
New Insights into the Crystal Chemistry of FeB-Type Compounds: The Case of CeGe


Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Crystal Structure and Thermal Analysis
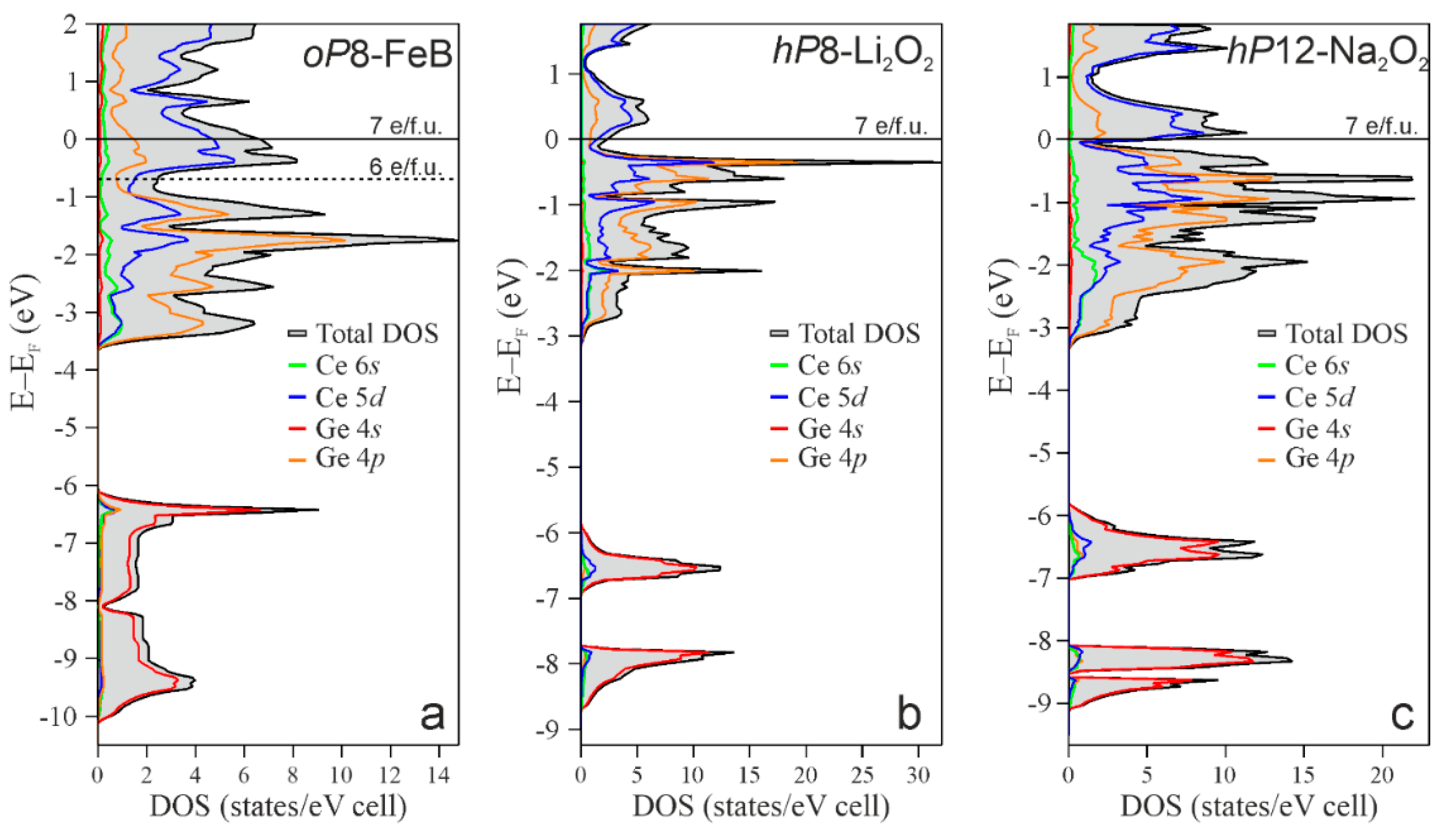
3.2. Stability and Electronic Structure
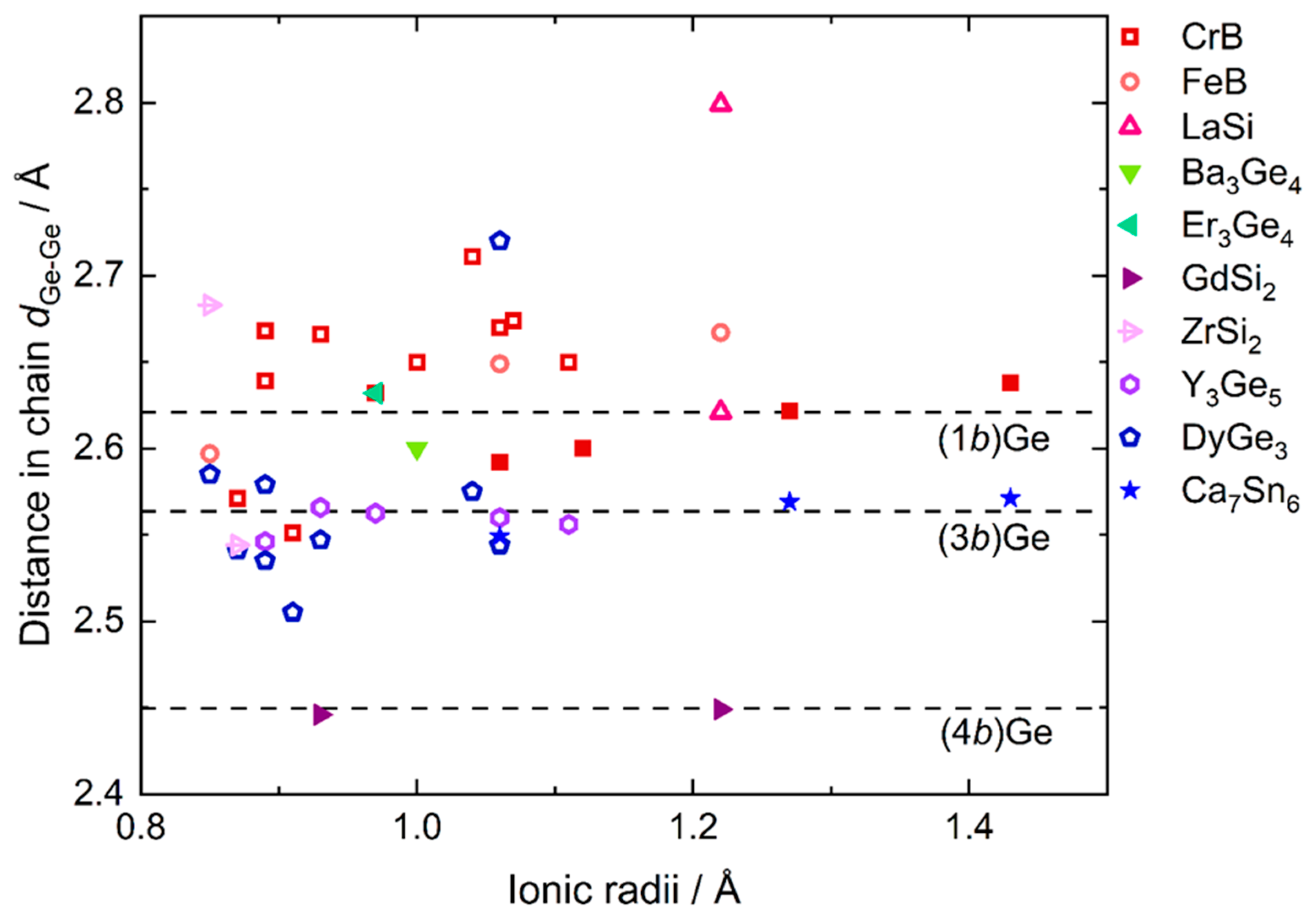
3.3. Crystal-Chemical Insights Based on Bonding Considerations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nesper, R. The Zintl-Klemm Concept—A Historical Survey. Z. Anorg. Allgem. Chem. 2014, 640, 2639–2648. [Google Scholar] [CrossRef]
- Wade, K. The structural significance of the number of skeletal bonding electron-pairs in carboranes, the higher boranes and borane anions, and various transition-metal carbonyl cluster compounds. J. Chem. Soc. D 1971, 15, 792–793. [Google Scholar] [CrossRef]
- Fredrickson, D.C.; Miller, G.J. Intermetallic Chemistry: New Advances in Humanity’s Age-Old Exploration of Metals and Alloys. Acc. Chem. Res. 2018, 51, 213. [Google Scholar] [CrossRef] [PubMed]
- Nesper, R. Bonding Patterns in Intermetallic Compounds. Angew. Chem. Int. Ed. 1991, 30, 789–817. [Google Scholar] [CrossRef]
- Kurylyshyn, I.M.; Fässler, T.F.; Fischer, A.; Hauf, C.; Eickerling, G.; Presnitz, M.; Scherer, W. Probing the Zintl–Klemm Concept: A Combined Experimental and Theoretical Charge Density Study of the Zintl Phase CaSi. Angew. Chem. Int. Ed. 2014, 53, 3029–3032. [Google Scholar] [CrossRef]
- Freccero, R.; Hübner, J.-M.; Prots, Y.; Schnelle, W.; Schmidt, M.; Wagner, F.R.; Schwarz, U.; Grin, Y. “Excess” electrons in LuGe. Angew. Chem. Int. Ed. 2021, 60, 6457. [Google Scholar] [CrossRef]
- Peierls, R. More Surprises in Theoretical Physics; Princeton University Press: Princeton, NJ, USA, 1991. [Google Scholar]
- Hohnke, D.; Parthe, E. AB compounds with Sc, Y and rare earth metals. II. FeB and CrB type structures of monosilicides and germanides. Acta Crystallogr. 1966, 20, 572–582. [Google Scholar] [CrossRef]
- Currao, A.; Curda, J.; Nesper, R. Kann man Arten von Zintl-Anionen steuern? Variationen ueber das Thema Si2− im System Sr/Mg/Si. Z. Anorg. Allg. Chem. 1996, 622, 85–94. [Google Scholar] [CrossRef]
- Eisenmann, B.; Schaefer, H.; Turban, K. On a new SrSi-modification and the new compound SrGe0.76. Z. Naturforsch. B 1974, 29, 464–468. [Google Scholar] [CrossRef]
- Schob, O.; Parthe, E. AB compounds with Sc, Y and rare earth metals. I. Scandium and yttrium compounds with Cr B and CsCl structure. Acta Crystallogr. 1965, 19, 214–224. [Google Scholar] [CrossRef]
- Mattausch, H.J.; Oeckler, O.; Simon, A. Eine neue Modifikation von Lanthanmonosilicid-IT-LaSi. Z. Anorg. Allg. Chem. 1999, 625, 1151–1154. [Google Scholar] [CrossRef]
- Duerr, I.; Bauer, B.; Roehr, C. Lanthan-Triel/Tetrel-ide La (Al, Ga)x (Si, Ge)1−x. Experimentelle und theoretische Studien zur Stabilitaet polarer 1:1-Phasen. Z. Naturforsch. B 2011, 66, 1107–1121. [Google Scholar] [CrossRef]
- Schobinger Papamantellos, P.; Buschow, K.H.J. Incommensurate magnetic structure of Ce Si as observed by neutron diffraction. J. Magn. Magn. Mater. 1994, 130, 242–246. [Google Scholar] [CrossRef]
- Gladyshevskii, E.I.; Kripyakevich, P.I. Monosilicides of rare earth metals and their crystal structures. J. Struct. Chem. 1965, 5, 789–794. [Google Scholar] [CrossRef]
- Nagaki, D.A.; Simon, A. Structure of gadolinium monosilicide. Acta Crystallogr. 1990, 46, 1197–1199. [Google Scholar] [CrossRef]
- Schobinger Papamantellos, P.; Janssen, T.; Buschow, K.H.J. Thermal variation of incommensurate magnetic phases in TbSi as observed by neutron diffraction. J. Magn. Magn. Mater. 1993, 127, 115–128. [Google Scholar] [CrossRef]
- Roger, J.; Babizhetskii, V.; Guizouarn, T.; Hiebl, K.; Guerin, R.; Halet, J.F. The ternary RE–Si–B systems (RE = Dy, Ho, Er and Y) at 1270 K: Solid state phase equilibria and magnetic properties of the solid solution REB2−xSix (RE = Dy and Ho). J. Alloys Compd. 2006, 417, 72–84. [Google Scholar] [CrossRef]
- Schobinger Papamantellos, P.; Brunelli, M.; Rodriguez Carvajal, J.; Buschow, K.H.J.; Ritter, C.; Gramm, F. Magneto structural transition in the DySi (Cr B)- and micro-structural changes in the (Fe B)-type compounds by XRPD and neutron diffraction. J. Magn. Magn. Mater. 2011, 323, 903–914. [Google Scholar] [CrossRef]
- Eckerlin, B.; Meyer, H.J.; Woelfel, E. Die Kristallstruktur von CaSn and CaGe. Z. Anorg. Allg. Chem. 1955, 281, 322–328. [Google Scholar] [CrossRef]
- Rieger, W.; Parthe, E. Alkaline earth silicides, germanides and stannides with CrB structure type. Acta Crystallogr. 1967, 22, 919–922. [Google Scholar] [CrossRef]
- Mattausch, H.J.; Simon, A. Eine neue Modifikation von Lanthanmonogermanid—IT-LaGe. Z. Naturforsch. B 2004, 59, 559–561. [Google Scholar] [CrossRef]
- Schobinger Papamantellos, P.; Buschow, K.H.J. Ferromagnetism of NdGe and PrGe studied by neutron diffraction and magnetic measurements. J. Less Common Met. 1985, 111, 125–138. [Google Scholar] [CrossRef]
- Gladyshevskii, E.I.; Uhryn, N.S. Monogermanides of the rare-earth metals and their crystal structures. Dop. Akad. Nauk. Ukrain. 1965, 10, 1326–1329. [Google Scholar]
- Tharp, A.G.; Smith, G.S.; Johnson, Q. Structures of the rare earth germanides at or near equiatomic proportions. Acta Crystallogr. 1966, 20, 583–585. [Google Scholar] [CrossRef]
- Merlo, F.; Fornasini, M.L. CrB-type equiatomic compounds of europium, ytterbium and alkaline-earth metals with Si, Ge, Sn, Pb. J. Less-Common Met. 1967, 13, 603–610. [Google Scholar] [CrossRef]
- Schobinger Papamantellos, P.; Buschow, K.H.J. A neutron diffraction and magnetic study of the first-order phase transition in TbGe1−xSix (0 = x = 0.4). J. Magn. Magn. Mater. 1986, 62, 15–28. [Google Scholar] [CrossRef]
- Buschow, K.H.J.; Schobinger Papamantellos, P.; Fischer, P. Magnetic structure and properties of equiatomic rare earth germanides. J. Less-Common Met. 1988, 139, 221–231. [Google Scholar] [CrossRef]
- Schobinger Papamantellos, P.; Buschow, K.H.J. Magnetic structure and incommensurate phase transition in HoGe. J. Magn. Magn. Mater. 1984, 44, 149–157. [Google Scholar] [CrossRef]
- Eremenko, V.N.; Meleshevich, K.A.; Buyanov, Y.I.; Martsenyuk, P.S. Structure of the alloys and phase diagram of the thulium-germanium system. Powder Metall. Met. Ceram. 1989, 28, 543–547. [Google Scholar] [CrossRef]
- Agilent. CrysAlis PRO; Agilent Technologies Ltd.: Yarnton, Oxfordshire, UK, 2014. [Google Scholar]
- Palatinus, L.; Chapuis, G. SUPERFLIP—A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Petříček, V.; Dusek, M.; Palatinus, L. Crystallographic Computing System JANA2006: General features. Z. Kristallogr.—Cryst. Mater. 2014, 229, 345–352. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter. 2009, 21, 395502. Available online: https://www.quantum-espresso.org/ (accessed on 5 April 2022). [CrossRef] [PubMed]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Dal Corso, A. Pseudopotentials periodic table: From H to Pu. Comput. Mater. Sci. 2014, 95, 337. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Maintz, S.; Deringer, V.L.; Tchougreeff, A.L.; Dronskowski, R. Analytic projection from plane-wave and PAW wavefunctions and application to chemical-bonding analysis in solids. J. Comput. Chem. 2013, 34, 2557–2567. [Google Scholar] [CrossRef] [PubMed]
- Maintz, S.; Deringer, V.L.; Tchougreeff, A.L.; Dronskowski, R. LOBSTER: A tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem. 2016, 37, 1030–1035. Available online: http://www.cohp.de/ (accessed on 25 October 2021). [CrossRef] [PubMed]
- Maintz, S.; Esser, M.; Dronskowski, R. Efficient Rotation of Local Basis Functions Using Real Spherical Harmonics. Acta Phys. Pol. B 2016, 47, 1165–1175. [Google Scholar] [CrossRef]
- Nelson, R.; Ertural, C.; George, J.; Deringer, V.L.; Hautier, G.; Dronskowski, R. LOBSTER: Local orbital projections, atomic charges, and chemical-bonding analysis from projector-augmented-wave-based density-functional theory. J. Comput. Chem. 2020, 41, 1931–1940. [Google Scholar] [CrossRef]
- Eck, B. wxDragon Program, v-2.1.8. 2019. Available online: http://wxdragon.de (accessed on 10 January 2022).
- Haschke, H.; Nowotny, H.; Benesovsky, F. Untersuchungen im System Cer-Silicium-Germanium. Mon. Chem. 1966, 97, 1452–1458. [Google Scholar] [CrossRef]
- Das, P.K.; Kumar, N.; Kulkarni, R.; Dhar, S.K.; Thamizhavel, A. Anisotropic magnetic properties and superzone gap formation in CeGe single crystal. J. Phys. Condens. Matter. 2012, 24, 146003. [Google Scholar] [CrossRef] [PubMed]
- Bjurström, T.; Arnfelt, H. Röntgenanalyse des Eisen-Bor-Systems. Z. Phys. Chem. B 1929, 4, 469–474. [Google Scholar] [CrossRef]
- Straumanis, M.E.; Aka, E.Z. Lattice parameters, coefficients of thermal expansion and atomic weights of purest silicon and germanium. J. Appl. Phys. 1952, 23, 330–334. [Google Scholar] [CrossRef]
- Pauling, L. Atomic Radii and Interatomic Distances in Metals. J. Am. Chem. Soc. 1947, 69, 542–553. [Google Scholar] [CrossRef]
- Zürcher, F.; Nesper, R. Ba3Ge4: Polymerization of Zintl Anions in the Solid and Bond Stretching Isomerism. Angew. Chem. 1998, 37, 3314–3318. [Google Scholar] [CrossRef]
- Tobash, P.H.; DiFilippo, G.; Bobev, S.; Hur, N.; Thompson, J.D.; Sarrao, J.L. Structure and properties of Gd3Ge4: The orthorhombic RE3Ge4 structures revisited (RE = Y, Tb–Tm). Inorg. Chem. 2007, 46, 8690–8697. [Google Scholar] [CrossRef]
- Schobinger Papamantellos, P.; de Mooij, D.B.; Buschow, K.H.J. Crystallographic and magnetic structure of TbGe2. J. Less Common Met. 1988, 144, 265–274. [Google Scholar] [CrossRef]
- Guloy, A.M.; Corbett, J.D. Syntheses and structures of lanthanum germanide, LaGe2−x, and lanthanum aluminum germanide, LaAlGe: Interrelationships among the α-ThSi2, α-GdSi2, and LaPtSi structure types. Inorg. Chem. 1991, 30, 4789–4794. [Google Scholar] [CrossRef]
- Tobash, P.H.; Meyers, J.J.; DiFilippo, G.; Bobev, S.; Ronning, F.; Thompson, J.D.; Sarrao, J.L. Structure and properties of a new family of nearly equiatomic rare-earth metal-tin-germanides RESn1+xGe1−x (RE = Y, Gd–Tm): An unusual example of site preferences between elements from the same group. Chem. Mater. 2008, 20, 2151–2159. [Google Scholar] [CrossRef]
- Francois, M.; Venturini, G.; Malaman, B.; Roques, B. Nouveaux isotypes de CeNiSi2 dans les systemes R-M-X (R ident to La-Lu, M ident to metaux des groupes 7 A 11 et X ident to Ge, Sn). I compositions et parametres cristallins. J. Less Common Met. 1990, 160, 197–213. [Google Scholar] [CrossRef]
- Bruskov, V.A.; Bodak, O.I.; Pecharskii, V.K.; Gladyshevskii, E.I.; Muratova, L.A. Crystal structure of Y3Ge5 (“YGe1.7”). Kristallografiya 1983, 28, 260–263. [Google Scholar]
- Tobash, P.H.; Lins, D.; Bobev, S.; Hur, N.; Thompson, J.D.; Sarrao, J.L. Vacancy Ordering in SmGe2−x and GdGe2−x (x = 0.33): Structure and Properties of Two Sm3Ge5 Polymorphs and of Gd3Ge5. Inorg. Chem. 2006, 45, 7286–7294. [Google Scholar] [CrossRef] [PubMed]
- Schobinger-Papamantellos, P.; de Mooij, D.B.; Buschow, K.H.J. Crystallographic and magnetic structure of Dy3Ge5 and DyGe1.9. J. Less-Common Met. 1990, 163, 319–330. [Google Scholar] [CrossRef]
- Zaharko, O.; Schobinger Papamantellos, P.; Ritter, C. Antiferromagnetic ordering in the novel Ho3Ge5 and HoGe1.85 compounds studied by X-ray and neutron diffraction. J. Alloys Compd. 1998, 280, 4–15. [Google Scholar] [CrossRef]
- Belyavina, N.M.; Markiv, V.Y.; Speka, M.V. Crystal structure of YGe3, YGe1.9 and a novel Y3Ge4 compound. J. Alloys Compd. 1999, 283, 162–168. [Google Scholar] [CrossRef]
- Schobinger-Papamantellos, P.; Andre, G.; Rodriguez-Carvajal, J.; de Groot, C.H.; Buschow, K.H.J. The magnetic ordering of the novel compound TbGe3. J. Alloys Compd. 1996, 232, 165–168. [Google Scholar] [CrossRef]
- Schobinger-Papamantellos, P.; de Mooij, D.B.; Buschow, K.H.J. Crystal structure of the compound DyGe3. J. Alloys Compd. 1992, 183, 181–186. [Google Scholar] [CrossRef]
- Schobinger-Papamantellos, P.; Rodriguez Carvajal, J.; Tung, L.D.; Ritter, C.; Buschow, K.H.J. Competing multiple-q magnetic structures in HoGe3: I. The magnetic phase diagram of HoGe3. J. Phys. Condens. Matter. 2008, 20, 195201. [Google Scholar] [CrossRef]
- Eremenko, V.N.; Obushenko, I.M. Phase Diagram of the Erbium-Germanium System. Sov. Non-Ferr. Met. Res. 1981, 9, 216–220. [Google Scholar]
- Fukuoka, H.; Yoshikawa, M.; Baba, K.; Yamanaka, S. Preparation and Structures of Lanthanoid Germanides, PrGe3.36, NdGe3.25, and TmGe3 with Double Square Ge-Mesh Structures. Bull. Chem. Soc. Jpn. 2010, 83, 323–327. [Google Scholar] [CrossRef]
- Harada, M.; Fukuoka, H.; Matsumura, D.; Inumaru, K. Structure and Chemical Bonding of Binary Ytterbium Germanides, Yb3Ge5 and YbGe3, Prepared by High-Pressure and High-Temperature Reactions. J. Phys. Chem. C 2012, 116, 2153–2158. [Google Scholar] [CrossRef]
- Hübner, J.-M.; Bobnar, M.; Akselrud, L.; Prots, Y.; Grin, Y.; Schwarz, U. Lutetium Trigermanide LuGe3: High-Pressure Synthesis, Superconductivity, and Chemical Bonding. Inorg. Chem. 2018, 57, 10295–10302. [Google Scholar] [CrossRef] [PubMed]
- Palenzona, A.; Manfrinetti, P.; Fornasini, M.L. The phase diagram of the Ca-Ge system. J. Alloys Compd. 2002, 345, 144–147. [Google Scholar] [CrossRef]
- Siggelkow, L.; Hlukhyy, V.; Fässler, T.F. Sr7Ge6, Ba7Ge6 and Ba3Sn2—Three new binary compounds containing dumbbells and four-membered chains of tetrel atoms with considerable Ge–Ge π-bonding character. J. Solid State Chem. 2012, 191, 76–89. [Google Scholar] [CrossRef]
- Emsley, J. The Elements; Clarendon Press: Oxford, UK, 1989. [Google Scholar]
- Witte, J.; von Schnering, H.G.; Hagenmuller, P. Das Verhalten der Alkalimetalle zu Halbmetallen. XI. Die Kristallstruktur von NaSi und NaGe. Z. Anorg. Allgem. Chem. 1964, 327, 260–273. [Google Scholar] [CrossRef]
- Hopf, V.; Mueller, W.; Schaefer, H. Die Struktur der Phase Li7Ge2. Z. Naturforsch. B 1972, 27, 1157–1160. [Google Scholar] [CrossRef]
- Busmann, E. Die Kristallstruktur von KGe und isotypen Germaniden und Siliciden. Naturwissenschaften 1960, 47, 82. [Google Scholar] [CrossRef]
- Arguilla, M.Q.; Cultrara, N.D.; Scudder, M.R.; Jiang, S.; Ross, R.D.; Goldberger, J.E. Optical properties and Raman-active phonon modes of two-dimensional honeycomb Zintl phases. J. Mater. Chem. C 2017, 5, 11259–11266. [Google Scholar] [CrossRef]
- Iandelli, A.; Franceschi, E. On the Crystal Structure of the Compounds CaP, SrP, CaAs, SrAs and EuAs. J. Less Common Met. 1973, 30, 211–216. [Google Scholar] [CrossRef]
- Tallman, R.L.; Margrave, J.L.; Bailey, S.W. The crystal structure of sodium peroxide. J. Am. Chem. Soc. 2002, 79, 2979–2980. [Google Scholar] [CrossRef]
- Freccero, R.; Solokha, P.; de Negri, S.; Saccone, A.; Grin, Y.; Wagner, F.R. Polar-Covalent Bonding beyond the Zintl Picture in Intermetallic Rare-Earth Germanides. Chem. Eur. J. 2019, 25, 6600–6612. [Google Scholar] [CrossRef] [PubMed]
- Mudring, A.V.; Corbett, J.D. Unusual Electronic and Bonding Properties of the Zintl Phase Ca5Ge3 and Related Compounds. A Theoretical Analysis. J. Am. Chem. Soc. 2004, 126, 5277–5281. [Google Scholar] [CrossRef] [PubMed]
- Freccero, R.; De Negri, S.; Rogl, G.; Binder, G.; Michor, H.; Rogl, P.F.; Saccone, A.; Solokha, P. La2Pd3Ge5 and Nd2Pd3Ge5 Compounds: Chemical Bonding and Physical Properties. Inorg. Chem. 2021, 60, 3345–3354. [Google Scholar] [CrossRef]
- Solokha, P.; De Negri, S.; Proserpio, D.M.; Blatov, V.A.; Saccone, A. Vacancy Ordering as a Driving Factor for Structural Changes in Ternary Germanides: The New R2Zn1−x Ge6 Series of Polar Intermetallics (R = Rare-Earth Metal). Inorg. Chem. 2015, 54, 2411–2424. [Google Scholar] [CrossRef] [PubMed]
- Miedema, A.R. The electronegativity parameter for transition metals: Heat of formation and charge transfer in alloys. J. Less-Common Met. 1973, 32, 117–136. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mg | Ca | Sr | Ba | Sc | Y | La | Ce | Pr | Nd | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | Lu | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Si | C2 | C2 | C2 | C2 | C2 | C1 | C1 | C1 | C1 | C1 | C2 | C1 | C1 | C1 | C1 | C1 | C2 | C2 | C2 | |
| P | C3 | C2 | C2 | C2 | ||||||||||||||||
| Ge | C2 | C2 | C2 | C2 | C2 | C1 | C1 | C1 | C2 | C2 | C2 | C2 | C2 | C2 | C2 | C2 | C2 | C1 | ||
| P | C3 | C2 |
| Crystal Structure | k-Mesh |
|---|---|
| oP8–FeB | (6, 14, 10) |
| oS8–CrB | (12, 4, 14) |
| oS16–LaSi | (12, 4, 8) |
| hP12–Na2O2 | (8, 8, 10) |
| hP8–Li2O2-β | (10, 10, 6) |
| Composition | CeGe |
| Space group, Pearson symbol | Pnma (No. 62), oP8 |
| Unit cell parameters | |
| a [Å] | 8.3524(4) |
| b [Å] | 4.0852(2) |
| c [Å] | 6.0322(3) |
| V [Å3] | 205.86(2) |
| Formula units Z | 4 |
| Diffractometer | Rigaku Xcalibur 3, CCD detector, graphite monochromator, Mo Kα radiation, λ = 0.71073 Å |
| Reflections collected/independent within F > 4σ(F) | 3250/303 |
| Measurement range | −10 ≤ h ≤ 11, −5 ≤ k ≤ 5, −8 ≤ l ≤ 8 |
| Fourier difference ρmin/ρmax (electrons/Å3) | −6.56/5.58 |
| Residuals and GOF | R = 0.0352, wR = 0.0321, GOF = 1.37 |
| Atom | Site | a/x | b/y | c/z | Uani |
|---|---|---|---|---|---|
| Ce | 4c | 0.1806(1) | 0.25 | 0.6164(2) | 0.0077(3) |
| Ge | 4c | 0.0372(2) | 0.25 | 0.1334(3) | 0.0104(6) |
| Atom | U11 | U22 | U33 | U12 | U13 | U23 |
|---|---|---|---|---|---|---|
| Ce | 0.0073(5) | 0.0084(5) | 0.0073(5) | 0 | 0.0013(4) | 0 |
| Ge | 0.010(1) | 0.009(1) | 0.012(1) | 0 | 0.0003(8) | 0 |
| Atom | Distance/Å | Atom | Distance/Å |
|---|---|---|---|
| Ce-2 Ge | 3.1186(4) | Ge-2 Ge | 2.6739(4) |
| Ce-2 Ge | 3.1232(4) | Ge-2 Ce | 3.1186(4) |
| Ce-1 Ge | 3.1495(3) | Ge-2 Ce | 3.1232(4) |
| Ce-1 Ge | 3.3358(4) | Ge-1 Ce | 3.1495(3) |
| Ce-1 Ge | 3.3403(4) | Ge-1 Ce | 3.3358(4) |
| Ce-4 Ce | 3.8222(5) | Ge-1 Ce | 3.3403(4) |
| Ce-2 Ce | 3.9028(5) | ||
| Ce-2 Ce | 4.0852(7) |
| Experimental | Calculated | |||||
|---|---|---|---|---|---|---|
| Ge Fragments | (2b)Ge | (2b)Ge | (1b)Ge | |||
| Pearson Symbol Prototype | oP8 FeB | oP8 FeB | oS8 CrB | oS16 LaSi | hP8 β-Li2O2 | hP12 Na2O2 |
| a (Å) | 8.3529(5) | 8.4324 | 4.5573 | 4.5402 | 4.7252 | 8.0172 |
| b (Å) | 4.0878(3) | 4.1185 | 11.275 | 13.757 | 4.7252 | 8.0172 |
| c (Å) | 6.0346(3) | 6.0816 | 4.1198 | 6.7556 | 12.2202 | 6.3456 |
| ΔE (eV/at) | - | 0.000 | 0.000 | 0.002 | −0.101 | −0.068 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freccero, R.; Frick, E.; Wilthorn, C.; Hübner, J.-M. New Insights into the Crystal Chemistry of FeB-Type Compounds: The Case of CeGe. Materials 2022, 15, 9089. https://doi.org/10.3390/ma15249089
Freccero R, Frick E, Wilthorn C, Hübner J-M. New Insights into the Crystal Chemistry of FeB-Type Compounds: The Case of CeGe. Materials. 2022; 15(24):9089. https://doi.org/10.3390/ma15249089
Chicago/Turabian StyleFreccero, Riccardo, Emmelina Frick, Caroline Wilthorn, and Julia-Maria Hübner. 2022. "New Insights into the Crystal Chemistry of FeB-Type Compounds: The Case of CeGe" Materials 15, no. 24: 9089. https://doi.org/10.3390/ma15249089
APA StyleFreccero, R., Frick, E., Wilthorn, C., & Hübner, J.-M. (2022). New Insights into the Crystal Chemistry of FeB-Type Compounds: The Case of CeGe. Materials, 15(24), 9089. https://doi.org/10.3390/ma15249089