
Exceptional Thermoelectric Properties of Bilayer GeSe: First Principles Calculation



Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
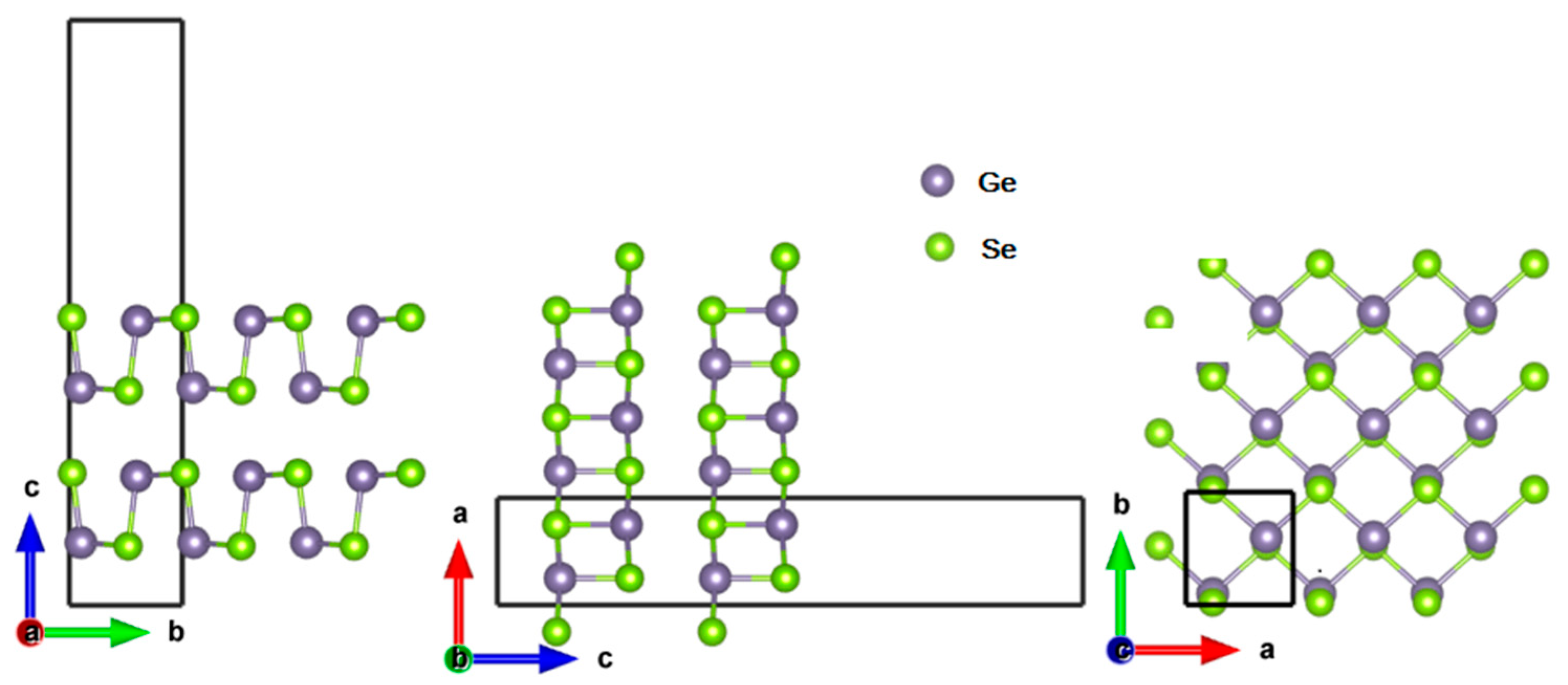
3.1. Geometry Optimization and Electronic Structure
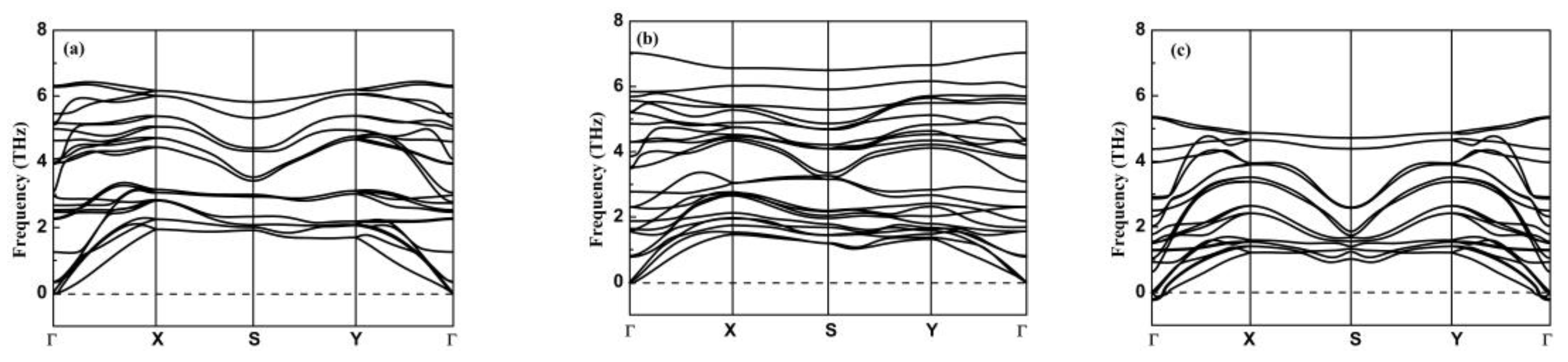
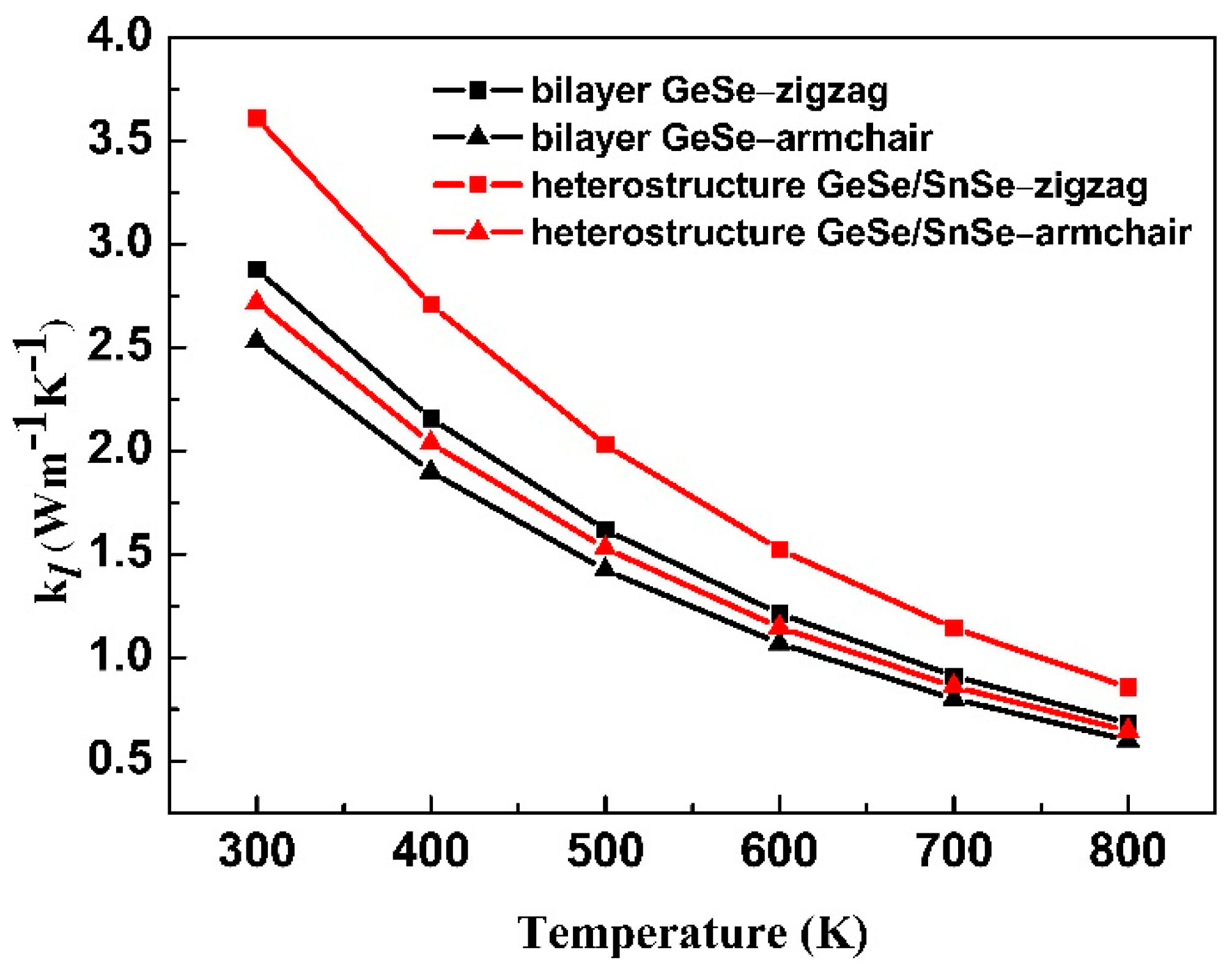
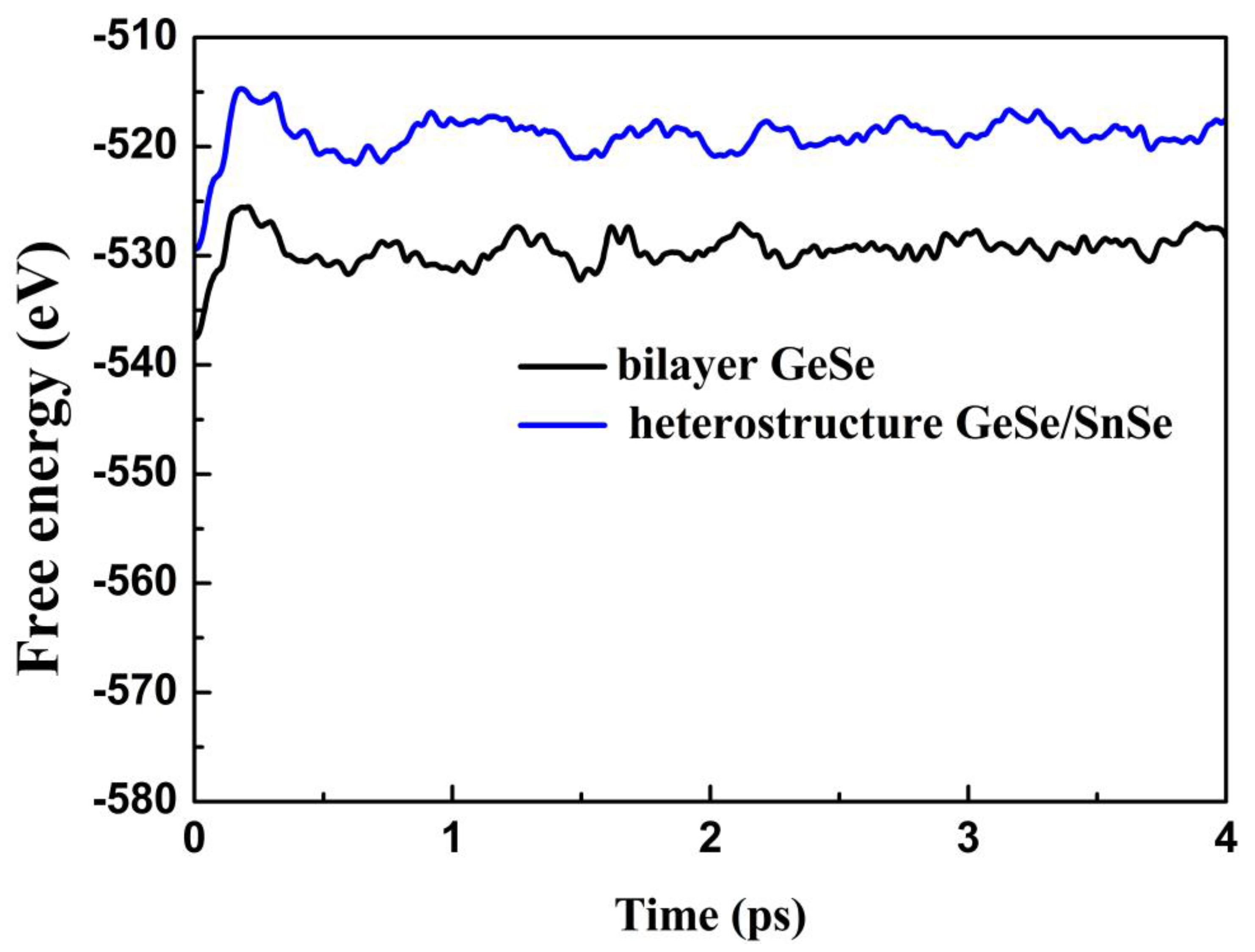
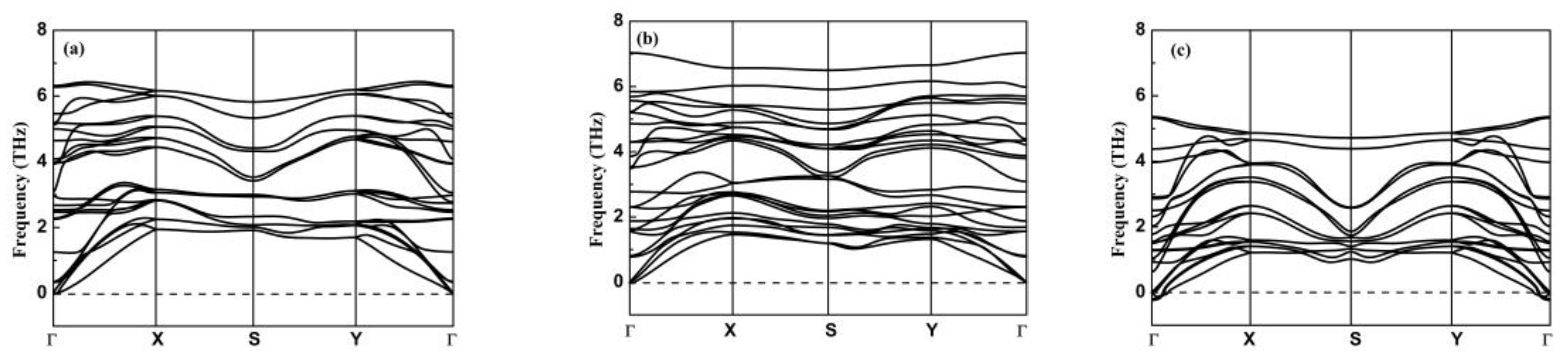
3.2. Stability, Lattice Thermal Conductivity
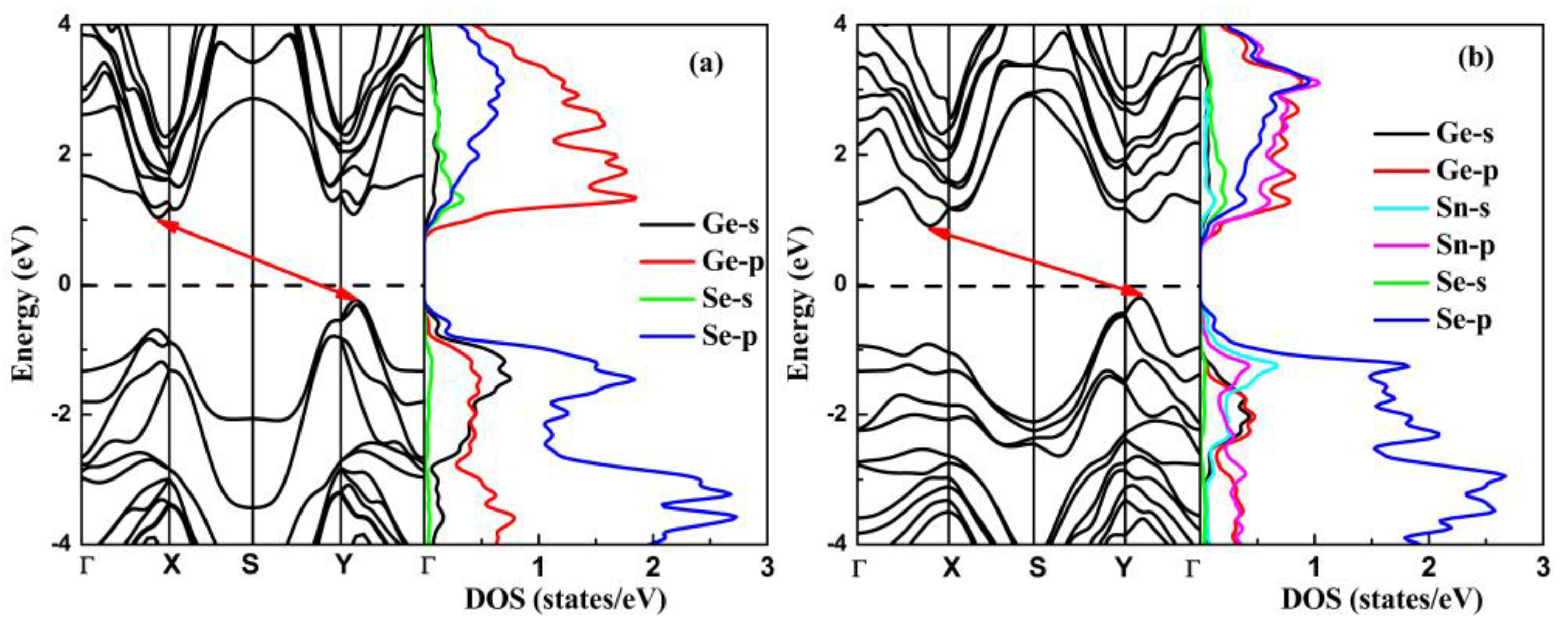
3.3. Electronic Structure
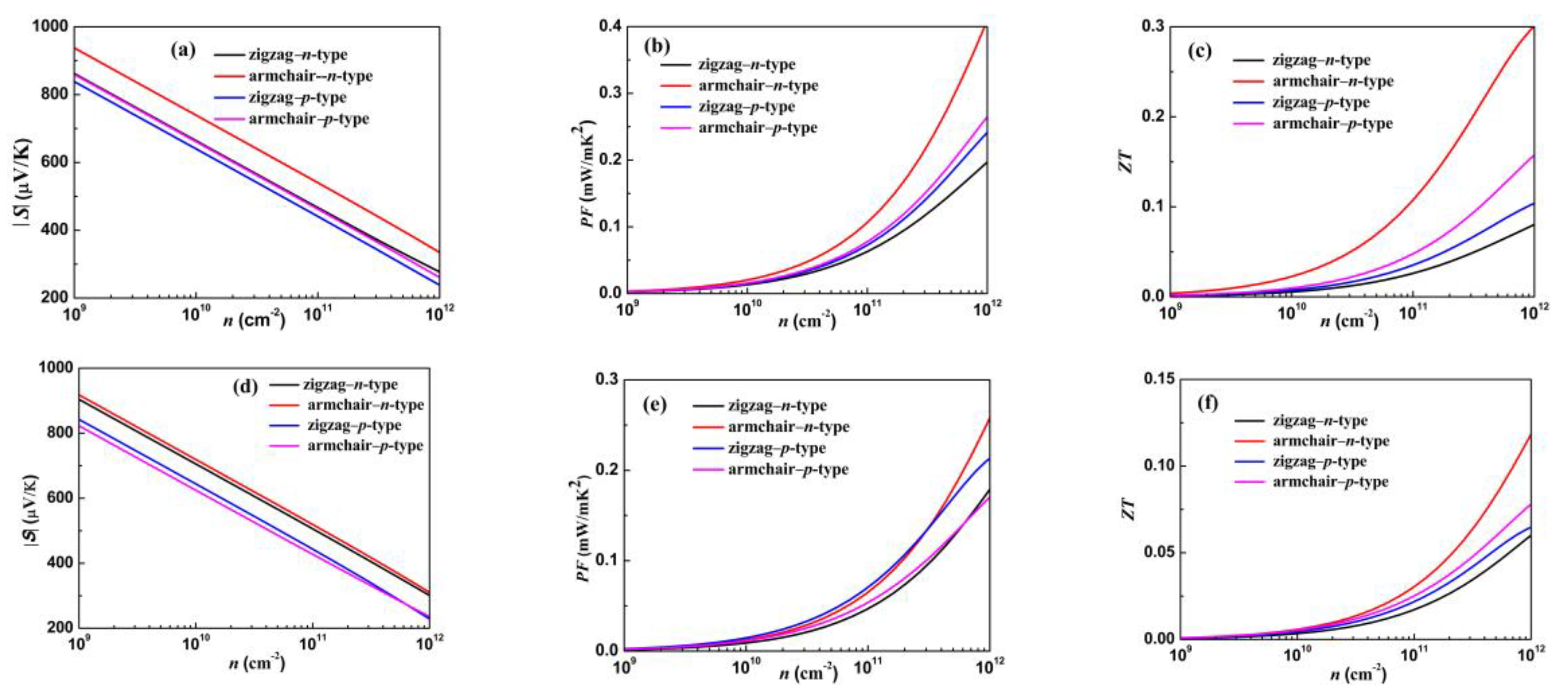
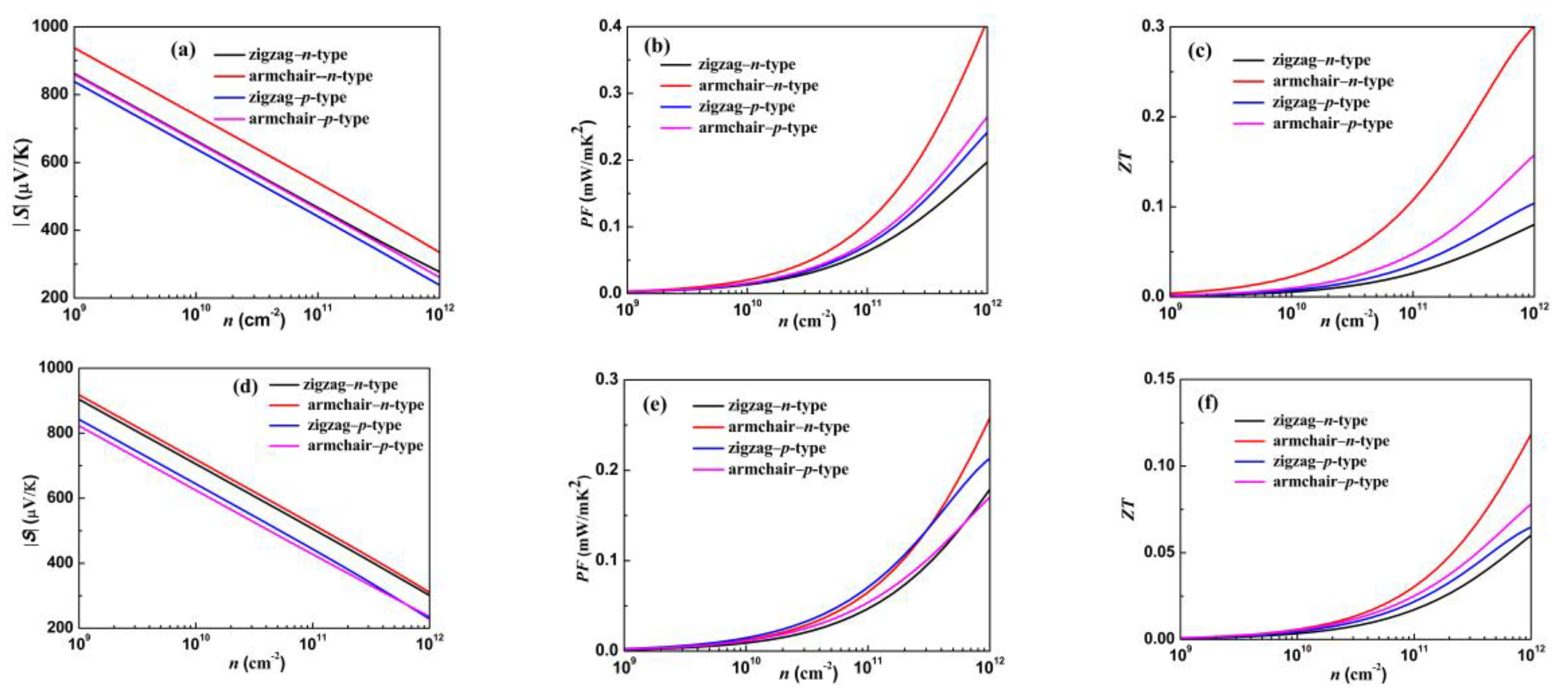
3.4. Electronic Transport and Thermoelectric Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ma, Z.; Wei, J.; Song, P.; Zhang, M.; Yang, L.; Ma, J.; Liu, W.; Yang, F.; Wang, X. Review of experimental approaches for improving zT of thermoelectric materials. Mat. Sci. Semicon. Proc. 2021, 121, 105303. [Google Scholar] [CrossRef]
- Cai, B.; Hu, H.; Zhuang, H.-L.; Li, J.-F. Promising materials for thermoelectric applications. J. Alloy. Compd. 2019, 806, 471–486. [Google Scholar] [CrossRef]
- Huang, H.H.; Xing, G.; Fan, X.; Singh, D.J.; Zheng, W.T.; Xing, G. Layered Tl2O: A model thermoelectric material. J. Mater. Chem. C 2019, 7, 5094–5103. [Google Scholar] [CrossRef]
- Wang, N.; Li, M.; Xiao, H.; Gao, Z.; Liu, Z.; Zu, X.; Li, S.; Qiao, L. Band degeneracy enhanced thermoelectric performance in layered oxyselenides by first-principles calculations. NPJ Comput. Mater. 2021, 7, 18. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, W.; Yang, J.; Li, S. Recent Advances of Layered Thermoelectric Materials. Adv. Sustain. Syst. 2018, 2, 1800046. [Google Scholar] [CrossRef]
- Xie, L.; He, D.; He, J. SnSe, the rising star thermoelectric material: A new paradigm in atomic blocks, building intriguing physical properties. Mater. Horiz. 2021, 8, 1847–1865. [Google Scholar] [CrossRef]
- Zhao, L.-D.; Tan, G.; Hao, S.; He, J.; Pei, Y.; Chi, H.; Wang, H.; Gong, S.; Xu, H.; Dravid, V.P.; et al. Ultrahigh power factor and thermoelectric performance in hole-doped single-crystal SnSe. Science 2016, 351, 141–144. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.-Z.; Ma, Z.; He, C.; Wu, K. Enhanced thermoelectric performance of layered SnS crystals: The synergetic effect of temperature and carrier concentration. RSC Adv. 2015, 5, 56382–56390. [Google Scholar] [CrossRef]
- Ding, G.; Gao, G.; Yao, K. High-efficient thermoelectric materials: The case of orthorhombic IV-VI compounds. Sci. Rep. 2015, 5, srep09567. [Google Scholar] [CrossRef] [Green Version]
- Hao, S.Q.; Shi, F.Y.; Dravid, V.P.; Kanatzidis, M.G.; Wolverton, C. Computational Prediction of High Thermoelectric Performance in Hole Doped Layered GeSe. Chem. Mater. 2016, 28, 3218–3226. [Google Scholar] [CrossRef]
- Khan, A.A.; Khan, I.; Ahmad, I.; Ali, Z. Thermoelectric studies of IV–VI semiconductors for renewable energy resources. Mater. Sci. Semicond. Process. 2016, 48, 85–94. [Google Scholar] [CrossRef]
- Hou, X.Y.; Cheng, Y.; Hu, C.E.; Piao, C.; Geng, H. Thermoelectric properties of strontium sulfide via first-principles calculations. Solid State Commun. 2020, 305, 113755. [Google Scholar] [CrossRef]
- Sassi, S.; Candolfi, C.; Vaney, J.B.; Ohorodniichuk, V.; Masschelein, P.; Dauscher, A.; Lenoir, B. Assessment of the thermoelectric performance of polycrystalline p-type SnSe. Appl. Phys. Lett. 2014, 104, 212105. [Google Scholar] [CrossRef]
- Wei, T.-R.; Tan, G.; Zhang, X.; Wu, C.-F.; Li, J.-F.; Dravid, V.P.; Snyder, G.J.; Kanatzidis, M.G. Distinct Impact of Alkali-Ion Doping on Electrical Transport Properties of Thermoelectric p-Type Polycrystalline SnSe. J. Am. Chem. Soc. 2016, 138, 8875–8882. [Google Scholar] [CrossRef]
- Wei, W.; Chang, C.; Yang, T.; Liu, J.; Tang, H.; Zhang, J.; Li, Y.; Xu, F.; Zhang, Z.; Li, J.; et al. Achieving High Thermoelectric Figure of Merit in Polycrystalline SnSe via Introducing Sn Vacancies. J. Am. Chem. Soc. 2018, 140, 499–505. [Google Scholar] [CrossRef]
- Qin, B.; Zhang, Y.; Wang, D.; Zhao, Q.; Gu, B.; Wu, H.; Zhang, H.; Ye, B.; Pennycook, S.J.; Zhao, L.-D. Ultrahigh Average ZT Realized in p-Type SnSe Crystalline Thermoelectrics through Producing Extrinsic Vacancies. J. Am. Chem. Soc. 2020, 142, 5901–5909. [Google Scholar] [CrossRef]
- Gowthamaraju, S.; Deshpande, U.P.; Bhobe, P.A. Understanding the role of defects in influencing the thermoelectric properties of SnSe. Curr. Appl. Phys. 2021, 24, 19–23. [Google Scholar] [CrossRef]
- Guan, J.; Zhang, Z.; Dou, M.; Ji, J.; Song, Y.; Liu, J.; Li, Z.; Wang, F. Thermoelectric properties of Bi-doped SnS: First-principle study. J. Phys. Chem. Solids 2020, 137, 109182. [Google Scholar] [CrossRef]
- Su, L.; Hong, T.; Wang, D.; Wang, S.; Qin, B.; Zhang, M.; Gao, X.; Chang, C.; Zhao, L.-D. Realizing high doping efficiency and thermoelectric performance in n-type SnSe polycrystals via bandgap engineering and vacancy compensation. Mater. Today Phys. 2021, 20, 100452. [Google Scholar] [CrossRef]
- Asfandiyar; Cai, B.; Zhuang, H.; Tang, H.; Li, J. Polycrystalline SnSe–Sn1–vS solid solutions: Vacancy engineering and nanostructuring leading to high thermoelectric performance. Nano Energy 2020, 69, 104393. [Google Scholar] [CrossRef]
- Wu, Y.; Xia, W.; Gao, W.; Ren, W.; Zhang, P. Engineering the Near-Edge Electronic Structure of SnSe through Strains. Phys. Rev. Appl. 2017, 8, 034007. [Google Scholar] [CrossRef] [Green Version]
- Shang, P.-P.; Dong, J.; Pei, J.; Sun, F.-H.; Pan, Y.; Tang, H.; Zhang, B.-P.; Zhao, L.-D.; Li, J.-F. Highly Textured N-Type SnSe Polycrystals with Enhanced Thermoelectric Performance. Research 2019, 2019, 9253132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Wang, H.; Zhou, Y.; Liao, L.; Jiang, Y.; Yang, X.; Chen, G.; Lin, M.; Wang, Y.; Peng, H.; et al. Controlled synthesis of single-crystal SnSe nanoplates. Nano Res. 2015, 8, 288–295. [Google Scholar] [CrossRef]
- Ramasamy, P.; Kwak, D.; Lim, D.; Ra, H.; Lee, J. Solution synthesis of GeS and GeSe nanosheets for high-sensitivity photodetectors. J. Mater. Chem. C. 2016, 4, 479–485. [Google Scholar] [CrossRef]
- Qin, G.; Qin, Z.; Fang, W.-Z.; Zhang, L.-C.; Yue, S.-Y.; Yan, Q.-B.; Hu, M.; Su, G. Diverse anisotropy of phonon transport in two-dimensional group IV–VI compounds: A comparative study. Nanoscale 2016, 8, 11306–11319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafique, A.; Shin, Y.-H. Thermoelectric and phonon transport properties of two-dimensional IV–VI compounds. Sci. Rep. 2017, 7, 506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.-D.; Wang, Y.-H. Thermoelectric properties of orthorhombic group IV–VI monolayers from the first-principles calculations. J. Appl. Phys. 2017, 121, 034302. [Google Scholar] [CrossRef]
- Wang, F.Q.; Zhang, S.; Yu, J.; Wang, Q. Thermoelectric properties of single-layered SnSe sheet. Nanoscale 2015, 7, 15962–15970. [Google Scholar] [CrossRef]
- Nag, S.; Saini, A.; Singh, R.; Kumar, R. Influence of vacancy defects on the thermoelectric performance of SnSe sheet. Phys. E Low Dimens. Syst. Nanostruct. 2021, 134, 114814. [Google Scholar] [CrossRef]
- Sharma, G.; Datta, S.; Ghosh, P. First Principles Investigations of Structural, Electronic and Transport Properties of BiI3/ZrS2van der Waals Heterostructure: A Thermoelectric Perspective. J. Electron. Mater. 2021, 50, 1644–1654. [Google Scholar] [CrossRef]
- Ahmad, S.; Khan, F.; Amin, B.; Ahmad, I. Effect of strain on structural and electronic properties, and thermoelectric response of MXY (M = Zr, Hf and Pt; X/Y = S, Se) vdW heterostructures; A fist principles study. J. Solid State Chem. 2021, 299, 122189. [Google Scholar] [CrossRef]
- Kouaydi, N.; Zemzemi, M. Electronic, Band Offset, and Thermoelectric Properties of ZnO/GaN Heterostructure from First-Principles Study. J. Electron. Mater. 2020, 49, 5773–5781. [Google Scholar] [CrossRef]
- Khan, F.; Din, H.U.; Khan, S.A.; Rehman, G.; Bilal, M.; Nguyen, C.V.; Ahmad, I.; Gan, L.; Amin, B. Theoretical investigation of electronic structure and thermoelectric properties of MX2 (M = Zr, Hf; X = S, Se) van der Waals heterostructures. J. Phys. Chem. Solids 2019, 126, 304–309. [Google Scholar] [CrossRef]
- Ding, G.; Wang, C.; Gao, G.; Yao, K.; Dun, C.; Feng, C.; Li, D.; Zhang, G. Engineering of charge carriers via a two-dimensional heterostructure to enhance the thermoelectric figure of merit. Nanoscale 2018, 10, 7077–7084. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Xu, C.; Yuan, J.; Zhao, H. Effect of stacking order and in-plane strain on the electronic properties of bilayer GeSe. Phys. Chem. Chem. Phys. 2018, 20, 6929–6935. [Google Scholar] [CrossRef]
- Nag, S.; Saini, A.; Singh, R.; Kumar, R. Ultralow lattice thermal conductivity and anisotropic thermoelectric performance of AA stacked SnSe bilayer. Appl. Surf. Sci. 2020, 512, 145640. [Google Scholar] [CrossRef]
- Sun, D.; Schaak, R.E. Solution-Mediated Growth of Two-Dimensional SnSe@GeSe Nanosheet Heterostructures. Chem. Mater. 2017, 29, 817–822. [Google Scholar] [CrossRef]
- Ni, H.; Li, M.; Hu, Y.; Mao, C.; Xue, L.; Zeng, H.; Yan, Z.; Wu, Y.; Zheng, C. Two-dimensional SnSe/GeSe van der Waals heterostructure with strain-tunable electronic and optical properties. J. Phys. Chem. Solids 2019, 131, 223–229. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef]
- Bučko, T.; Hafner, J.; Lebègue, S.; Ángyán, J.G. Improved Description of the Structure of Molecular and Layered Crystals: Ab Initio DFT Calculations with van der Waals Corrections. J. Phys. Chem. A 2010, 114, 11814–11824. [Google Scholar] [CrossRef] [PubMed]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Slack, G.A. The Thermal Conductivity of Nonmetallic Crystals. Solid State Physics 1979, 34, 1–71. [Google Scholar] [CrossRef]
- Belomestnykh, V.N. The acoustical Grüneisen constants of solids. Tech. Phys. Lett. 2004, 30, 91–93. [Google Scholar] [CrossRef]
- Sanditov, D.; Mashanov, A.A.; Darmaev, M.D. Propagation velocity of longitudinal and transverse acoustic waves and anharmonicity of lattice oscillations. Tech. Phys. 2009, 54, 1398–1401. [Google Scholar] [CrossRef]
- Madsen, G.K.; Carrete, J.; Verstraete, M. BoltzTraP2, a program for interpolating band structures and calculating semi-classical transport coefficients. Comput. Phys. Commun. 2018, 231, 140–145. [Google Scholar] [CrossRef] [Green Version]
- Haleoot, R.; Hamad, B. Thermoelectric properties of doped β-InSe by Bi: First principle calculations. Phys. B Condens. Matter 2020, 587, 412105. [Google Scholar] [CrossRef]
- Nguyen, H.T.T.; Vu, T.V.; Binh, N.T.T.; Hoat, D.M.; Hieu, N.V.; Anh, N.T.T.; Nguyen, C.V.; Phuc, H.V.; Jappor, H.R.; Obeid, M.M.; et al. Strain-tunable electronic and optical properties of monolayer GeSe: Promising for photocatalytic water splitting applications. Chem. Phys. 2020, 529, 110543. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Liu, M.-Y.; Huang, Y.; Chen, Q.-Y.; Cao, C.; He, Y. Tuning the electronic properties of bilayer group-IV monochalcogenides by stacking order, strain and an electric field: A computational study. Phys. Chem. Chem. Phys. 2018, 20, 214–220. [Google Scholar] [CrossRef]
- Zhong, Q.; Dai, Z.; Liu, J.; Zhao, Y.; Meng, S. A comprehensive study of phonon thermal transport in 2D IV-VI semiconductors MX (M = Ge, Sn; X = S, Se). Phys. Lett. A 2020, 384, 126676. [Google Scholar] [CrossRef]
- Zhou, M.; Chen, X.; Li, M.; Du, A. Widely tunable and anisotropic charge carrier mobility in monolayer tin (ii) selenide using biaxial strain: A first-principles study. J. Mater. Chem. C 2017, 5, 1247–1254. [Google Scholar] [CrossRef]
- Li, Y.; Ma, K.; Fan, X.; Liu, F.; Li, J.; Xie, H. Enhancing thermoelectric properties of monolayer GeSe via strain-engineering: A first principles study. Appl. Surf. Sci. 2020, 521, 146256. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Direction | ν (m/s) | γ | (K) | ||
|---|---|---|---|---|---|---|
| ZA | TA | LA | ||||
| GeSe | zigzag | 592 | 778 | 1425 | 1.698 | 183 |
| armchair | 249 | 452 | 855 | 1.810 | 183 | |
| GeSe/SnSe | zigzag | 626 | 870 | 1591 | 1.694 | 188 |
| armchair | 246 | 583 | 1149 | 1.952 | 188 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Q.; Zhang, W.; Qing, H.; Yang, J. Exceptional Thermoelectric Properties of Bilayer GeSe: First Principles Calculation. Materials 2022, 15, 971. https://doi.org/10.3390/ma15030971
Fan Q, Zhang W, Qing H, Yang J. Exceptional Thermoelectric Properties of Bilayer GeSe: First Principles Calculation. Materials. 2022; 15(3):971. https://doi.org/10.3390/ma15030971
Chicago/Turabian StyleFan, Qiang, Weibin Zhang, Haiyin Qing, and Jianhui Yang. 2022. "Exceptional Thermoelectric Properties of Bilayer GeSe: First Principles Calculation" Materials 15, no. 3: 971. https://doi.org/10.3390/ma15030971
APA StyleFan, Q., Zhang, W., Qing, H., & Yang, J. (2022). Exceptional Thermoelectric Properties of Bilayer GeSe: First Principles Calculation. Materials, 15(3), 971. https://doi.org/10.3390/ma15030971