
Synthesis, DFT Molecular Geometry and Anticancer Activity of Symmetrical 2,2′-(2-Oxo-1H-benzo[d]imidazole-1,3(2H)-diyl) Diacetate and Its Arylideneacetohydrazide Derivatives


 , , , , and
, , , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. General
2.2. Preparation of 1H-Benzo[a]imidazol-2(3H)-2-one (2)
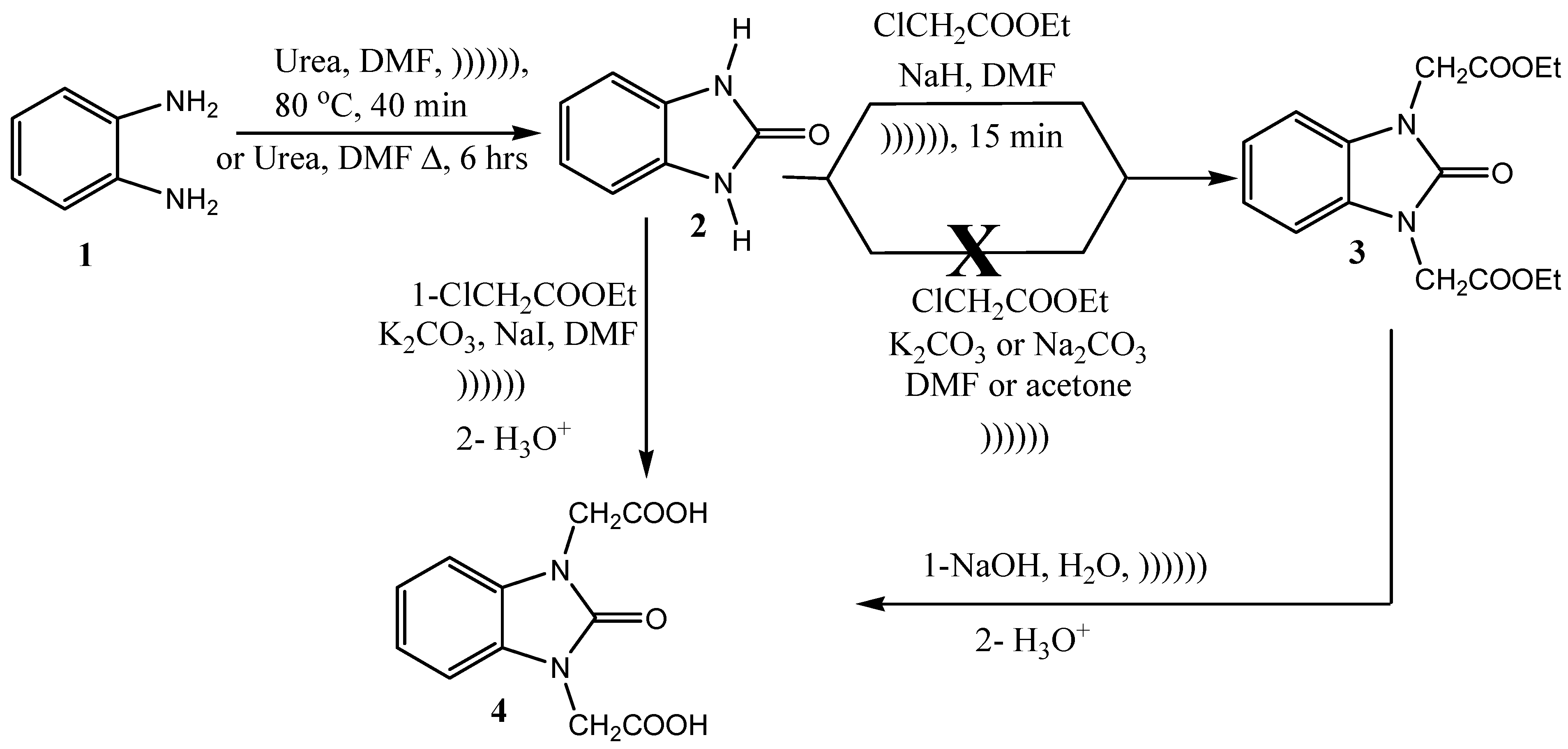
2.3. General Method for Alkylation Reaction
2.4. Preparation of Diethyl N,N′-2,2′-(2-Oxo-1H-benzo[d]imidazole-1,3(2H)-diyl)diacetate (3)
2.5. Preparation of N,N′-2,2′-(2-Oxo-1H-benzo[d]imidazole-1,3(2H)-diyl)diacetic Acid (4)
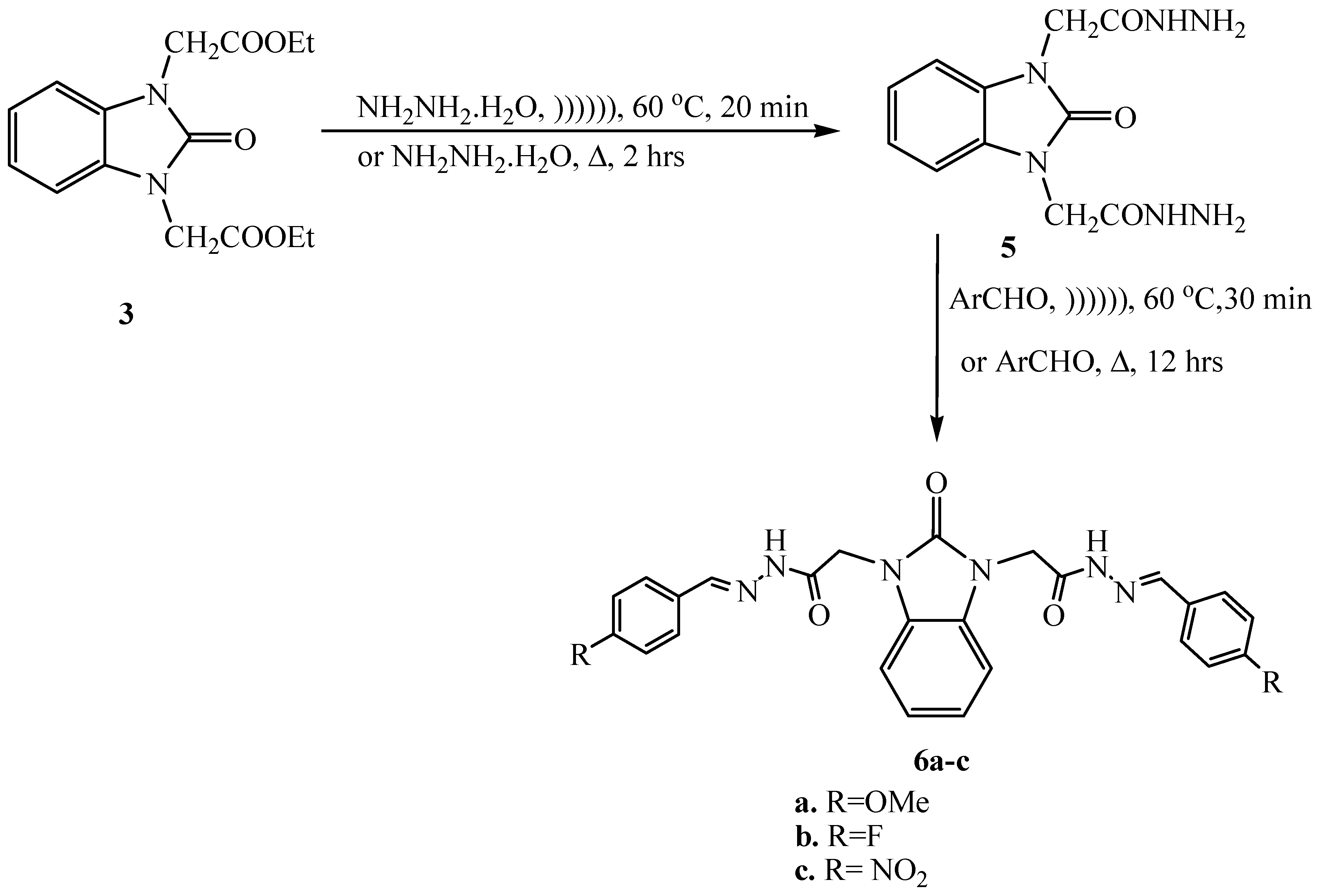
2.6. Synthesis of 2,2′-(2-Oxo-1H-benzo[d]imidazole-1,3(2H)-diyl)diacetohydrazide (5)
2.7. Synthesis of N,N′-2,2′-(2-Oxo-1H-benzo[d]imidazole-1,3(2H)-diyl)bis(arylidene) Acetohydrazide (6a–c)
2.8. N,N′-2,2′-(2-Oxo-1H-benzo[d]imidazole-1,3(2H)-diyl)bis(4-methoxybenzylidene) Acetohydrazide) (6a)
2.9. N,N′-2,2′-(2-Oxo-1H-benzo[d]imidazole-1,3(2H)-diyl)bis(4-flourobenzylidene) Acetohydrazide) (6b)
2.10. N,N′-2,2′-(2-Oxo-1H-benzo[d]imidazole-1,3(2H)-diyl)bis(4-nitrobenzylidene) Acetohydrazide) (6c)
2.11. Computational Details
2.12. In Vitro Cell Culture
2.13. MTT Assay
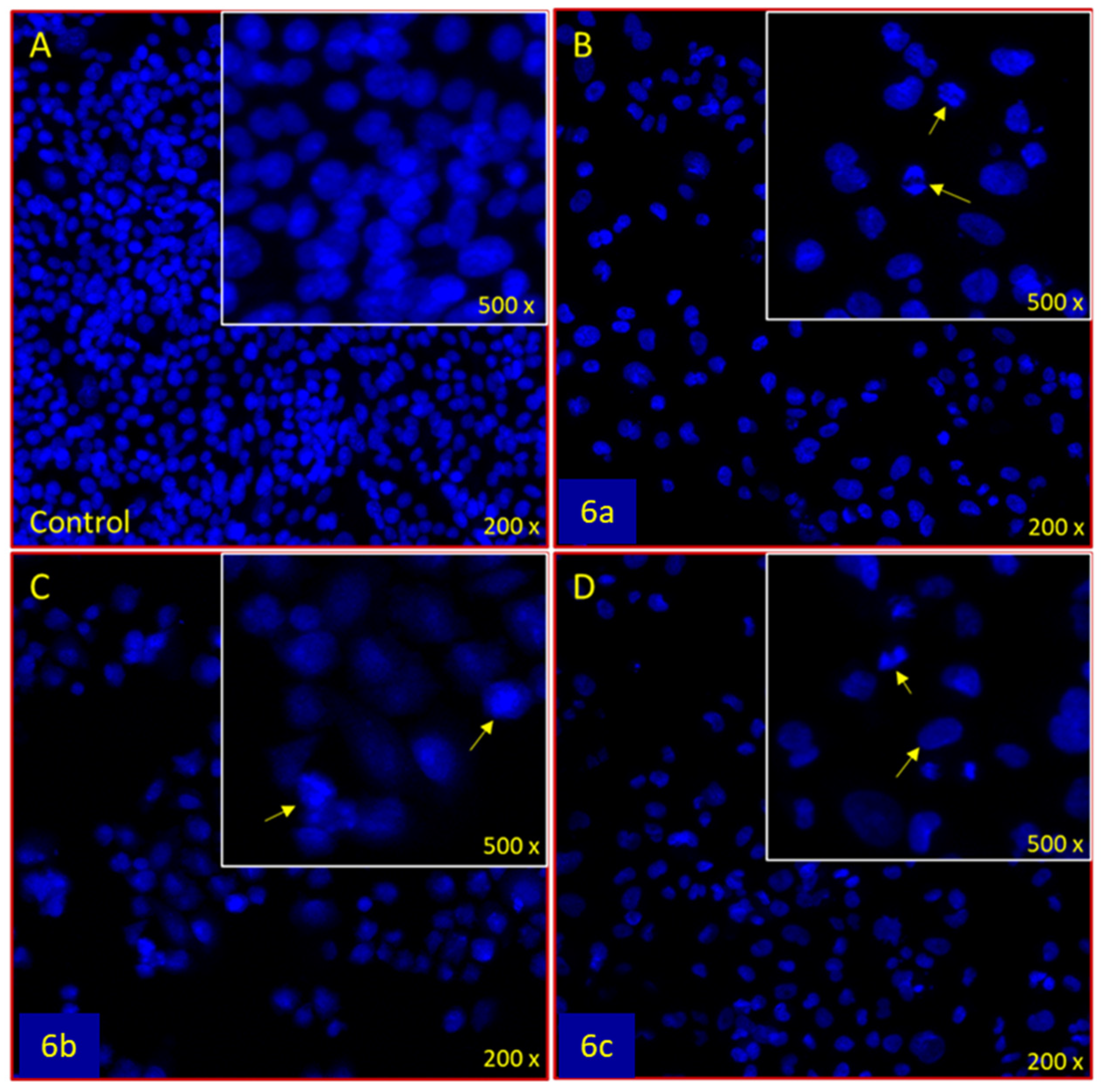
2.14. Apoptotic DAPI Staining
3. Results and Discussion
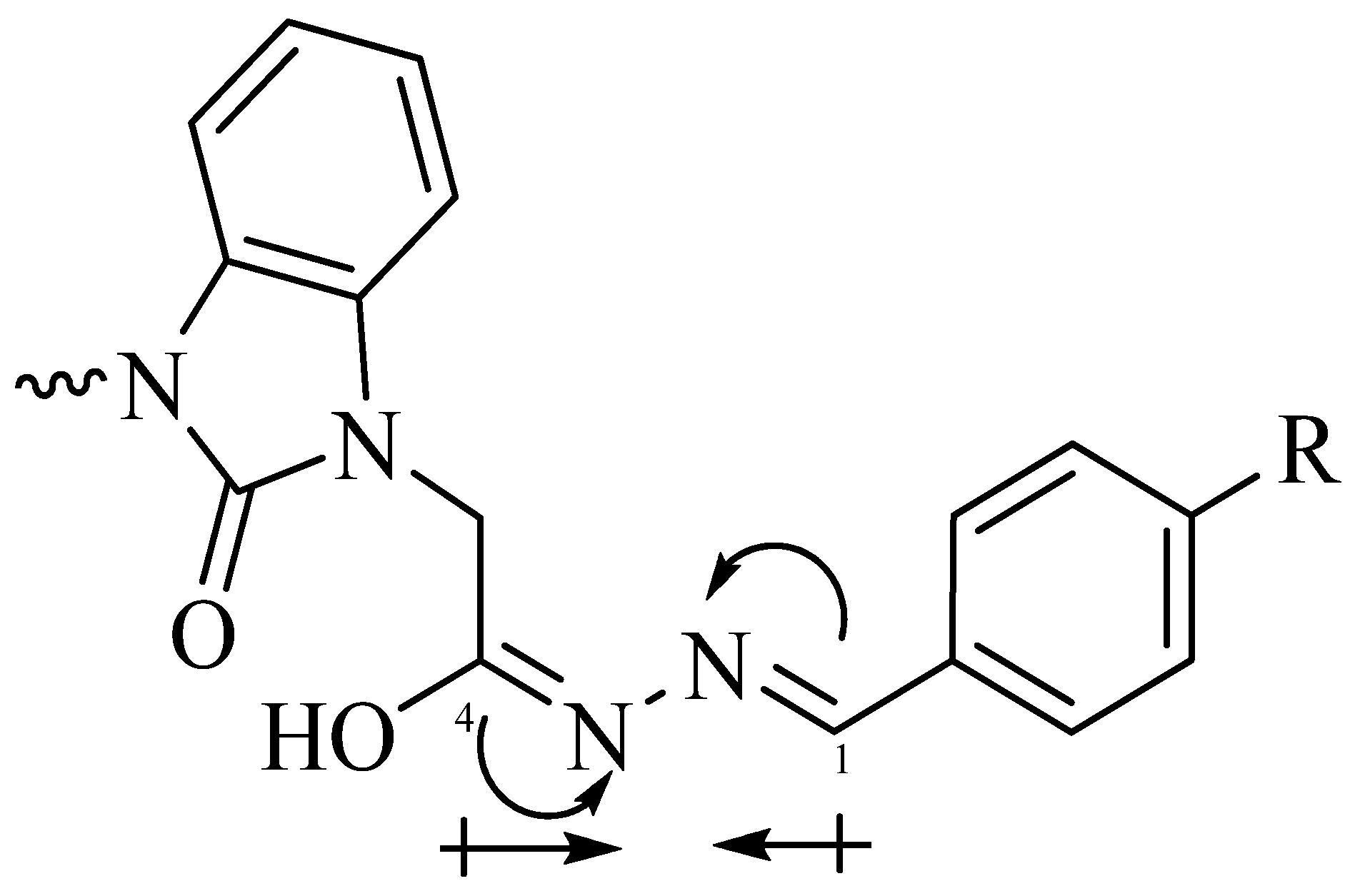
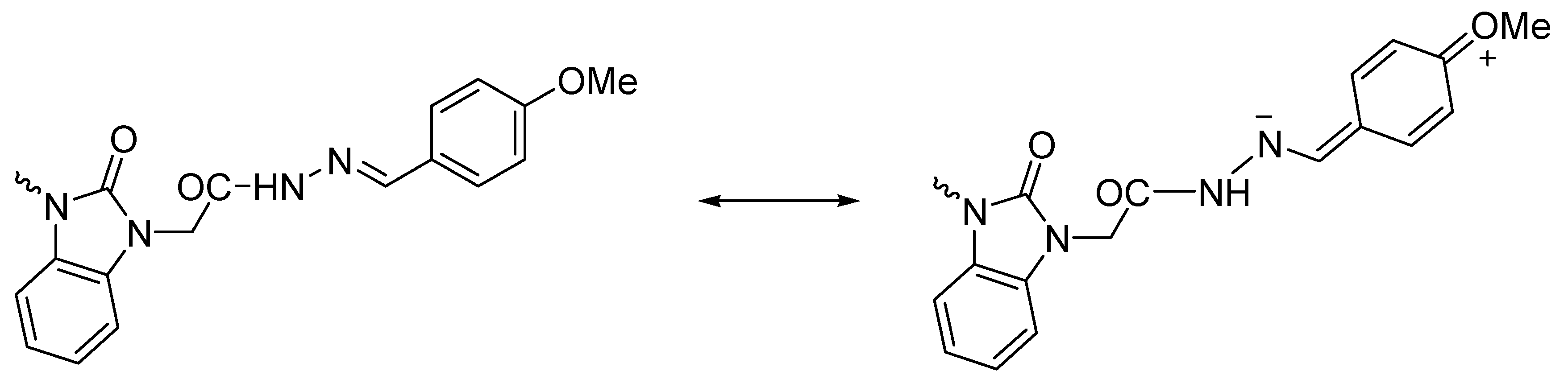
3.1. Ultrasonic Synthesis of 1H-Benzo[d]imidazol-2(3H)-one and Its Derivatives
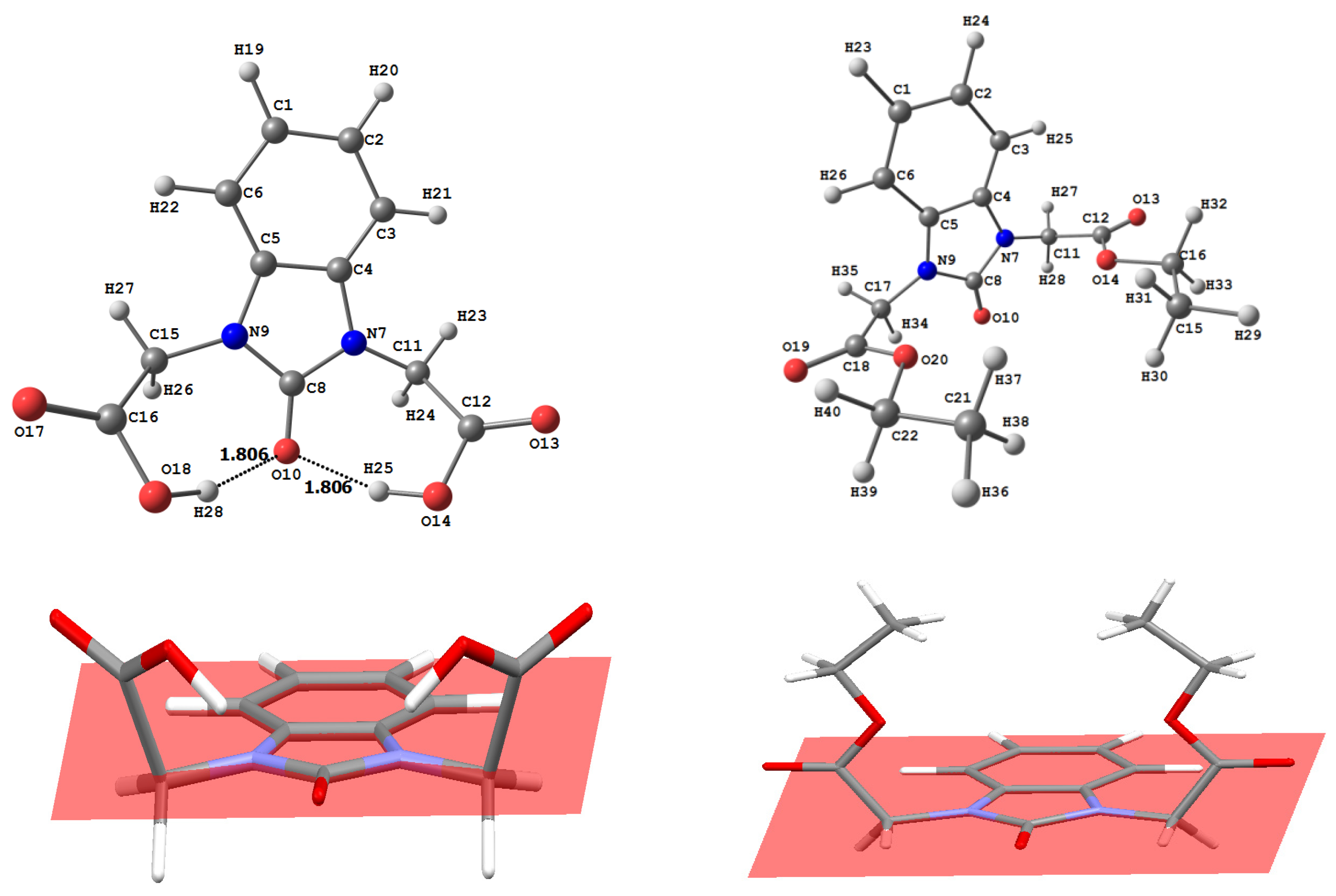
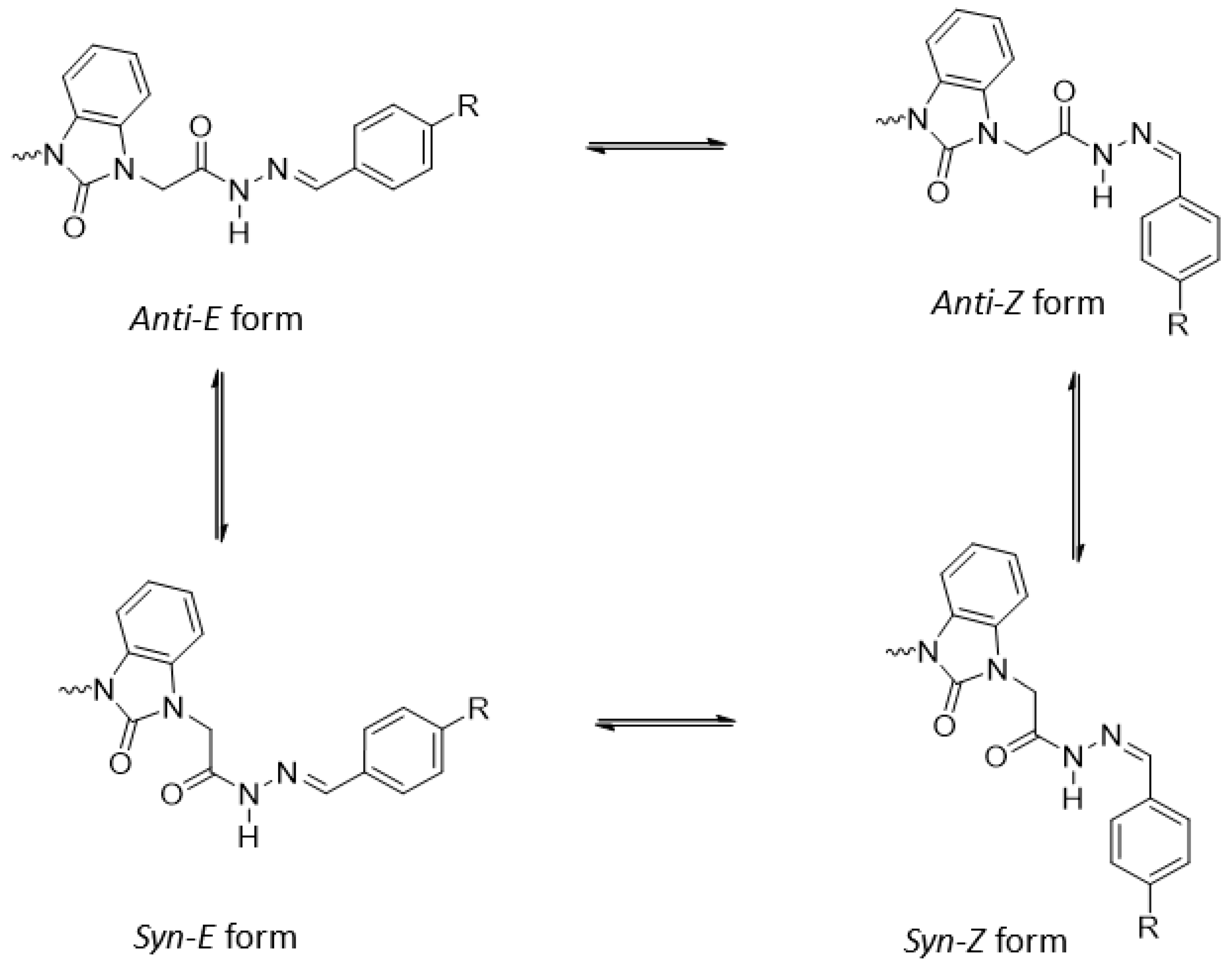
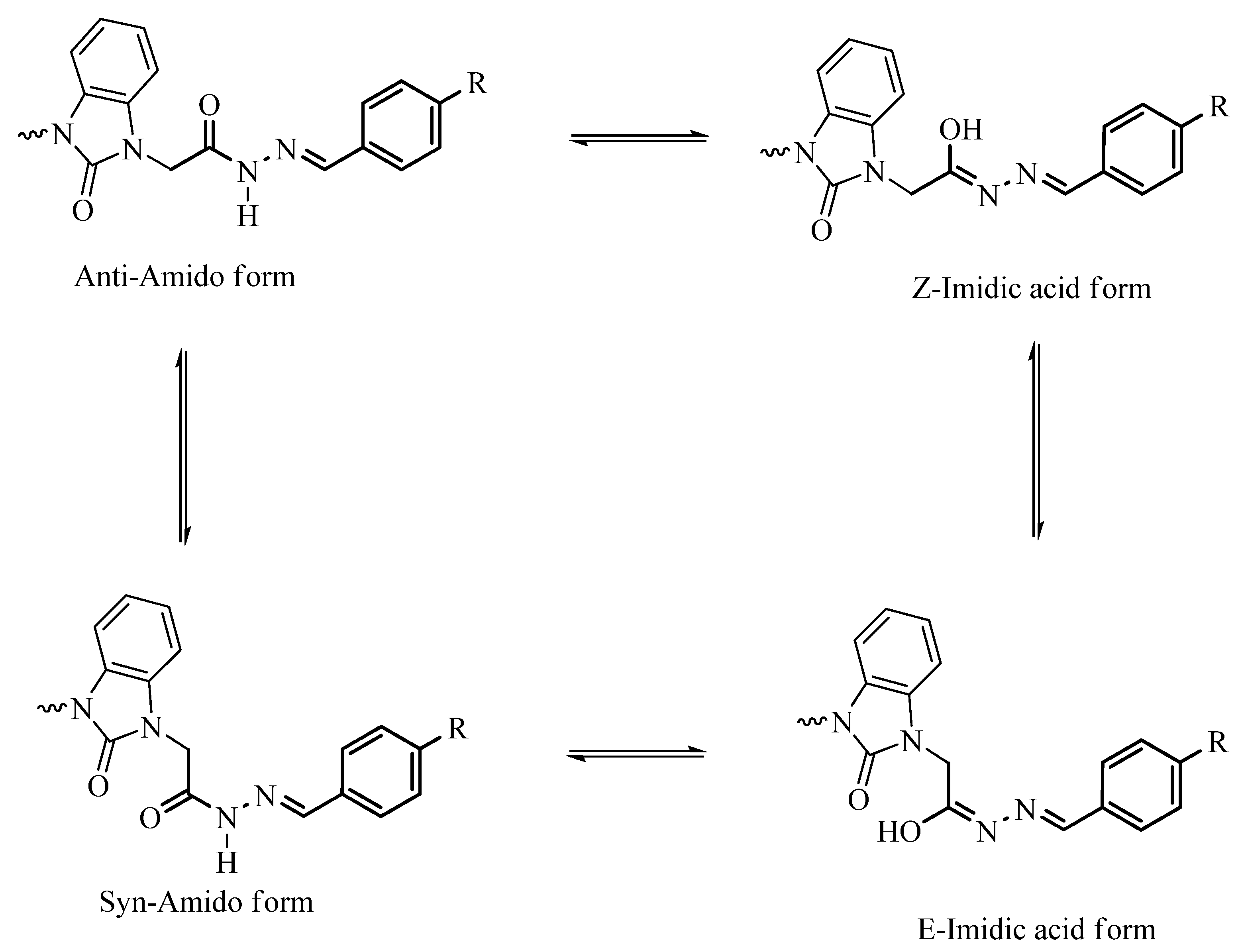
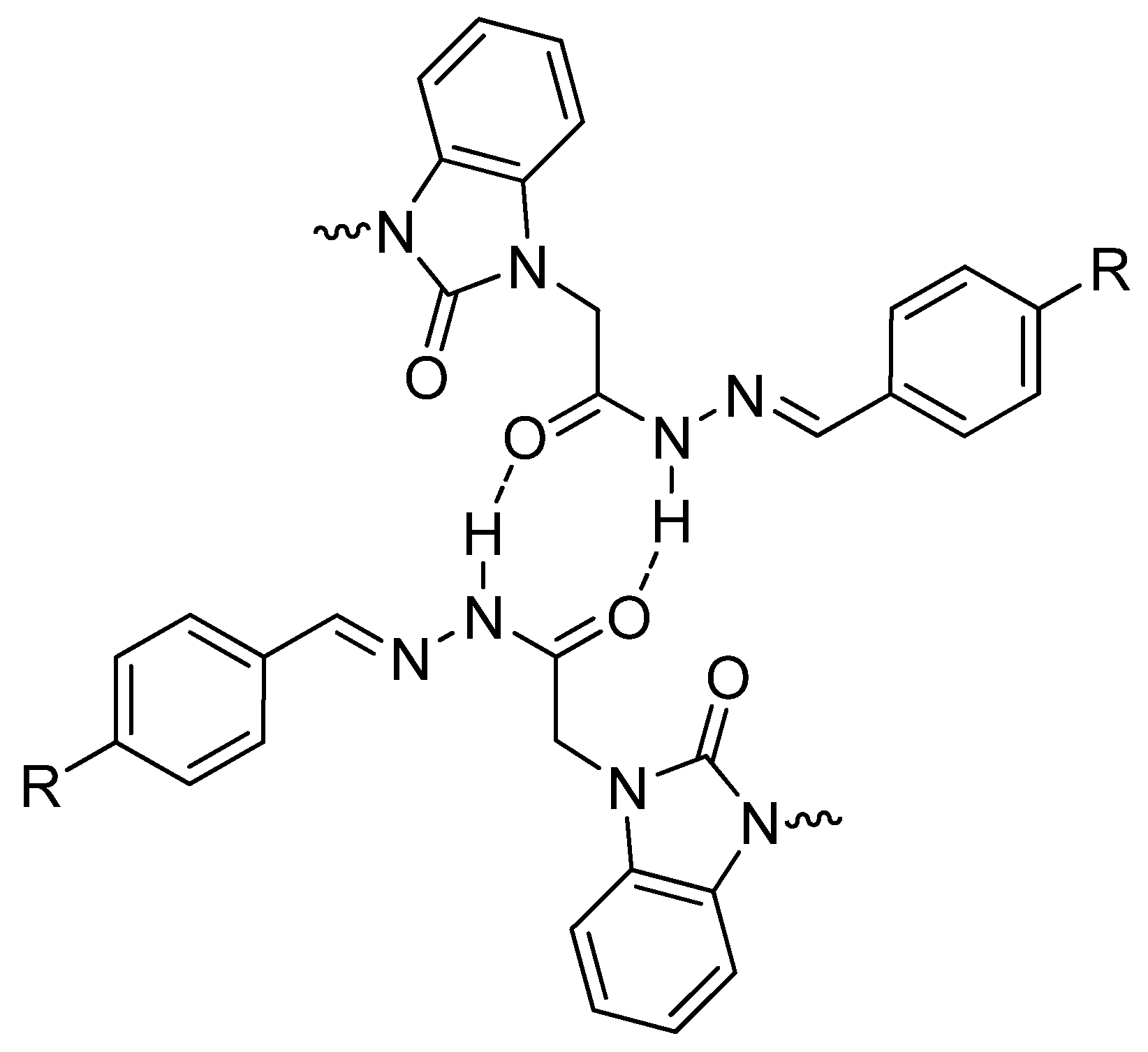
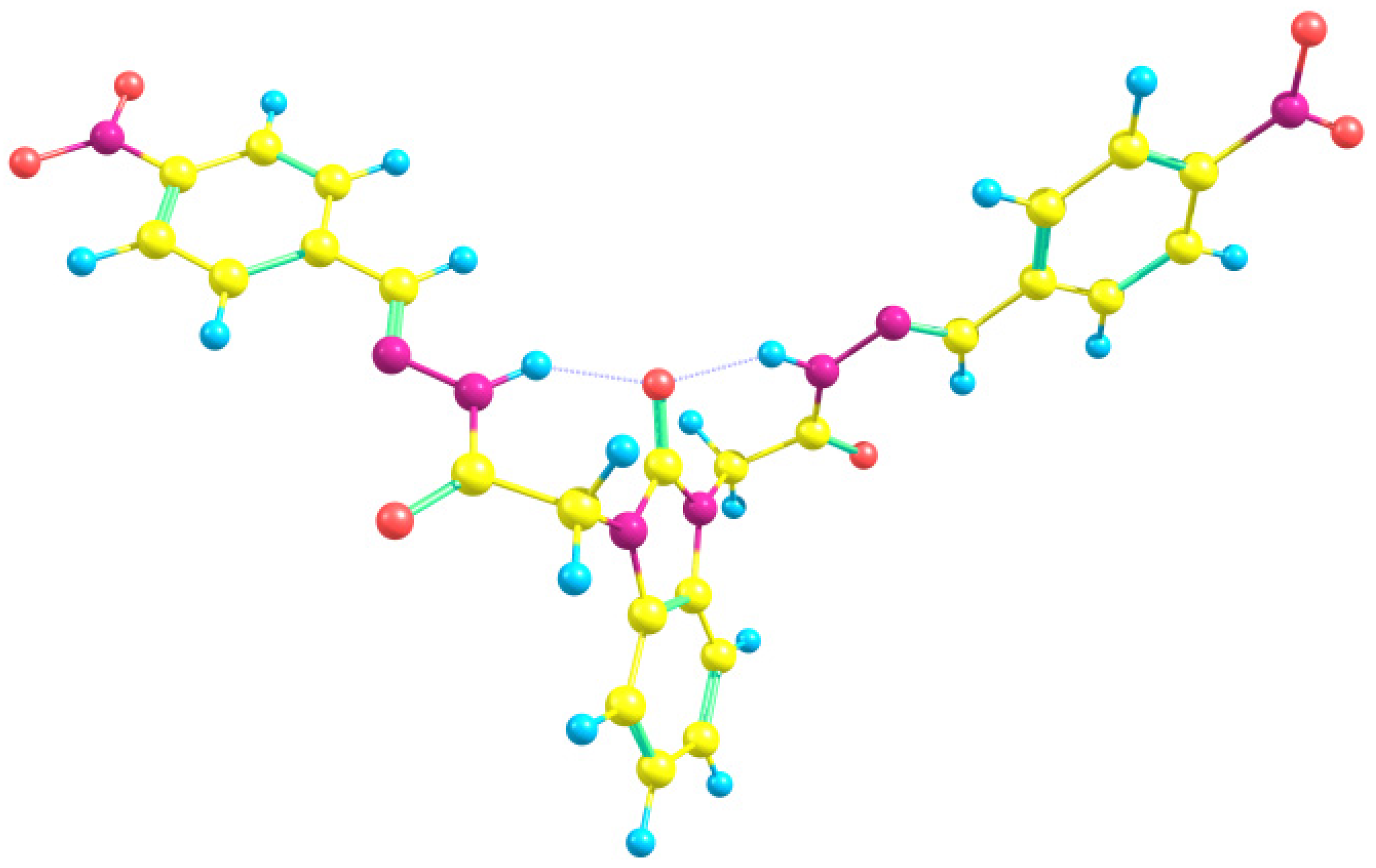
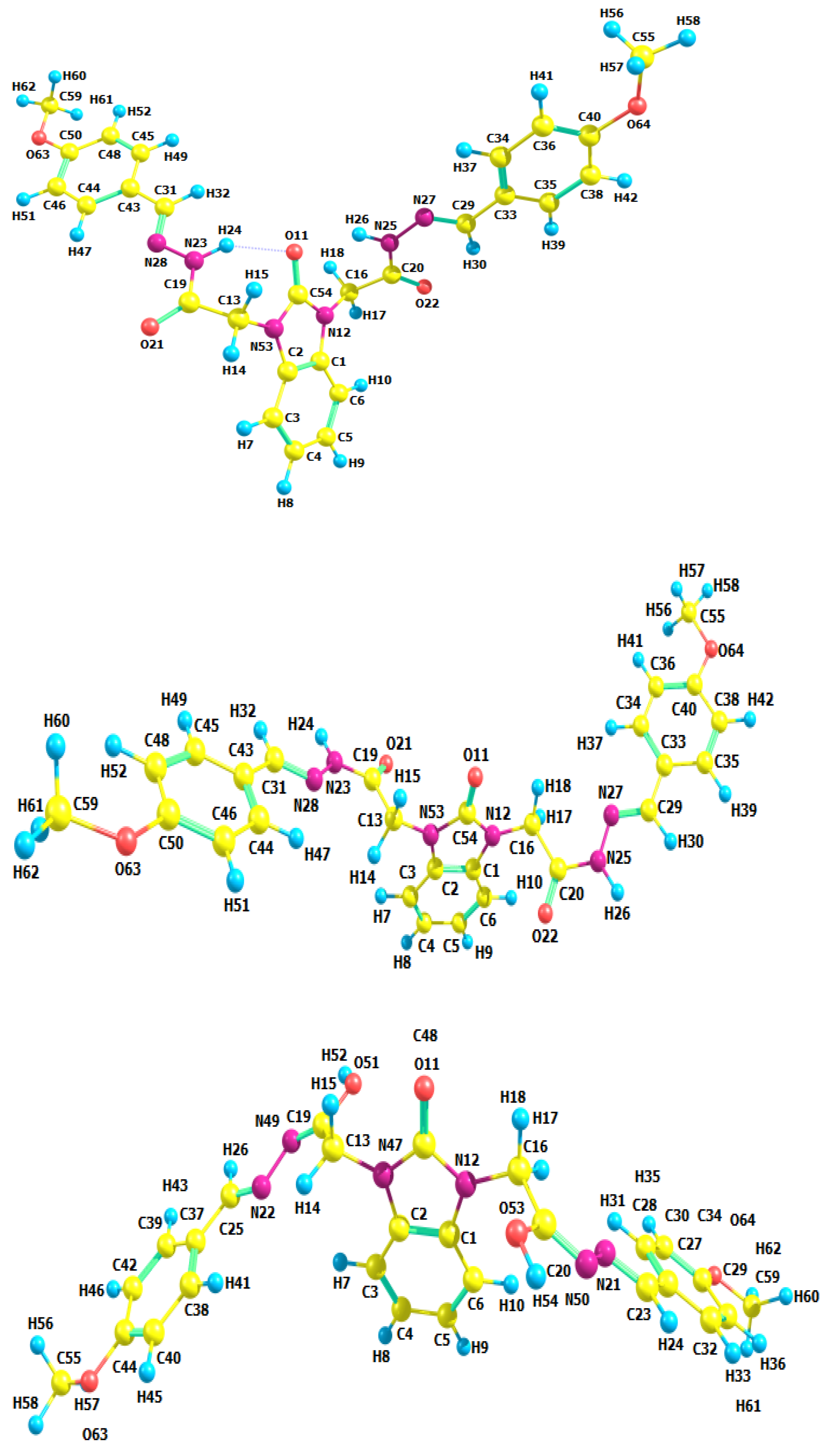
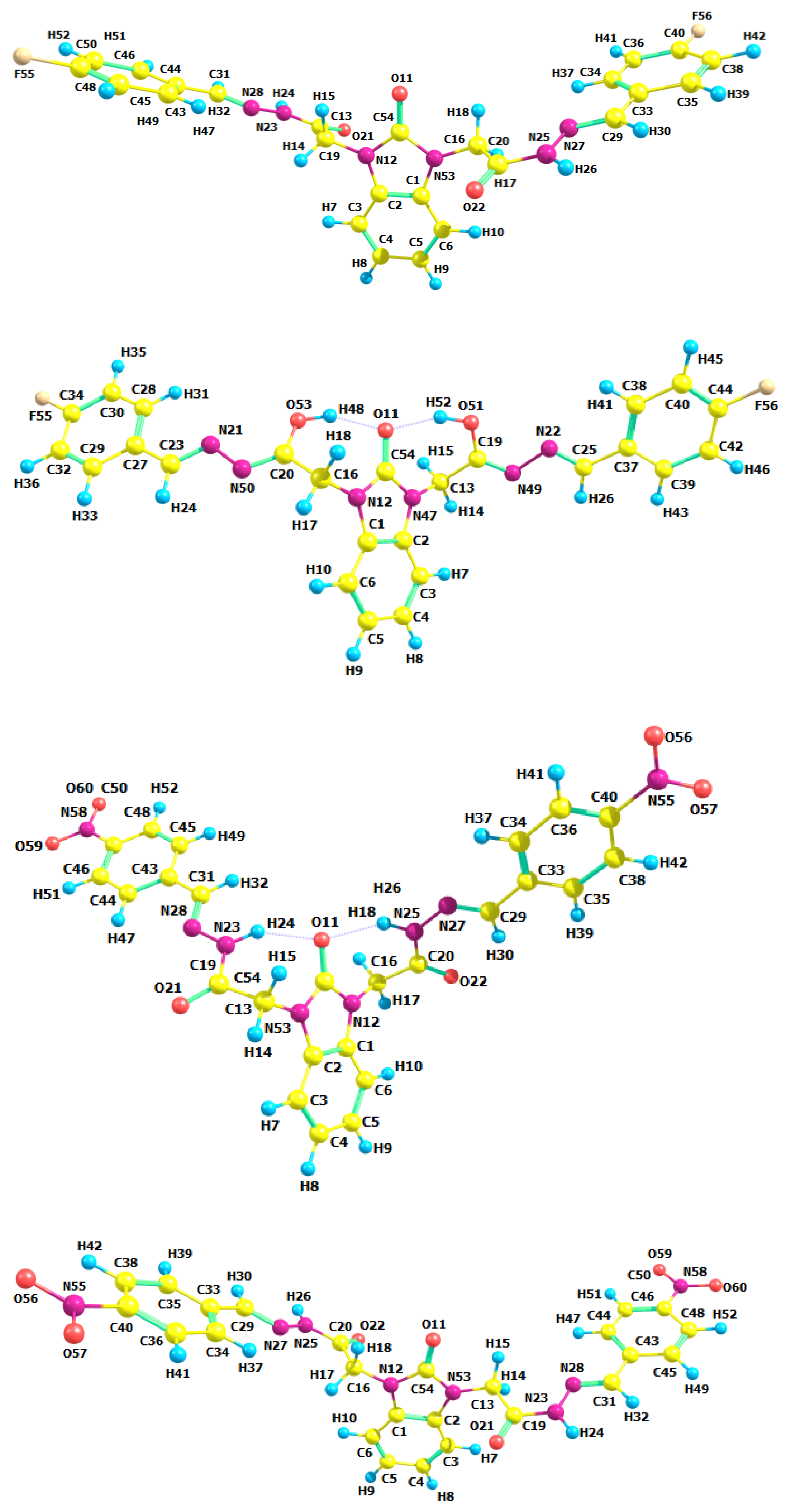
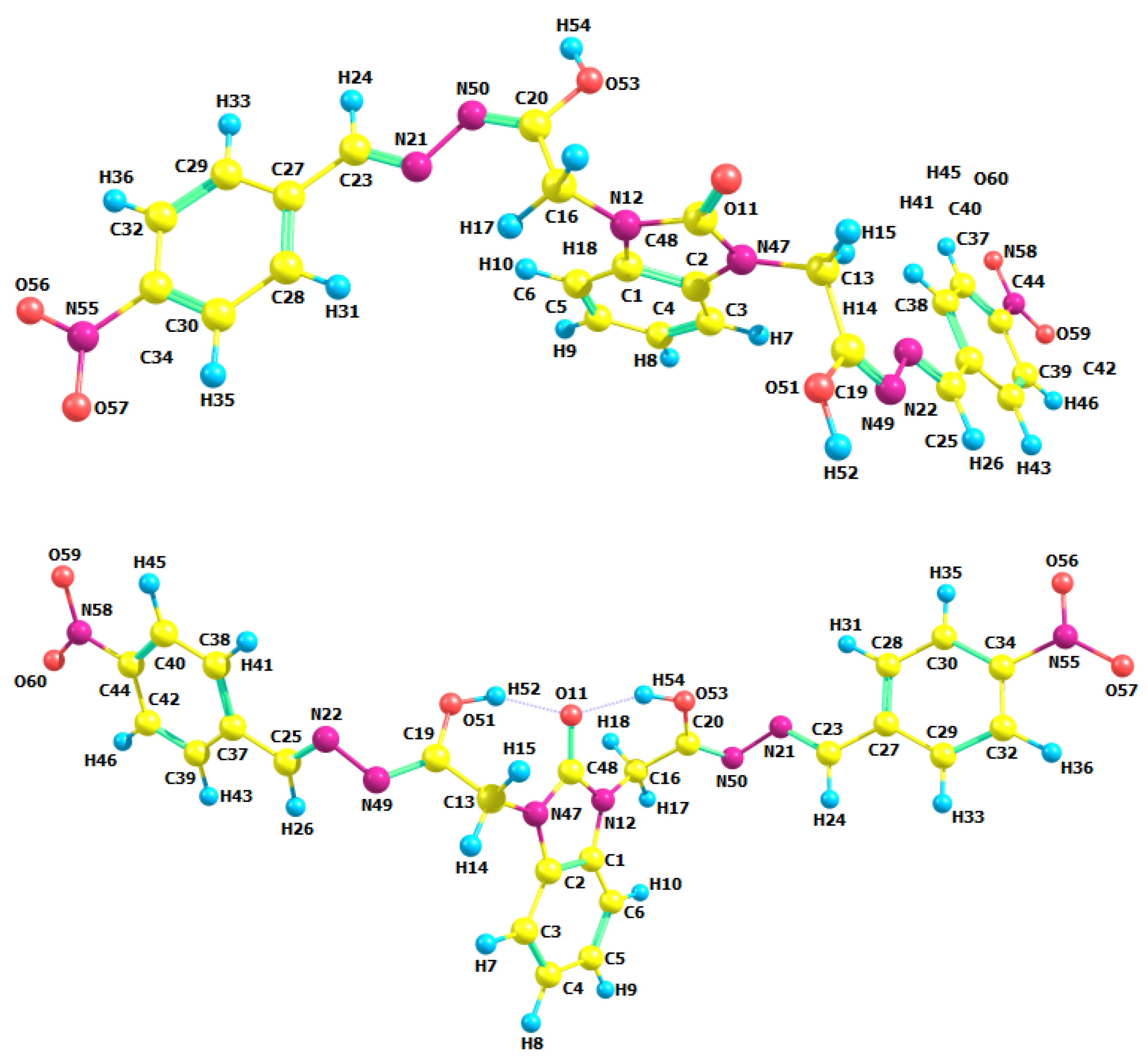
3.2. Optimized Molecular Geometry
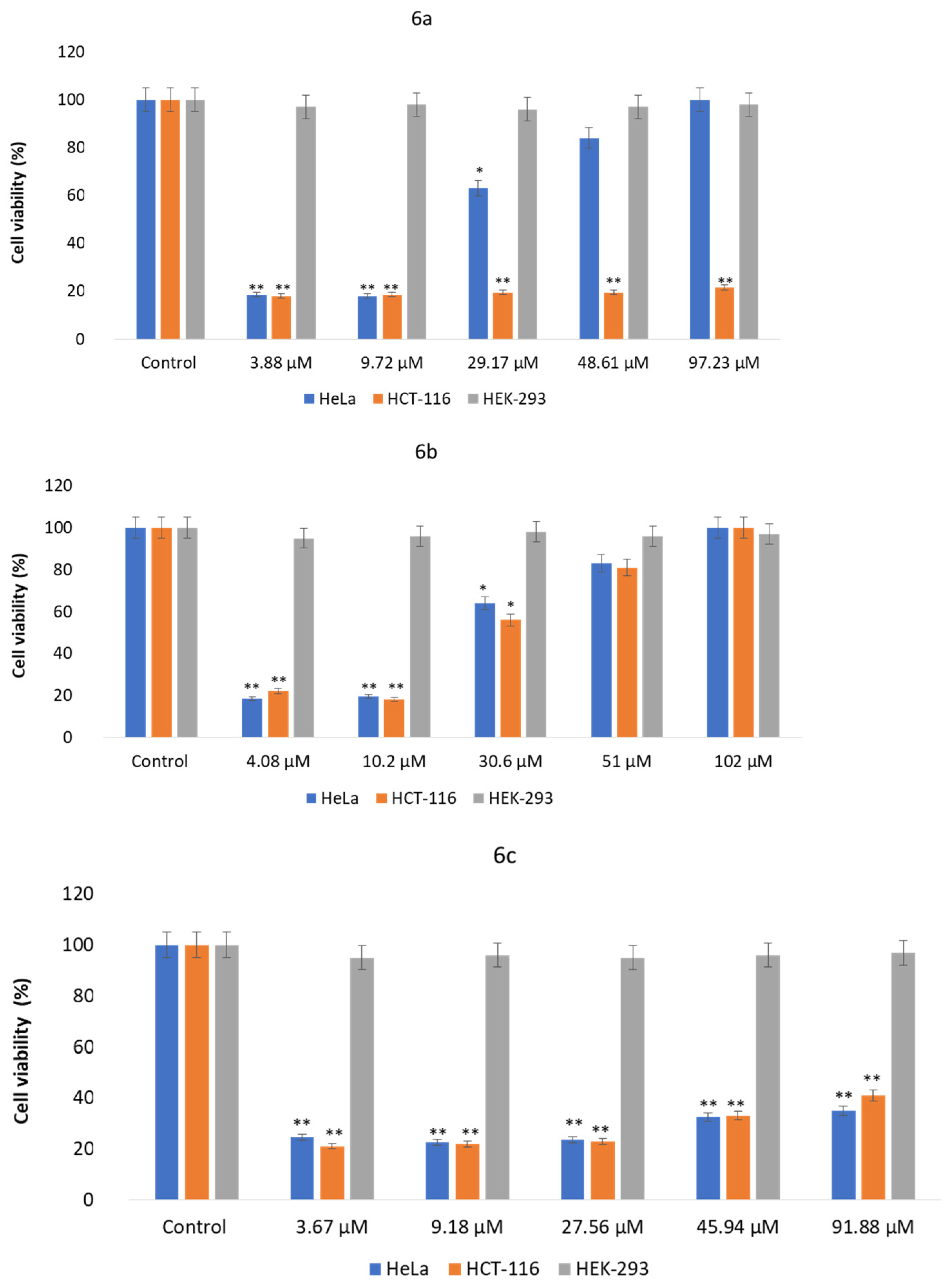
3.3. Antiproliferative Activity
3.3.1. MTT Assay
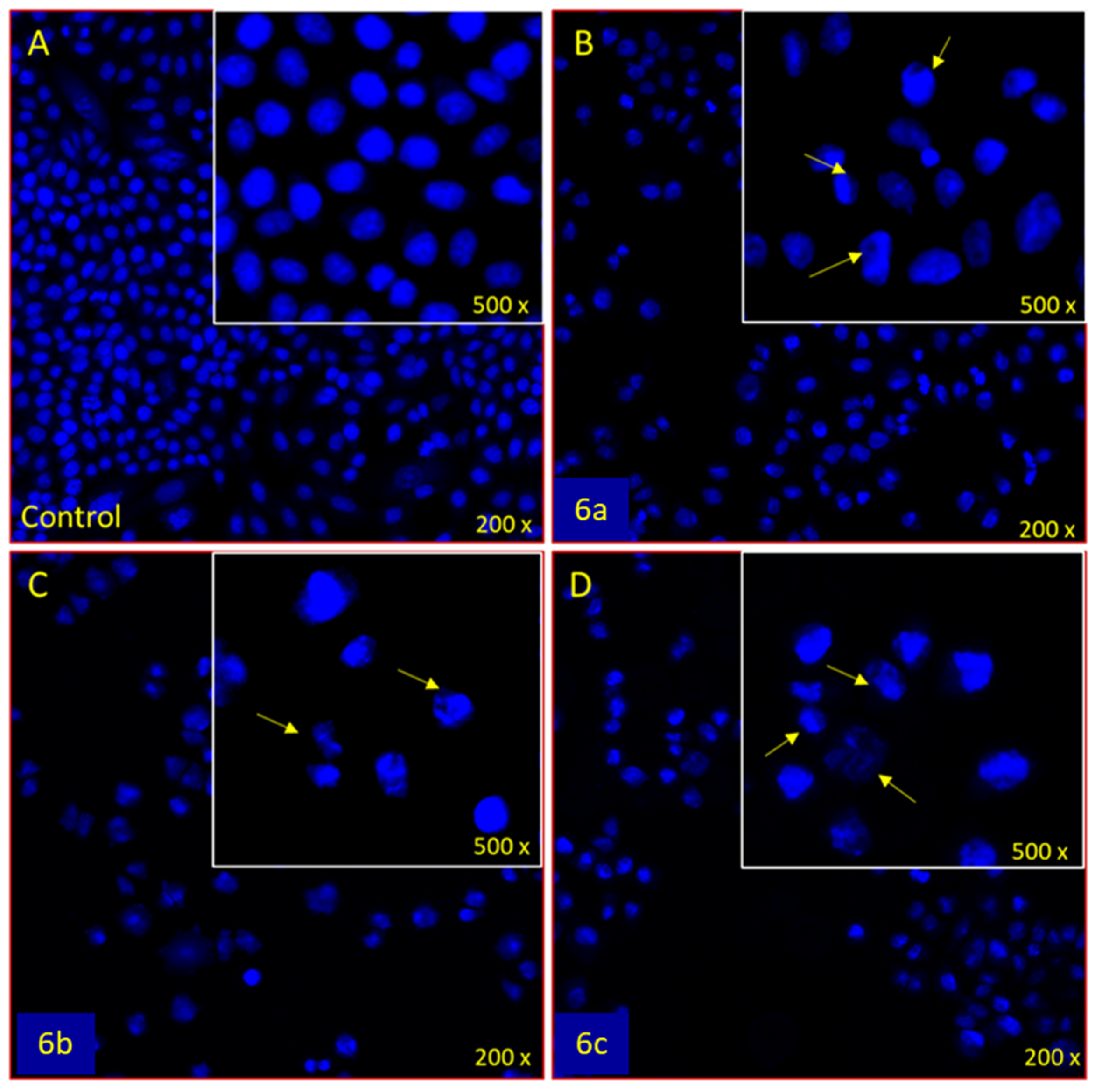
3.3.2. Apoptotic DAPI Staining
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Morcoss, M.M.; Abdelhafez, E.S.; Abdel-Rahman, H.M.; Abdel-Aziz, M.; El-Ella, A.; Dalal, A.; Sciences, P. Novel Benzimidazole/Hydrazone Derivatives as Promising Anticancer Lead Compounds: Design, Synthesis, and Molecular Docking Study. JABPS 2020, 3, 45–52. [Google Scholar] [CrossRef]
- Tahlan, S.; Kumar, S.; Kakkar, S.; Narasimhan, B. Benzimidazole scaffolds as promising antiproliferative agents: A review. BMC Chem. 2019, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Atmaca, H.; İlhan, S.; Batır, M.B.; Pulat, Ç.Ç.; Güner, A.; Bektaş, H. Novel benzimidazole derivatives: Synthesis, in vitro cytotoxicity, apoptosis and cell cycle studies. Chem.-Biol. Interact. 2020, 327, 109163. [Google Scholar] [CrossRef] [PubMed]
- El Rashedy, A.A.; Aboul-Enein, H.Y. Benzimidazole derivatives as potential anticancer agents. Mini-Rev. Med. Chem. 2013, 13, 399–407. [Google Scholar]
- Zengin-Karadayi, F.; Yaman, M.; Kisla, M.M.; Konu, Ö.; Ates-Alagoz, Z. Design, Synthesis, Anticancer Activity, Molecular Docking and ADME Studies of Novel Methylsulfonyl Indole-benzimidazoles in Comparison with Ethylsulfonyl Counterparts. New J. Chem. 2021, 45, 9010–9019. [Google Scholar] [CrossRef]
- Mashkovskii, M. Lekarstvennye Sredstva (Drugs), in 2 vols., (New); Kharkov, Ukraine, 1997; Volume 1. [Google Scholar]
- Lagorce, J.; Fatimi, J.; Lakhdar, M.; Chabernaud, M.; Buxeraud, J.; Raby, C. Synthesis and inhibitory effects of 2-pyridyl-2-thiobenzoxazole and 2-pyridyl-2-thiobenzimidazole derivatives on arachidonic acid metabolism. Arzneim.-Forsch. 1995, 45, 1207–1210. [Google Scholar]
- Garaliene, V.; Labanauskas, L.; Brukstus, A.; Dauksas, V. Synthesis and positive inotropic effects of 1-acyl-5, 6-diethoxy-2-methylthiobenzimidazoles. Arzneim.-Forsch. 1998, 48, 1137–1142. [Google Scholar] [CrossRef]
- Sammaiah, B.; Sumalatha, D.; Reddy, G.S.; Rajeswari, M.; Sharada, L. Cadmium chloride (CdCl 2): A mild and efficient catalyst for the synthesis of benzimidazoles. Int. J. Ind. Chem. 2012, 3, 11. [Google Scholar] [CrossRef][Green Version]
- Nakano, H.; Inoue, T.; Kawasaki, N.; Miyataka, H.; Matsumoto, H.; Taguchi, T.; Inagaki, N.; Nagai, H.; Satoh, T. Synthesis and biological activities of novel antiallergic agents with 5-lipoxygenase inhibiting action. Bioorg. Med. Chem. 2000, 8, 373–380. [Google Scholar] [CrossRef]
- Barnard, E.; Stein, W.; Centers, A.A.; by Organo-Phosphates, B.I.; Role, T.C.; Bonding, V.H.; Binding, A.P.M.; Nucleoproteins, V. The roles of imidazole in biological systems. Adv. Enzymol. 2006, 51–110. [Google Scholar] [CrossRef]
- Nannapaneni, D.T.; Gupta, A.V.; Reddy, M.I.; Sarva, R.C. Synthesis, Characterization, and Biological Evaluation of Benzimidazole Derivatives as Potential Anxiolytics. J. Young Pharm. JYP 2010, 2, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.; Ganguly, S.; Surana, S. A systematic review of benzimidazole derivatives as an antiulcer agent. Rasayan J. Chem. 2008, 1, 447–460. [Google Scholar]
- Valdez, J.; Cedillo, R.; Hernandez-Campos, A.; Yepez, L.; Hernandez-Luis, F.; Navarrete-Vazquez, G.; Tapia, A.; Cortes, R.; Hernández, M.; Castillo, R. Synthesis and antiparasitic activity of 1H-benzimidazole derivatives. Bioorg. Med. Chem. Lett. 2002, 12, 2221–2224. [Google Scholar] [CrossRef]
- Cochrane, J.C.; Lipchock, S.V.; Strobel, S.A. Structural investigation of the GlmS ribozyme bound to its catalytic cofactor. Chem. Biol. 2007, 14, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.J.; Been, M.D.; Ferré-D’Amaré, A.R. Essential role of an active-site guanine in glmS ribozyme catalysis. J. Am. Chem. Soc. 2007, 129, 14858–14859. [Google Scholar] [CrossRef]
- Gilbert, S.D.; Reyes, F.E.; Edwards, A.L.; Batey, R.T. Adaptive ligand binding by the purine riboswitch in the recognition of guanine and adenine analogs. Structure 2009, 17, 857–868. [Google Scholar] [CrossRef]
- Singh, V.; Peng, C.S.; Li, D.; Mitra, K.; Silvestre, K.J.; Tokmakoff, A.; Essigmann, J.M. Direct observation of multiple tautomers of oxythiamine and their recognition by the thiamine pyrophosphate riboswitch. ACS Chem. Biol. 2013, 9, 227–236. [Google Scholar] [CrossRef]
- Feng, G.S.; Chen, M.W.; Shi, L.; Zhou, Y.G. Facile Synthesis of Chiral Cyclic Ureas through Hydrogenation of 2-Hydroxypyrimidine/Pyrimidin-2 (1H)-one Tautomers. Angew. Chem. Int. Ed. 2018, 57, 5853–5857. [Google Scholar] [CrossRef]
- Abele, E.; Abele, R. Oximes of Nucleosides and Related Compounds: Synthesis, Reactions and Biological Activity. Curr. Org. Synth. 2018, 15, 650–665. [Google Scholar] [CrossRef]
- Hagar, M.; Soliman, S.M.; Ibid, F.; El Sayed, H. Quinazolin-4-yl-sulfanylacetyl-hydrazone derivatives; Synthesis, molecular structure and electronic properties. J. Mol. Struct. 2013, 1049, 177–188. [Google Scholar] [CrossRef]
- Soliman, S.M.; Hagar, M.; Ibid, F.; El Sayed, H. Experimental and theoretical spectroscopic studies, HOMO–LUMO, NBO analyses and thione–thiol tautomerism of a new hybrid of 1, 3, 4-oxadiazole-thione with quinazolin-4-one. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 145, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Hagar, M.; Soliman, S.M.; Ibid, F.; El Sayed, H. Synthesis, molecular structure and spectroscopic studies of some new quinazolin-4 (3H)-one derivatives; an account on the N-versus S-Alkylation. J. Mol. Struct. 2016, 1108, 667–679. [Google Scholar] [CrossRef]
- Aboelnaga, A.; Hagar, M.; Soliman, S.M. Ultrasonic Synthesis, Molecular Structure and Mechanistic Study of 1, 3-Dipolar Cycloaddition Reaction of 1-Alkynylpyridinium-3-olate and Acetylene Derivatives. Molecules 2016, 21, 848. [Google Scholar] [CrossRef] [PubMed]
- Kotzebue, L.R.; de Oliveira, J.s.R.; da Silva, J.B.; Mazzetto, S.E.; Ishida, H.; Lomonaco, D. Development of Fully Biobased High-Performance Bis-Benzoxazine under Environmentally Friendly Conditions. ACS Sustain. Chem. Eng. 2018, 6, 5485–5494. [Google Scholar] [CrossRef]
- Soltani, R.; Shahvar, A.; Dinari, M.; Saraji, M. Environmentally-friendly and ultrasonic-assisted preparation of two-dimensional ultrathin Ni/Co-NO3 layered double hydroxide nanosheet for micro solid-phase extraction of phenolic acids from fruit juices. Ultrason. Sonochem. 2018, 40, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-J.; Hong, S.-B.; Eom, T.-J. Preparation of Eucalyptus pulp by mild condition of low-temperature, atmospheric pressure, and short-reaction-time with high-boiling-point solvent and pulp properties. Cellulose 2018, 25, 753–761. [Google Scholar] [CrossRef]
- Maleki, A. Green oxidation protocol: Selective conversions of alcohols and alkenes to aldehydes, ketones and epoxides by using a new multiwall carbon nanotube-based hybrid nanocatalyst via ultrasound irradiation. Ultrason. Sonochem. 2018, 40, 460–464. [Google Scholar] [CrossRef]
- Yadav, J.; Reddy, B.; Reddy, K.S. Ultrasound-accelerated synthesis of chiral allylic alcohols promoted by indium metal. Tetrahedron 2003, 59, 5333–5336. [Google Scholar] [CrossRef]
- Wang, S.Y.; Ji, S.J.; Su, X.M. A meldrum’s acid catalyzed synthesis of Bis (indolyl) methanes in water under ultrasonic condition. Chin. J. Chem. 2008, 26, 22–24. [Google Scholar] [CrossRef]
- Pasha, M.; Jayashankara, V. Reduction of arylnitro compounds to azoarenes and/or arylamines by Al/NaOH in methanol under ultrasonic conditions. Ultrason. Sonochem. 2005, 12, 433–435. [Google Scholar] [CrossRef]
- Li, J.-T.; Li, X.-L.; Li, T.-S. Synthesis of oximes under ultrasound irradiation. Ultrason. Sonochem. 2006, 13, 200–202. [Google Scholar] [CrossRef] [PubMed]
- Disselkamp, R.S.; Hart, T.R.; Williams, A.M.; White, J.; Peden, C.H. Ultrasound-assisted hydrogenation of cinnamaldehyde. Ultrason. Sonochem. 2005, 12, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Shaabani, A.; Hooshmand, S.E. Diversity-oriented catalyst-free synthesis of pseudopeptides containing rhodanine scaffolds via a one-pot sequential is ocyanide-based six-component reactions in water using ultrasound irradiation. Ultrason. Sonochem. 2018, 40, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Abdel Hafez, N.A.; Hassaneen, H.M.; Farghaly, T.A.; Riyadh, S.M.; Elzahabi, H.S. Synthesis of New Bis-Spiropyrazoles as Antitumor Agents under Ultrasound Irradiation. Mini Rev. Med. Chem. 2018, 18, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Cintas, P.; Luche, J.-L. Green chemistry. The sonochemical approach. Green Chem. 1999, 1, 115–125. [Google Scholar] [CrossRef]
- Qiao, S.Z.; Liu, J.; Lu, G.Q.M. Synthetic chemistry of nanomaterials. In Modern Inorganic Synthetic Chemistry; Elsevier: Amsterdam, The Netherlands, 2011; pp. 479–506. [Google Scholar]
- Brahmachari, G. Green Synthetic Approaches for Biologically Relevant Heterocycles: Advanced Synthetic Techniques—An Overview; Elsevier: Amsterdam, The Netherlands, 2021; pp. 1–8. [Google Scholar]
- Mou, L.; Liang, B.; Liu, G.; Jiang, J.; Liu, J.; Zhou, B.; Huang, J.; Zang, N.; Liao, Y.; Ye, L.; et al. Berbamine exerts anticancer effects on human colon cancer cells via induction of autophagy and apoptosis, inhibition of cell migration and MEK/ERK signalling pathway. J. BUON Off. J. Balk. Union Oncol. 2019, 24, 1870–1875. [Google Scholar]
- Khorsandi, L.; Orazizadeh, M.; Niazvand, F.; Abbaspour, M.; Mansouri, E.; Khodadadi, A.J.B.M.J. Quercetin induces apoptosis and necroptosis in MCF-7 breast cancer cells. Bratisl. Med. J. 2017, 118, 123–128. [Google Scholar] [CrossRef]
- Mettu, A.; Talla, V.; Thumma, S.; Prameela, S.N. Mechanistic investigations on substituted benzene sulphonamides as apoptosis inducing anticancer agents. Bioorg. Chem. 2020, 95, 103539. [Google Scholar] [CrossRef]
- EL-Sayed, T.; Aboelnaga, A.; Hagar, M. Ball Milling Assisted Solvent and Catalyst Free Synthesis of Benzimidazoles and Their Derivatives. Molecules 2016, 21, 1111. [Google Scholar] [CrossRef]
- El Ashry, E.S.H.; El Kilany, Y.; Nahas, N.M.; Barakat, A.; Al-Qurashi, N.; Ghabbour, H.A.; Fun, H.-K.J.M. Synthesis and crystal structures of benzimidazole-2-thione derivatives by alkylation reactions. Molecules 2016, 21, 12. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01. 2009. Available online: https://www.scirp.org/%28S%28vtj3fa45qm1ean45vvffcz55%29%29/reference/referencespapers.aspx?referenceid=1399207 (accessed on 16 March 2022).
- Dennington, R.; Keith, T.; Millam, J. Shawnee Mission KS GaussView Version; Semichem Inc.: Shawnee, KS, USA, 2009; Volume 5. [Google Scholar]
- Khan, F.A.; Akhtar, S.; Almohazey, D.; Alomari, M.; Almofty, S.A.; Eliassari, A. Fluorescent magnetic submicronic polymer (FMSP) nanoparticles induce cell death in human colorectal carcinoma cells. Artif. Cells Nanomed. Biotechnol. 2018, 46, S247–S253. [Google Scholar] [CrossRef] [PubMed]
- Baig, U.; Ansari, M.A.; Gondal, M.; Akhtar, S.; Khan, F.A.; Falath, W. Single step production of high-purity copper oxide-titanium dioxide nanocomposites and their effective antibacterial and anti-biofilm activity against drug-resistant bacteria. Mater. Sci. Eng. C 2020, 113, 110992. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.; Asiri, S.M.; Khan, F.A.; Jermy, B.R.; Khan, H.; Akhtar, S.; Jindan, R.A.; Khan, K.M.; Qurashi, A. Biocompatible tin oxide nanoparticles: Synthesis, antibacterial, anticandidal and cytotoxic activities. ChemistrySelect 2019, 4, 4013–4017. [Google Scholar] [CrossRef]
- El Rayes, S.M.; Aboelmagd, A.; Gomaa, M.S.; Ali, I.A.; Fathalla, W.; Pottoo, F.H.; Khan, F.A. Convenient Synthesis and Anticancer Activity of Methyl 2-[3-(3-Phenyl-quinoxalin-2-ylsulfanyl) propanamido] alkanoates and N-Alkyl 3-((3-Phenyl-quinoxalin-2-yl) sulfanyl) propanamides. ACS Omega 2019, 4, 18555–18566. [Google Scholar] [CrossRef]
- Thakuria, H.; Das, G. An expeditious one-pot solvent-free synthesis of benzimidazole Derivatives. Arkivoc 2008, 15, 321–328. [Google Scholar] [CrossRef]
- Anbarasu, G.; Srividhya, C.; Maheswaran, P.; Rajavel, R. Isatin Based Schiff Base And Its Complexes-Synthesis, Spectral Investigation And Antibacterial Studies. J. Appl. Chem. 2013, 2, 1315–1323. [Google Scholar]
- D’yakonov, V.A.; Finkelshtein, E.S.; Ibragimov, A.G. Dzhemilev reaction for the synthesis of spiro [3.3] heptane and spiro [3.4]octanes. Tetrahedron Lett. 2007, 48, 8583–8586. [Google Scholar] [CrossRef]
- Elassar, A.-Z.A.; Dib, H.H.; Al-Awadi, N.A.; Elnagdi, M.H. Chemistry of carbofunctionally substituted hydrazones. Arkivoc 2007, 2, 272–315. [Google Scholar] [CrossRef]
- Rutavlcius, A.; Valiulene, S.; Kuodis, Z. Isomerism of hydrazones of (2-benzothiazolylthio)-and (2-benzoxazolylthio) acetic acids. Chem. Heterocycl. Compd. 1995, 31, 629–633. [Google Scholar] [CrossRef]
- Rutavichyus, A.; Valyulene, S.; Kuodis, Z. Synthesis and structure of dihydrazones obtained from the dihydrazide of 1,3,4-thiadiazole-2,5-dithioglycolic acid. Chem. Heterocycl. Compd. 1997, 33, 118–124. [Google Scholar] [CrossRef]
- Palla, G.; Predieri, G.; Domiano, P.; Vignali, C.; Turner, W. Conformational behaviour and E/Z isomerization of N-acyl and N-aroylhydrazones. Tetrahedron 1986, 42, 3649–3654. [Google Scholar] [CrossRef]
- Tavakol, H. Computational study of simple and water-assisted tautomerism of hydroxamic acids. J. Mol. Struct. THEOCHEM 2009, 916, 172–179. [Google Scholar] [CrossRef]
- Sałdyka, M.; Mielke, Z. Keto–iminol tautomerism in acetohydroxamic and formohydroxamic acids: Experimental and theoretical study. Vib. Spectrosc. 2007, 45, 46–54. [Google Scholar] [CrossRef]
- Tolosa, S.; Mora-Diez, N.; Hidalgo, A.; Sansón, J. Amide-imide tautomerism of acetohydroxamic acid in aqueous solution: Quantum calculation and SMD simulations. RSC Adv. 2014, 4, 44757–44768. [Google Scholar] [CrossRef]
- Kakkar, R.; Grover, R.; Chadha, P. Conformational behavior of some hydroxamic acids. Org. Biomol. Chem. 2003, 1, 2200–2206. [Google Scholar] [CrossRef]
- Emamian, S.R.; Domingo, L.R.; Tayyari, S.F. Tautomerism in pyridazin-3 (2H)-one: A theoretical study using implicit/explicit solvation models. J. Mol. Graph. Model. 2014, 49, 47–54. [Google Scholar] [CrossRef]
- Grosch, A.A.; van der Lubbe, S.C.C.; Fonseca Guerra, C. Nature of Intramolecular Resonance Assisted Hydrogen Bonding in Malonaldehyde and Its Saturated Analogue. J. Phys. Chem. A 2018, 122, 1813–1820. [Google Scholar] [CrossRef]
- Dhanishta, P.; Sai Siva kumar, P.; Mishra, S.K.; Suryaprakash, N. Intramolecular hydrogen bond directed stable conformations of benzoyl phenyl oxalamides: Unambiguous evidence from extensive NMR studies and DFT-based computations. RSC Adv. 2018, 8, 11230–11240. [Google Scholar] [CrossRef]
- Clow, A. Resonance in urea and its derivatives—Part I—Diamagnetics. Trans. Faraday Soc. 1937, 33, 381–388. [Google Scholar] [CrossRef]
- Ren, B.; Liu, R.-C.; Ji, K.; Tang, J.-J.; Gao, J.-M.J.B.; Letters, M.C. Design, synthesis and in vitro antitumor evaluation of novel pyrazole-benzimidazole derivatives. Bioorg. Med. Chem. Lett. 2021, 43, 128097. [Google Scholar] [CrossRef]
- Saeed, S.; Rashid, N.; Jones, P.G.; Ali, M.; Hussain, R. Synthesis, characterization and biological evaluation of some thiourea derivatives bearing benzothiazole moiety as potential antimicrobial and anticancer agents. Eur. J. Med. Chem. 2010, 45, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Taherian, E.; Khodarahmi, G.; Khajouei, M.R.; Hassanzadeh, F.; Dana, N. Synthesis and cytotoxic evaluation of novel quinozalinone derivatives with substituted benzimidazole in position 3. Res. Pharm. Sci. 2019, 14, 247. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Meguid, E.A.; El-Deen, E.M.M.; Nael, M.A.; Anwar, M.M. Novel benzimidazole derivatives as anti-cervical cancer agents of potential multi-targeting kinase inhibitory activity. Arab. J. Chem. 2020, 13, 9179–9195. [Google Scholar] [CrossRef]
- Hoffman, J.M.; Margolis, K.G. Building community in the gut: A role for mucosal serotonin. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 6–8. [Google Scholar] [CrossRef]
- Khan, F.A.; Akhtar, S.; Almohazey, D.; Alomari, M.; Almofty, S.A. Extracts of clove (Syzygium aromaticum) potentiate FMSP-nanoparticles induced cell death in MCF-7 cells. Int. J. Biomater. 2018, 2018. [Google Scholar] [CrossRef]
- Qi, F.; Yan, Q.; Zheng, Z.; Liu, J.; Chen, Y.; Zhang, G.J.J.B. Geraniol and geranyl acetate induce potent anticancer effects in colon cancer Colo-205 cells by inducing apoptosis, DNA damage and cell cycle arrest. J. BUON Off. J. Balk. Union Oncol. 2018, 23, 346–352. [Google Scholar]
- Wang, Y.; Shi, L.-Y.; Qi, W.-H.; Yang, J.; Qi, Y. Anticancer activity of sugiol against ovarian cancer cell line SKOV3 involves mitochondrial apoptosis, cell cycle arrest and blocking of the RAF/MEK/ERK signalling pathway. Arch. Med. Sci. 2020, 16, 428. [Google Scholar] [CrossRef] [PubMed]
- Mettu, A.; Talla, V.; Naikal, S.J.P.; Letters, M.C. Novel anticancer Hsp90 inhibitor disubstituted pyrazolyl 2-aminopyrimidine compound 7t induces cell cycle arrest and apoptosis via mitochondrial pathway in MCF-7 cells. Bioorg. Med. Chem. Lett. 2020, 30, 127470. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Etot (hartrees) | ZPVE (hartrees) | Ecorr (hartrees) | H (hartrees) | G (hartrees) | µ Deby | ΔE (kcal/mol) | ΔH (kcal/mol) | ΔG (kcal/mol) | |
|---|---|---|---|---|---|---|---|---|---|---|
| 6a | syn | −1748.6260 | 0.507655 | −1748.1184 | −1748.0833 | −1748.1922 | 3.619 | 0.50 | 0.25 | 1.7 |
| anti | −1748.6271 | 0.509456 | −1748.1176 | −1748.0829 | −1748.1895 | 3.031 | ||||
| Z | −1748.5762 | 0.506670 | −1748.0696 | −1748.0348 | −1748.1419 | 6.193 | 1.20 | 0.94 | 1.82 | |
| E | −1748.5783 | 0.506799 | −1748.0715 | −1748.0363 | −1748.1448 | 5.801 | ||||
| 6b | syn | −1718.0825 | 0.425877 | −1717.6566 | −1717.6251 | −1717.7250 | 0.873 | 0.56 | 0.82 | 0.82 |
| anti | −1718.0852 | 0.427737 | −1717.6575 | −1717.6264 | −1717.7263 | 3.721 | ||||
| Z | −1718.0343 | 0.424811 | −1717.6095 | −1717.5782 | −1717.6784 | 7.045 | 0.25 | 0.31 | 1.38 | |
| E | −1718.0334 | 0.424245 | −1717.6091 | −1717.5777 | −1717.6762 | 2.419 | ||||
| 6c | syn | −1928.4927 | 0.446261 | −1928.0465 | −1928.0116 | −1928.1217 | 4.038 | 1.69 | 1.95 | 0.25 |
| anti | −1928.4972 | 0.448051 | −1928.0492 | −1928.0147 | −1928.1221 | 8.282 | ||||
| Z | −1928.4466 | 0.445125 | −1928.0015 | −1927.9669 | −1928.0754 | 11.412 | 1.44 | 1.63 | 0.63 | |
| E | −1928.4444 | 0.445191 | −1927.9992 | −1927.9643 | −1928.0744 | 0.9046 | ||||
| ΔE (kcal/mol) | ΔH (kcal/mol) | |||||||
|---|---|---|---|---|---|---|---|---|
| Syn-Z | Syn-E | Anti-Z | Anti-E | Syn-Z | Syn-E | Anti-Z | Anti-E | |
| 6a | 30.62 | 29.93 | 30.12 | 28.92 | 30.43 | 29.49 | 30.18 | 29.24 |
| 6b | 29.56 | 29.81 | 30.12 | 30.37 | 29.43 | 29.74 | 30.25 | 30.56 |
| 6c | 28.24 | 29.86 | 29.93 | 31.38 | 28.05 | 29.68 | 30.00 | 31.62 |
| Structural Parameters | 6a | 6b | 6c | |||
|---|---|---|---|---|---|---|
| Syn | Anti | Syn | Anti | Syn | Anti | |
| R(a) | ||||||
| C54-O11 | 1.246 | 1.269 | 1.247 | 1.270 | 1.247 | 1.271 |
| N53-C54 | 1.400 | 1.384 | 1.400 | 1.385 | 1.399 | 1.384 |
| C19-O21 | 1.243 | 1.242 | 1.242 | 1.241 | 1.240 | 1.250 |
| C19-N23 | 1.381 | 1.376 | 1.384 | 1.378 | 1.389 | 1.375 |
| N23-H24 | 1.019 | 1.027 | 1.027 | 1.027 | 1.028 | 1.020 |
| N23-N28 | 1.378 | 1.381 | 1.374 | 1.378 | 1.366 | 1.383 |
| C31-N28 | 1.297 | 1.297 | 1.296 | 1.296 | 1.297 | 1.301 |
| A (a) | ||||||
| N12-C16-C20 | 112.14 | 114.05 | 112.08 | 113.92 | 112.00 | 113.96 |
| C16-C20-N25 | 115.41 | 113.87 | 115.60 | 113.92 | 115.72 | 112.90 |
| N12-C54-N53 | 105.68 | 107.22 | 105.67 | 107.26 | 105.67 | 107.34 |
| C20-N25-N27 | 121.82 | 129.82 | 121.78 | 120.54 | 121.76 | 129.36 |
| N27-C29-C33 | 122.78 | 119.94 | 122.14 | 121.291 | 121.63 | 119.13 |
| D (a) | ||||||
| C19-C13-N53-C54 | 100.18 | 80.87 | 99.46 | 80.09 | 98.26 | 79.45 |
| N28-C31-C43-C44 | 0.75 | 0.346 | 1.03 | 0.22 | 0.59 | 0.03 |
| C1-C2-N53-C54 | 1.30 | 0.729 | 1.29 | 1.12 | 1.28 | 1.22 |
| Compound | HCT-116 (IC50) | HeLa (IC50) | HEK-293 (IC50) |
|---|---|---|---|
| 6a | 29.5 + 4.53 µM | 57.1 + 6.7 µM | NI |
| 6b | 57.9 + 7.01 µM | 65.6 + 6.63 µM | NI |
| 6c | 40.6 + 5.42 µM | 33.8 + 3.54 µM | NI |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhahri, M.; Khan, F.A.; Emwas, A.-H.; Alnoman, R.B.; Jaremko, M.; Rezki, N.; Aouad, M.R.; Hagar, M. Synthesis, DFT Molecular Geometry and Anticancer Activity of Symmetrical 2,2′-(2-Oxo-1H-benzo[d]imidazole-1,3(2H)-diyl) Diacetate and Its Arylideneacetohydrazide Derivatives. Materials 2022, 15, 2544. https://doi.org/10.3390/ma15072544
Dhahri M, Khan FA, Emwas A-H, Alnoman RB, Jaremko M, Rezki N, Aouad MR, Hagar M. Synthesis, DFT Molecular Geometry and Anticancer Activity of Symmetrical 2,2′-(2-Oxo-1H-benzo[d]imidazole-1,3(2H)-diyl) Diacetate and Its Arylideneacetohydrazide Derivatives. Materials. 2022; 15(7):2544. https://doi.org/10.3390/ma15072544
Chicago/Turabian StyleDhahri, Manel, Firdos Alam Khan, Abdul-Hamid Emwas, Rua B. Alnoman, Mariusz Jaremko, Nadjet Rezki, Mohamed Reda Aouad, and Mohamed Hagar. 2022. "Synthesis, DFT Molecular Geometry and Anticancer Activity of Symmetrical 2,2′-(2-Oxo-1H-benzo[d]imidazole-1,3(2H)-diyl) Diacetate and Its Arylideneacetohydrazide Derivatives" Materials 15, no. 7: 2544. https://doi.org/10.3390/ma15072544
APA StyleDhahri, M., Khan, F. A., Emwas, A.-H., Alnoman, R. B., Jaremko, M., Rezki, N., Aouad, M. R., & Hagar, M. (2022). Synthesis, DFT Molecular Geometry and Anticancer Activity of Symmetrical 2,2′-(2-Oxo-1H-benzo[d]imidazole-1,3(2H)-diyl) Diacetate and Its Arylideneacetohydrazide Derivatives. Materials, 15(7), 2544. https://doi.org/10.3390/ma15072544