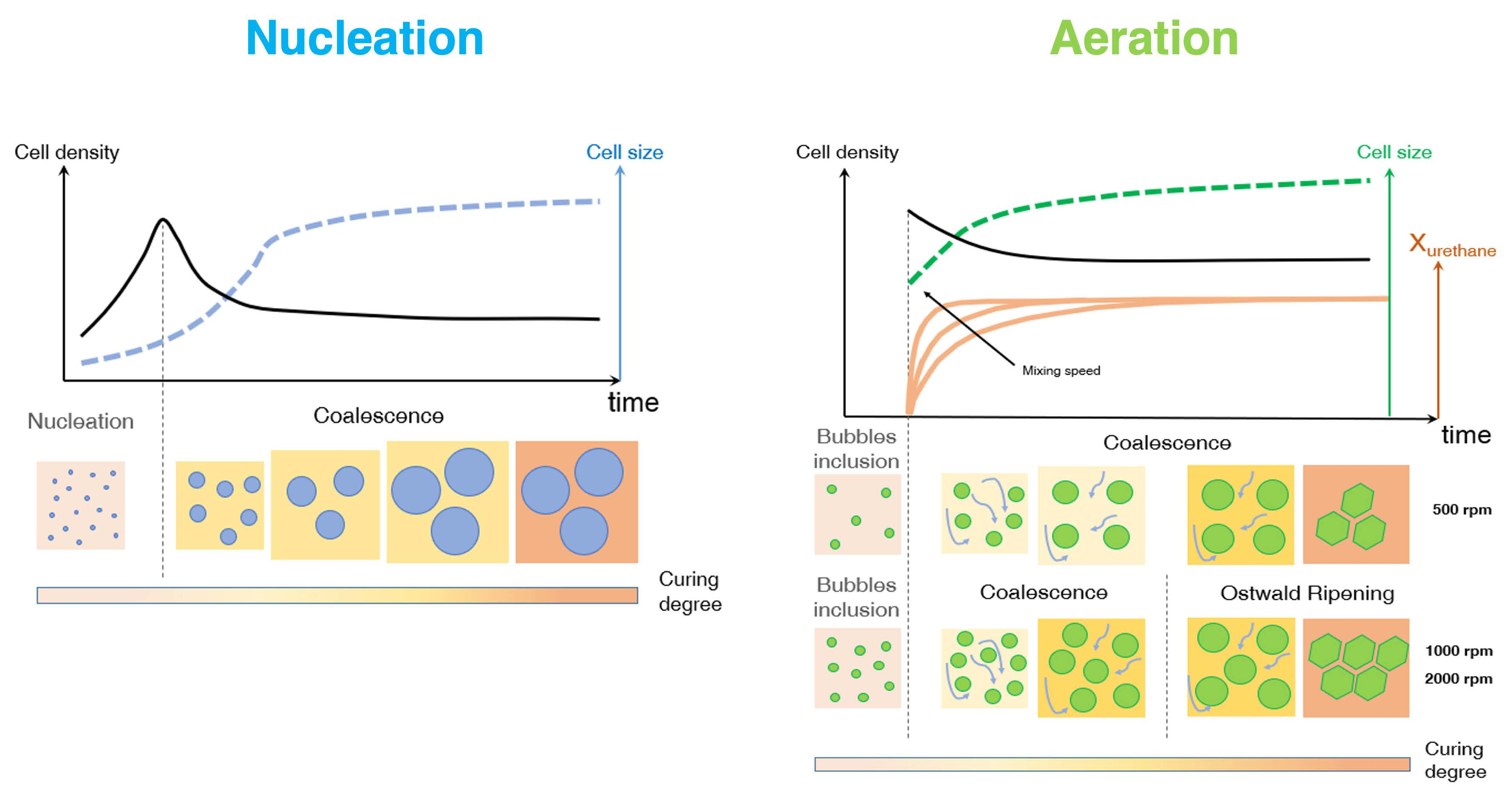
The polymer expansion is regulated by two bubble formation mechanisms during the early stage of the foaming process, after which bubbles grow, impinge and may eventually merge into each other, inducing coarsening on the ongoing morphology. The bubble formation mechanisms are aeration and nucleation. For aeration (also known as frothing), the gas phase (commonly air) is dispersed into the starting reactants via vigorous stirring, entrapping gas bubbles within the polymeric matrix [
8]. For nucleation, according to the classical homogeneous nucleation theories, the system must overcome a thermodynamical (surface) energy barrier in order to let nucleation occur. For gas–liquid systems, once the solubility limit is exceeded, the gas dissolved into the liquid matrix precipitates in the form of metastable nuclei that may re-dissolve or grow beyond a critical radius [
9]. Bubble stabilization (or, on the contrary, their collapse) may occur after the growth stage [
9,
10]. Aeration and nucleation may be considered competitive mechanisms: in fact, if pre-existing bubbles are already present in the reaction system (e.g., air bubbles entrapped by mechanical stirring), there is no surface energy barrier to overcome and the blowing agent evolving from CBA (or PBA) may simply inflate the pre-existing bubbles via diffusion, with no formation of additional bubbles formed via nucleation.
PU’s final properties, such as the thermal conductivity, mechanical properties, dimensional stability and fire resistance, are tightly related to the foam density and morphology. In order to tailor the final properties, it is key to study and understand the governing mechanisms in the morphology evolution, namely the balance between the gelling and blowing reaction and the physical aspects of the different stages of the process: nucleation, growing, coarsening and coalescence. In addition, the bubble size distribution and the degree of open and closed bubbles, again largely affecting the properties, should also be considered [
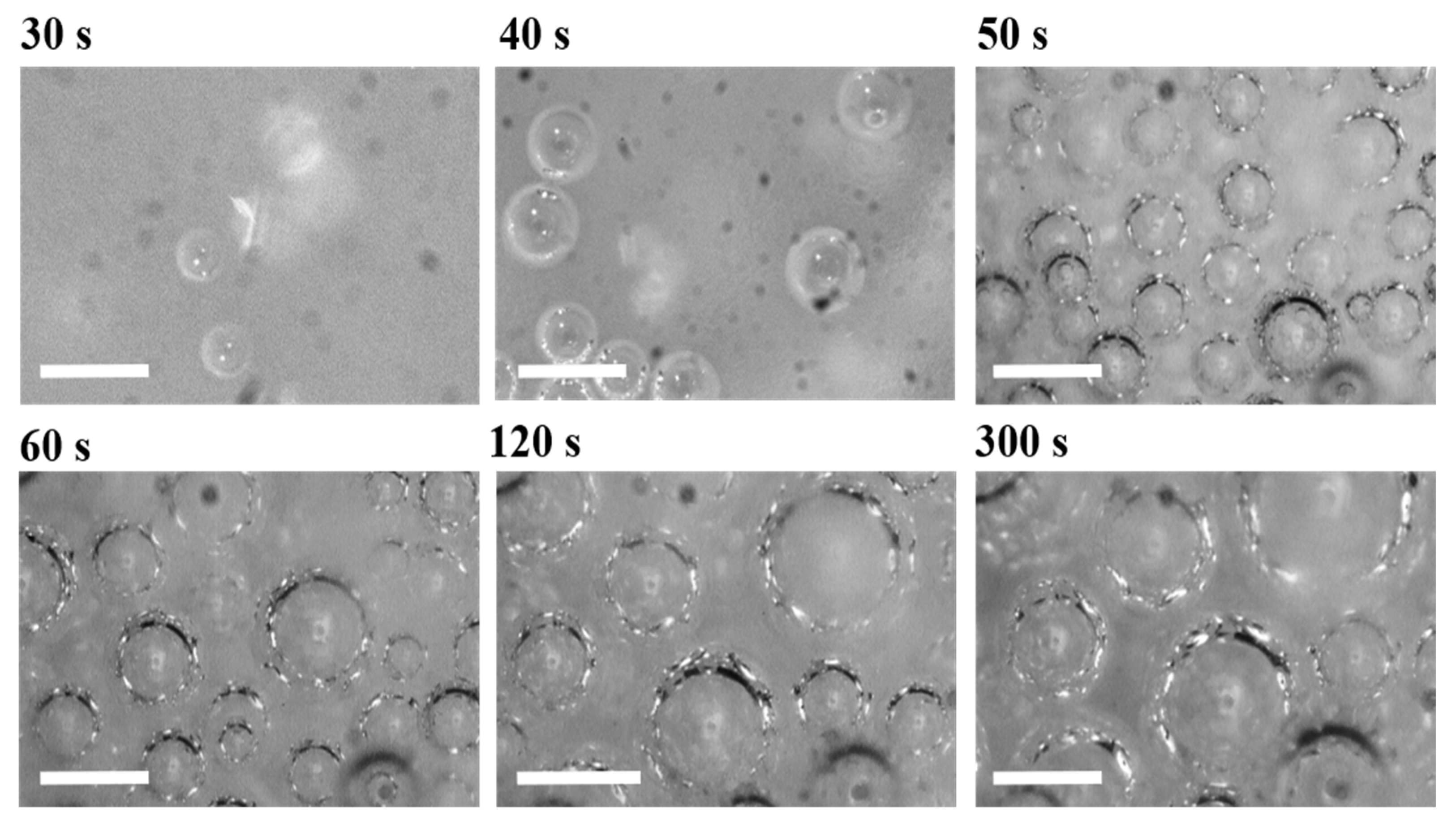
11]. Recently, Pardo et al. used time-resolved X-ray radioscopy in order to study the bubble nucleation and growth mechanisms taking place in a small droplet (used to limit the number of cells to 2–3 maximum in the depth direction) [
12]. By using this technique in combination with image analysis (IA), they were able to monitor the bubble development during the PU foaming process. In our previous work [
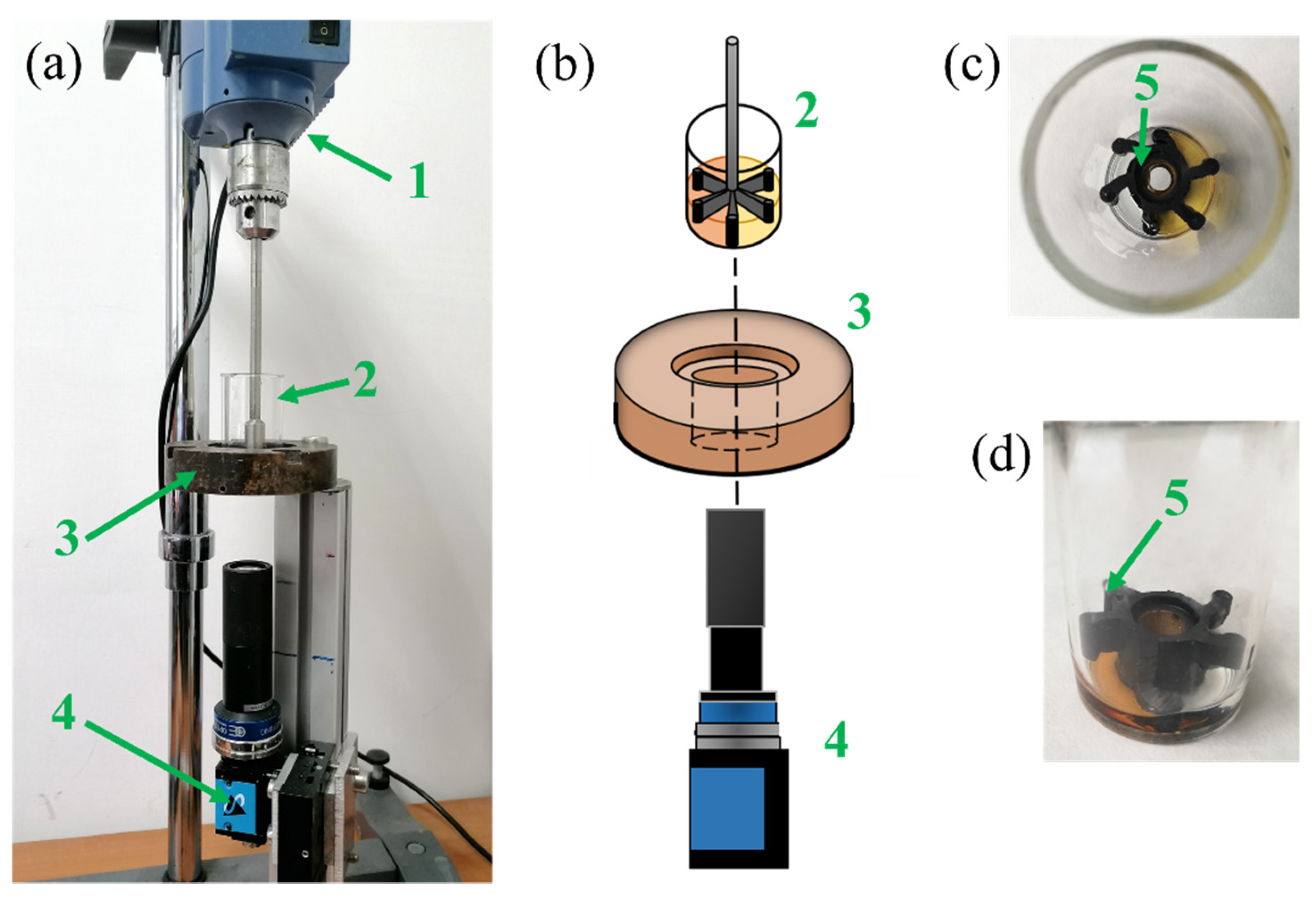
13], we developed equipment that allowed us to monitor the different stages of the expansion process with different blowing agents. We were able to monitor the bubble size distribution as well as the bubble number density during the PU expansion. The aforementioned works clearly elucidate the progress made so far in developing equipment suitable to monitor these physical phenomena and characterize them. However, knowledge of the effects of the mixing conditions and the introduction of air bubbles into the PU reactive system on the nucleation and growth mechanisms is still poor, as well as on the reaction kinetics [
14,
15]. Baumhäkel et al. [
10] studied the influence of the mixer stirring velocity on the final morphology of a flexible PU foam. Air bubbles were introduced in an open container by varying the mixing speed up to 3000 rpm. In this way, the authors concluded that the air bubbles introduced into the system by the mixing process accounted for all bubbles present in the final foam. A similar result was observed by Reignier et al. [
15] on PU foams studied by cryogenic scanning electron microscopy. They also observed that the number of air bubbles per unit volume (~10
6 bubbles·cm
−3) was similar to the bubble population density of the final foam.
In the recent literature, several additives have been extensively studied and used to reduce the bubble size distribution in PU foams. For instance, solid-type nucleating agents such as talc and organoclay particles induced significant reductions in cell size, which induced complications due to the precipitation of solid particles during the foaming process and the difficulty in reaching a good dispersion [
16]. Liquid-type additives such as tetramethylsilane compounds (TEMs) were also used and PU foams with a more uniform and finer cellular structure were obtained. However, these additives presented several limitations, such as high flammability, high vapor pressure and high costs [
17]. In this context, this work is part of a larger project aimed at obtaining PU foams with reduced bubble sizes obtained via the introduction of novel additives. In a previous paper [
18], we studied the effects of organofluorine additives (OFAs) on PU’s cellular structure, along with their relative properties. It was shown that the smaller bubble size in these foams leads to reduced thermal conductivity and slightly improved mechanical properties. These results, combined with the relative low cost of OFAs and their non-flammability [
17], persuaded us to investigate the possible mechanisms induced by these compounds during the PU foaming process. To this aim, we first studied the effects of the air bubble inclusion on the nucleation and growth mechanisms during the PU foaming process in the absence of the aforementioned additives [
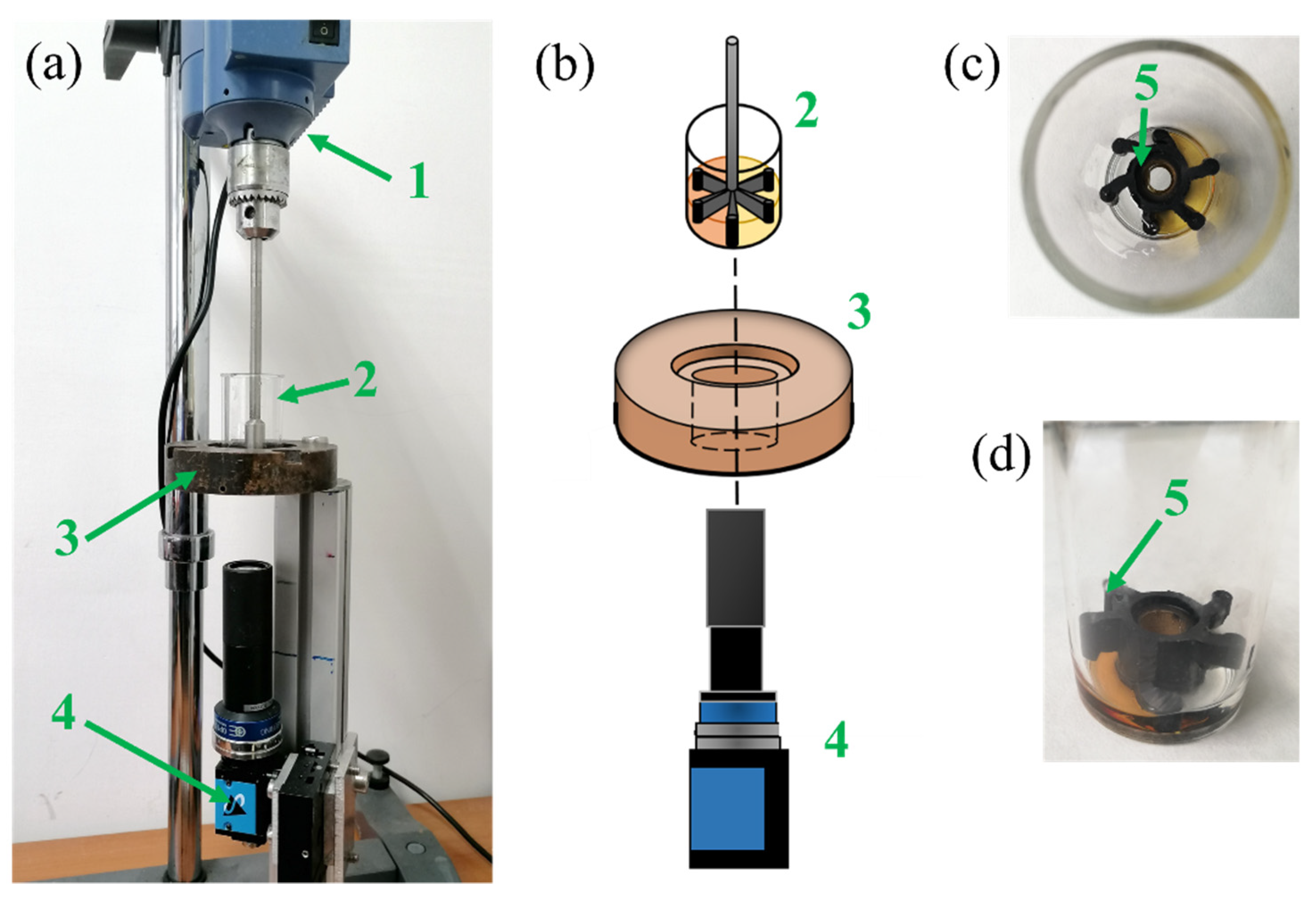
13]. For air bubble inclusion (obtained at a high stirring velocity of 1000 rpm for 8 s), we observed that the blowing agent molecules diffuse toward these pre-existing air bubbles rather than nucleating new gas bubbles. On the contrary, in the absence of air bubbles (at a low stirring velocity of 50 rpm for 20 s), bubble nucleation was detected. Besides the investigation of these physical mechanisms, further study is needed to understand how the chemical processes are affected by the air bubble inclusion.
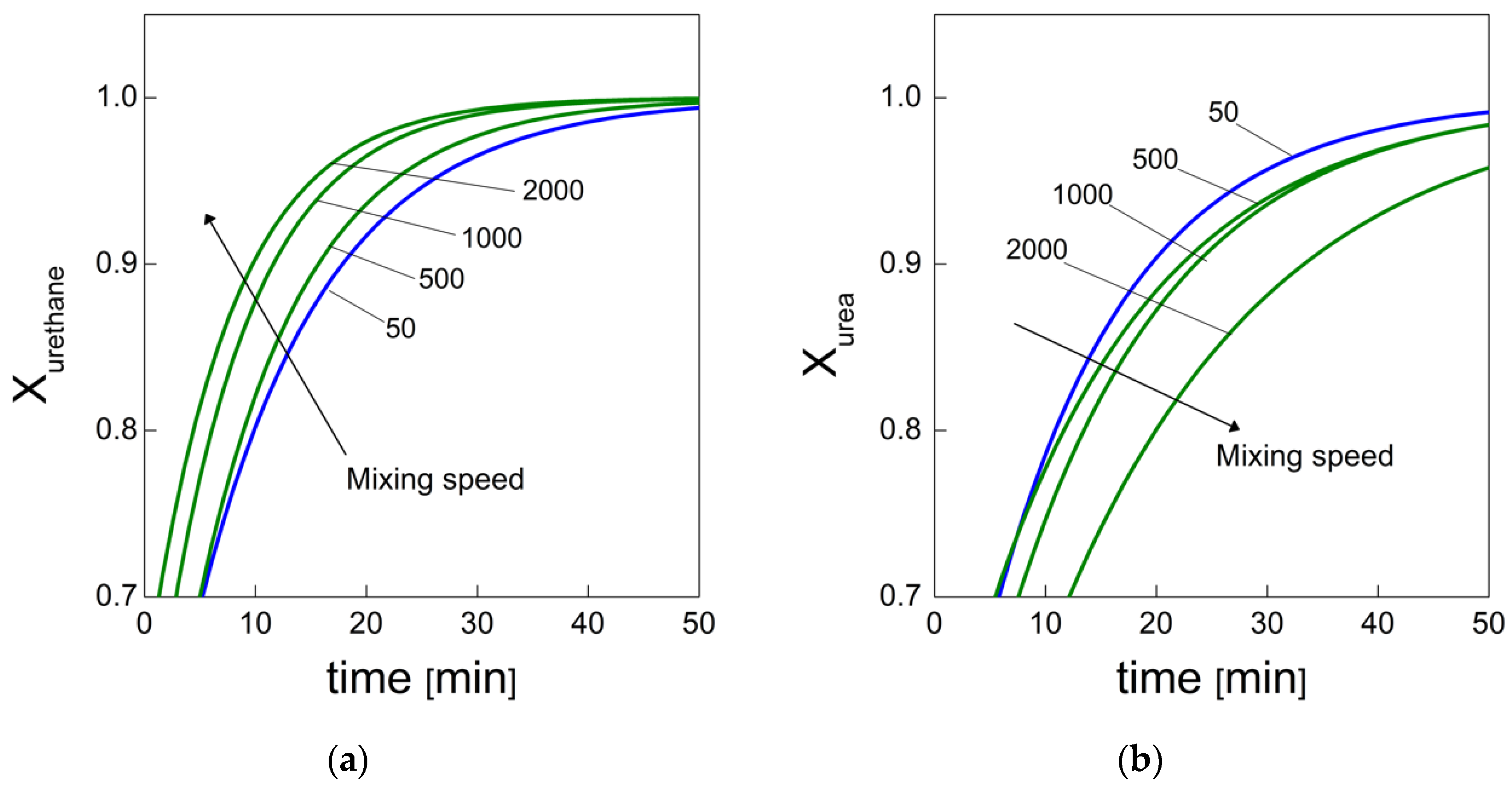
In the context of this project, we are interested in evaluating the effects of the air bubble inclusion on the reaction kinetics of rigid water-blown PU foams and how these affect the bubble formation and degeneration mechanisms. In the work performed by Santiago-Calvo et al. [
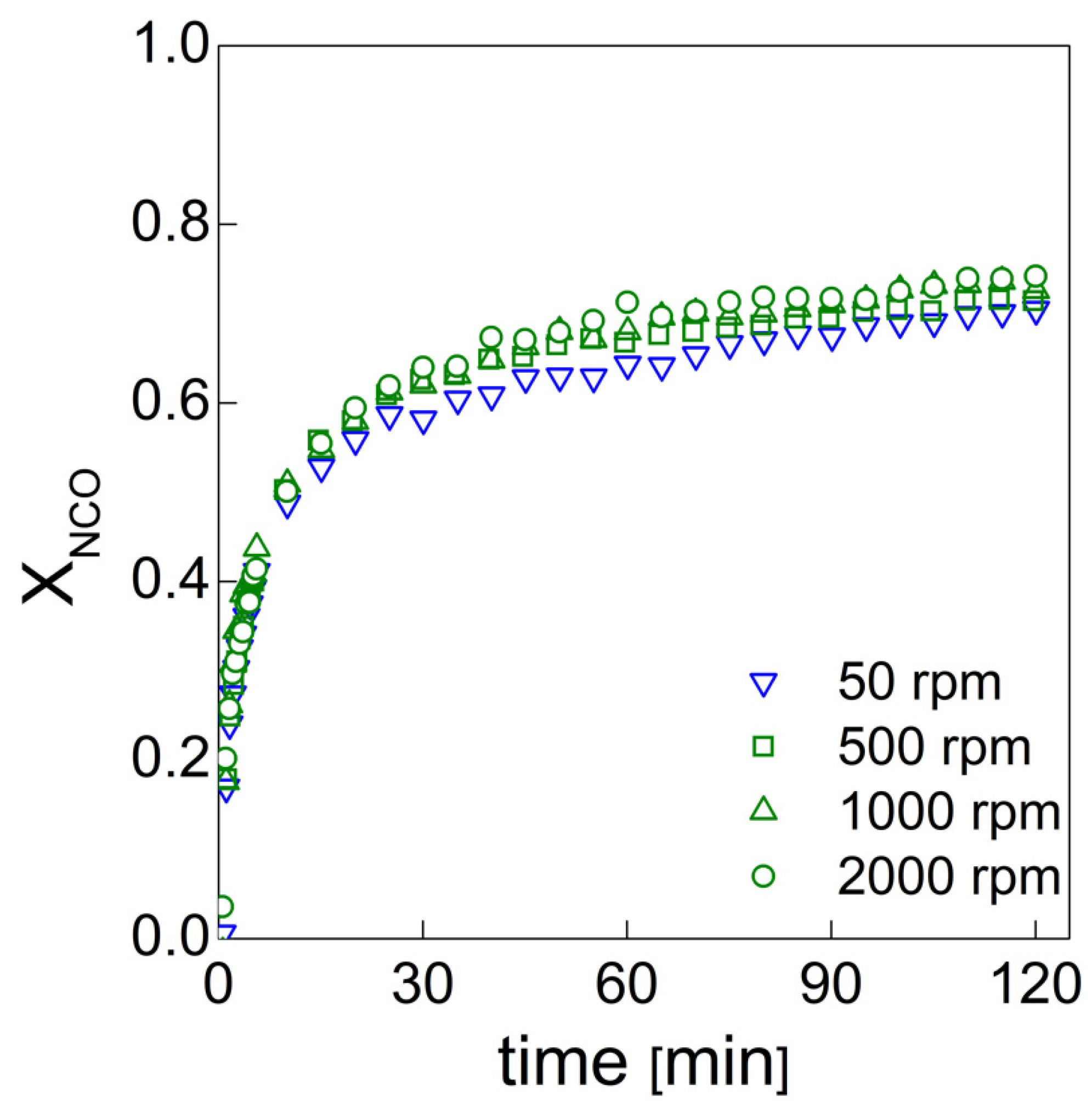
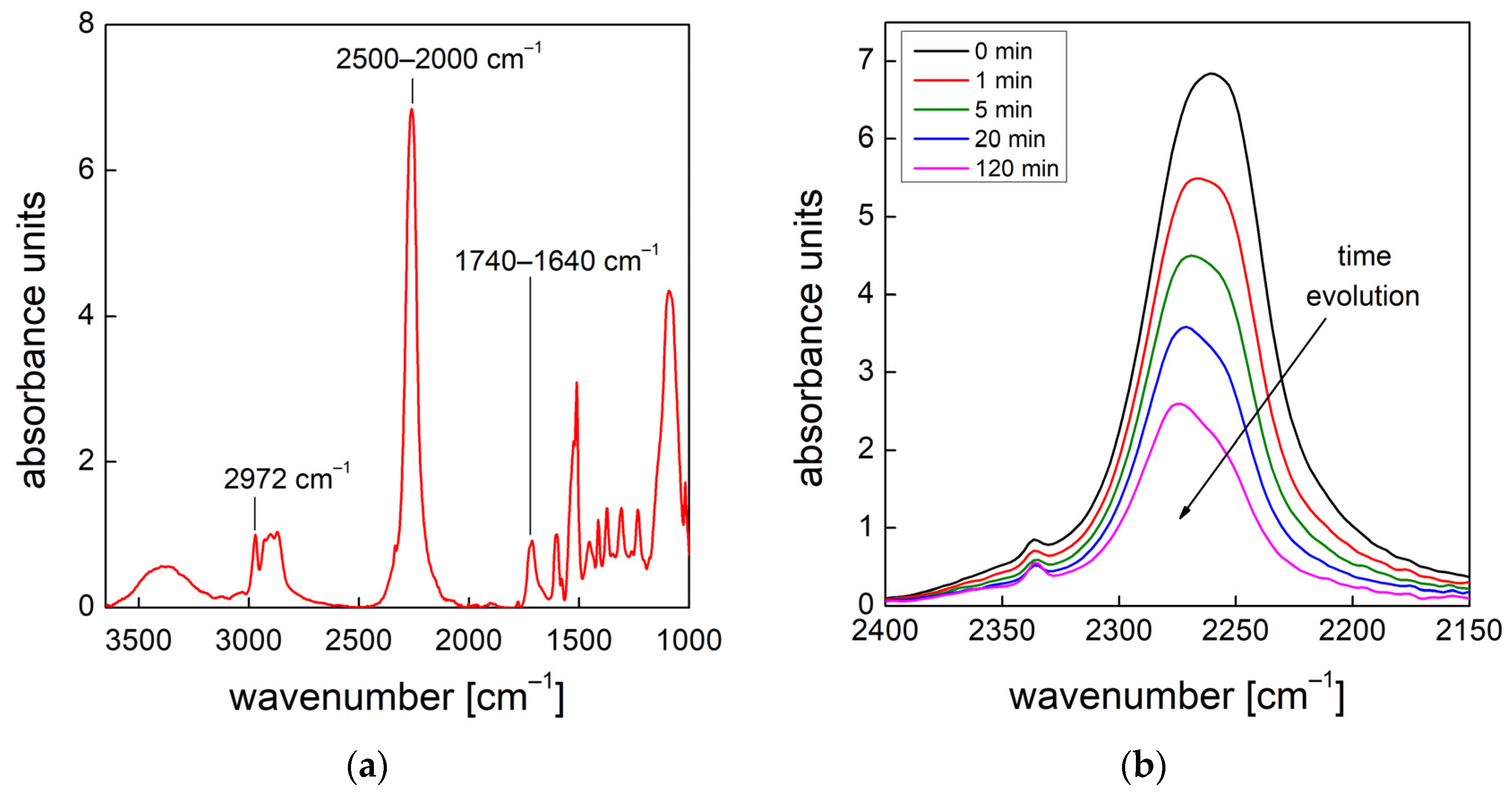
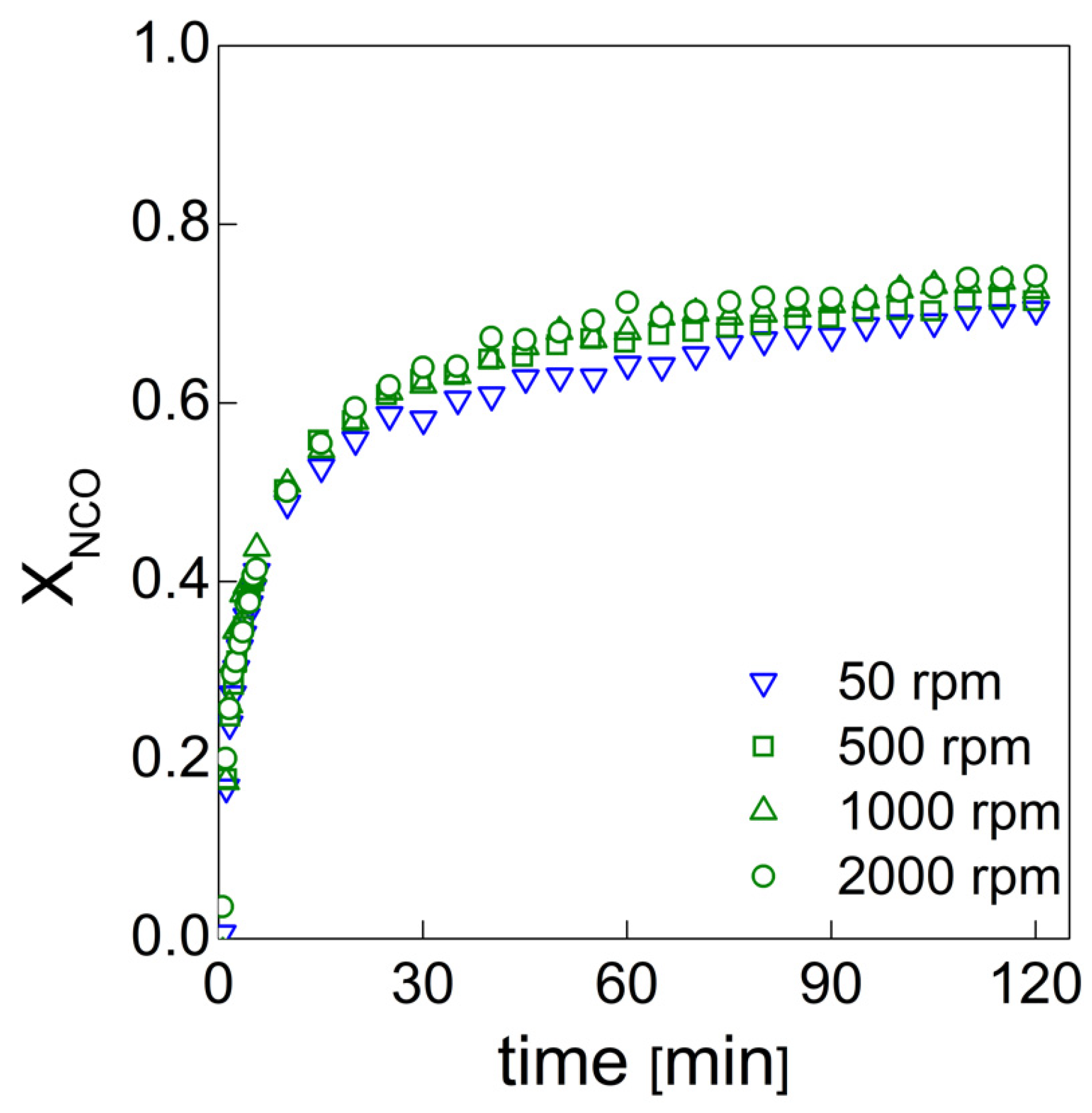
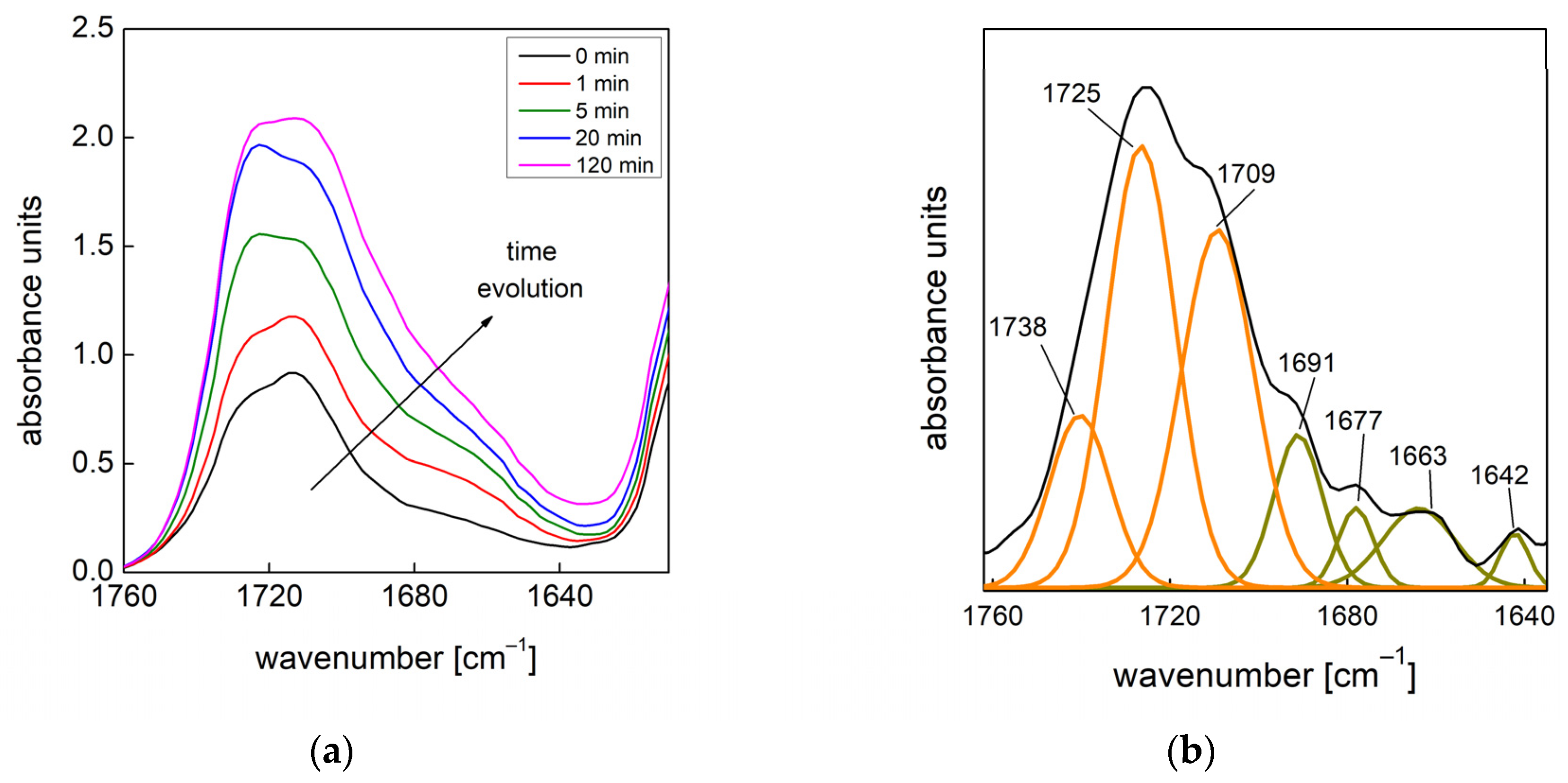
19], the effects of functional nanofillers on the reaction kinetics of water blown PU foams were studied. To this end, the authors developed a method that allowed them to evaluate the formation of the urethane and urea groups by deconvolution of via amide I region in the infrared. In this way, it was possible to quantify the products that are representative of the gelling and blowing reactions during the foaming process. In this paper, we used this approach to follow the chemical reaction kinetics [
19] combined with the bubble formation monitoring approach [
13] to give a full picture of both the chemical and physical processes taking place during the foaming processes, in which different amounts of air bubbles are entrapped during the mixing stage.
4. Conclusions
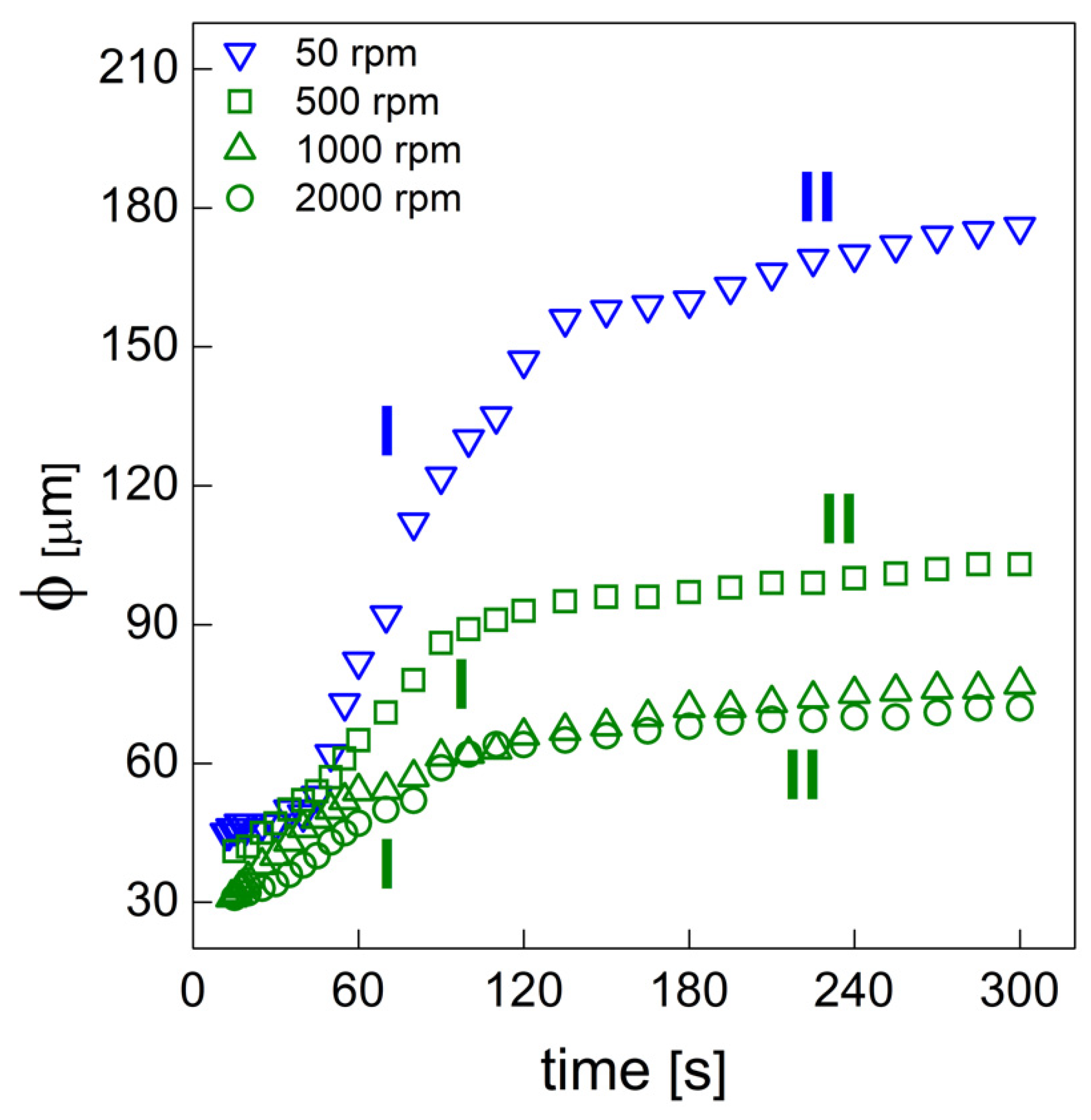
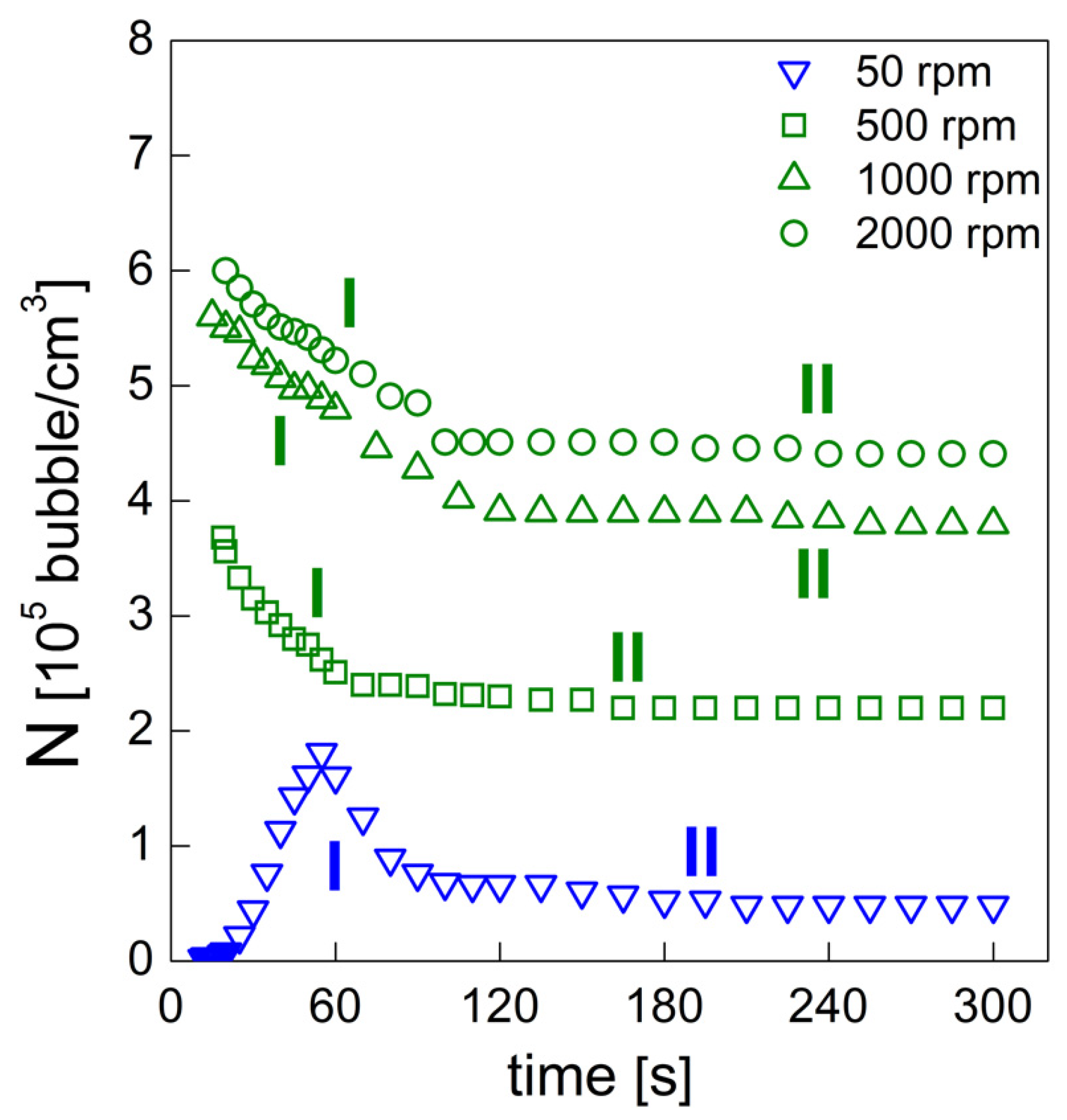
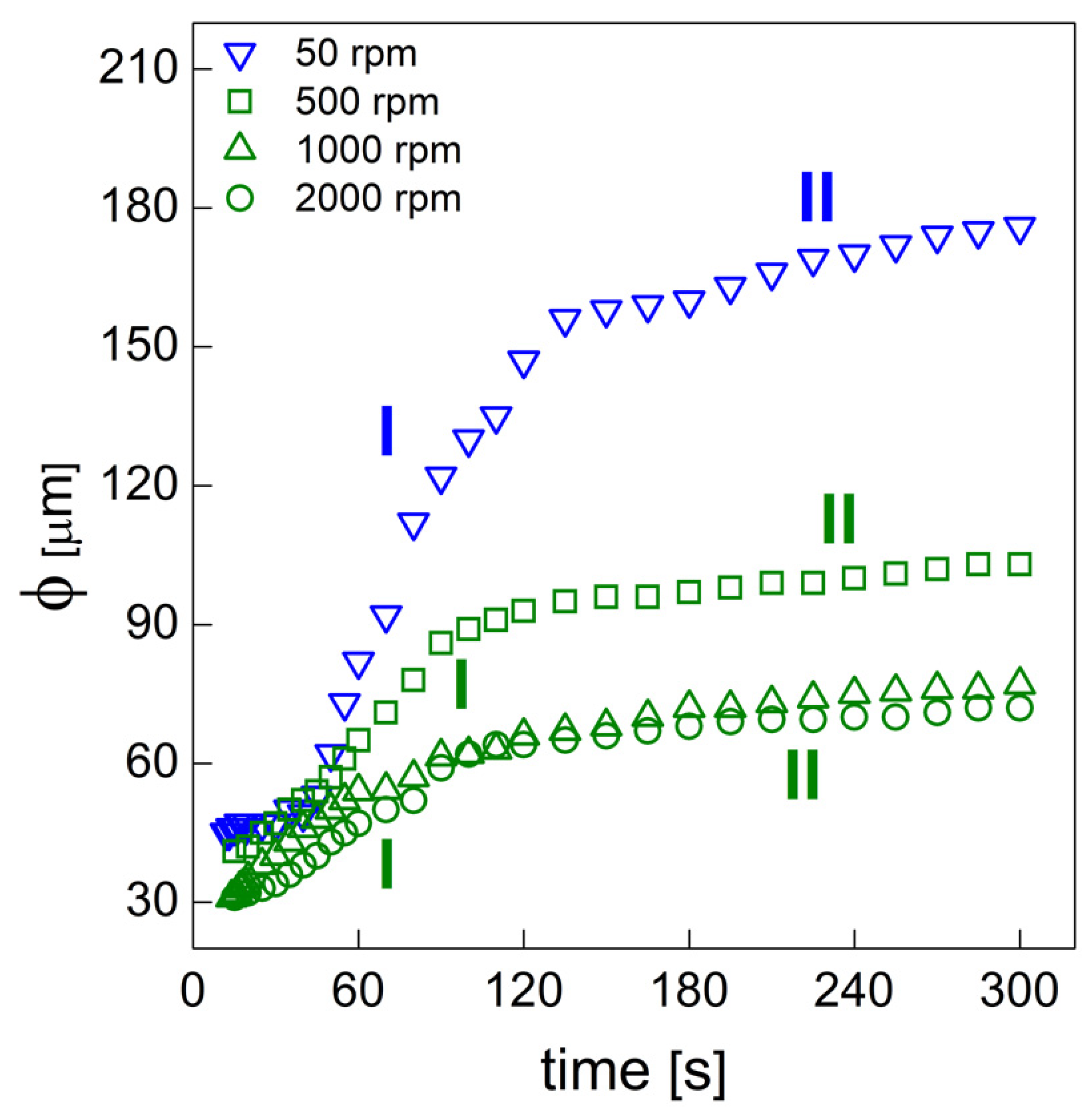
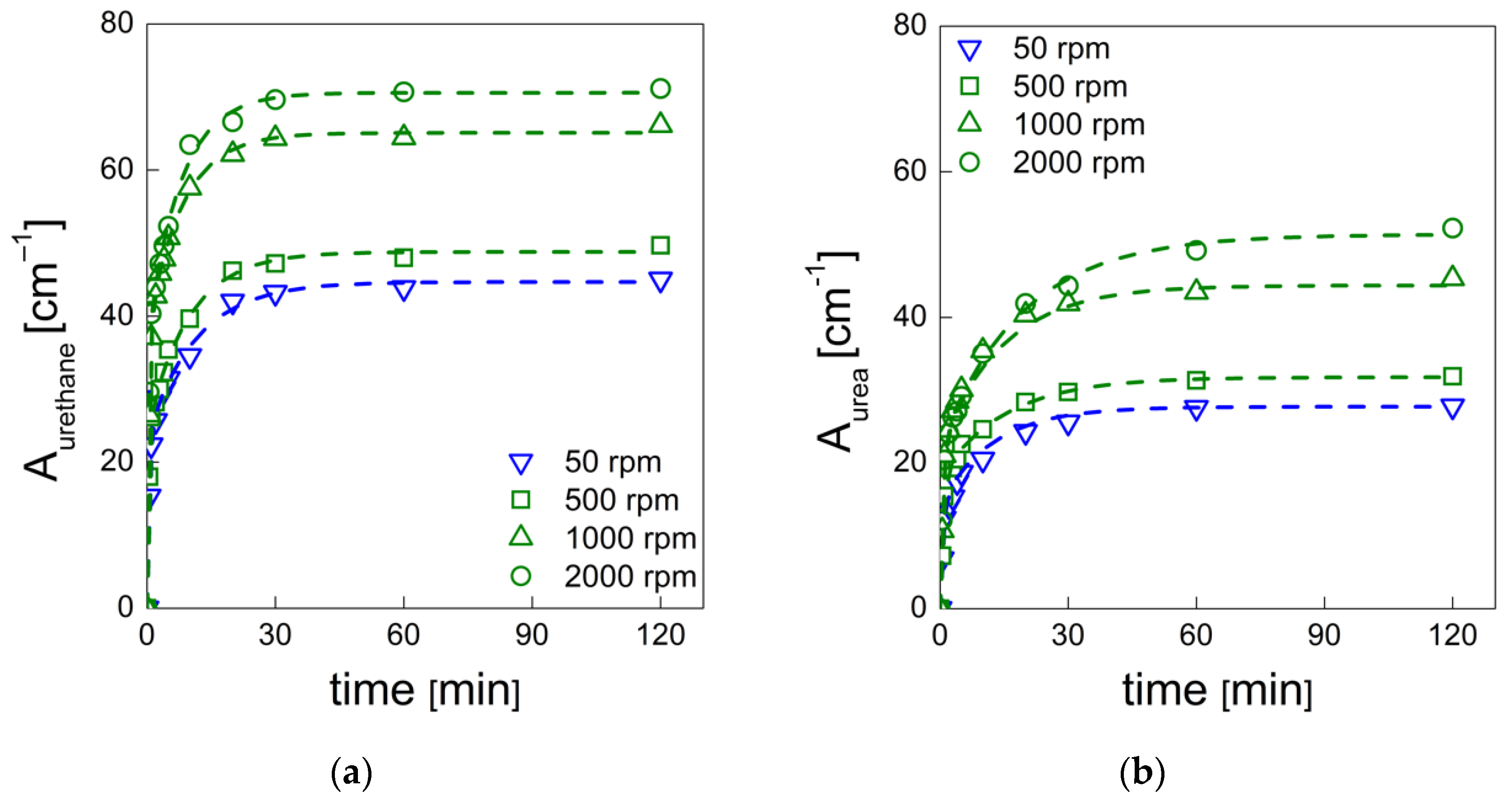
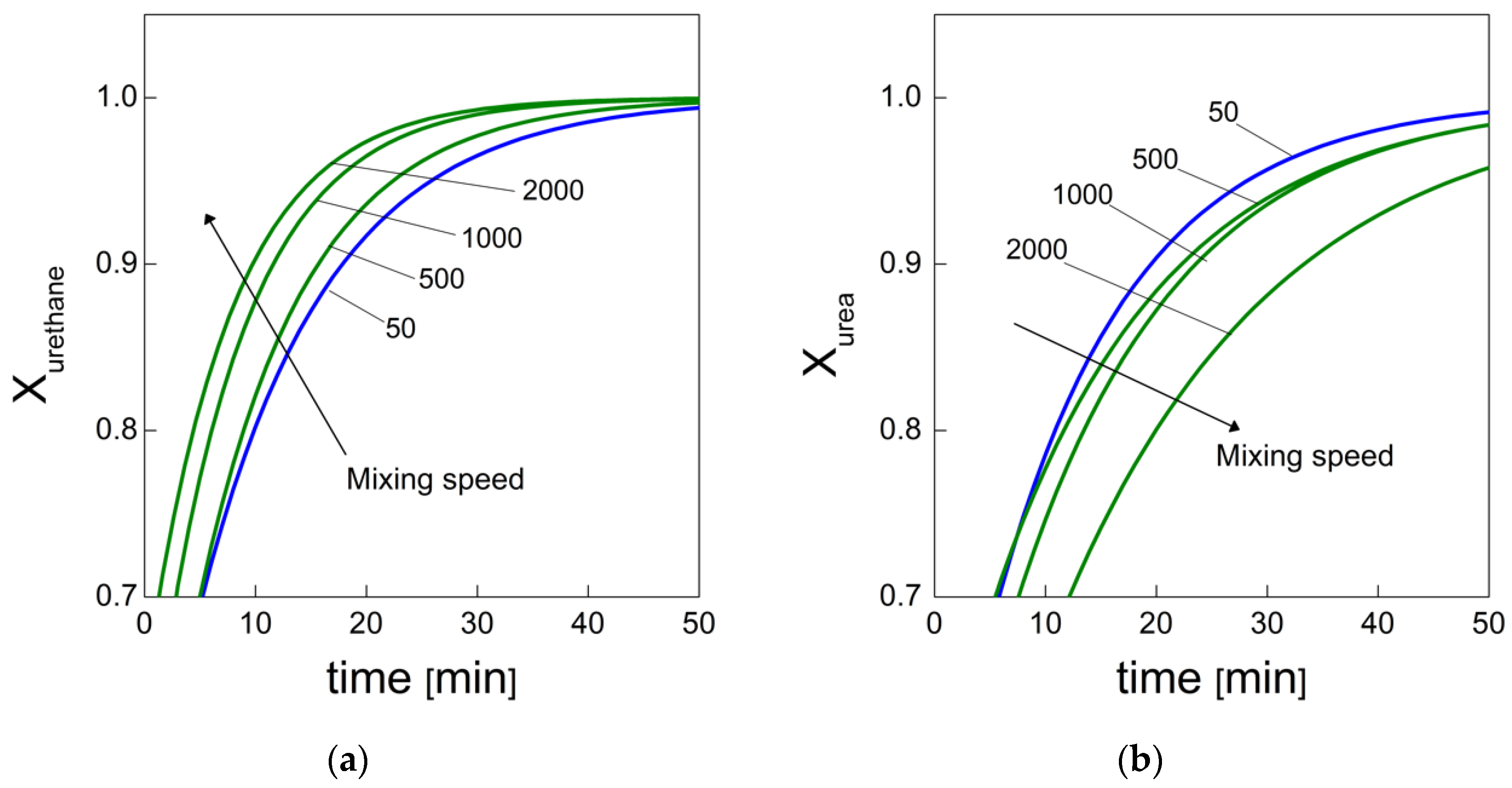
PU bubble formation mechanisms and reaction kinetics at several mixing speeds were studied using in situ micro-optical observations and FTIR spectroscopy, respectively. The bubble size, evolution and density were determined via image analysis, while gelling and blowing reactions were quantified by deconvolution analysis of the amide I region. At 50 rpm, right after the mixing stage, no air bubbles are included in the PU reaction system. New CO2 bubbles appear to nucleate during the early foaming process. In this case, as with the product formation rate given via FTIR data, the gelling reaction is inhibited by the indigent mixing conditions, meaning the polymer cannot withstand the bubble expansion and the relative coarsening. Indeed, after the nucleation stage, these bubbles undergo coalescence and this degeneration mechanism affects the overall foaming process. At 500 rpm, air bubbles are included in the PU reaction system and no formation of new bubbles was observed during the foaming process. After the mixing stage, these bubbles undergo growing caused by CO2 diffusion. For this PU system, the gelling reaction is not strongly depleted, as in the case at 50 rpm, but still the polymer is not capable of withstanding the bubble expansion, meaning the coalescence still affects the overall foaming process. At 1000 and 2000 rpm, bubble degeneration was affected by coalescence during the early foaming stage and by Ostwald Ripening in the late foaming process. In any case, PU samples obtained at 1000 and 2000 rpm were characterized by a finer bubble size distribution when compared to samples obtained at 500 rpm, and were also affected by coalescence. For these samples, the polymerization was enhanced by the higher stirring velocity, which allowed us to obtain more suitable mixing conditions. In this way, the PU foaming results in a balanced process between the bubbles inflation and the viscosity increase.
In summary, these observations reflect how the different mixing conditions can affect the overall reaction kinetics. The final cellular structure of the product can also be modulated as a consequence of the different selected mixing speeds. Based on this approach, by coupling optical observation and FTIR spectroscopy processes, one can adequately choose the process conditions in order to obtain the desired PU foam that is suitable for the appropriate industrial application.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}