

Atomic Diffusion and Crystal Structure Evolution at the Fe-Ti Interface: Molecular Dynamics Simulations
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Simulation Method
2.1. Potential Function
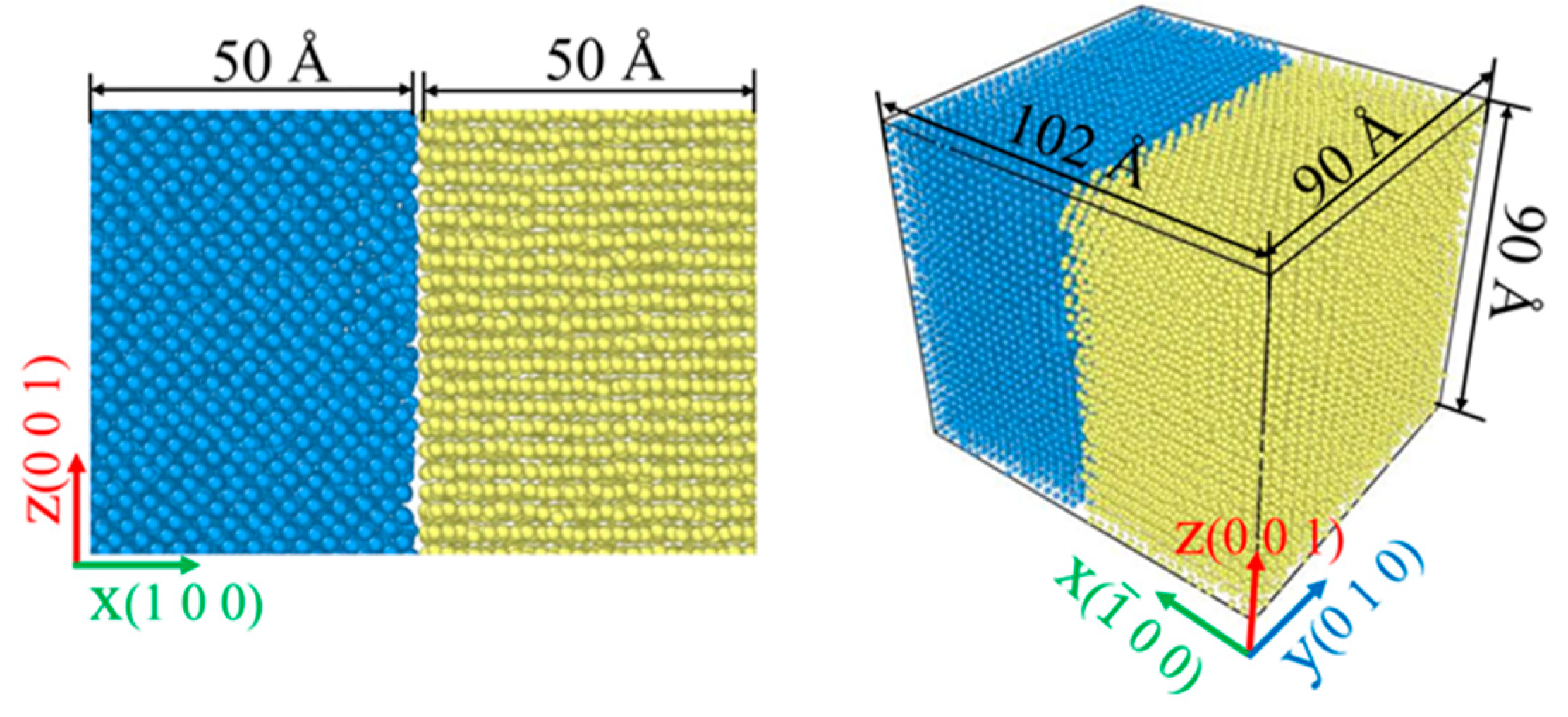
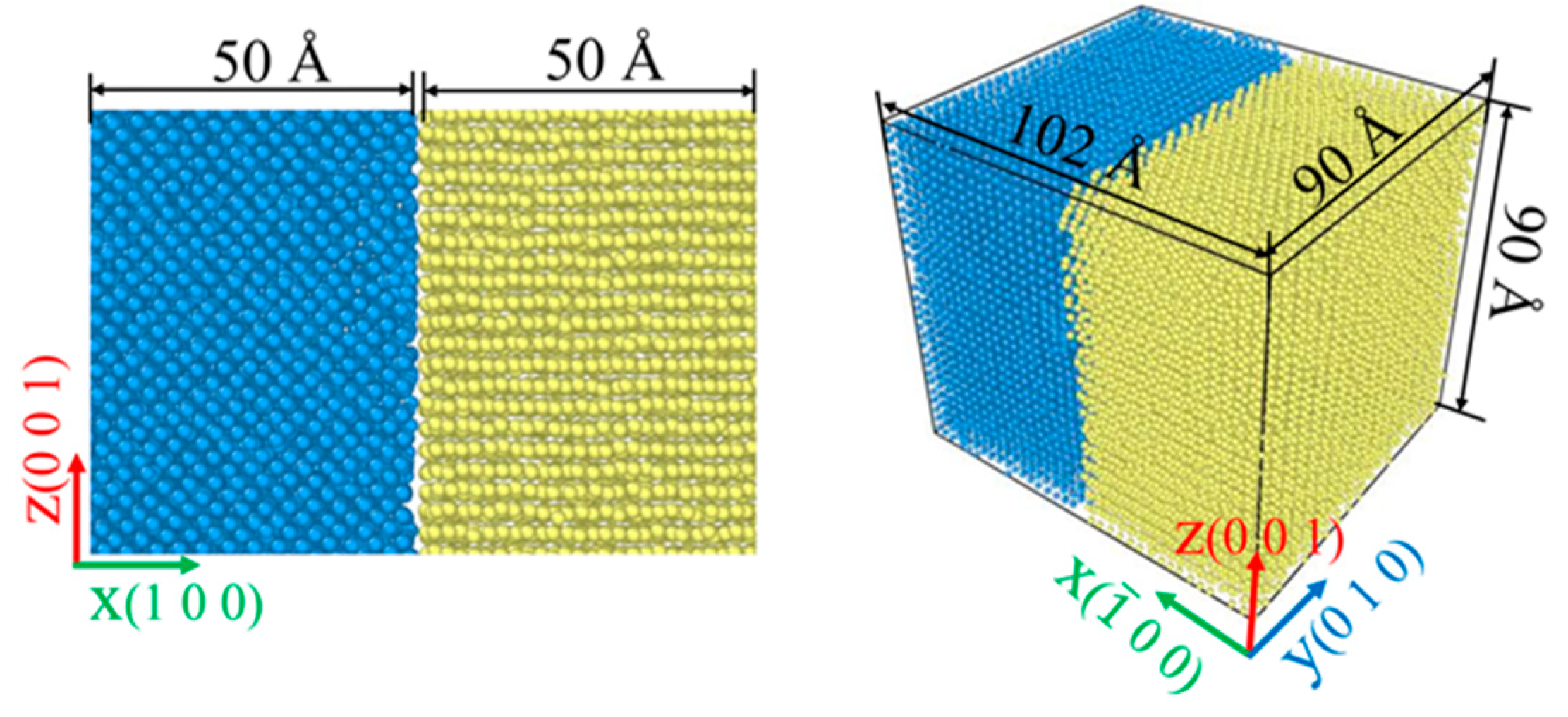
2.2. Simulation Model
3. Result and Discussion
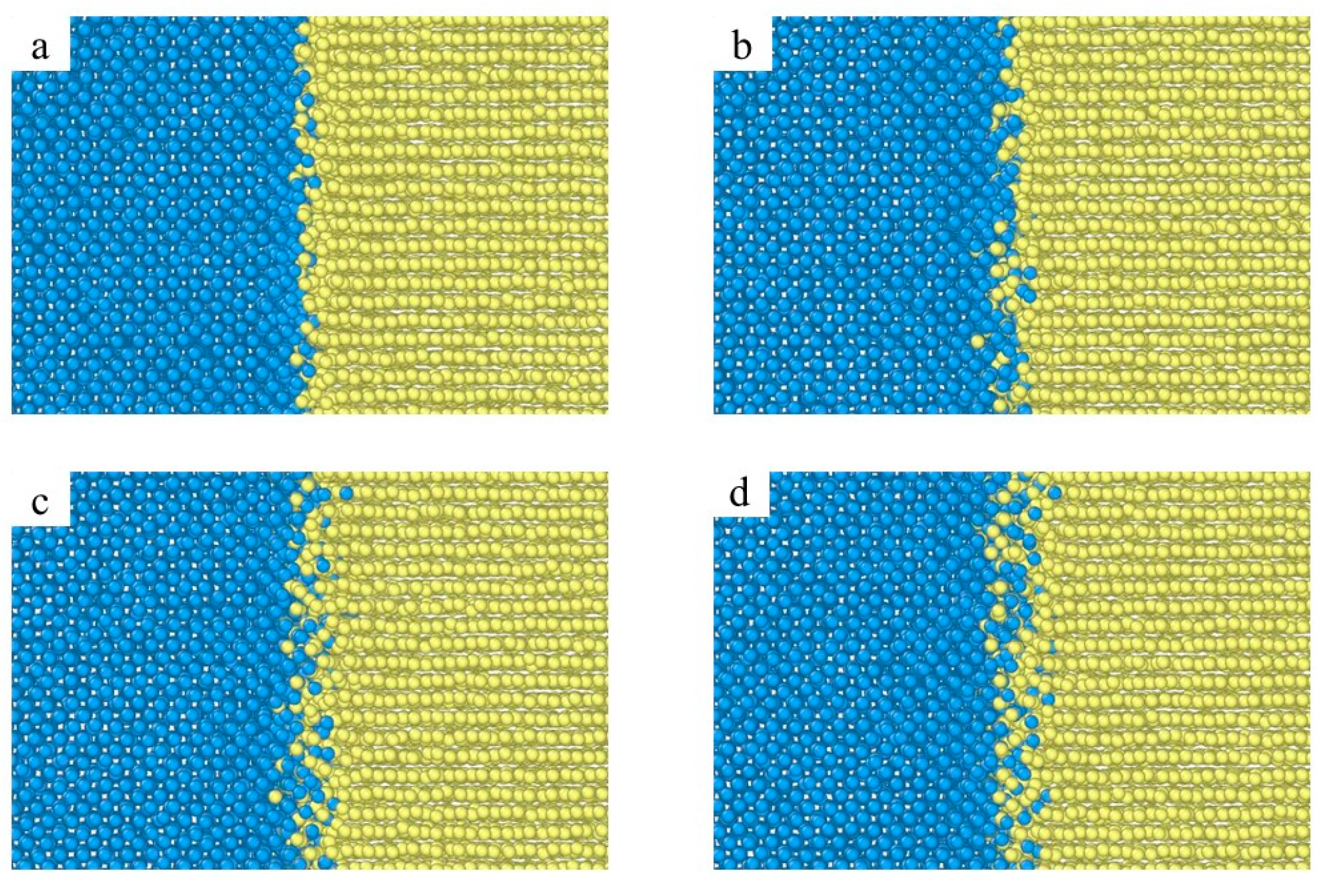
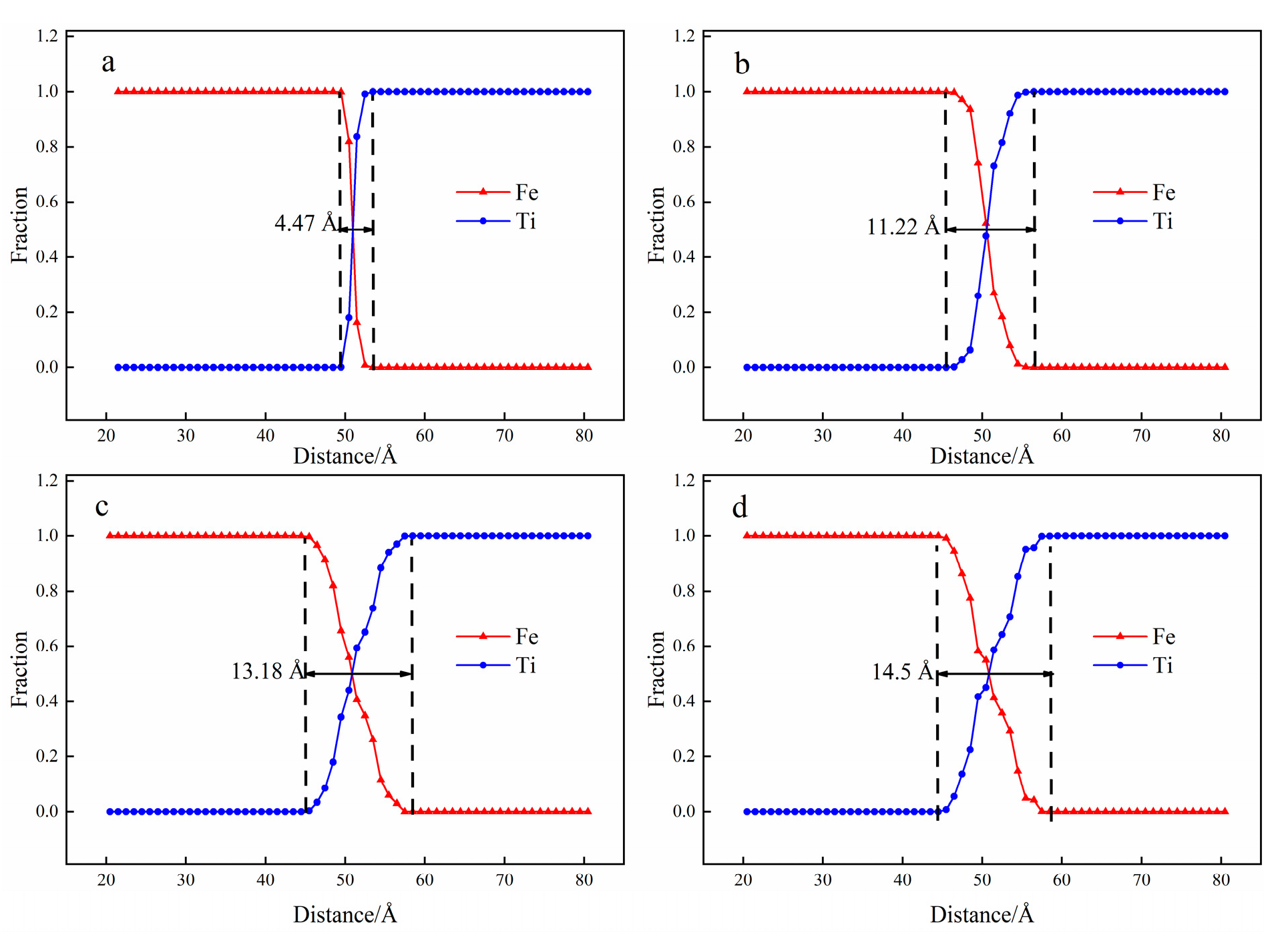
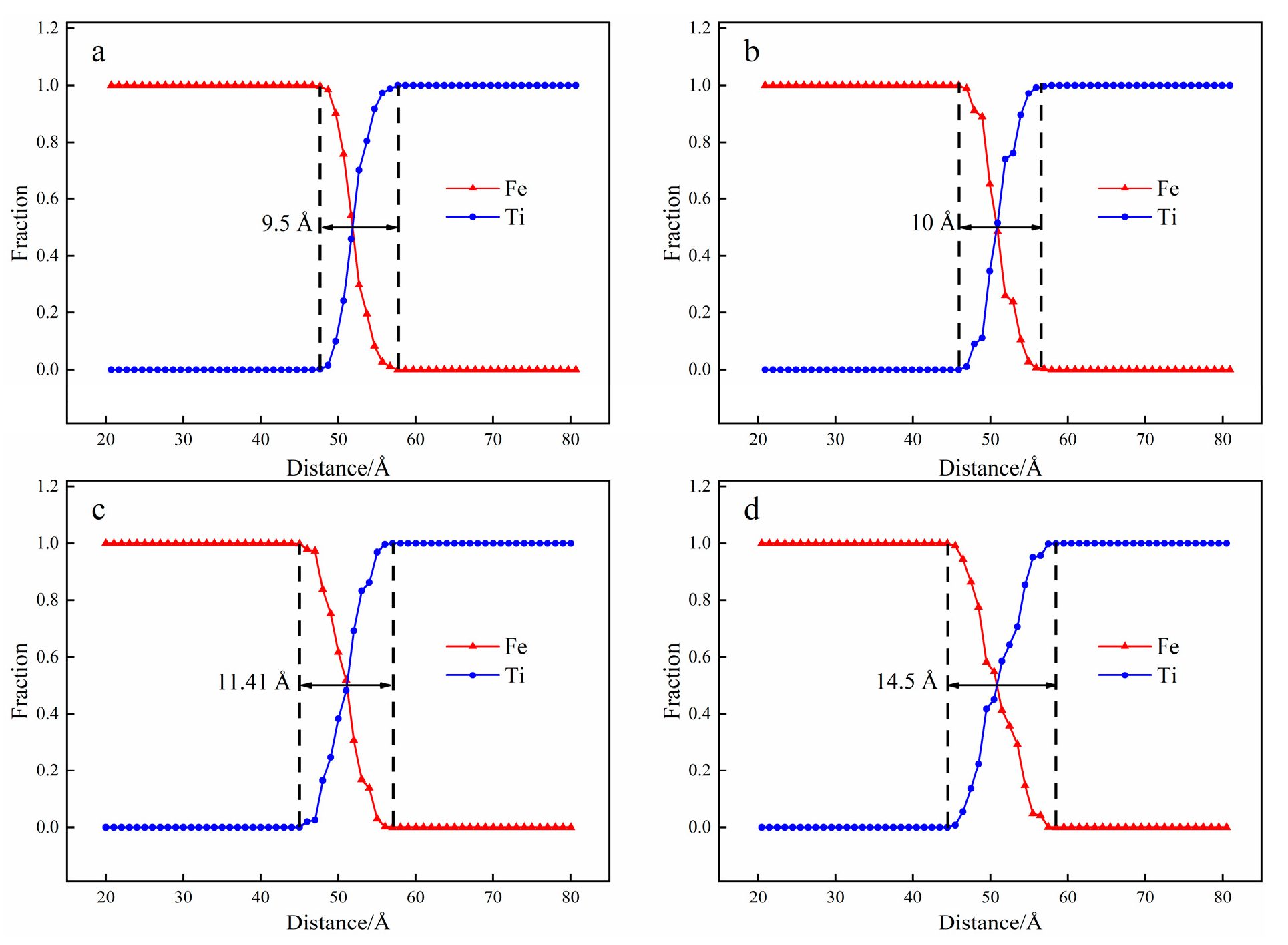
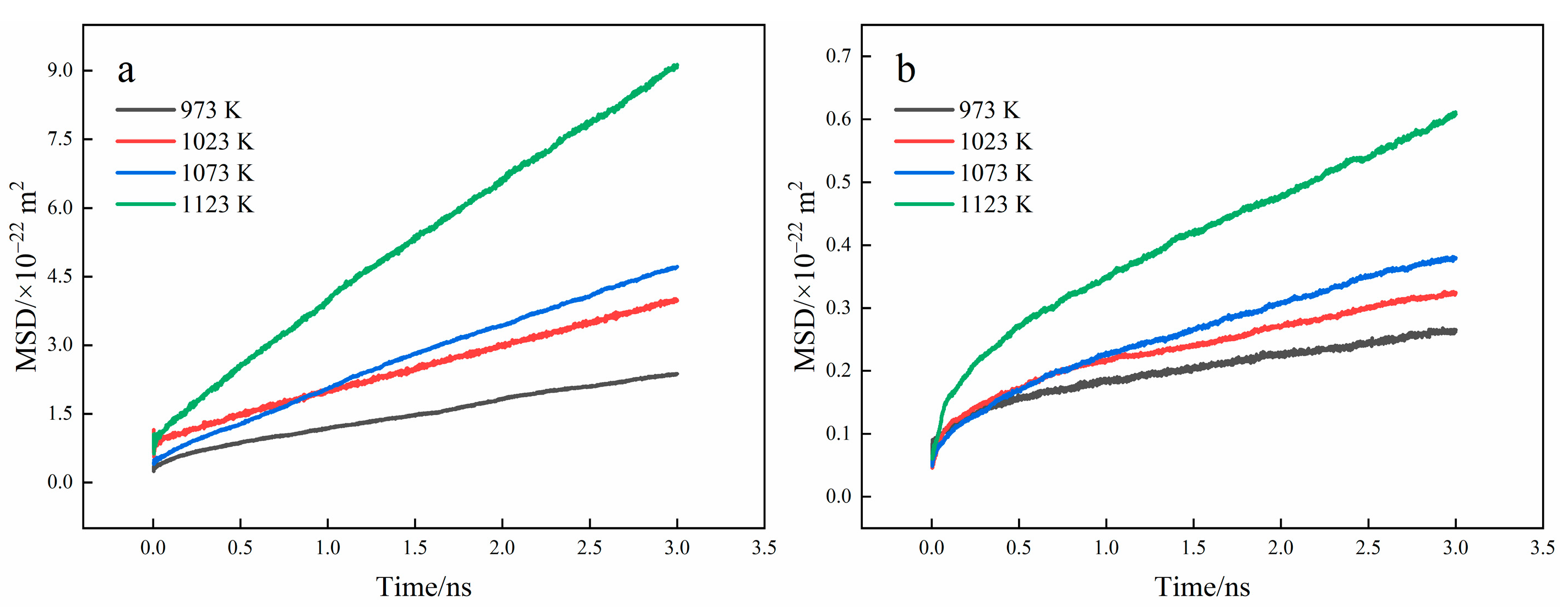
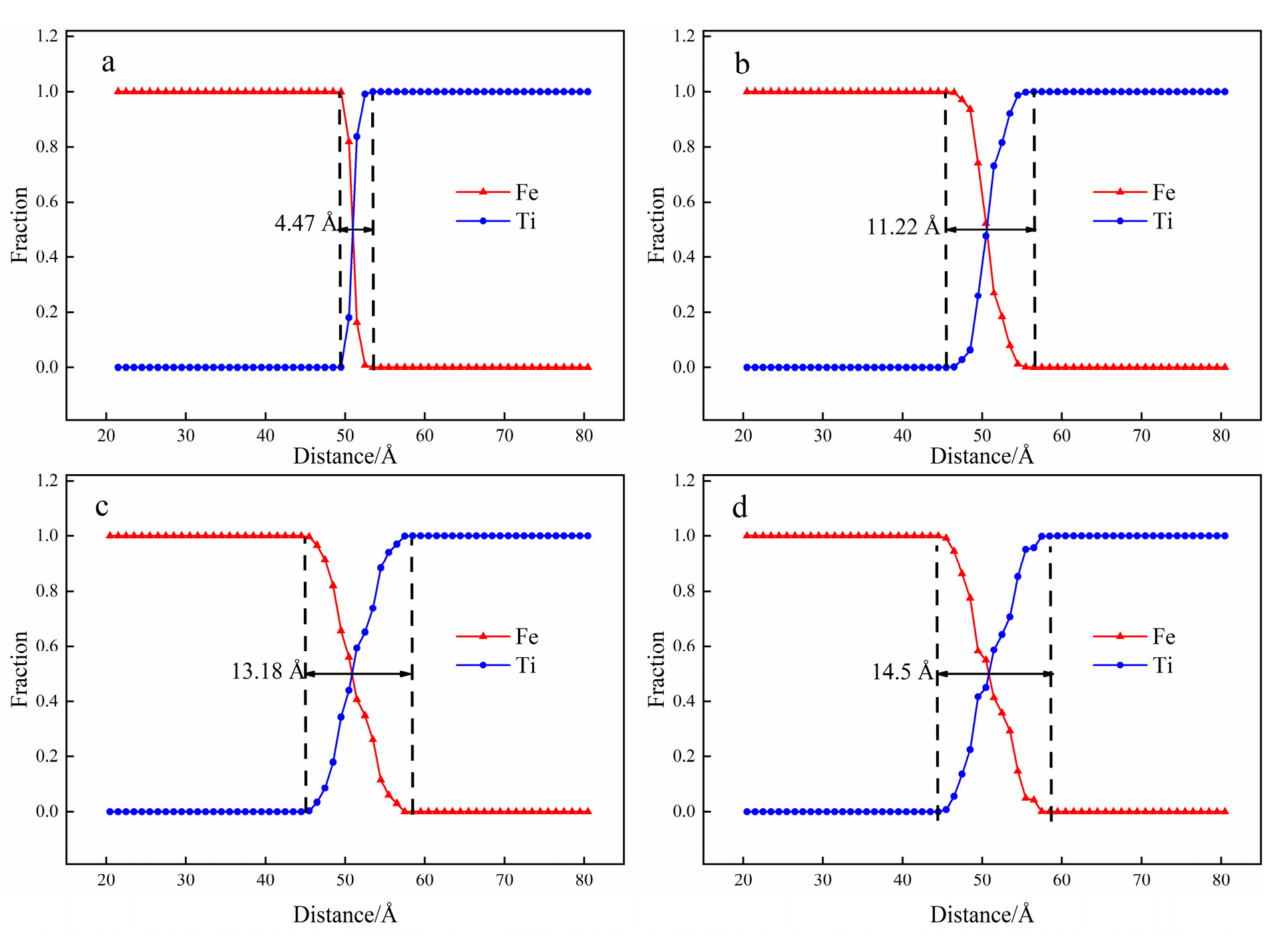
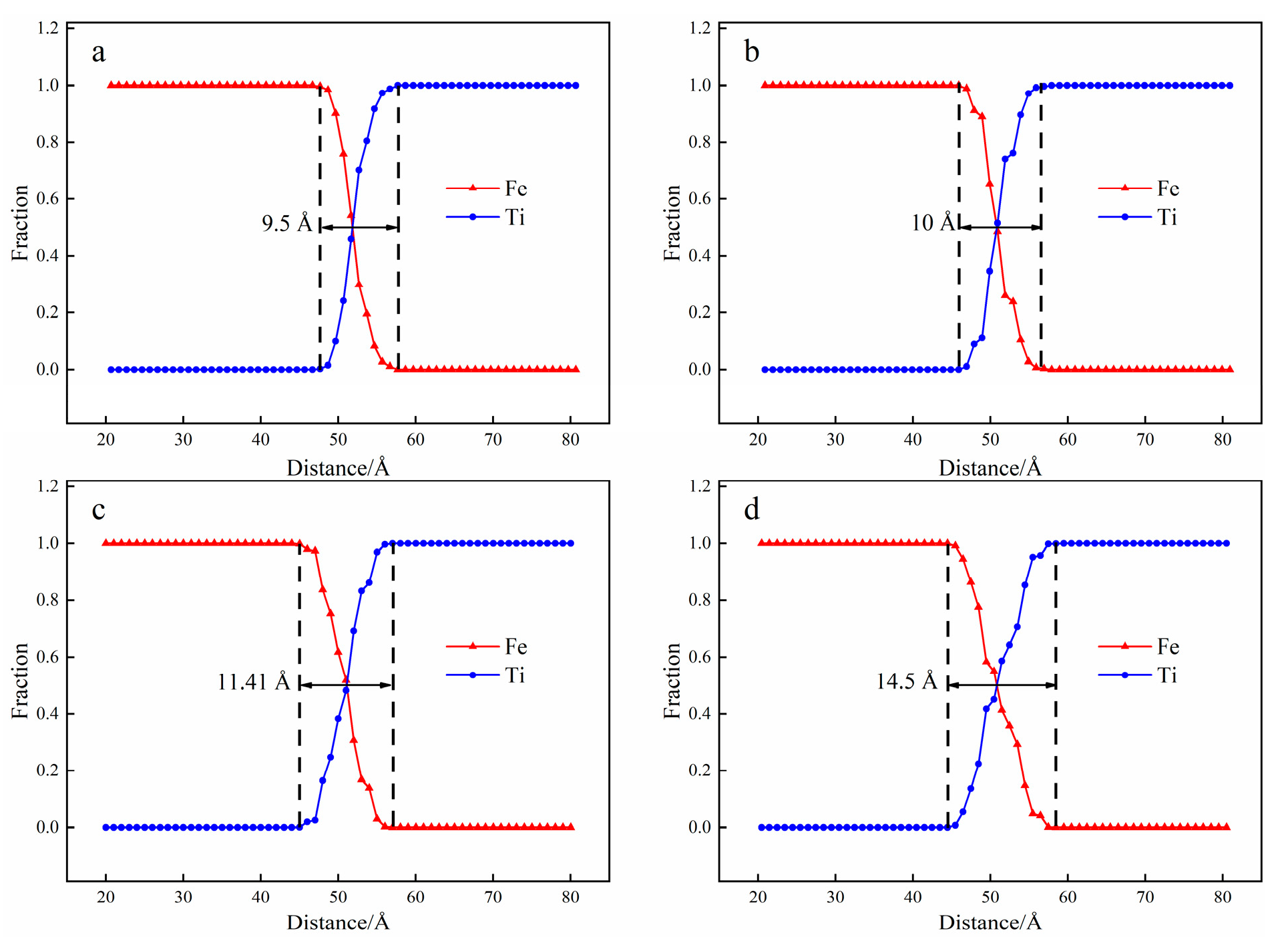
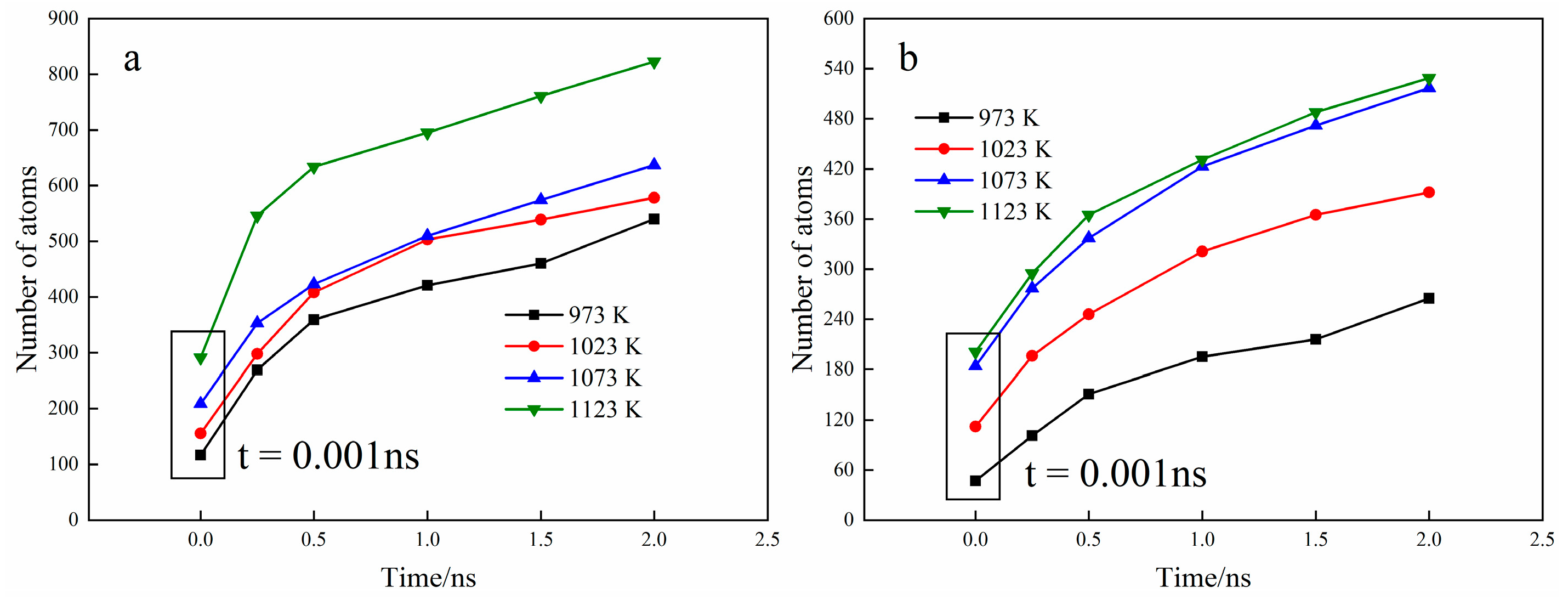
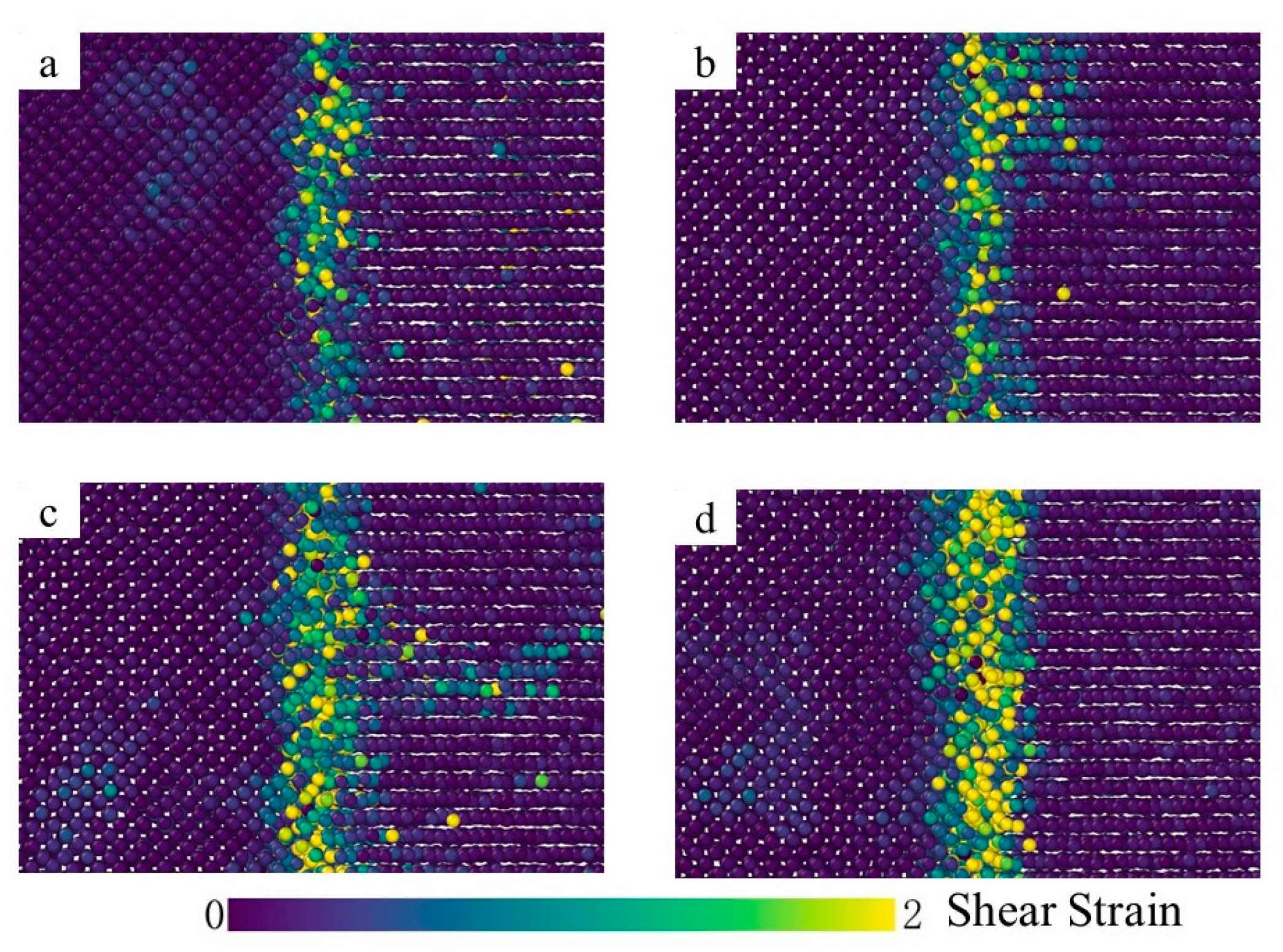
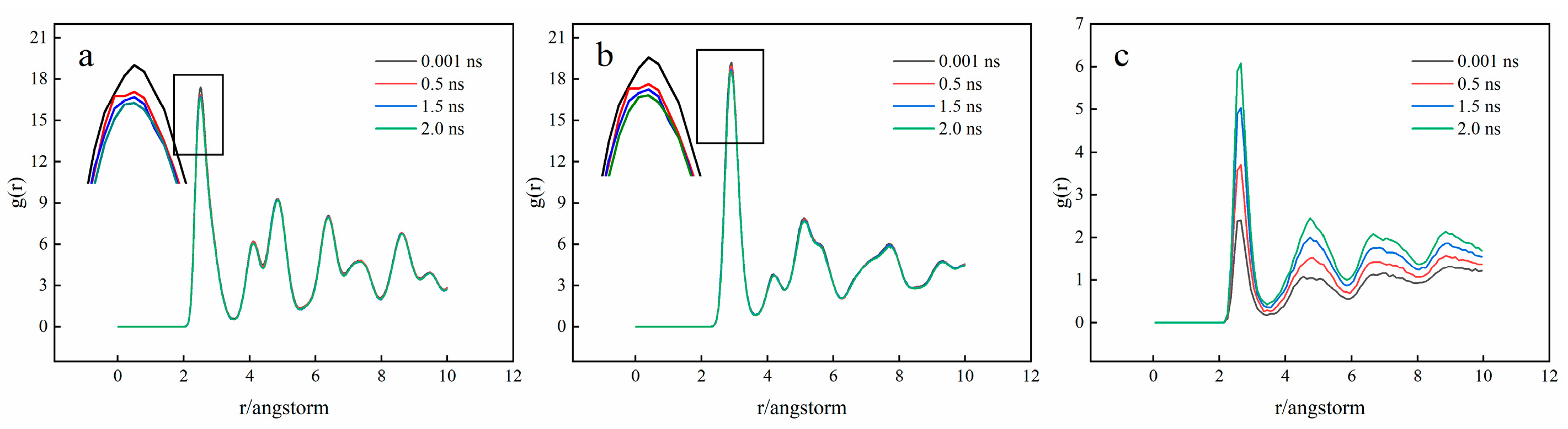
3.1. Interface Diffusion Behavior
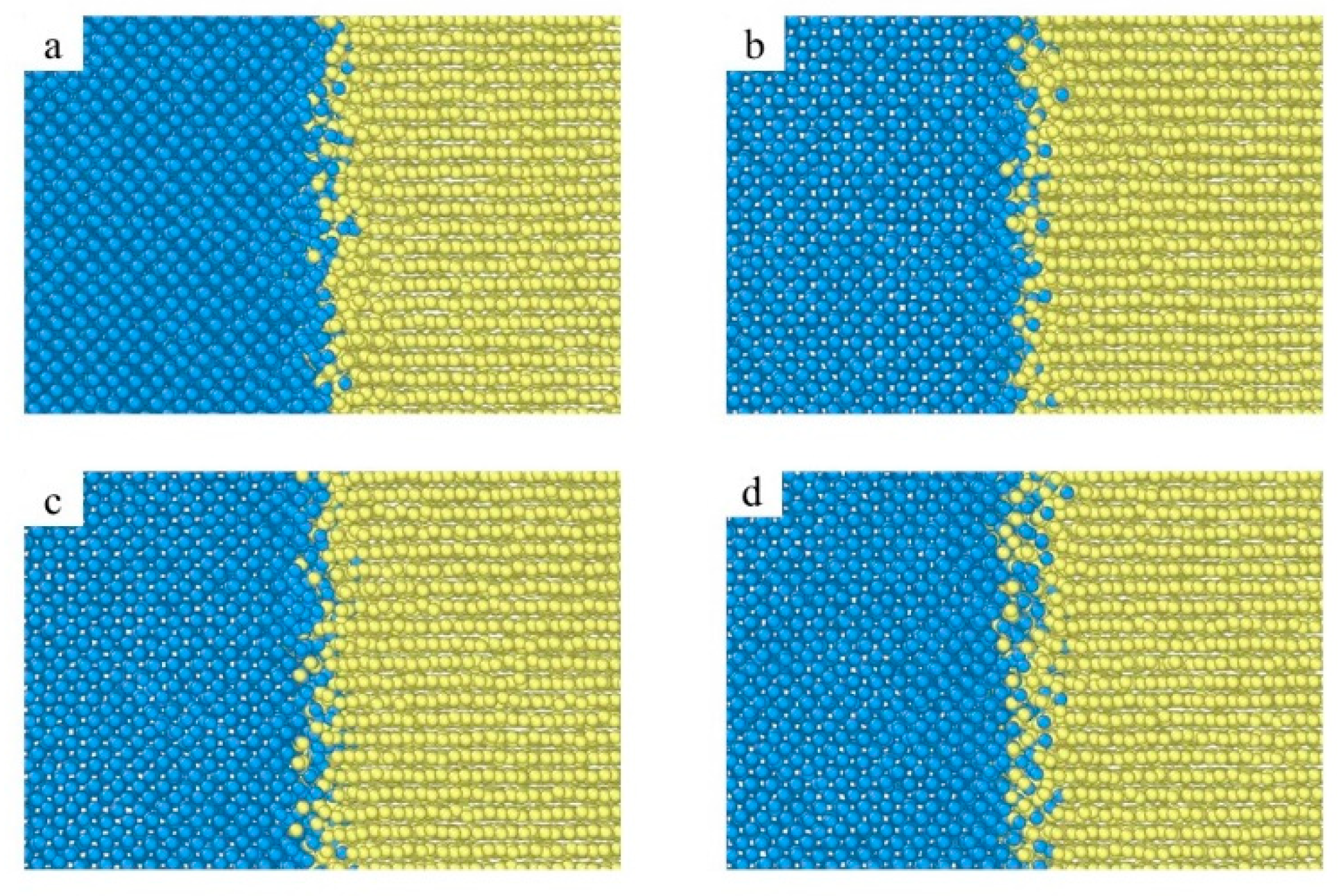
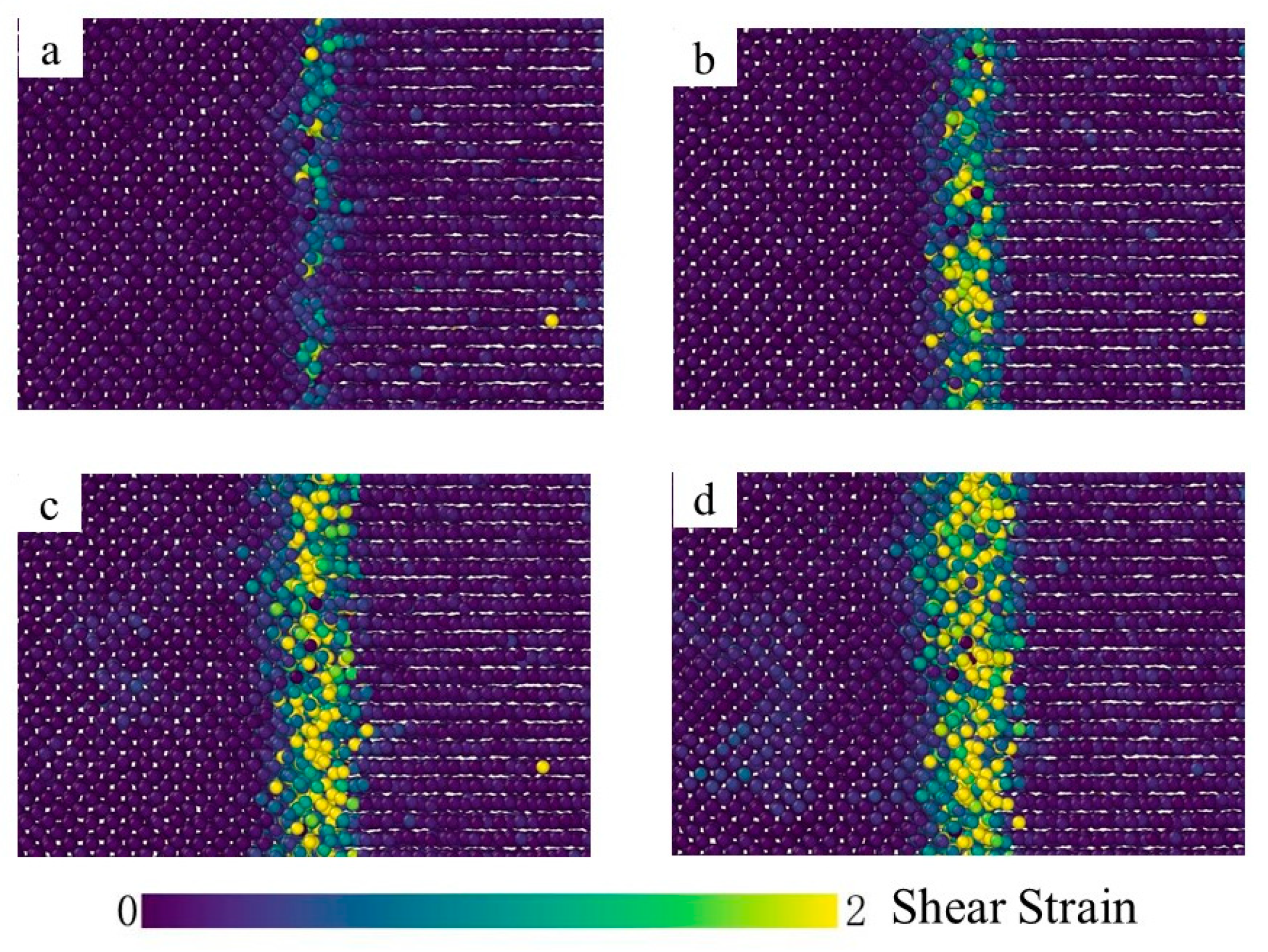
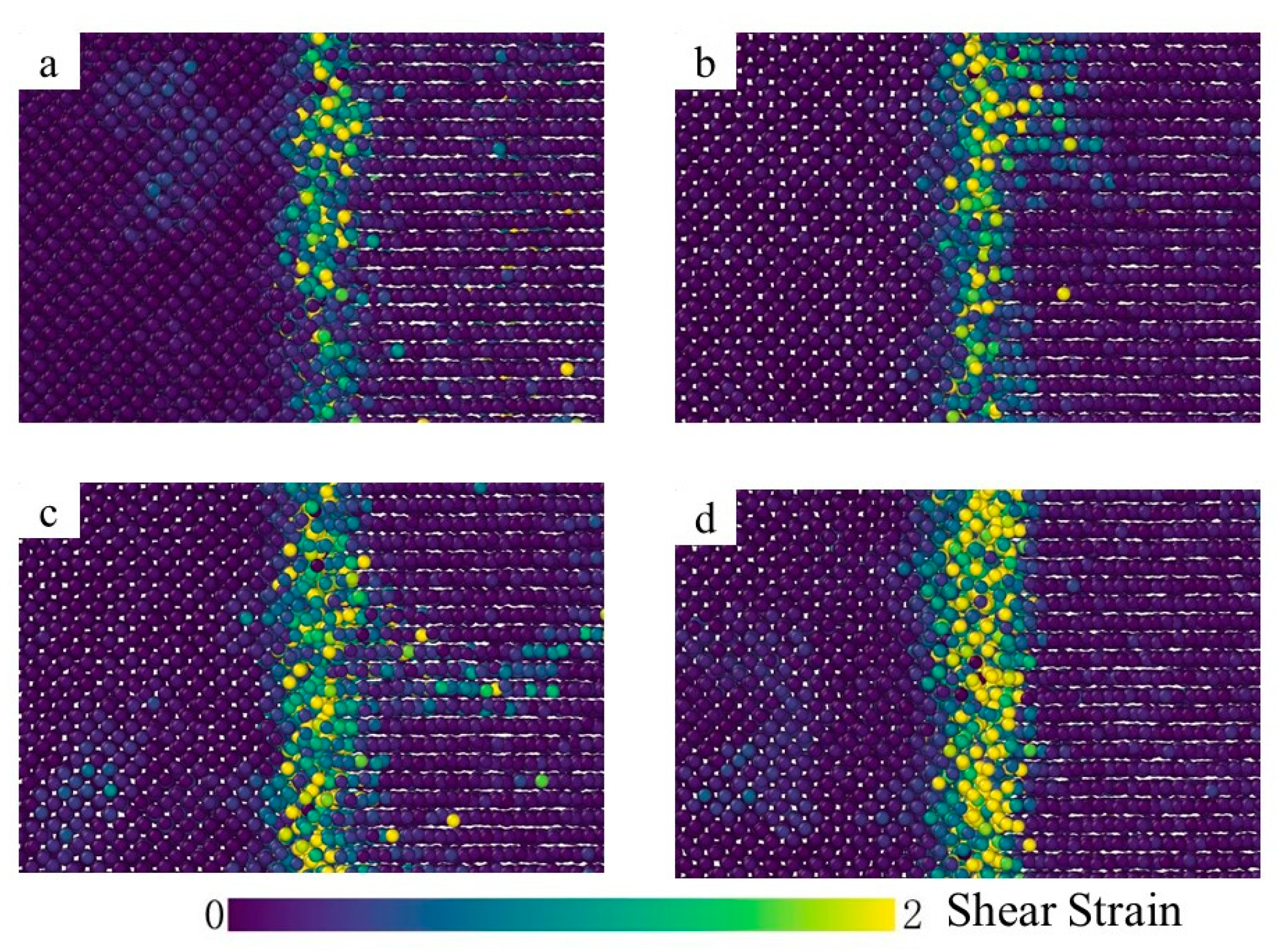
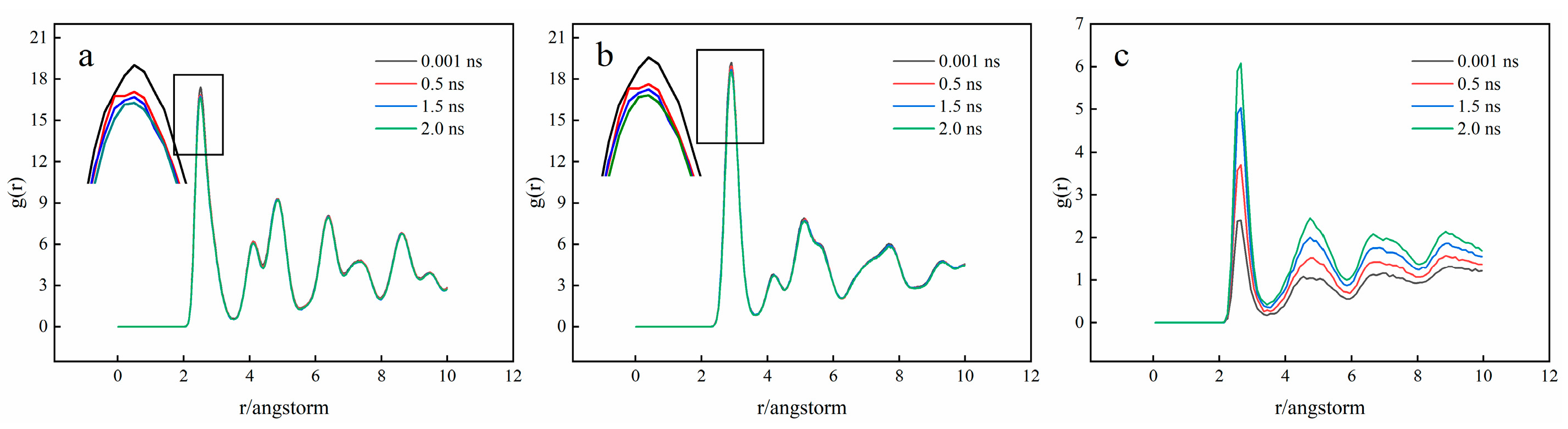
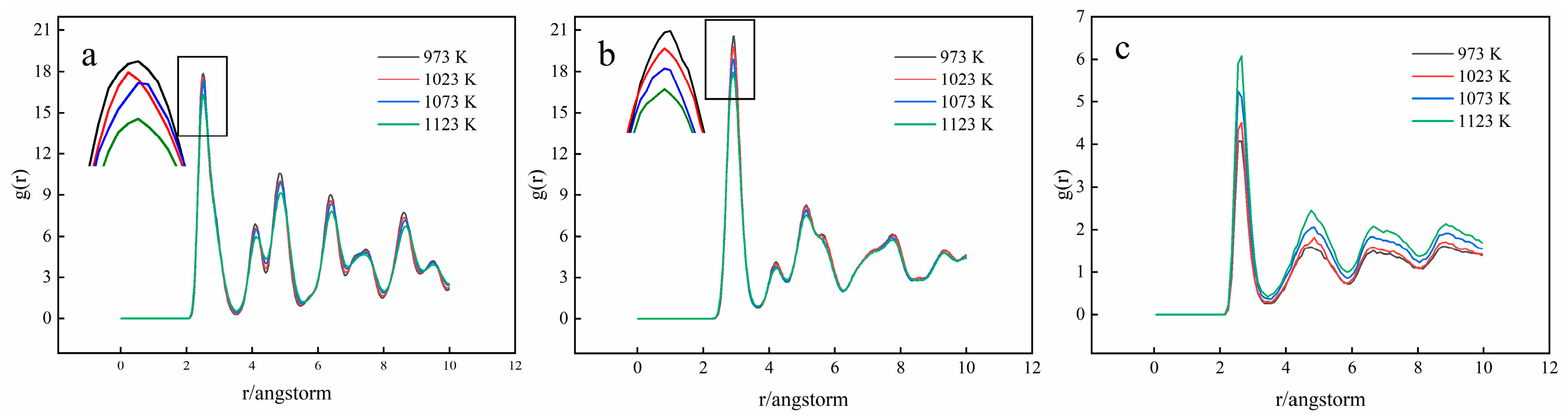
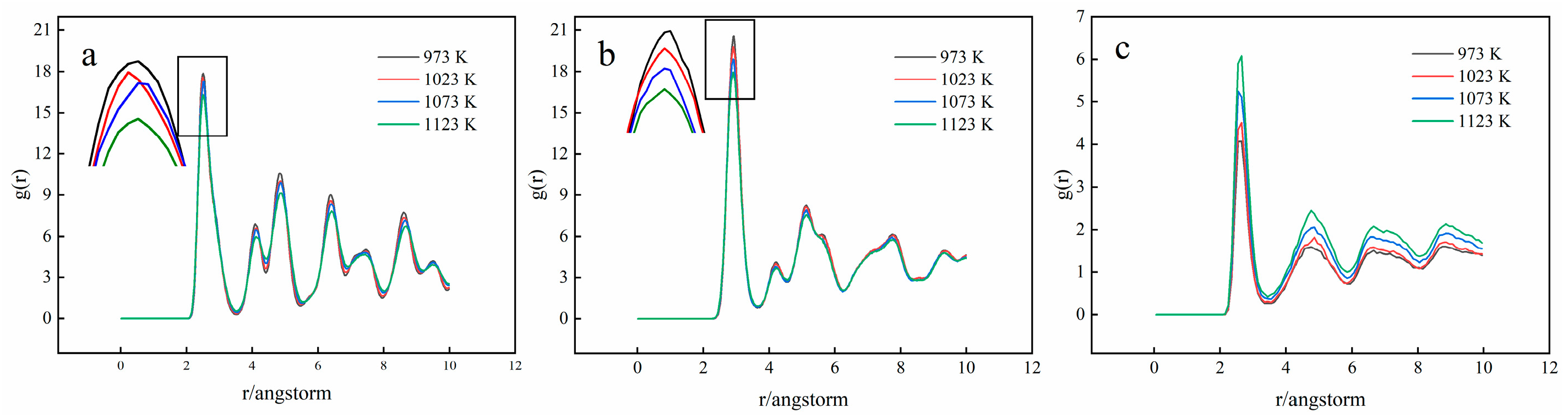
3.2. Crystal Structure Analysis
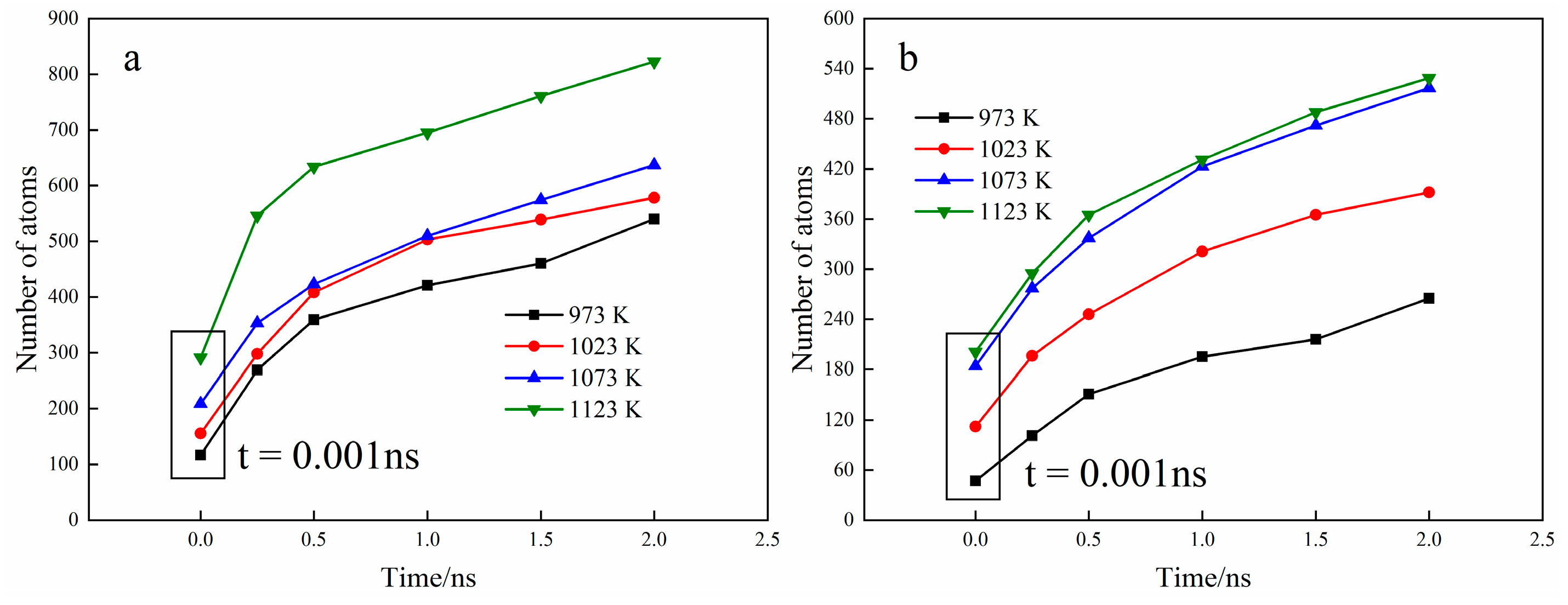
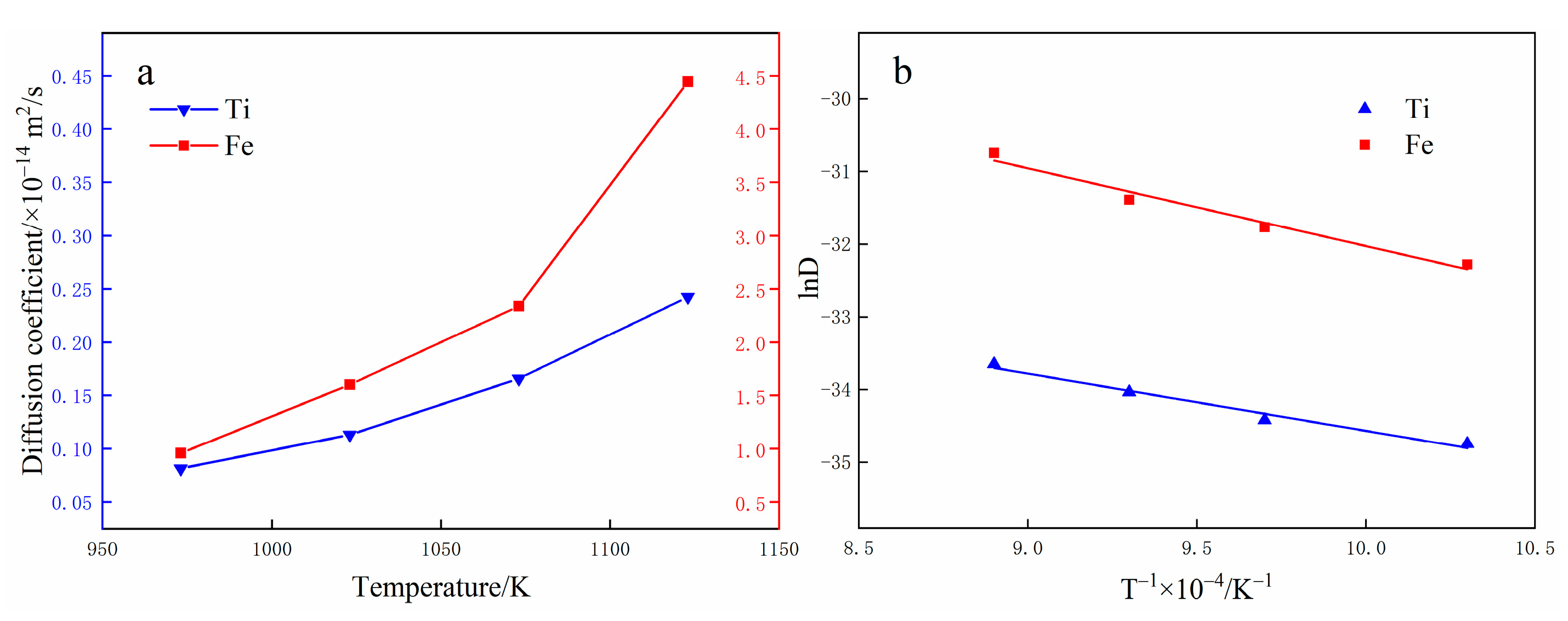
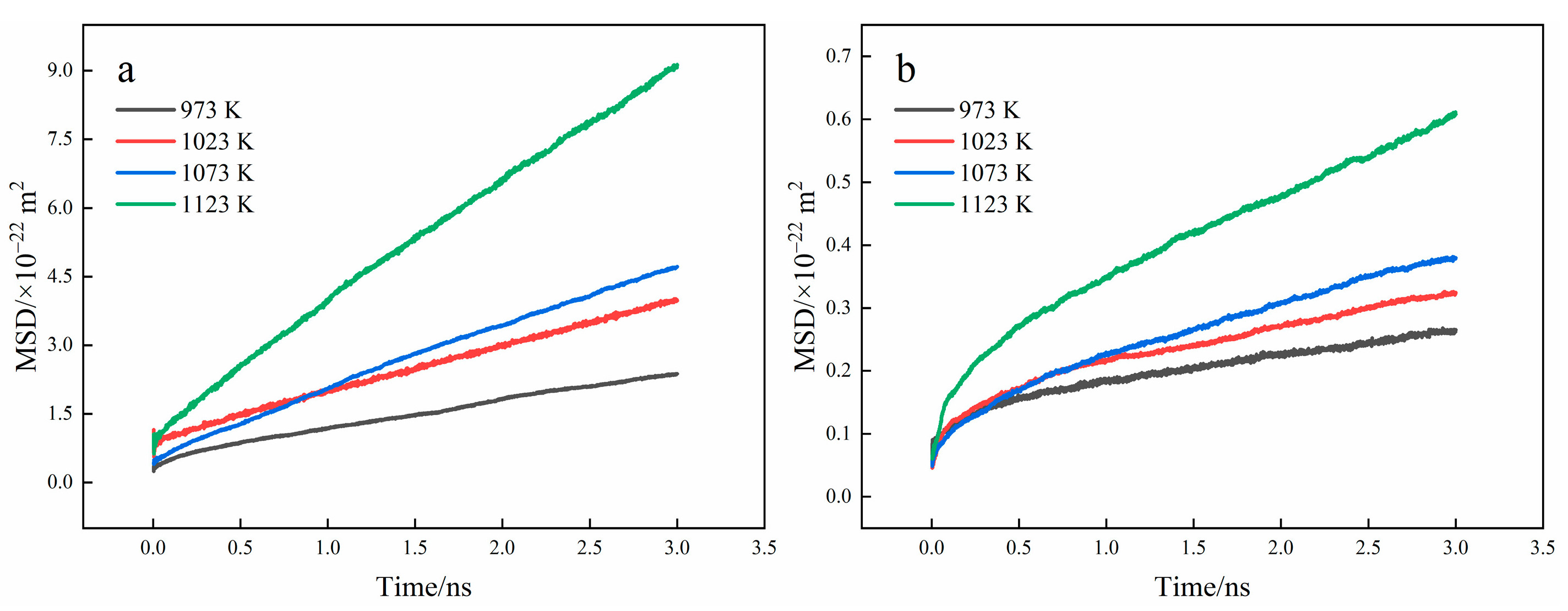
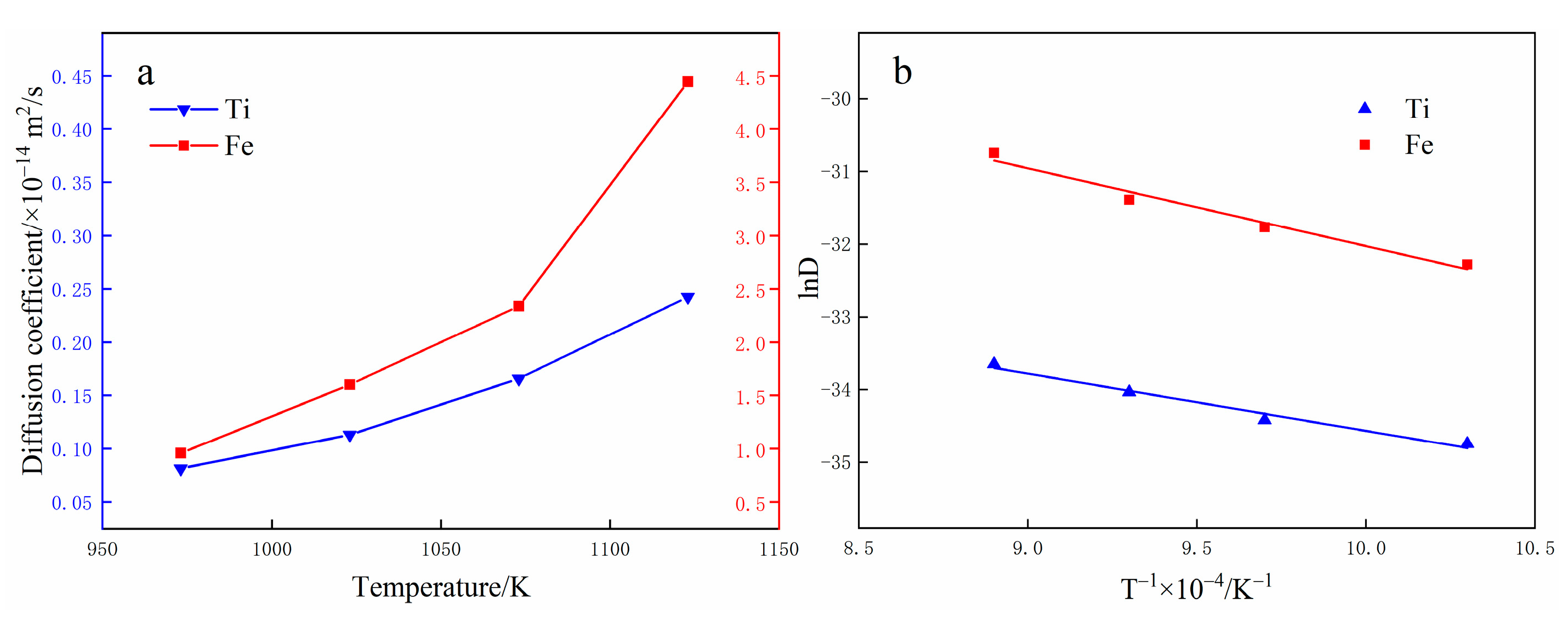
3.3. Diffusion Coefficient Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Luo, Z.; Wang, G.; Xie, G.; Wang, L.; Zhao, K. Effect of niobium interlayer on microstructure and properties of vacuum rolling composite Ti-stainless steel plate. J. Nonferrous Met. China 2013, 23, 3335–3340. [Google Scholar]
- Yadegari, M.; Ebrahimi, A.R.; Karami, A. Effect of Heat Treatment on Interface Microstructure and Bond Strength in Explosively Welded Ti/304L Stainless Steel Clad. Mater. Sci. Technol. 2013, 29, 69–75. [Google Scholar] [CrossRef]
- Jiang, H.; Yan, X.; Liu, J.; Zeng, S.; Duan, X. Diffusion behavior and mathematical model of titanium-steel explosive composite plate during heat treatment. Rare Met. Mater. Eng. 2015, 44, 972–976. [Google Scholar]
- Momono, T.; Enjo, T.; Ikeuchi, K. Effect of Carbon Content on the Diffusion Bonding of Iron and Steel to Titanium. ISIJ Int. 1990, 30, 978–984. [Google Scholar] [CrossRef]
- Li, B.; Chen, Z.; He, W.; Zhou, T.; Wang, Y.; Peng, L.; Li, J.; Liu, Q. Effect of titanium grain orientation on the growth of compounds at diffusion bonded titanium/steel interfaces. Mater. Charact. 2019, 148, 243–251. [Google Scholar] [CrossRef]
- Miglio, L.; Iannuzzi, M.; Raiteri, P.; Celino, M. Silicon diffusion in competitive TiSi2 phases by molecular dynamics simulations. Microelectron. Eng. 2001, 55, 83–92. [Google Scholar] [CrossRef]
- Chen, S.D.; Soh, A.K.; Ke, F.J. Molecular dynamics modeling of diffusion bonding. Scr. Mater. 2005, 52, 1135–1140. [Google Scholar] [CrossRef]
- Chen, S.Y.; Wu, Z.W.; Liu, K.X. Atomic diffusion across Ni50Ti50—Cu explosive welding interface: Diffusion layer thickness and atomic concentration distribution. Chin. Phys. B 2014, 23, 066802. [Google Scholar] [CrossRef]
- Xiu, L.; Wu, J.F. Atomic diffusion behavior in W/Cu diffusion bonding process. J. Fusion Energy 2015, 34, 769–773. [Google Scholar] [CrossRef]
- Samsonov, V.M.; Bembel, A.G.; Popov, I.V.; Vasilyev, S.A.; Talyzin, I.V. Solid-state wetting at the nanoscale: Molecular dynamics and surface diffusion approach. Surf. Innov. 2017, 5, 161–169. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, S. Atomistic investigation on diffusion welding between stainless steel and pure Ni based on molecular dynamics simulation. Mater. 2018, 11, 1957. [Google Scholar] [CrossRef] [PubMed]
- Soltani, S.; Abdolrahim, N.; Sepehrband, P. Molecular dynamics study of self-diffusion in the core of a screw dislocation in face centered cubic crystals. Scr. Mater. 2017, 133, 101–104. [Google Scholar] [CrossRef]
- Béjaud, R.; Durinck, J.; Brochard, S. Twin-interface interactions in nanostructured Cu/Ag: Molecular dynamics study. Acta Mater. 2018, 144, 314–324. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, J.; Qiao, J. Molecular dynamics simulations of atomic diffusion during the Al–Cu ultrasonic welding process. Mater. 2019, 12, 2306. [Google Scholar] [CrossRef]
- Zhou, X.; Zhu, J.; Wu, H. Molecular dynamics studies of the grain-size dependent hydrogen diffusion coefficient of nanograined Fe. Int. J. Hydrogen Energy 2021, 46, 5842–5851. [Google Scholar] [CrossRef]
- Mao, A.; Zhang, J.; Yao, S.; Wang, A.; Wang, W.; Li, Y.; Qiao, C.; Xie, J.; Jia, Y. The diffusion behaviors at the Cu-Al solid-liquid interface: A molecular dynamics study. Results Phys. 2020, 16, 102998. [Google Scholar] [CrossRef]
- Chen, J.; Chen, W.; Wang, C. Modeling and investigation for atomic diffusion and mechanical properties of TiAl/Ti3Al interface: Temperature effect. Appl. Phys. A 2020, 126, 1–9. [Google Scholar] [CrossRef]
- Wang, K.; Bai, H.; Hu, W.; Wu, S.; Wang, H.; Fan, H. Investigation of atomic diffusion behavior of Mo/Au interface. Mater. Chem. Phys. 2021, 257, 123839. [Google Scholar] [CrossRef]
- Zhan, Y.; Wu, H.; Song, W.; Zhu, L. Molecular Dynamics Study of the Diffusion between Virgin and Aged Asphalt Binder. Coat 2022, 12, 403. [Google Scholar] [CrossRef]
- Zurhelle, A.F.; Stehling, W.; Waser, R.; De Souza, R.A.; Menzel, S. Oxygen Diffusion in Platinum Electrodes: A Molecular Dynamics Study of the Role of Extended Defects. Adv. Mater. Interfaces 2022, 9, 2101257. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, J.; Yu, X.; Liu, G.; Fan, D. Atomic-level diffusion at the amorphous Zr50Cu50/crystalline Cu interface: A molecular dynamics study. J. Adv. Join. Processes 2022, 6, 100120. [Google Scholar] [CrossRef]
- Daw, M.S.; Baskes, M.I. Embedded-atom method: Derivation and application to impurities, surfaces, and other defects in metals. Phys. Rev. B 1984, 29, 6443. [Google Scholar] [CrossRef] [Green Version]
- Baskes, M.I. Modified embedded-atom potentials for cubic materials and impurities. Phys. Rev. B 1992, 46, 2727. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Jung, W.S.; Lee, B.J. Modified embedded-atom method interatomic potentials for the Fe–Ti–C and Fe–Ti–N ternary systems. Acta Mater. 2009, 57, 3140–3147. [Google Scholar] [CrossRef]
- Prasanthi, T.N.; Sudha, C.; Saroja, S. Molecular Dynamics Simulation of Diffusion of Fe in HCP Ti Lattice. Trans. Indian Inst. Met. 2018, 71, 1951–1955. [Google Scholar] [CrossRef]
- Ledbetter, H.M.; Reed, R.P. Elastic properties of metals and alloys, I. Iron, nickel, and iron-nickel alloys. J. Phys. Chem. Ref. Data 1973, 2, 531–618. [Google Scholar] [CrossRef]
- Song, Y.; Yang, R.; Li, D.; Guo, Z. A first-principles study of the theoretical strength and bulk modulus of hcp metals. Philos. Mag. A 2001, 81, 321–330. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2009, 18, 015012. [Google Scholar] [CrossRef]
- Terban, M.W.; Billinge, S.J.L. Structural analysis of molecular materials using the pair distribution function. Chem. Rev. 2021, 122, 1208–1272. [Google Scholar] [CrossRef]
- Sa, I.; Lee, B.J. Modified embedded-atom method interatomic potentials for the Fe-Nb and Fe-Ti binary systems. Scr. Mater. 2008, 59, 595–598. [Google Scholar] [CrossRef]
- Trong, D.N.; Long, V.C.; Ţălu, Ş. Effects of Number of Atoms and Doping Concentration on the Structure, Phase Transition, and Crystallization Process of Fe1-x-yNixCoy Alloy: A Molecular Dynamic Study. Appl. Sci. 2022, 12, 8473. [Google Scholar] [CrossRef]
- Yuan, J.; Yu, A.; Zou, P.; Zhao, S.; Li, Y.; Wu, Q. Molecular dynamics simulation of hot pressing diffusion behavior between diamond and titanium. Rare Met. Mater. Eng. 2022, 51, 159–165. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, G.; Luo, X.; Cao, T.; Zhang, A.; Yu, H. Atomic Diffusion and Crystal Structure Evolution at the Fe-Ti Interface: Molecular Dynamics Simulations. Materials 2022, 15, 6302. https://doi.org/10.3390/ma15186302
Xiang G, Luo X, Cao T, Zhang A, Yu H. Atomic Diffusion and Crystal Structure Evolution at the Fe-Ti Interface: Molecular Dynamics Simulations. Materials. 2022; 15(18):6302. https://doi.org/10.3390/ma15186302
Chicago/Turabian StyleXiang, Guojin, Xu Luo, Tianxu Cao, Ankang Zhang, and Hui Yu. 2022. "Atomic Diffusion and Crystal Structure Evolution at the Fe-Ti Interface: Molecular Dynamics Simulations" Materials 15, no. 18: 6302. https://doi.org/10.3390/ma15186302
APA StyleXiang, G., Luo, X., Cao, T., Zhang, A., & Yu, H. (2022). Atomic Diffusion and Crystal Structure Evolution at the Fe-Ti Interface: Molecular Dynamics Simulations. Materials, 15(18), 6302. https://doi.org/10.3390/ma15186302