
Autophagy Plays Multiple Roles in the Soft-Tissue Healing and Osseointegration in Dental Implant Surgery—A Narrative Review

 , ,
, ,
Abstract
:1. Introduction
2. Dental Implant Surgery Lesion Healing and Implant Osseointegration
- 1.
- Wounding is immediately followed by blood vessel constriction and coagulation cascade triggering clot generation. Fibrin clots have hemostatic effects and also expose a temporary extracellular matrix (ECM) that encourages cell migration [39] (Figure 1). Fibrin clots also release chemical signals in order to recruit the inflammatory cells to the wound scene. The neutrophils that reach the injury site remove necrotic tissue. The recruited monocytes differentiate into MFs that act as phagocytes and initiate the secretion of inflammatory cytokines. These cytokines will trigger local immune reactions [39] (Figure 1).
- 2.
- Following the inflammatory phase, the next step of the wound healing process is the proliferative phase, which includes: (1) vascular network remodeling, granulation tissues formation, and epithelial regeneration; (2) MFs, important sources of growth factors, release of vascular endothelial growth factor (VEGF) to initiate vascular remodeling by ECs activation; (3) FBs migrate to the injury site and synthesize collagen, fibronectin, consequently initiating the ECM organization. KCs proliferation and their migration from the edges of the wound to the wound center are essential for wound closure (re-epithelialization) [39,40,41,42] (Figure 1).
- 3.
- Remodeling is the final phase of wound healing. This step is characterized by type III collagen replacement with type I collagen within the granulation tissue. FBs and the FBs derived MFBs play key roles in further wound sealing. Additionally, granulation tissue degradation and blood vessel degeneration generate the avascular and acellular mature wound [39] (Figure 1).
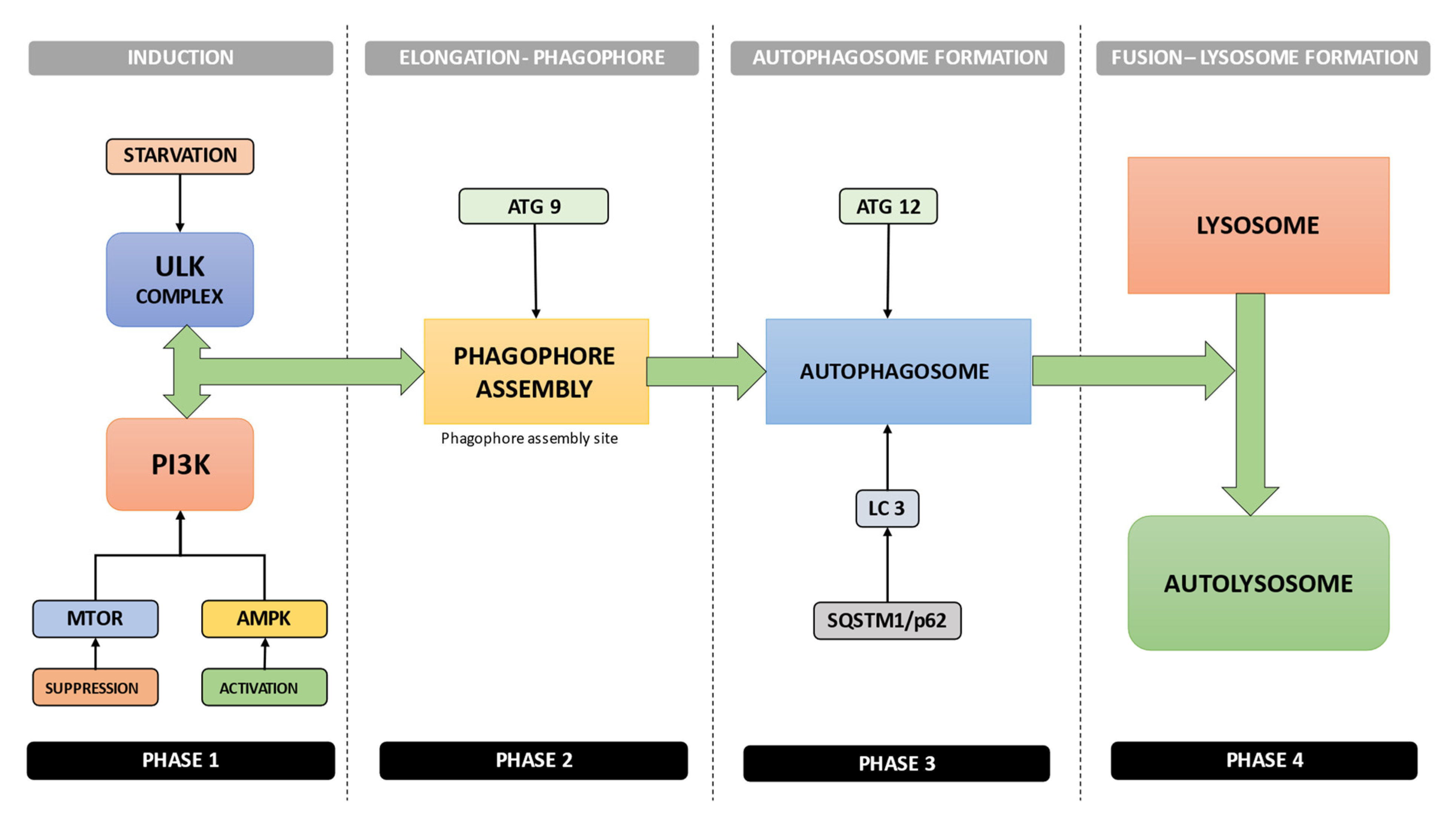
3. Autophagy Mechanism
- 1.
- 2.
- 3.
- The conjugation systems ATG12 and LC3 are key factors in the regulation of the phagophore elongation to the autophagosome. Autophagy is suppressed by mTOR, the major autophagy inhibitory factor, as a response to abundant nutrient conditions. Class I PI3K and AKT signaling mediates this suppressive action [44] (Figure 2);
- 4.
- 5.
- Subsequently, the autophagosome will fuse with a lysosome, resulting in the formation of the autolysosome. The autophagosome constituents placed inside the autolysosome will be hydrolytically degraded. The engulfed SQSTM1 complex will be degraded inside the autolysosome, which emphasizes SQSTM1’s role as an autophagy flux marker [44] (Figure 2).
4. Autophagy in the Main Cellular Types Involved in Dental Implant Surgery Lesion Healing and Implant Osseointegration
4.1. Autophagy in Macrophages (MFs)
4.2. Autophagy in Endothelial Cells (ECs)
4.3. Autophagy in Osteoclasts (OCs)
4.4. Autophagy in Osteoblasts (OBs)
4.5. Autophagy in Fibroblasts (FBs) and Myofibroblasts (MFBs)
4.6. Autophagy in Keratinocytes (KCs)
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Branemark, R.; Adell, U.; Breine, B.O.; Hansson, J.; Strom, L.; Ohlsson, A. Intra-osseous anchorage of dental prostheses: I. Experimental studies. Scand. J. Plast. Reconstr. Surg. 1969, 3, 81–100. [Google Scholar] [CrossRef]
- Searson, L.J. History and Development of Dental Implants. In Implantology in General Dental Practice; Narim, L., Wilson, H.F., Eds.; Quintessence Publishing Co.: London, UK, 2005; pp. 19–41. [Google Scholar]
- Branemark, R.; Adell, T.; Albrektsson, U.; Lekholm, S.; Lundkvist; Rockler, B. Osseointegrated titanium fixtures in the treatment of edentulousness. Biomaterials 1983, 4, 25–28. [Google Scholar] [CrossRef]
- Ehrenfest, D.M.D.; Coelho, P.G.; Kang, B.-S.; Sul, Y.-T.; Albrektsson, T. Classification of osseointegrated implant surfaces: Materials, chemistry and topography. Trends Biotechnol. 2010, 28, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Smeets, R.; Henningsen, A.; Jung, O.; Heiland, M.; Hammacher, C.; Stein, J.M. Definition, etiology, prevention and treatment of peri-implantitis—A review. Head Face Med. 2014, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.J.; Kim, J.H.; Byun, S. Modulation of Autophagy for Controlling Immunity. Cells 2019, 8, 138. [Google Scholar] [CrossRef]
- Liu, K.; Zhao, E.; Ilyas, G.; Lalazar, G.; Lin, Y.; Haseeb, M.; Tanaka, K.E.; Czaja, M.J. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015, 11, 271–284. [Google Scholar] [CrossRef]
- Zhang, Q.; Sun, J.; Wang, Y.; He, W.; Wang, L.; Zheng, Y.; Wu, J.; Zhang, Y.; Jiang, X. Antimycobacterial and Anti-inflammatory Mechanisms of Baicalin via Induced Autophagy in Macrophages Infected with Myco-bacterium tuberculosis. Front. Microbiol. 2017, 8, 2142. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef]
- Zhang, J.; Song, X.; Cao, W.; Lu, J.; Wang, X.; Wang, G.; Wang, Z.; Chen, X. Autophagy and mitochondrial dysfunction in adjuvant-arthritis rats treatment with resveratrol. Sci. Rep. 2016, 6, 32928. [Google Scholar] [CrossRef]
- Simu, S.Y.; Ahn, S.; Castro-Aceituno, V.; Singh, P.; Mathiyalagan, R.; Jimenez-Perez, Z.E.; Hurh, J.; Oi, L.Z.; Hun, N.J.; Kim, Y.J.; et al. Gold Nanoparticles Synthesized with Fresh Panax ginseng Leaf Extract Suppress Adipogenesis by Downregulating PPARgamma/CEBPalpha Signaling in 3T3-L1 Mature Adipocytes. J. Nanosci. Nanotechnol. 2019, 19, 701–708. [Google Scholar] [CrossRef]
- Na Yun, S.; Moon, S.J.; Ko, S.K.; Im, B.O.; Chung, S.H. Wild ginseng prevents the onset of high-fat diet induced hyperglycemia and obesity in icr mice. Arch. Pharmacal Res. 2004, 27, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.-F.; Yu, T.; Chu, X.-M. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020, 11, 776. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V. Autophagy in Inflammation, Infection, and Immunometabolism. Immunity 2021, 54, 437–453. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Li, Y.; Ji, T.; Chao, Y.; Li, J.; Fu, Y.; Wang, S.; Chen, Q.; Chen, W.; Huang, F.; et al. Estrogen prevent atherosclerosis by attenuating endothelial cell pyroptosis via activation of estrogen receptor α-mediated autophagy. J. Adv. Res. 2020, 28, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ding, H.; Yan, Y.; Chen, Q.; Zhang, J.; Chen, B.; Cao, J. Intermittent hypoxia-induced autophagy via AMPK/mTOR signaling pathway attenuates endothelial apoptosis and dysfunction in vitro. Sleep Breath. 2021, 25, 1859–1865. [Google Scholar] [CrossRef]
- Zha, S.; Li, Z.; Chen, S.; Liu, F.; Wang, F. MeCP2 inhibits cell functionality through FoxO3a and autophagy in endothelial progenitor cells. Aging 2019, 11, 6714–6733. [Google Scholar] [CrossRef]
- Liang, P.; Jiang, B.; Li, Y.; Liu, Z.; Zhang, P.; Zhang, M.; Huang, X.; Xiao, X. Autophagy promotes angiogenesis via AMPK/Akt/mTOR signaling during the recovery of heat-denatured endothelial cells. Cell Death Dis. 2018, 9, 1152. [Google Scholar] [CrossRef]
- Stolarz, A.J.; Mu, S.; Zhang, H.; Fouda, A.Y.; Rusch, N.J.; Ding, Z. Opinion: Endothelial Cells-Macrophage-Like Gatekeepers? Front. Immunol. 2022, 13, 902945. [Google Scholar] [CrossRef]
- Chung, Y.H.; Jang, Y.; Choi, B.; Song, D.H.; Lee, E.J.; Kim, S.M.; Song, Y.; Kang, S.W.; Yoon, S.Y.; Chang, E.J. Beclin-1 is required for RANKL-induced OCsdifferentiation. J. Cell. Physiol. 2014, 229, 1963–1971. [Google Scholar] [CrossRef]
- DeSelm, C.J.; Miller, B.C.; Zou, W.; Beatty, W.L.; van Meel, E.; Takahata, Y.; Klumperman, J.; Tooze, S.A.; Teitelbaum, S.L.; Virgin, H.W. Autophagyproteins regulate the secretory component of OCsic bone resorption. Dev. Cell 2011, 21, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.H.; Yoon, S.Y.; Choi, B.; Sohn, D.H.; Yoon, K.H.; Kim, W.J.; Kim, D.H.; Chang, E.J. Microtu-bule-associated protein light chain 3 regulatesCdc42-dependent actin ring formation in OCs. Int. J. Biochem. Cell Biol. 2012, 44, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Wang, J.; Huang, S.; Sun, S.; Zhang, Z.; Bao, N. Overactive autophagy is a pathological mechanism underlying premature suture ossification in nonsyndromic craniosynostosis. Sci. Rep. 2018, 8, 6525. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y.; Cao, Z.; Dou, C.; Bai, Y.; Liu, C.; Dong, S.; Fei, J. Staphylococcal lipoteichoic acid promotes osteogenic differentiation of mouse mesenchymal stem cells by increasing autophagic activity. Biochem. Biophys. Res. Commun. 2017, 485, 421–426. [Google Scholar] [CrossRef]
- Xu, R.; Shi, G.; Xu, L.; Gu, Q.; Fu, Y.; Zhang, P.; Cheng, J.; Jiang, H. Simvastatin improves oral implant osseointegration via enhanced autophagy and osteogenesis of BMSCs and inhibited osteoclast activity. J. Tissue Eng. Regen. Med. 2018, 12, 1209–1219. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, Z.; Chen, S.; Qiu, J.; Li, Q.; Wang, S.; Zhou, W.; Chen, D.; Yang, G.; Guo, L. Transcription factor EB-mediated autophagy promotes dermal fibroblast differentiation and collagen production by regulating endoplasmic reticulum stress and autophagy-dependent secretion. Int. J. Mol. Med. 2020, 47, 547–560. [Google Scholar] [CrossRef]
- Cao, C.; Wang, W.; Lu, L.; Wang, L.; Chen, X.; Guo, R.; Li, S.; Jiang, J. Inactivation of Beclin-1-dependent autophagy promotes ursolic acid-induced apoptosis in hypertrophic scar fibroblasts. Exp. Dermatol. 2018, 27, 58–63. [Google Scholar] [CrossRef]
- Migneault, F.; Hébert, M.-J. Autophagy, tissue repair, and fibrosis: A delicate balance. Matrix Biol. 2021, 100–101, 182–196. [Google Scholar] [CrossRef]
- Li, R.; Kato, H.; Taguchi, Y.; Umeda, M. Intracellular glucose starvation affects gingival homeostasis and autophagy. Sci. Rep. 2022, 12, 1–10. [Google Scholar] [CrossRef]
- Gump, J.M.; Thorburn, A. Autophagy and apoptosis: What is the connection? Trends Cell Biol. 2011, 21, 387–392. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yamamoto, A.; Hatano, M.; Kobayashi, Y.; Kabeya, Y.; Suzuki, K.; Tokuhisa, T.; Ohsumi, Y.; Yoshimori, T. Dissection of Autophagosome Formation Using Apg5-Deficient Mouse Embryonic Stem Cells. J. Cell Biol. 2001, 152, 657–668. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; White, E. Autophagy and Metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Vescarelli, E.; Pilloni, A.; Dominici, F.; Pontecorvi, P.; Angeloni, A.; Polimeni, A.; Ceccarelli, S.; Marchese, C. Autophagy activation is required for myofibroblast differentiation during healing of oral mucosa. J. Clin. Periodontol. 2017, 44, 1039–1050. [Google Scholar] [CrossRef]
- Qiang, L.; Yang, S.; Cui, Y.H.; He, Y.Y. Keratinocyte autophagy enables the activation of keratinocytes and fibroblasts and facilitates wound healing. Autophagy 2020, 17, 2128–2143. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, C.; Jiang, X.; Li, L.; Zhang, D.; Tang, D.; Yan, T.; Zhang, Q.; Yuan, H.; Jia, J.; et al. Involvement of autophagy in hypoxia-BNIP3 signaling to promote epidermal keratinocyte migration. Cell Death Dis. 2019, 10, 234. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Wang, J.; Kaplan, N.; Wang, S.; Yang, W.; Wang, L.; He, C.; Peng, H. Autophagy plays a positive role in induction of epidermal proliferation. FASEB J. 2020, 34, 10657–10667. [Google Scholar] [CrossRef]
- Ren, H.; Zhao, F.; Zhang, Q.; Huang, X.; Wang, Z. Autophagy and skin wound healing. Burn. Trauma 2022, 10, tkac003. [Google Scholar] [CrossRef] [PubMed]
- Lindhe, J.; Berglundh, T. The interface between the mucosa and the implant. Periodontology 1998, 17, 47–54. [Google Scholar] [CrossRef]
- Bosshardt, D.D.; Chappuis, V.; Buser, D. Osseointegration of titanium, titanium alloy and zirconia dental im-plants: Current knowledge and open questions. Periodontol. 2000 2017, 73, 22–40. [Google Scholar] [CrossRef] [PubMed]
- Berglundh, T.; Abrahamsson, I.; Welander, M.; Lang, N.P.; Lindhe, J. Morphogenesis of the peri-implant mucosa: An experimental study in dogs. Clin. Oral Implant. Res. 2007, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Gao, C.; Li, W.; Zhu, J.; Novakovic, V.; Wang, J.; Ma, R.; Zhou, J.; Gilbert, G.E.; Shi, J. Phagocytosis by macrophages and endothelial cells inhibits procoagulant and fibrinolytic activity of acute promyelocytic leukemia cells. Blood 2012, 119, 2325–2334. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.-Q.; Zhang, J.; Zhou, G. Autophagy and its implication in human oral diseases. Autophagy 2016, 13, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Reggiori, F. ATG proteins: Are we always looking at autophagy? Autophagy 2016, 12, 2502–2503. [Google Scholar] [CrossRef]
- Martin, P. Wound healing–aiming for perfect skin regeneration. Science 1997, 276, 75–81. [Google Scholar] [CrossRef]
- Mattsson, M.O.; Simkó, M. Grouping of experimental conditions as an approach to evaluate effects of extremely low-frequency magnetic fields on oxidative response in in vitro studies. Front. Public Health 2014, 2, 132. [Google Scholar] [CrossRef]
- Skendros, P.; Mitroulis, I.; Ritis, K. Autophagy in Neutrophils: From Granulopoiesis to Neutrophil Extracellular Traps. Front. Cell Dev. Biol. 2018, 6, 109. [Google Scholar] [CrossRef]
- Rodrigues, M.; Kosaric, N.; Bonham, C.A.; Gurtner, G.C. Wound Healing: A Cellular Perspective. Physiol. Rev. 2019, 99, 665–706. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Wei, Q.; Shin, J.N.; Fattah, E.A.; Bonilla, D.L.; Xiang, Q.; Eissa, N.T. Autophagy is required for neutrophil-mediated inflammation. Cell Rep. 2015, 12, 1731–1739. [Google Scholar] [CrossRef]
- Ullah, I.; Ritchie, N.D.; Evans, T.J. The interrelationship between phagocytosis, autophagy and formation of neutrophil extracellular traps following infection of human neutrophils by Streptococcus pneumoniae. Innate Immun. 2017, 23, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Shrestha, S.; Youn, Y.-J.; Kim, J.-K.; Kim, S.-Y.; Kim, H.J.; Ahn, W.-G.; Lee, M.G.; Jung, K.-S.; Park, Y.B.; et al. Autophagy Primes Neutrophils for Neutrophil Extracellular Trap Formation during Sepsis. Am. J. Respir. Crit. Care Med. 2017, 196, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Qu, L.; Farragher, C.; Vella, A.; Zhou, B. MicroRNA regulated macrophage activation in obesity. J. Transl. Intern. Med. 2019, 7, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, H.; Ding, S.; Wang, Y. Autophagy inhibition promotes phagocytosis of macrophage and protects mice from methicillin-resistant staphylococcus aureus pneumonia. J. Cell. Biochem. 2018, 119, 4808–4814. [Google Scholar] [CrossRef] [PubMed]
- Kawano, A.; Ariyoshi, W.; Yoshioka, Y.; Hikiji, H.; Nishihara, T.; Okinaga, T. Docosahexaenoic acid enhances M2 macrophage polarization via the p38 signaling pathway and autophagy. J. Cell. Biochem. 2019, 120, 12604–12617. [Google Scholar] [CrossRef]
- Das, L.M.; Binko, A.M.; Traylor, Z.P.; Peng, H.; Lu, K.Q. Vitamin D improves sunburns by increasing autophagy in M2 macrophages. Autophagy 2019, 15, 813–826. [Google Scholar] [CrossRef]
- Lackington, W.A.; Fleyshman, L.; Schweizer, P.; Elbs-Glatz, Y.; Guimond, S.; Rottmar, M. The response of soft tissue cells to Ti implants is modulated by blood-implant interactions. Mater. Today Bio 2022, 15, 100303. [Google Scholar] [CrossRef]
- Kuang, M.; Cen, Y.; Qin, R.; Shang, S.; Zhai, Z.; Liu, C.; Pan, X.; Zhou, H. Artesunate Attenuates Pro-Inflammatory Cytokine Release from Macrophages by Inhibiting TLR4-Mediated Autophagic Activation via the TRAF6-Beclin1-PI3KC3 Pathway. Cell. Physiol. Biochem. 2018, 47, 475–488. [Google Scholar] [CrossRef]
- Paul, S.; Kashyap, A.K.; Jia, W.; He, Y.-W.; Schaefer, B.C. Selective Autophagy of the Adaptor Protein Bcl10 Modulates T Cell Receptor Activation of NF-κB. Immunity 2012, 36, 947–958. [Google Scholar] [CrossRef]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy Controls IL-1β Secretion by Targeting Pro-IL-1β for Degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef]
- Lupfer, C.; Thomas, P.G.; Anand, P.K.; Vogel, P.; Milasta, S.; Martinez, J.; Huang, G.; Green, M.; Kundu, M.; Chi, H.; et al. Receptor interacting protein kinase 2–mediated mitophagy regulates inflammasome activation during virus infection. Nat. Immunol. 2013, 14, 480–488. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Verma, S.; Seranova, E.; Sarkar, S.; Kumar, D. Selective Autophagy and Xenophagy in Infection and Disease. Front. Cell Dev. Biol. 2018, 6, 147. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhuang, J.; Jiang, Y.; Sun, J.; Prinz, R.A.; Sun, J.; Jiao, X.; Xu, X. Toll-like receptor signalling cross-activates the autophagic pathway to restrict Salmonella Typhimurium growth in macrophages. Cell. Microbiol. 2019, 21, e13095. [Google Scholar] [CrossRef] [PubMed]
- Trindade, R.; Albrektsson, T.; Wennerberg, A. Current concepts for the biological basis of dental implants: Foreign body equilibrium and osseointegration dynamics. Oral Maxillofac. Surg. Clin. N. Am. 2015, 27, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Jiang, M.; Yin, X.; Yao, P.; Sun, H. The role of autophagy in the process of osseointegration around titanium implants with micro-nano topography promoted by osteoimmunity. Sci. Rep. 2021, 11, 18418. [Google Scholar] [CrossRef]
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2014, 235, 288–297. [Google Scholar] [CrossRef]
- Yang, P.; Ling, L.; Sun, W.; Yang, J.; Zhang, L.; Chang, G.; Guo, J.; Sun, J.; Sun, L.; Lu, D. Ginsenoside Rg1 inhibits apoptosis by increasing autophagy via the AMPK/mTOR signaling in serum deprivation macrophages. Acta Biochim. Biophys. Sin. 2018, 50, 144–155. [Google Scholar] [CrossRef]
- Qiao, L.; Zhang, X.; Liu, M.; Liu, X.; Dong, M.; Cheng, J.; Zhang, X.; Zhai, C.; Song, Y.; Lu, H.; et al. Ginsenoside Rb1 Enhances Atherosclerotic Plaque Stability by Improving Autophagy and Lipid Metabolism in Macrophage Foam Cells. Front. Pharmacol. 2017, 8, 727. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Houbaert, D.; Meçe, O.; Agostinis, P. Autophagy in endothelial cells and tumor angiogenesis. Cell Death Differ. 2019, 26, 665–679. [Google Scholar] [CrossRef]
- Reinke, J.; Sorg, H. Wound Repair and Regeneration. Eur. Surg. Res. 2012, 49, 35–43. [Google Scholar] [CrossRef]
- Sprott, D.; Poitz, D.M.; Korovina, I.; Ziogas, A.; Phieler, J.; Chatzigeorgiou, A.; Mitroulis, I.; Deussen, A.; Chavakis, T.; Ameln, A.K.-V. Endothelial-Specific Deficiency of ATG5 (Autophagy Protein 5) Attenuates Ischemia-Related Angiogenesis. Arter. Thromb. Vasc. Biol. 2019, 39, 1137–1148. [Google Scholar] [CrossRef]
- Jeong, I.H.; Bae, W.Y.; Choi, J.S.; Jeong, J.W. Ischemia induces autophagy of endothelial cells and stimulates angiogenic effects in a hindlimb ischemia mouse model. Cell Death Dis. 2020, 11, 624. [Google Scholar] [CrossRef] [PubMed]
- Chandel, S.; Manikandan, A.; Mehta, N.; Nathan, A.A.; Tiwari, R.K.; Mohapatra, S.B.; Chandran, M.; Jaleel, A.; Manoj, N.; Dixit, M. Protein tyrosine phosphatase-PEST mediates hypoxia-induced endothelial autophagy and angiogenesis via AMPK activation. J. Cell Sci. 2020, 134, jcs250274. [Google Scholar] [CrossRef] [PubMed]
- Dini, L.; Lentini, A.; Diez, G.; Rocha, M.; Falasca, L.; Serafino, L.; Vidal-Vanaclocha, F. Phagocytosis of apoptotic bodies by liver endothelial cells. J. Cell Sci. 1995, 108, 967–973. [Google Scholar] [CrossRef]
- Fens, M.H.; Storm, G.; Pelgrim, R.C.; Ultee, A.; Byrne, A.T.; Gaillard, C.A.; van Solinge, W.W.; Schiffelers, R.M. Erythrophagocytosis by angiogenic endothelial cells is enhanced by loss of erythrocyte deformability. Exp. Hematol. 2010, 38, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Xie, R.; Li, W.; Zhou, J.; Liu, S.; Cao, F.; Liu, Y.; Ma, R.; Si, Y.; Liu, Y.; et al. Endothelial Cell Phagocytosis of Senescent Neutrophils Decreases Procoagulant Activity. Thromb. Haemost. 2013, 109, 1079–1090. [Google Scholar] [CrossRef]
- Zhou, T.; Zheng, Y.; Sun, L.; Badea, S.R.; Jin, Y.; Liu, Y.; Rolfe, A.J.; Sun, H.; Wang, X.; Cheng, Z.; et al. Microvascular endothelial cells engulf myelin debris and promote macrophage recruitment and fibrosis after neural injury. Nat. Neurosci. 2019, 22, 421–435. [Google Scholar] [CrossRef]
- Takeshita, S.; Kaji, K.; Kudo, A. Identification and Characterization of the New Osteoclast Progenitor with Macrophage Phenotypes Being Able to Differentiate into Mature Osteoclasts. J. Bone Miner. Res. 2000, 15, 1477–1488. [Google Scholar] [CrossRef]
- Bang, S.-M.; Moon, H.-J.; Kwon, Y.-D.; Yoo, J.-Y.; Pae, A.; Kwon, I.K. Osteoblastic and osteoclastic differentiation on SLA and hydrophilic modified SLA titanium surfaces. Clin. Oral Implant. Res. 2013, 25, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Mamalis, A.A.; Markopoulou, C.; Vrotsos, I.; Koutsilirieris, M. Chemical modification of an implant surface increases osteogenesis and simultaneously reduces osteoclastogenesis: An in vitro study. Clin. Oral Implant. Res. 2010, 22, 619–626. [Google Scholar] [CrossRef]
- Theill, L.E.; Boyle, W.J.; Penninger, J.M. RANK-L and RANK: T Cells, Bone Loss, and Mammalian Evolution. Annu. Rev. Immunol. 2002, 20, 795–823. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.-Y.; Yoshida, H.; Sarosi, I.; Tan, H.-L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-Dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N.; Takahashi, N.; Akatsu, T.; Tanaka, H.; Sasaki, T.; Nishihara, T.; Koga, T.; Martin, T.J.; Suda, T. Origin of osteoclasts: Mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc. Natl. Acad. Sci. USA 1990, 87, 7260–7264. [Google Scholar] [CrossRef]
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Luthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A Novel Secreted Protein Involved in the Regulation of Bone Density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef]
- Fonseca, J.; Cortez-Dias, N.; Francisco, A.; Sobral, M.; Canhão, H.; Resende, C.; Castelão, W.; Macieira, C.; Sequeira, G.; Saraiva, F.; et al. Inflammatory cell infiltrate and RANKL/OPG expression in rheumatoid synovium: Comparison with other inflammatory arthropathies and correlation with outcome. Clin. Exp. Rheumatol. 2005, 23, 185–192. [Google Scholar] [PubMed]
- Han, Y.; You, X.; Xing, W.; Zhang, Z.; Zou, W. Paracrine and endocrine actions of bone—The functions of secretory proteins from osteoblasts, osteocytes, and osteoclasts. Bone Res. 2018, 6, 16. [Google Scholar] [CrossRef]
- Felix, R.; Halasy-Nagy, J.; Wetterwald, A.; Cecchini, M.G.; Fleisch, H.; Hofstetter, W. Synthesis of membrane- and matrix-bound colony-stimulating factor-1 by cultured osteoblasts. J. Cell. Physiol. 1996, 166, 311–322. [Google Scholar] [CrossRef]
- Teitelbaum, S.L. Bone Resorption by Osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef]
- Glantschnig, H.; Fisher, J.E.; Wesolowski, G.; Rodan, G.A.; Reszka, A.A. M-CSF, TNFα and RANK ligand promote osteoclast survival by signaling through mTOR/S6 kinase. Cell Death Differ. 2003, 10, 1165–1177. [Google Scholar] [CrossRef]
- Elson, A.; Anuj, A.; Barnea-Zohar, M.; Reuven, N. The origins and formation of bone-resorbing osteoclasts. Bone 2022, 164, 116538. [Google Scholar] [CrossRef]
- Wei, S.; Kitaura, H.; Zhou, P.; Ross, F.P.; Teitelbaum, S. IL-1 mediates TNF-induced osteoclastogenesis. J. Clin. Investig. 2005, 115, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Zwerina, J.; Redlich, K.; Polzer, K.; Joosten, L.; Krönke, G.; Distler, J.; Hess, A.; Pundt, N.; Pap, T.; Hoffmann, O.; et al. TNF-induced structural joint damage is mediated by IL-1. Proc. Natl. Acad. Sci. USA 2007, 104, 11742–11747. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, M.; Hayakawa, N.; Mihara, M. IL-6 trans-signalling directly induces RANKL on fibroblast-like synovial cells and is involved in RANKL induction by TNF- and IL-17. Rheumatology 2008, 47, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, N.; Bertolini, D.; Suda, T.; Akiyama, Y.; Roodman, G.D. IL-6 stimulates osteoclast-like multinucleated cell formation in long term human marrow cultures by inducing IL-1 release. J. Immunol. 1990, 144, 4226–4230. [Google Scholar]
- Kitaura, H.; Sands, M.S.; Aya, K.; Zhou, P.; Hirayama, T.; Uthgenannt, B.; Wei, S.; Takeshita, S.; Novack, D.V.; Silva, M.J.; et al. Marrow Stromal Cells and Osteoclast Precursors Differentially Contribute to TNF-α-Induced Osteoclastogenesis In Vivo. J. Immunol. 2004, 173, 4838–4846. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Hakim, I.; Tschoep, K.; Endres, S.; Bar-Shavit, Z. Tumor necrosis factor-α mediates RANK ligand stimulation of osteoclast differentiation by an autocrine mechanism. J. Cell. Biochem. 2001, 83, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Ho, V.W.; Sly, L.M. Derivation and characterization of murine alternatively activated (M2) macrophages. Methods Mol. Biol. 2009, 531, 173–185. [Google Scholar] [CrossRef]
- Tjiu, J.-W.; Chen, J.-S.; Shun, C.-T.; Lin, S.-J.; Liao, Y.-H.; Chu, C.-Y.; Tsai, T.-F.; Chiu, H.-C.; Dai, Y.-S.; Inoue, H.; et al. Tumor-Associated Macrophage-Induced Invasion and Angiogenesis of Human Basal Cell Carcinoma Cells by Cyclooxygenase-2 Induction. J. Investig. Dermatol. 2009, 129, 1016–1025. [Google Scholar] [CrossRef]
- Hayashi, M.; Nakashima, T.; Taniguchi, M.; Kodama, T.; Kumanogoh, A.; Takayanagi, H. Osteoprotection by semaphorin 3A. Nature 2012, 485, 69–74. [Google Scholar] [CrossRef]
- Movérare-Skrtic, S.; Henning, P.; Liu, X.; Nagano, K.; Saito, H.; Börjesson, A.E.; Sjögren, K.; Windahl, S.H.; Farman, H.; Kindlund, B.; et al. Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat. Med. 2014, 20, 1279–1288. [Google Scholar] [CrossRef]
- Maeda, K.; Kobayashi, Y.; Udagawa, N.; Uehara, S.; Ishihara, A.; Mizoguchi, T.; Kikuchi, Y.; Takada, I.; Kato, S.; Kani, S.; et al. Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat. Med. 2012, 18, 405–412. [Google Scholar] [CrossRef]
- Takada, I.; Mihara, M.; Suzawa, M.; Ohtake, F.; Kobayashi, S.; Igarashi, M.; Youn, M.-Y.; Takeyama, K.-I.; Nakamura, T.; Mezaki, Y.; et al. A histone lysine methyltransferase activated by non-canonical Wnt signalling suppresses PPAR-γ transactivation. Nat. Cell Biol. 2007, 9, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Day, C.J.; Selinger, C.I.; Magno, C.L.; Stephens, S.R.; Morrison, N.A. MCP-1-induced human OCs-like cells are tartrate-resistant acidphosphatase, NFATc1, and calcitonin receptor-positive but require receptoractivator of NFkappaB ligand for bone resorption. J. Biol. Chem. 2006, 281, 1274–1285. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Magno, C.L.; Day, C.J.; Morrison, N.A. Induction of chemokinesand chemokine receptors CCR2b and CCR4 in authentic human OCs differentiated with RANKL and OCs like cells differentiated by MCP-1and RANTES. J. Cell. Biochem. 2006, 97, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Huitema, L.F.; Vaandrager, A.B. What triggers cell-mediated mineralization? Front. Biosci. 2007, 12, 2631–2645. [Google Scholar] [CrossRef] [PubMed]
- Smeets, R.; Stadlinger, B.; Schwarz, F.; Beck-Broichsitter, B.; Jung, O.; Precht, C.; Kloss, F.; Gröbe, A.; Heiland, M.; Ebker, T. Impact of Dental Implant Surface Modifications on Osseointegration. BioMed Res. Int. 2016, 2016, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Xiao, Y. The Autophagy in Osteoimmonology: Self-Eating, Maintenance, and Beyond. Front. Endocrinol. 2019, 10, 490. [Google Scholar] [CrossRef] [PubMed]
- Hocking, L.J.; Whitehouse, C.; Helfrich, M.H. Autophagy: A new player in skeletal maintenance? J. Bone Miner. Res. 2012, 27, 1439–1447. [Google Scholar] [CrossRef]
- Shapiro, I.M.; Layfield, R.; Lotz, M.; Settembre, C.; Whitehouse, C. Boning up on autophagy: The role of autophagy in skeletal biology. Autophagy 2014, 10, 7–19. [Google Scholar] [CrossRef]
- Li, Y.; Su, J.; Sun, W.; Cai, L.; Deng, Z. AMP-activated protein kinase stimulates OBs diferentiation and mineralization through autophagy induction. Int. J. Mol. Med. 2018, 41, 2535–2544. [Google Scholar]
- Klein-Nulend, J.; Bakker, A.D.; Bacabac, R.G.; Vatsa, A.; Weinbaum, S. Mechanosensation and transduction in osteocytes. Bone 2013, 54, 182–190. [Google Scholar] [CrossRef]
- Iglesias-Bartolome, R.; Uchiyama, A.; Molinolo, A.A.; Abusleme, L.; Brooks, S.R.; Callejas-Valera, J.L.; Edwards, D.; Doci, C.; Asselin-Labat, M.-L.; Onaitis, M.W.; et al. Transcriptional signature primes human oral mucosa for rapid wound healing. Sci. Transl. Med. 2018, 10, eaap8798. [Google Scholar] [CrossRef] [PubMed]
- Torres, P.; Castro, M.; Reyes, M.; Torres, V.A. Histatins, wound healing, and cell migration. Oral Dis. 2018, 24, 1150–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häkkinen, L.; Uitto, V.J.; Larjava, H. Cell biology of gingival wound healing. Periodontology 2000, 24, 127–152. [Google Scholar]
- Li, G.; Ma, F.; Guo, J.; Zhao, H. Case Study of Roadway Deformation Failure Mechanisms: Field Investigation and Numerical Simulation. Energies 2021, 14, 1032. [Google Scholar] [CrossRef]
- Buskermolen, J.K.; Roffel, S.; Gibbs, S. Stimulation of oral fibroblast chemokine receptors identifies CCR3 and CCR4 as potential wound healing targets. J. Cell. Physiol. 2017, 232, 2996–3005. [Google Scholar] [CrossRef]
- Singer, A.J.; Clark, R.A. Cutaneous wound healing. N. Engl. J. Med. 1999, 341, 738–746. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2022, 3, 349–363. [Google Scholar] [CrossRef]
- Tomasi, C.; Tessarolo, F.; Caola, I.; Wennstr€om, J.; Nollo, G.; Berglundh, T. Morphogenesis of peri-implant mucosa revisited: An experimental study in humans. Clin. Oral Implant. Res. 2014, 25, 997–1003. [Google Scholar] [CrossRef]
- Moon, I.-S.; Berglundh, T.; Abrahamsson, I.; Linder, E.; Lindhe, J. The barrier between the keratinized mucosa and the dental implant. J. Clin. Periodontol. 1999, 26, 658–663. [Google Scholar] [CrossRef]
- Yoon, J.-Y.; Park, C.-G.; Park, B.-S.; Kim, E.-J.; Byeon, G.-J.; Yoon, J.-U. Effects of Remifentanil Preconditioning Attenuating Oxidative Stress in Human Dermal Fibroblast. Tissue Eng. Regen. Med. 2017, 14, 133–141. [Google Scholar] [CrossRef]
- Asai, E.; Yamamoto, M.; Ueda, K.; Waguri, S. Spatiotemporal alterations of autophagy marker LC3 in rat skin fibroblasts during wound healing process. Fukushima J. Med. Sci. 2018, 64, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Kirsner, R.S.; Eaglstein, W.H. The wound healing process. Dermatol. Clin. 1993, 11, 629–640. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.L.; Zierath, J.R. AMP-activated protein kinase signaling in metabolic regulation. J. Clin. Investig. 2006, 116, 1776–1783. [Google Scholar]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis; Shackelford, D.B. & Shaw, R.J. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007, 17, 422–427. [Google Scholar] [CrossRef]
- Szpaderska, A.; Walsh, C.; Steinberg, M.; DiPietro, L. Distinct Patterns of Angiogenesis in Oral and Skin Wounds. J. Dent. Res. 2005, 84, 309–314. [Google Scholar] [CrossRef]
- Onesti, M.G.; Fioramonti, P.; Carella, S.; Fino, P.; Sorvillo, V.; Scuderi, N. A new association between hyaluronic acid and collagenase in wound repair: An open study. Eur. Rev. Med. Pharmacol. Sci. 2013, 2, 210–216. [Google Scholar]
- Mah, W.; Jiang, G.; Olver, D.; Cheung, G.; Kim, B.; Larjava, H.; Häkkinen, L. Human gingival fibroblasts display a non-fibrotic phenotype distinct from skin fibroblasts in three-dimensional cultures. PLoS ONE 2014, 9, e90715. [Google Scholar]
- Desmouliere, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming growth factor-beta 1 in-duces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef]
- Desmouliere, A.; Chaponnier, C.; Gabbiani, G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 2005, 13, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. Formation and Function of the Myofibroblast during Tissue Repair. J. Investig. Dermatol. 2007, 127, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Skalli, O.; Ropraz, P.; Trzeciak, A.; Benzonana, G.; Gillessen, D.; Gabbiani, G. A monoclonal antibody against alpha-smooth muscle actin: A new probe for smooth muscle differentiation. J. Cell Biol. 1986, 103, 2787–2796. [Google Scholar] [CrossRef] [PubMed]
- Darby, I.; Skalli, O.; Gabbiani, G. Alpha-smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab. Investig. 1990, 63, 21–29. [Google Scholar]
- Shannon, D.B.; McKeown, S.T.; Lundy, F.T.; Irwin, C.R. Phenotypic differences between oral and skin fibroblasts in wound contraction and growth factor expression. Wound Repair Regen. 2006, 14, 172–178. [Google Scholar] [CrossRef]
- Mallat, A.; Lodder, J.; Teixeira-Clerc, F.; Moreau, R.; Codogno, P.; Lotersztajn, S. Autophagy: A multifaceted partner in liver fibrosis. Biomed Res. Int. 2014, 2014, 869390. [Google Scholar] [CrossRef]
- Goktas, S.; Dmytryk, J.J.; McFetridge, P.S. Biomechanical Behavior of Oral Soft Tissues. J. Periodontol. 2011, 82, 1178–1186. [Google Scholar] [CrossRef]
- Gao, X.; Fraulob, M.; Haïat, G. Biomechanical behaviours of the bone–implant interface: A review. J. R. Soc. Interface 2019, 16, 20190259. [Google Scholar] [CrossRef]
- Rakic, M.; Galindo-Moreno, P.; Monje, A.; Radovanovic, S.; Wang, H.-L.; Cochran, D.; Sculean, A.; Canullo, L. How frequent does peri-implantitis occur? A systematic review and meta-analysis. Clin. Oral Investig. 2017, 22, 1805–1816. [Google Scholar] [CrossRef]
- Oh, S.L.; Shiau, H.J.; Reynolds, M.A. Survival of dental implants at sites after implant failure: A systematic review. J. Prosthet. Dent. 2020, 123, 54–60. [Google Scholar] [CrossRef]
- Apablaza, J.A.; Días, F.J.; Sánchez, K.G.; Navarro, P.; Venegas, C. Analysis of the Chemical Composition and Morphological Characterization of Tissue Osseointegrated to a Dental Implant after 5 Years of Function. Int. J. Mol. Sci. 2022, 23, 8882. [Google Scholar] [CrossRef] [PubMed]
- Nicolau, P.; Guerra, F.; Reis, R.; Krafft, T.; Benz, K.; Jackowski, J. 10-year outcomes with immediate and early loaded implants with a chemically modified SLA surface. Quintessence Int. 2019, 50, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.W. Titanium Alloys for Dental Implants: A Review. Prosthesis 2020, 2, 100–116. [Google Scholar] [CrossRef]
- Talwar, B.S. A focus on soft tissue in dental implantology. J. Indian Prosthodont. Soc. 2012, 12, 137–142. [Google Scholar] [CrossRef]
- Köunönen, M.; Hormia, M.; Kivilahti, J.; Hautaniemi, J.; Thesleff, I. Effect of surface processing on the attachment, orientation, and proliferation of human gingival fibroblasts on titanium. J. Biomed. Mater. Res. 1992, 26, 1325–1341. [Google Scholar] [CrossRef] [PubMed]
- Lauer, G.; Wiedmann-Al-Ahmad, M.; Otten, J.; Hübner, U.; Schmelzeisen, R.; Schilli, W. The titanium surface texture effects adherence and growth of human gingival keratinocytes and human maxillar osteoblast-like cells in vitro. Biomaterials 2001, 22, 2799–2809. [Google Scholar] [CrossRef]
- Zhao, B.; van der Mei, H.C.; Subbiahdoss, G.; de Vries, J.; Rustema-Abbing, M.; Kuijer, R.; Busscher, H.J.; Ren, Y. Soft tissue integration versus early biofilm formation on different dental implant materials. Dent. Mater. 2014, 30, 716–727. [Google Scholar] [CrossRef]
- Ferraris, S.; Warchomicka, F.; Ramskogler, C.; Tortello, M.; Cochis, A.; Scalia, A.; di Confiengo, G.G.; Keckes, J.; Rimondini, L.; Spriano, S. Surface structuring by Electron Beam for improved soft tissues adhesion and reduced bacterial contamination on Ti-grade 2. J. Mater. Process. Technol. 2018, 266, 518–529. [Google Scholar] [CrossRef]
- Zhang, Y.; Cheng, X.; Jansen, J.A.; Yang, F.; Beucken, J.J.V.D. Titanium surfaces characteristics modulate macrophage polarization. Mater. Sci. Eng. C 2018, 95, 143–151. [Google Scholar] [CrossRef]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in Wound Repair: Molecular and Cellular Mechanisms. J. Investig. Dermatol. 2007, 127, 514–525. [Google Scholar] [CrossRef]
- Werner, S.; Krieg, T.; Smola, H. Keratinocyte–Fibroblast Interactions in Wound Healing. J. Investig. Dermatol. 2007, 127, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Koh, T.J.; DiPietro, L.A. Inflammation and wound healing: The role of the macrophage. Expert Rev. Mol. Med. 2011, 13, e23. [Google Scholar] [CrossRef] [PubMed]
- Darby, I.A.; Laverdet, B.; Bonté, F.; Desmouliere, A. Fibroblasts and myofibroblasts in wound healing. Clin. Cosmet. Investig. Dermatol. 2014, 7, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Studied Cell Type | Roles of Autophagy | References |
|---|---|---|
| Macrophages (MFs) |
| [6] |
| [6,7] | |
| [6] | |
| [6,8,9,10] | |
| [11,12,13] | |
| Endothelial cells (ECs) |
| [14,15] |
| [14,15] | |
| [16,17] | |
| [18] | |
| [19] | |
| [20] | |
| Osteoclasts (OCs) |
| [21,22,23] |
| [21,22,23] | |
| [23] | |
| Osteoblasts (OBs) |
| [6] |
| [24,25,26] | |
| [25] | |
| Fibroblasts (FBs) and Myofibroblasts (MFBs) |
| [27] |
| [27] | |
| [28,29,30,31] | |
| [30,32,33] | |
| [34] | |
| [34] | |
| Keratinocytes (KCs) |
| [35,36] |
| [35] | |
| [35,36,37,38] | |
| [35,36,37,38] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ripszky Totan, A.; Imre, M.M.; Parvu, S.; Meghea, D.; Radulescu, R.; Enasescu, D.S.A.; Moisa, M.R.; Pituru, S.M. Autophagy Plays Multiple Roles in the Soft-Tissue Healing and Osseointegration in Dental Implant Surgery—A Narrative Review. Materials 2022, 15, 6041. https://doi.org/10.3390/ma15176041
Ripszky Totan A, Imre MM, Parvu S, Meghea D, Radulescu R, Enasescu DSA, Moisa MR, Pituru SM. Autophagy Plays Multiple Roles in the Soft-Tissue Healing and Osseointegration in Dental Implant Surgery—A Narrative Review. Materials. 2022; 15(17):6041. https://doi.org/10.3390/ma15176041
Chicago/Turabian StyleRipszky Totan, Alexandra, Marina Melescanu Imre, Simona Parvu, Daniela Meghea, Radu Radulescu, Dan Sebastian Alexandru Enasescu, Mihai Radu Moisa, and Silviu Mirel Pituru. 2022. "Autophagy Plays Multiple Roles in the Soft-Tissue Healing and Osseointegration in Dental Implant Surgery—A Narrative Review" Materials 15, no. 17: 6041. https://doi.org/10.3390/ma15176041
APA StyleRipszky Totan, A., Imre, M. M., Parvu, S., Meghea, D., Radulescu, R., Enasescu, D. S. A., Moisa, M. R., & Pituru, S. M. (2022). Autophagy Plays Multiple Roles in the Soft-Tissue Healing and Osseointegration in Dental Implant Surgery—A Narrative Review. Materials, 15(17), 6041. https://doi.org/10.3390/ma15176041