
Towards Optimized Photoluminescent Copper(I) Phenanthroline-Functionalized Complexes: Control of the Photophysics by Symmetry-Breaking and Spin–Orbit Coupling


Abstract
:1. Introduction
2. Computational Method
3. Results
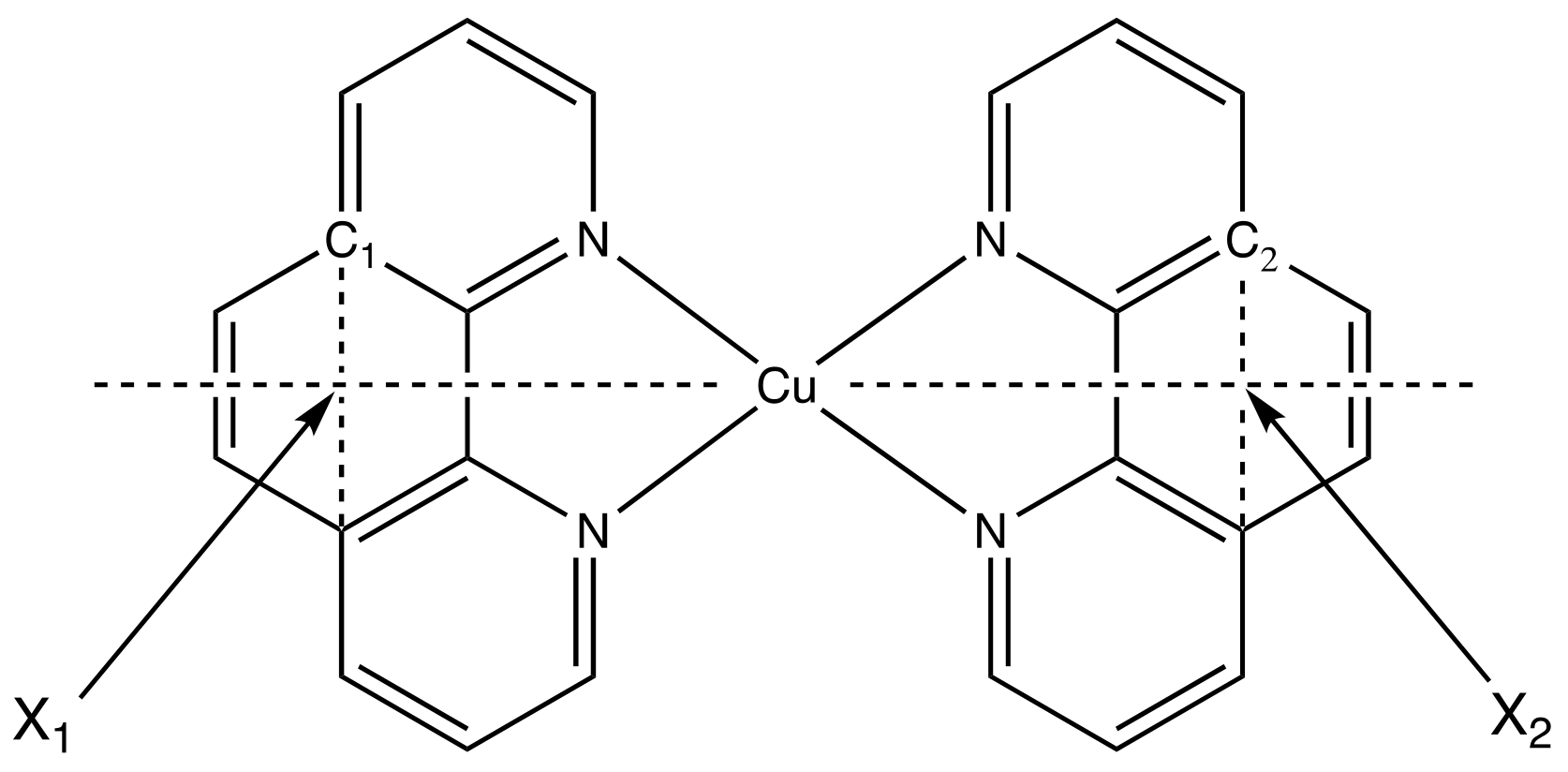
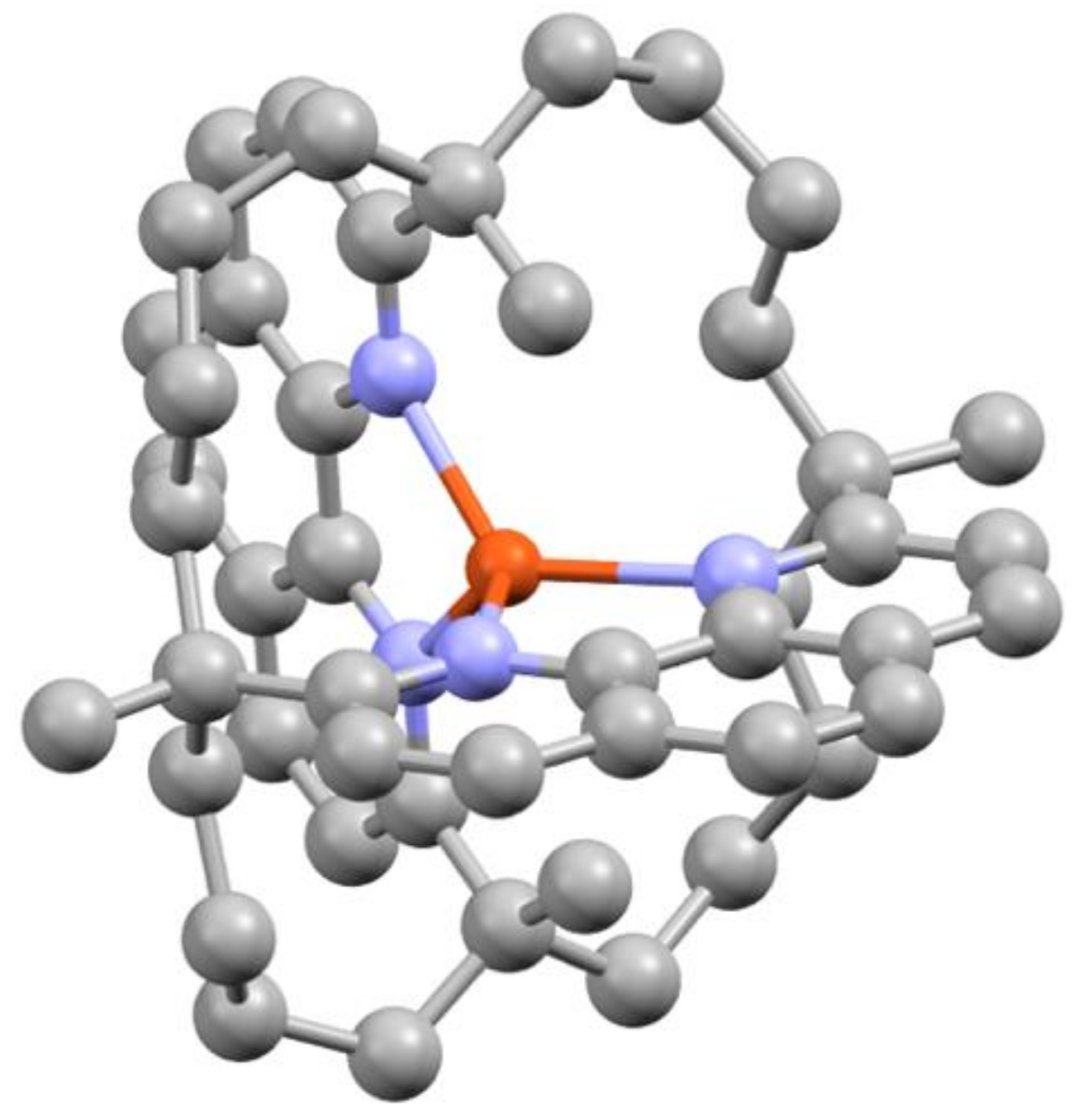
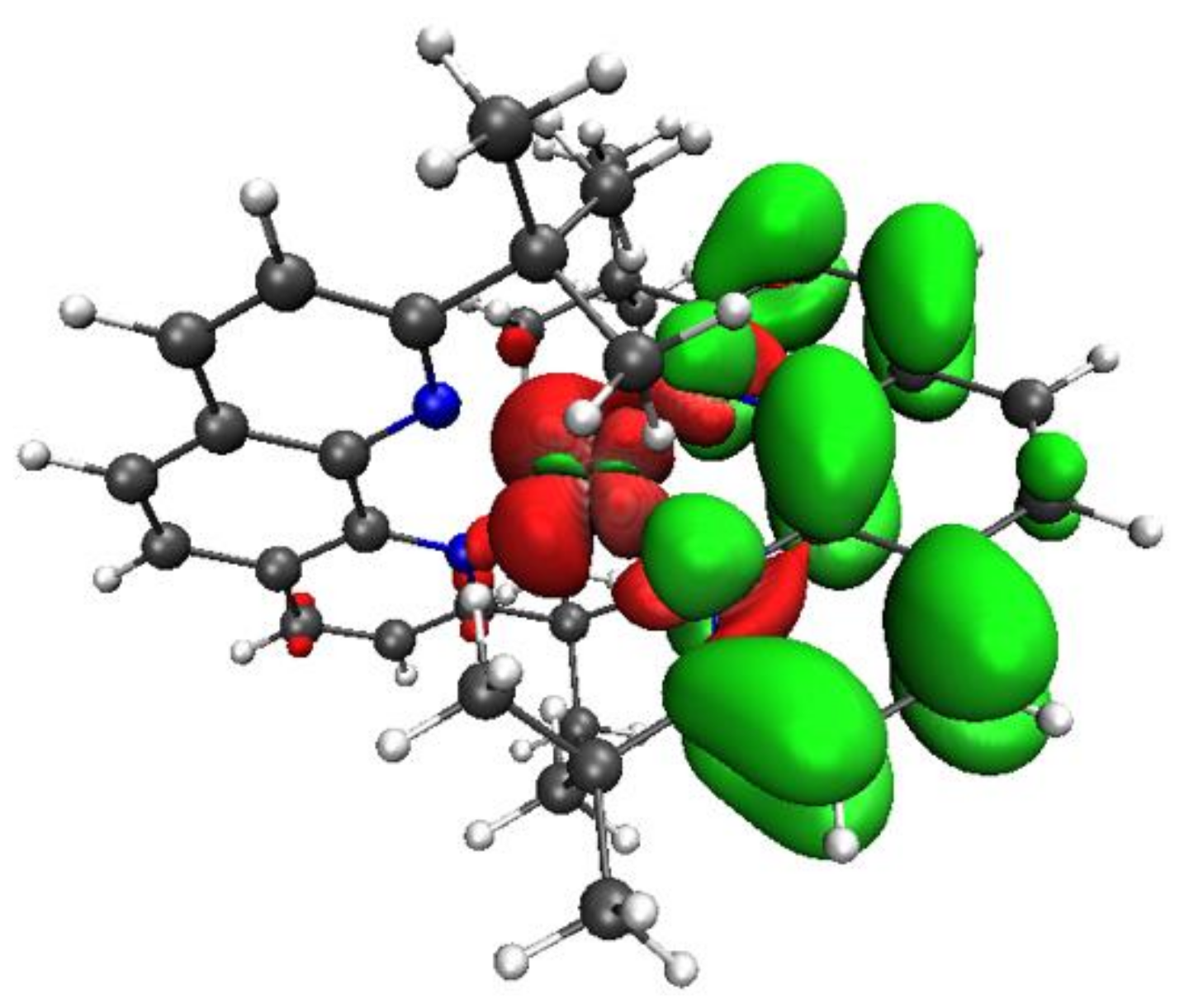
3.1. Ground State and Excited State Structural Properties
3.2. Low-Lying Singlet and Triplet Excited State Properties
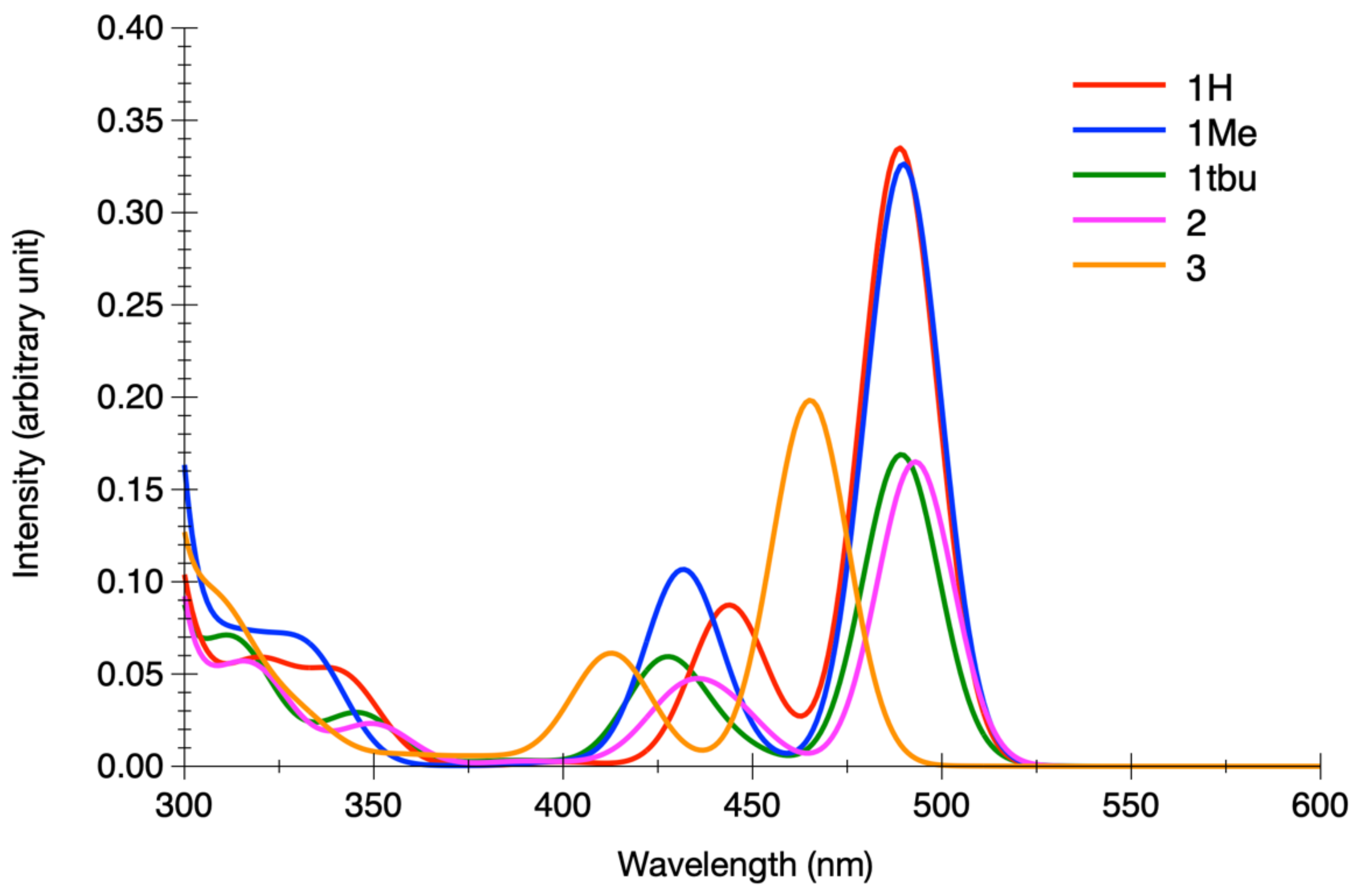
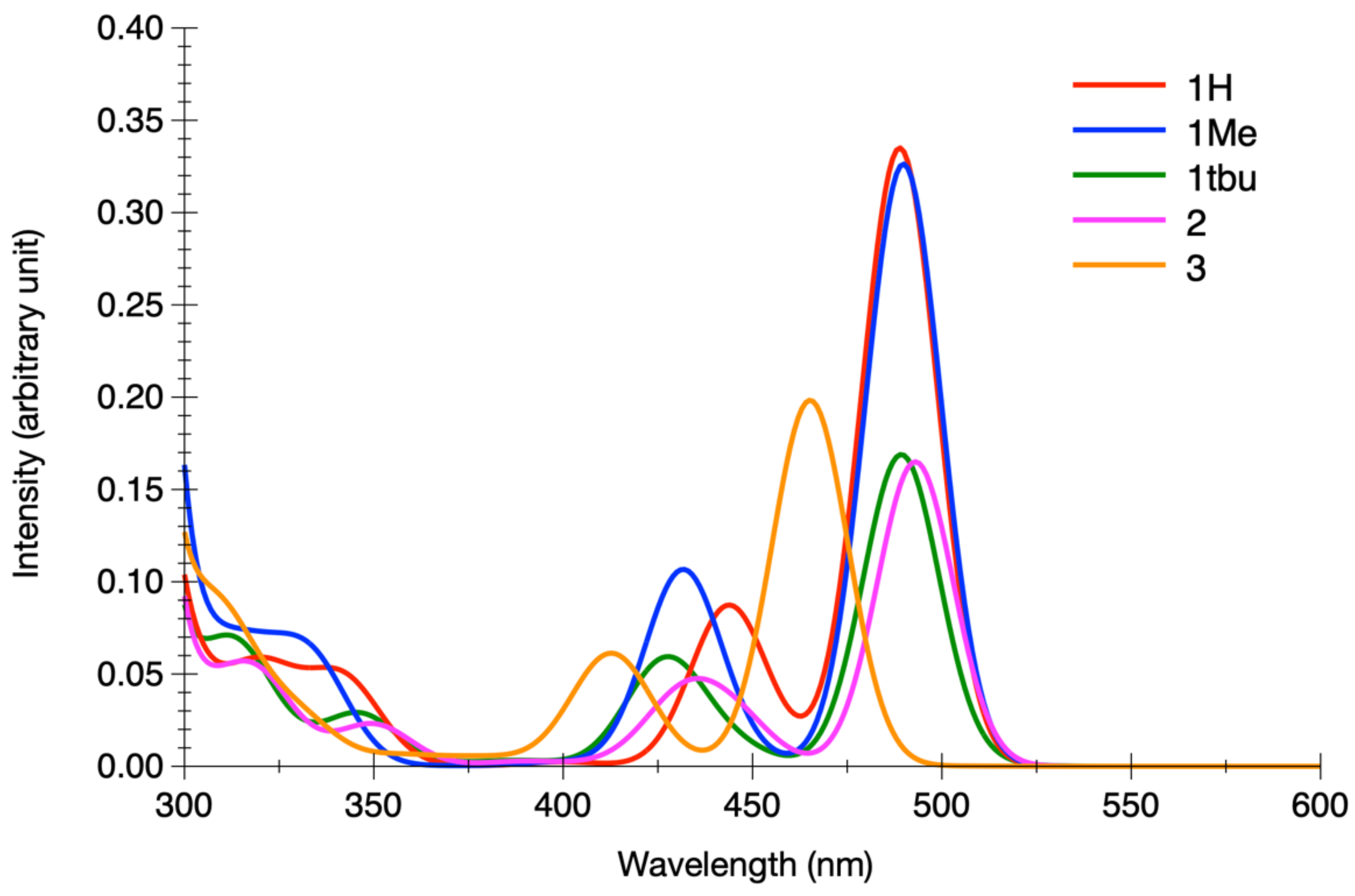
3.2.1. Energetics, Absorption Spectra and Spin–Orbit Coupling
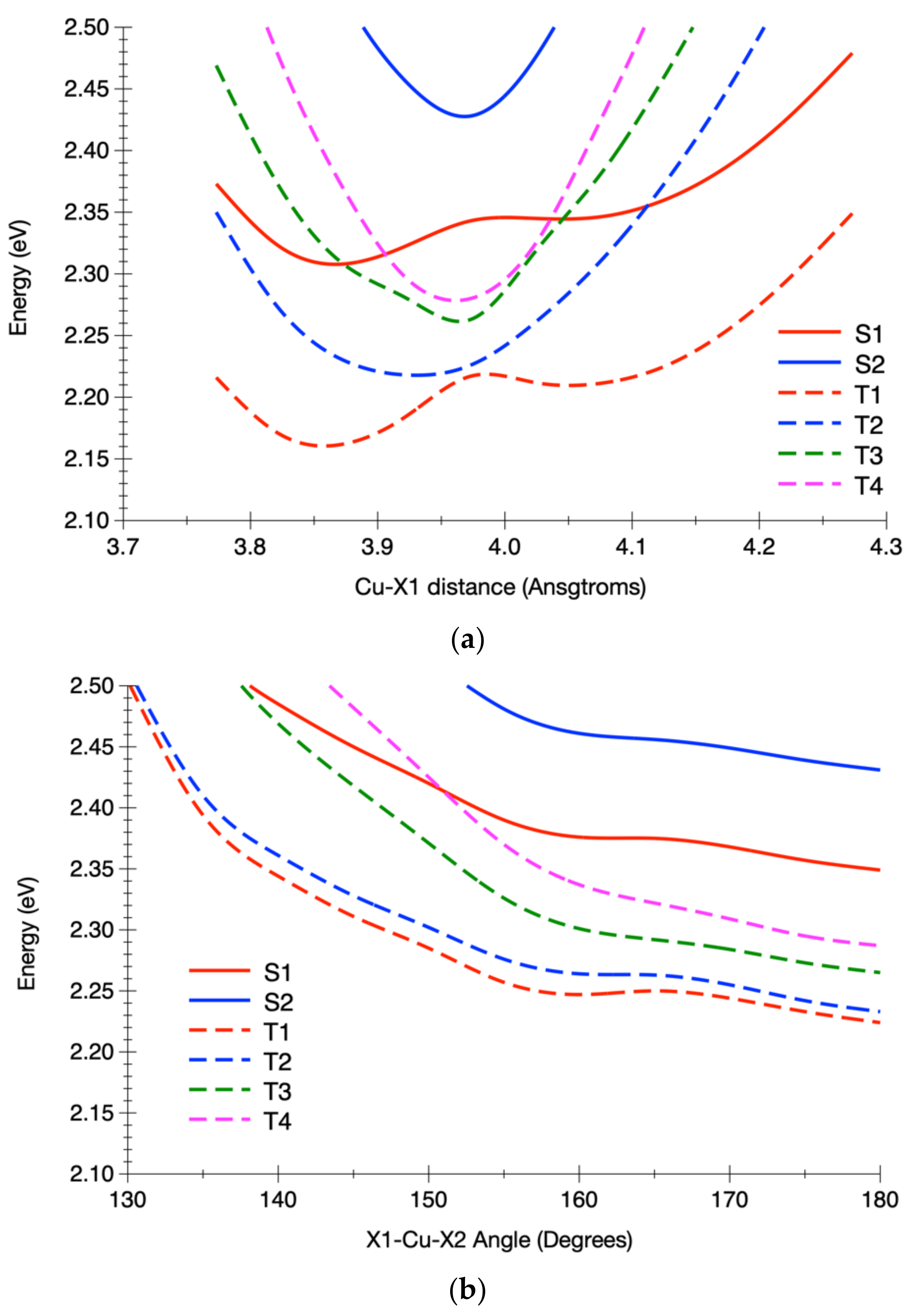
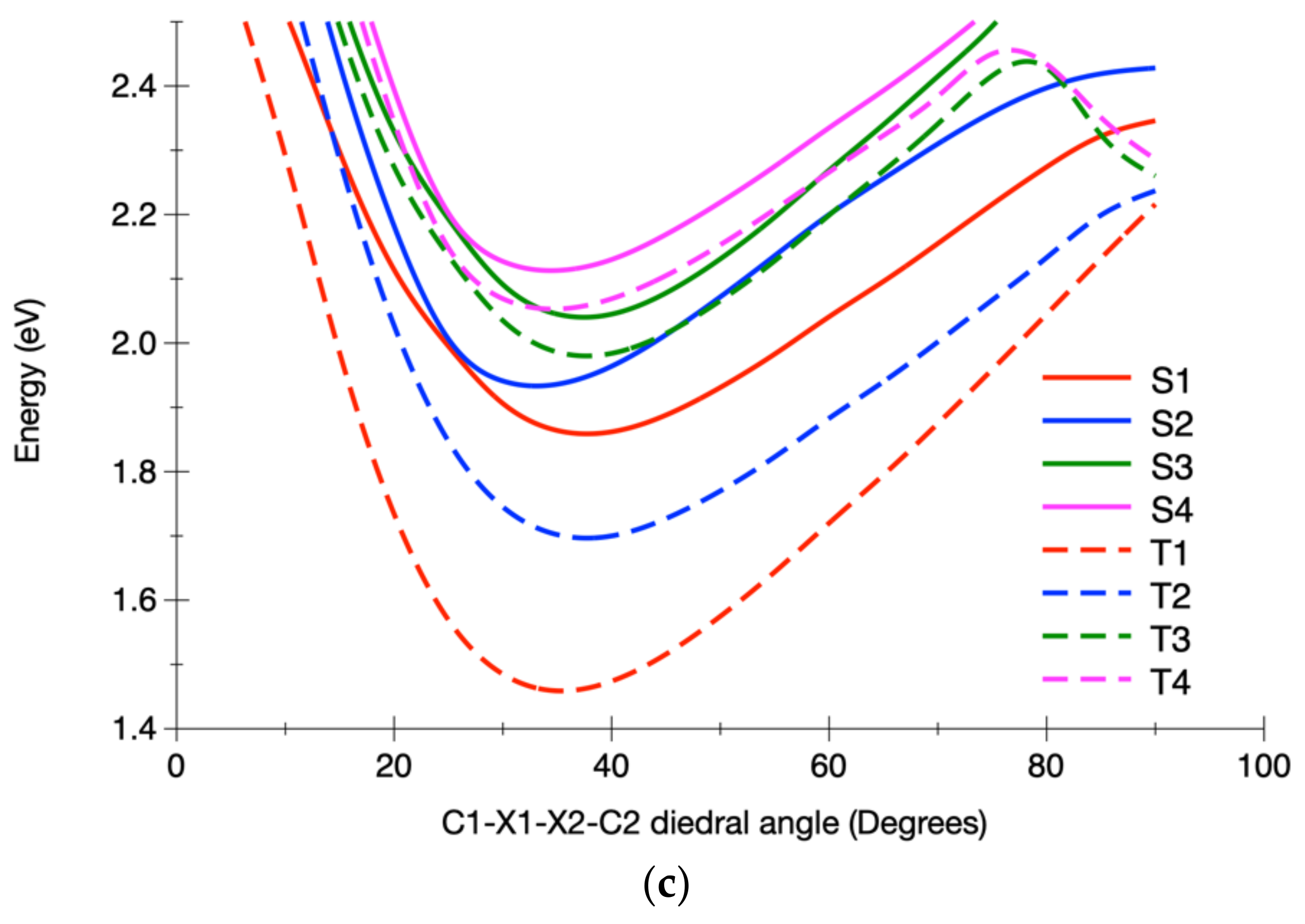
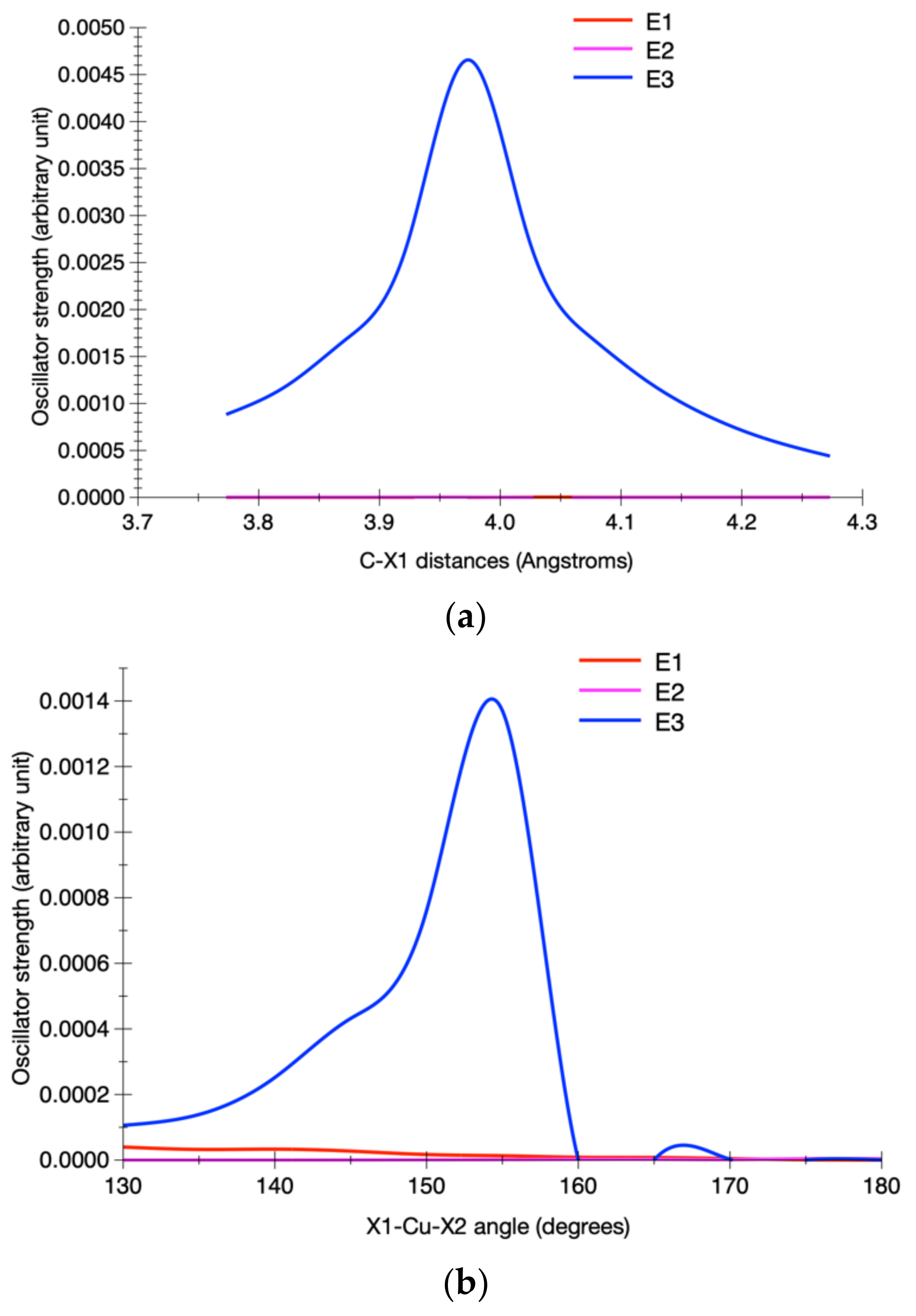
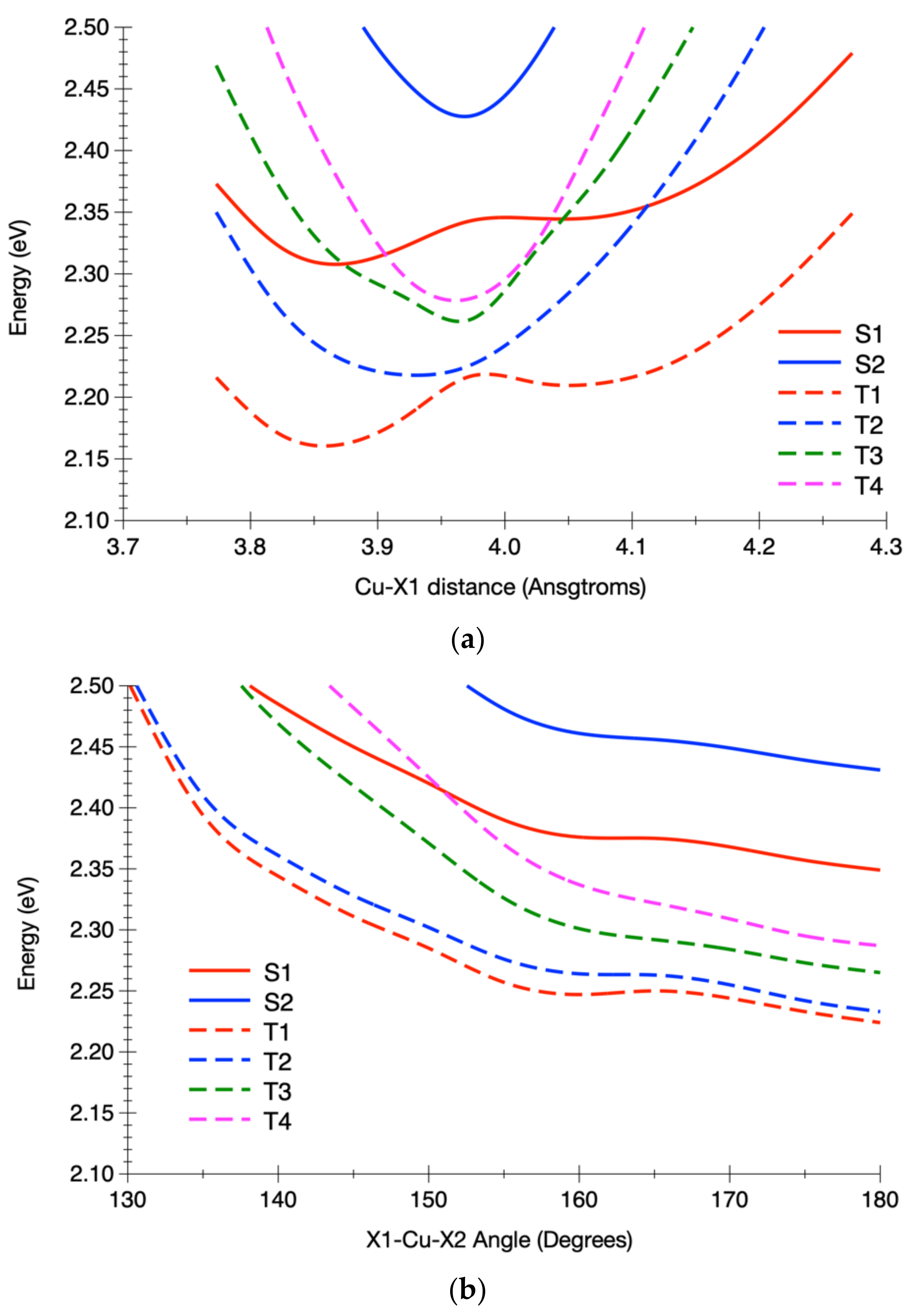
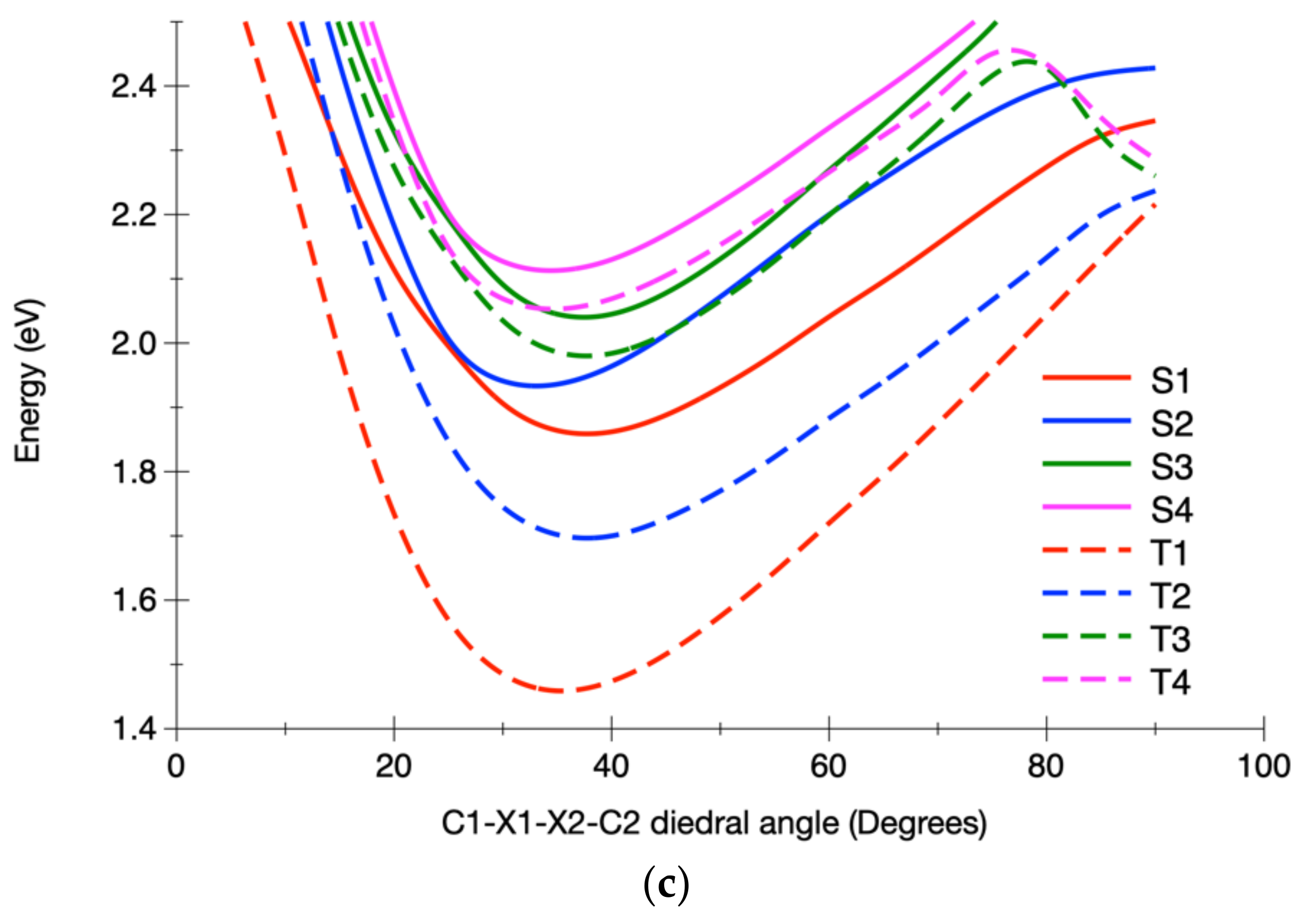
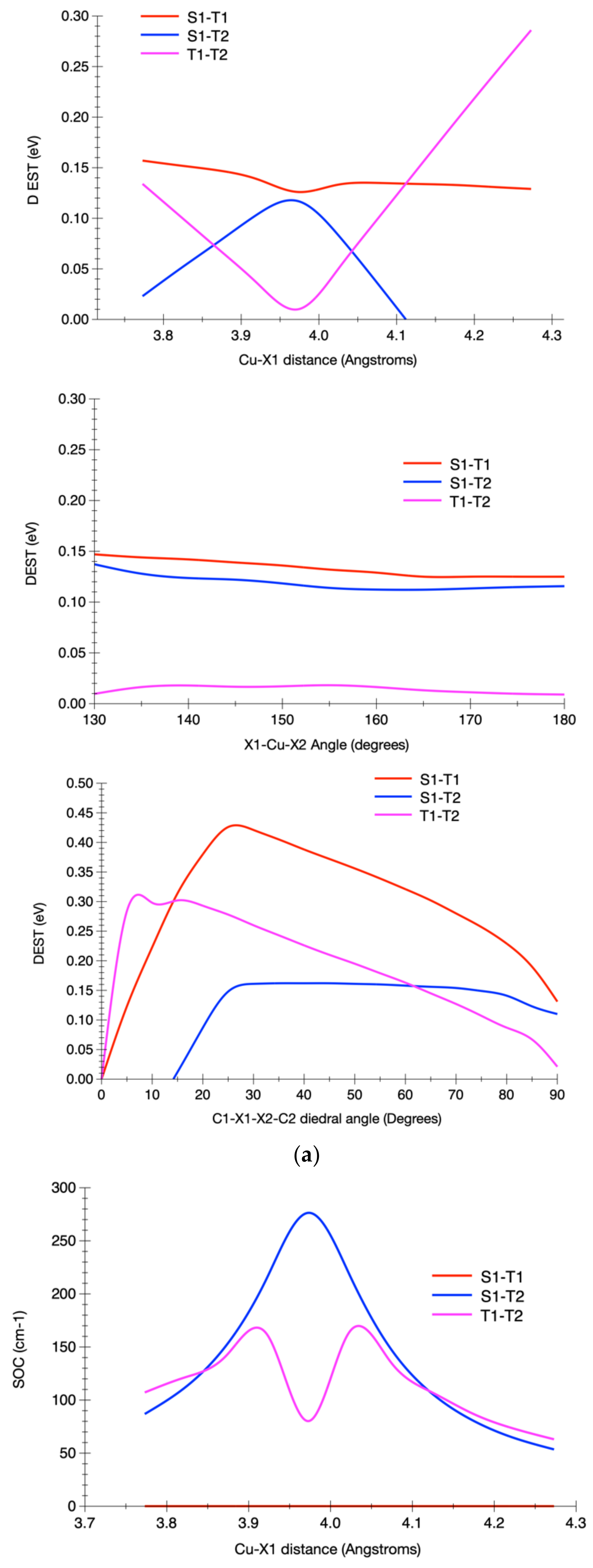
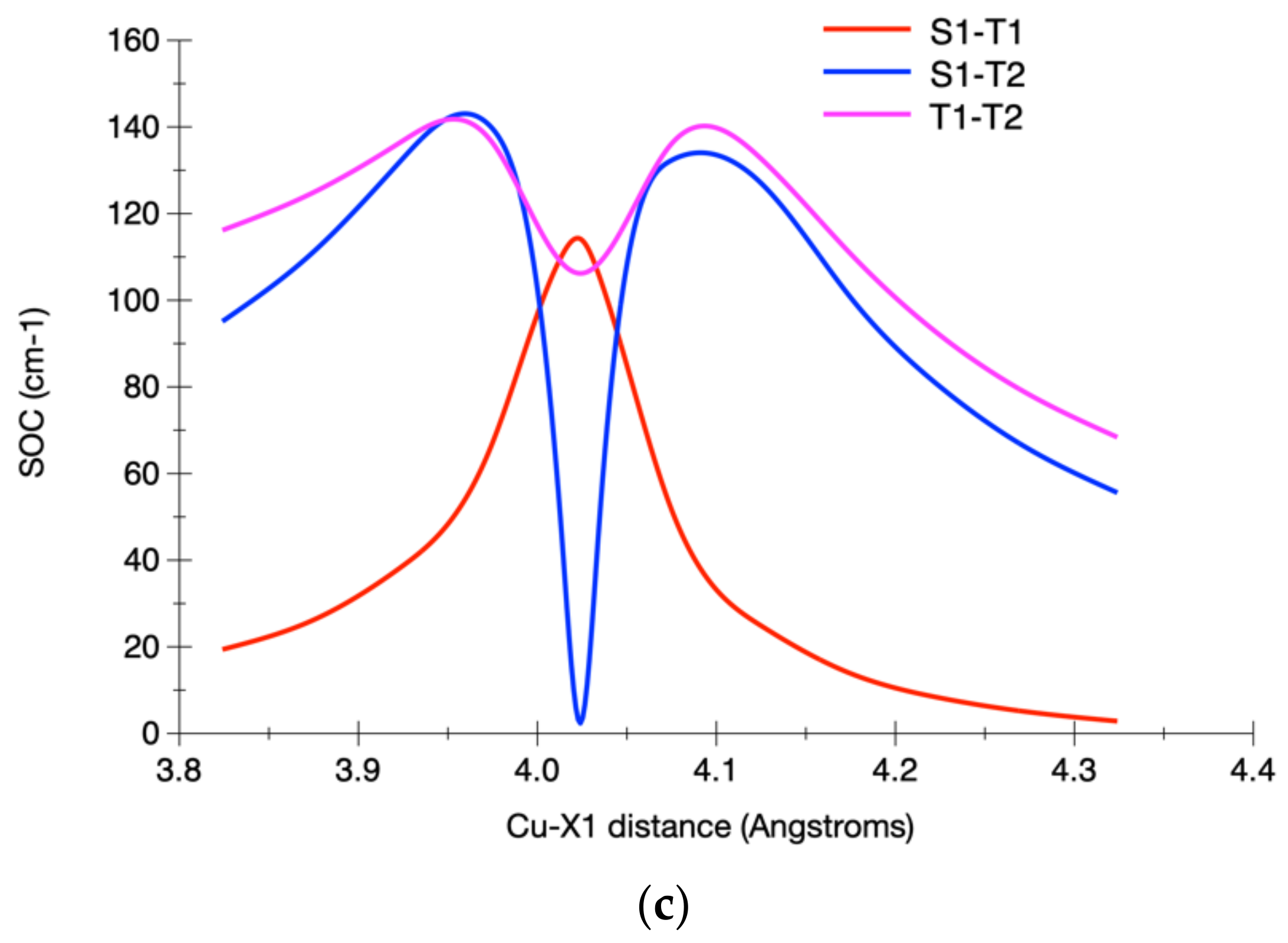
3.2.2. Calculated Potential Energy Curves Associated with the Low-Lying Excited States and Their Spin–Orbit Interactions
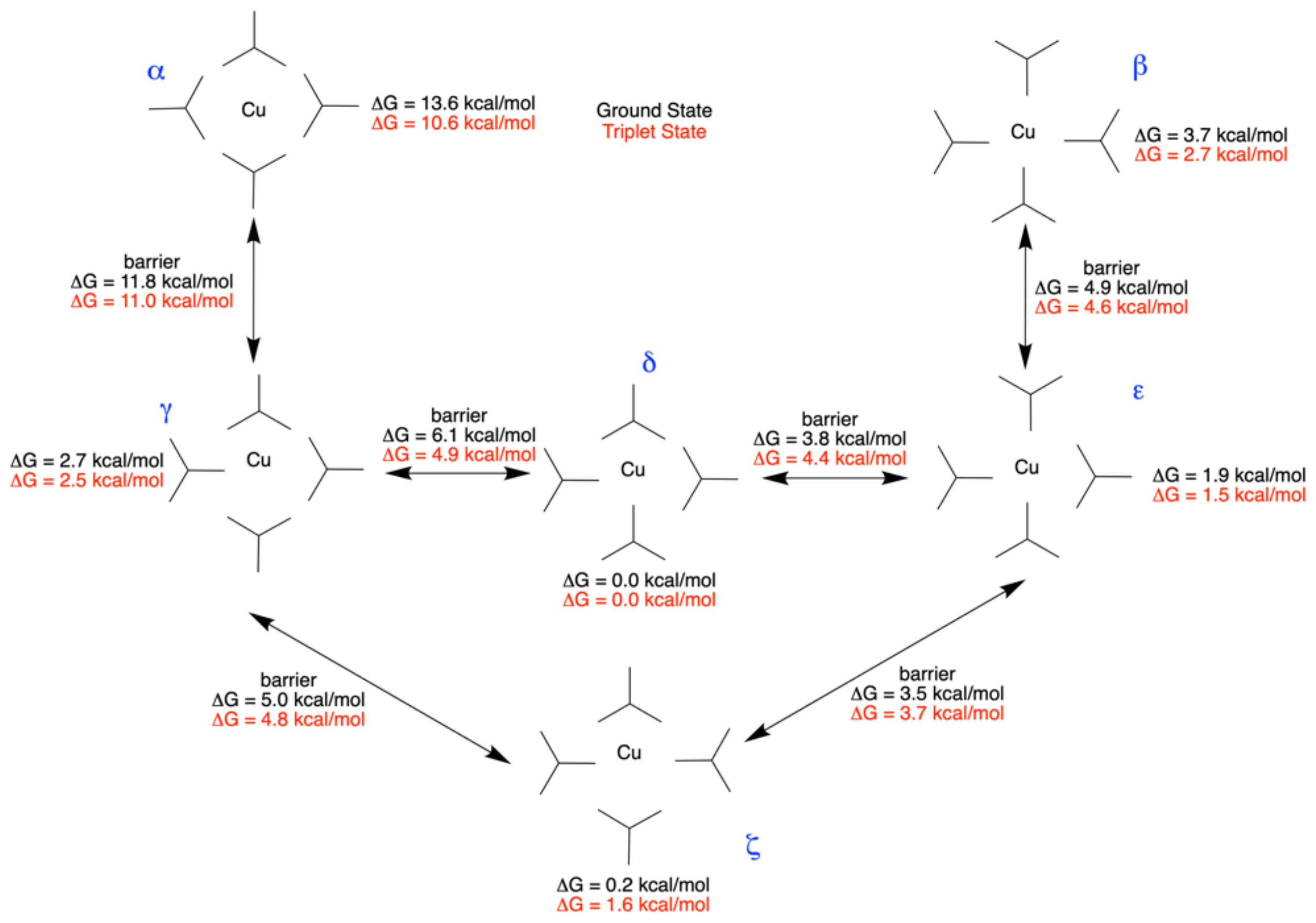
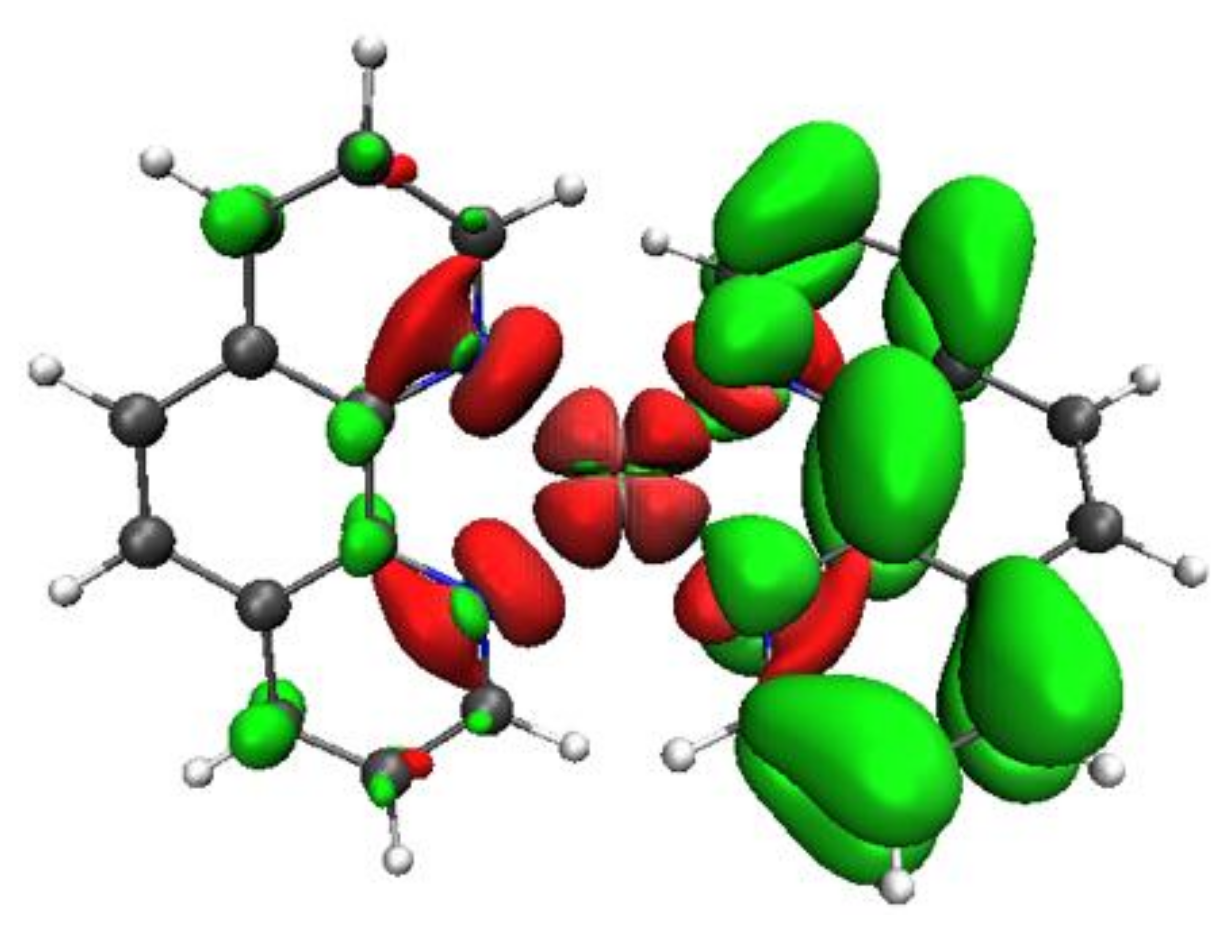
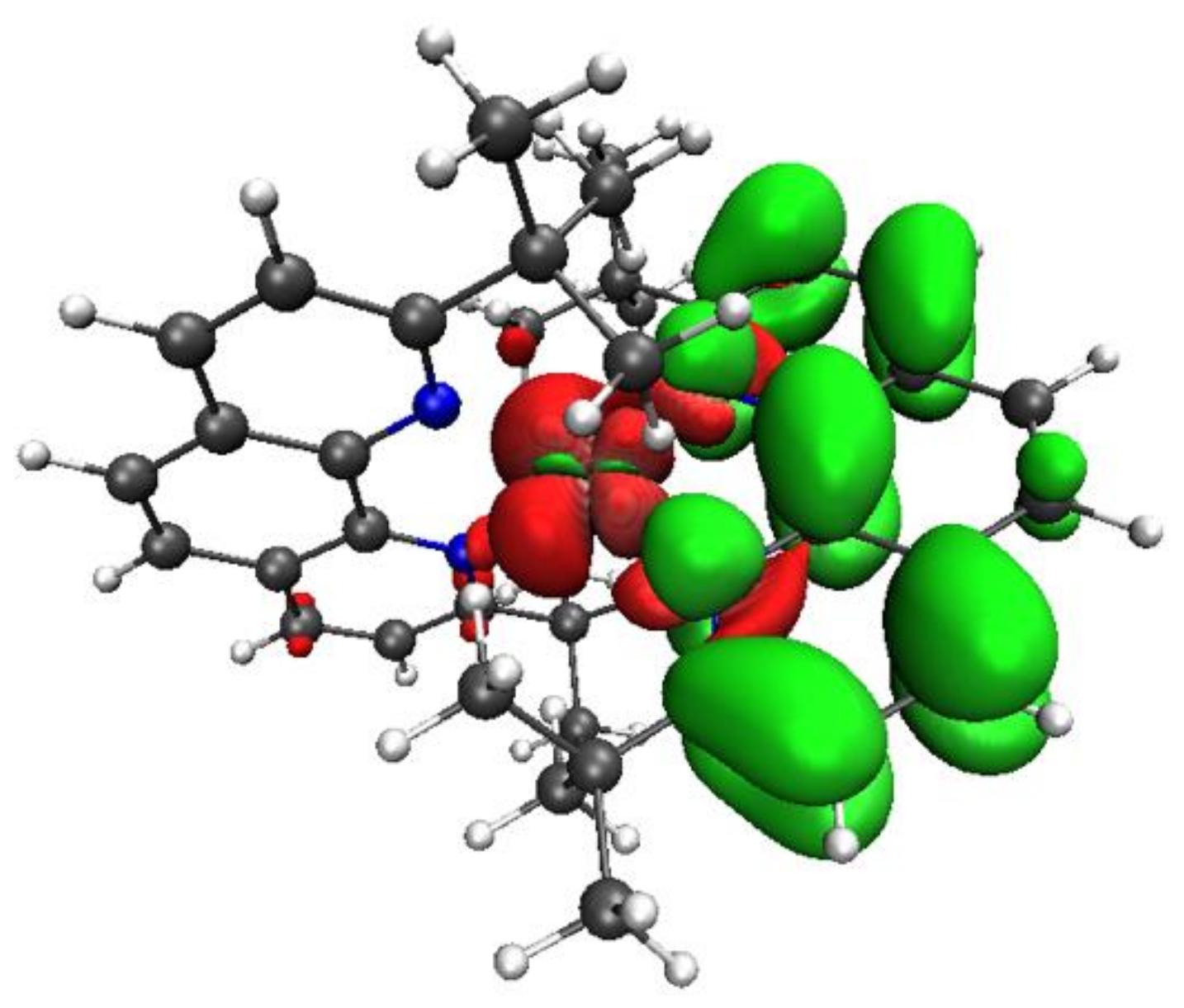
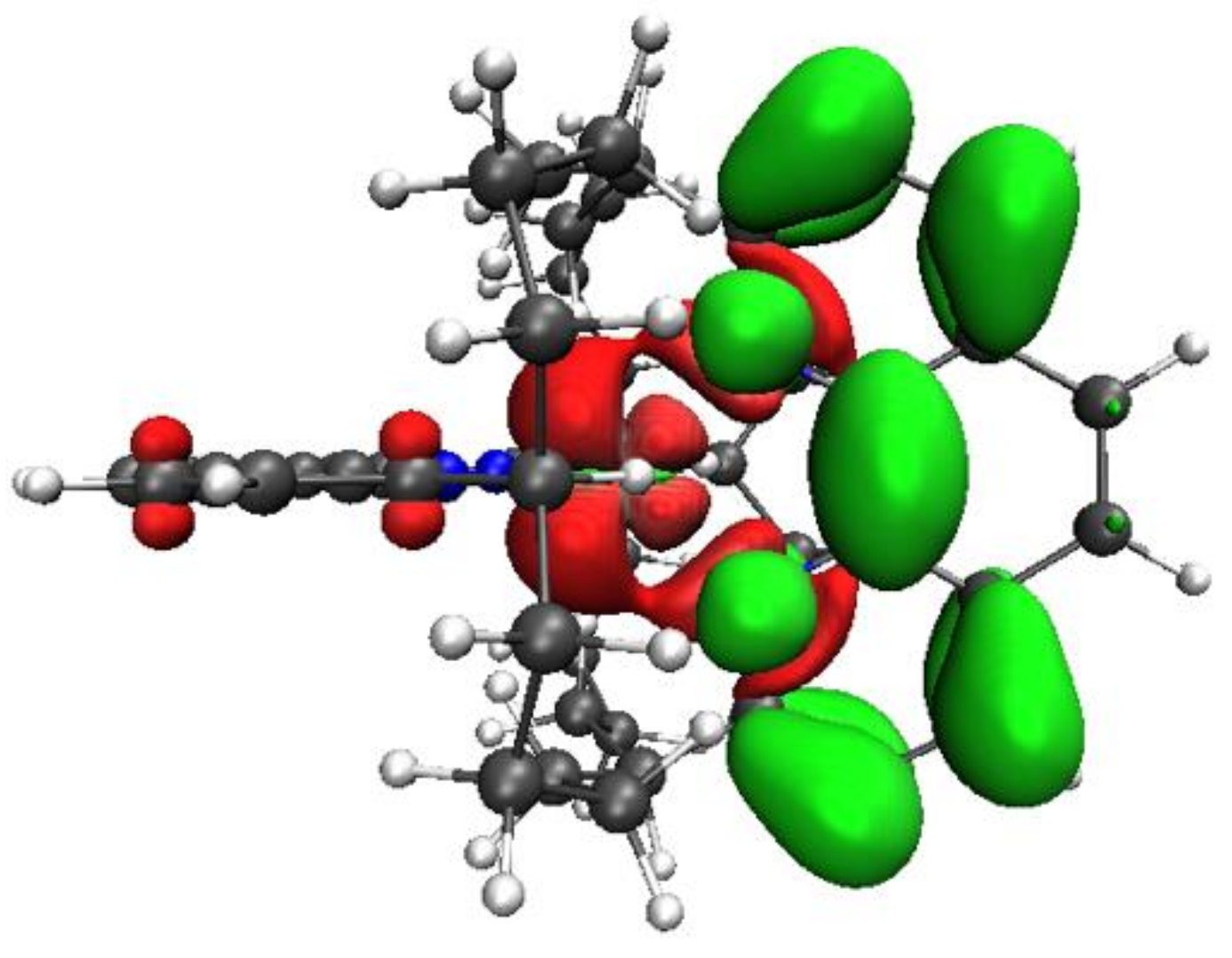
3.3. Towards Supramolecular Design
3.3.1. Complex 2
3.3.2. Complex 3
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Buckner, M.T.; McMillin, D.R. Photoluminescence from copper(I) complexes with low-lying metal-to-ligand charge transfer excited states. J. Chem. Soc. Chem. Commun. 1978, 759–761. [Google Scholar] [CrossRef]
- Blaskie, M.W.; McMillin, D.R. Photostudies of copper(I) systems. 6. Room-temperature emission and quenching studies of bis(2,9-dimethyl-1,10-phenanthroline)copper(I). Inorg. Chem. 1980, 19, 3519–3522. [Google Scholar] [CrossRef]
- Rader, R.A.; McMillin, D.R.; Buckner, M.T.; Matthews, T.G.; Casadonte, D.J.; Lengel, R.K.; Whittaker, S.B.; Darmon, L.M.; Lytle, F.E. Photostudies of [Cu(bpy)(PPh3)2]+, [Cu(phen)(PPh3)2]+, and [Cu(dmp)(PPh3)2]+ in Solution and in Rigid, Low-Temperature Glasses. Simultaneous Multiple Emissions from Intraligand and Charge-Transfer States. J. Am. Chem. Soc. 1981, 103, 5906–5912. [Google Scholar] [CrossRef]
- Kirchhoff, J.R.; Gamache, R.E., Jr.; Blaskie, M.W.; Del Paggio, A.A.; Lengel, R.K.; McMillin, D.R. Temperature dependence of luminescence from Cu(NN)2+ systems in fluid solution. Evidence for the participation of two excited states. Inorg. Chem. 1983, 22, 2380–2384. [Google Scholar] [CrossRef]
- Kutal, C. Spectroscopic and photochemical properties of d10 metal complexes. Coord. Chem. Rev. 1990, 99, 213–252. [Google Scholar] [CrossRef]
- Sabin, F.; Ryu, C.K.; Ford, P.C.; Vogler, A. Photophysical properties of hexanuclear copper(I) and silver(I) clusters. Inorg. Chem. 1992, 31, 1941–1945. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Che, C.-M.; Wong, W.T.; Shieh, S.J.; Peng, S.M. Luminescent three-co-ordinated dinuclear copper(I) complexes of bis(diphenylphosphino)methane: Photophysical properties and crystal structure. J. Chem. Soc. Dalton Trans. 1993, 653–654. [Google Scholar] [CrossRef]
- Yam, V.W.-W.; Fung, W.K.-M.; Cheung, K.-K. Synthesis, Structure, Photophysics, and Excited-State Redox Properties of the Novel Luminescent Tetranuclear Acetylidocopper(I) Complex [Cu4(μ-dppm)4(μ4-η1,η2-C{C-)](BF4)2. Angew. Chem. Int. Ed. 1996, 35, 1100–1102. [Google Scholar] [CrossRef]
- Yam, V.W.-W.; Lo, K.K.-W.; Fung, W.K.-M.; Wang, C.R. Design of luminescent polynuclear copper(I) and silver(I) complexes with chalcogenides and acetylides as the bridging ligands. Coord. Chem. Rev. 1998, 171, 17–41. [Google Scholar] [CrossRef]
- Miller, M.T.; Karpishin, T.B. Phenylethynyl substituent effects on the photophysics and electrochemistry of [Cu(dpp2)]2+ (dpp = 2,9-Diphenyl-1,10-phenanthroline). Inorg. Chem. 1999, 38, 5246–5249. [Google Scholar] [CrossRef]
- Miller, M.T.; Gantzel, P.K.; Karpishin, T.B. A highly emissive heteroleptic copper(I) bis(phenanthroline) complex: [Cu(dbp)(dmp)]+ (dbp = 2, 9-Di-tert-butyl-1,10-phenanthroline; dmp = 2, 9-Dimethyl-1,10-phenanthroline). J. Am. Chem. Soc. 1999, 121, 4292–4293. [Google Scholar] [CrossRef]
- Yam, V.W.-K.; Lo, K.K.-W. Luminescent polynuclear d10 metal complexes. Chem. Soc. Rev. 1999, 28, 323–334. [Google Scholar] [CrossRef]
- Ford, P.C.; Cariati, E.; Bourassa, J. Photoluminescence properties of multinuclear copper(I) compounds. Chem. Rev. 1999, 99, 3625–3647. [Google Scholar] [CrossRef] [PubMed]
- Scaltrito, D.V.; Thompson, D.W.; O’Calaghan, J.A.; Meyer, G.J. MLCT excited states of cuprous bis-phenanthroline coordination compounds. Coord. Chem. Rev. 2000, 208, 243–266. [Google Scholar] [CrossRef]
- Che, C.M.; Mao, Z.; Miskowski, V.M.; Tse, M.C.; Chan, C.K.; Cheung, K.K.; Phillips, D.L.; Leung, K.H. Cuprophilicity: Spectroscopic and Structural Evidence for Cu−Cu Bonding Interactions in Luminescent Dinuclear Copper(I) Complexes with Bridging Diphosphane Ligands. Angew. Chem. Int. Ed. 2000, 112, 4250–4254. [Google Scholar] [CrossRef]
- Mao, Z.; Chao, H.Y.; Hui, Z.; Che, C.-M.; Fu, W.-F.; Cheung, K.-K.; Zhu, N. Excited States of Binuclear Copper(I) Phosphine Complexes: Effect of Copper–Ligand and Copper–Copper Interactions on Excited State Properties and Photocatalytic Reductions of the 4,4′-Dimethyl-2,2′-bipyridinium Ion in Alcohols. Chem. Eur. J. 2003, 9, 2885–2894. [Google Scholar] [CrossRef]
- Pawlowski, V.; Knoer, G.; Lennartz, C.; Vogler, A. Luminescence and Theoretical Studies of Cu(tripod)X [tripod = 1,1,1-Tris(diphenylphosphanylmethyl)ethane; X− = Halide, Thiophenolate, Phenylacetylide]. Eur. J. Inorg. Chem. 2005, 15, 3167–3171. [Google Scholar] [CrossRef]
- Peng, R.; Li, D.; Wu, T.; Zhou, X.-P.; Ng, S.W. Increasing Structure Dimensionality of Copper(I) Complexes by Varying the Flexible Thioether Ligand Geometry and Counteranions. Inorg. Chem. 2006, 45, 4035–4046. [Google Scholar] [CrossRef]
- Armaroli, N.; Accorsi, G.; Cardinali, F.; Listorti, A. Photochemistry and Photophysics of Coordination Compounds: Copper. Top. Curr. Chem. 2007, 280, 69–115. [Google Scholar]
- Barbieri, A.; Accorsi, G.; Armaroli, N. Luminescent complexes beyond the platinumgroup: The d10 avenue. Chem. Commun. 2008, 2185–2193. [Google Scholar] [CrossRef]
- Tsubomura, T.; Tsukuda, T.; Matsumoto, K. Luminescent d10 transition metal complexes. Bull. Jpn. Soc. Coord. Chem. 2008, 52, 29–42. [Google Scholar] [CrossRef]
- Lavie-Combot, A.; Cantuel, M.; Leydet, Y.; Jonusauskas, G.; Bassani, D.M.; McClenaghan, N.D. Improving the photophysical properties of copper(I) bis(phenanthroline) complexes. Coord. Chem. Rev. 2008, 252, 2572–2584. [Google Scholar] [CrossRef]
- Tsuge, K. Luminescent Complexes Containing Halogeno-bridged Dicopper(I) Unit {Cu2(µ-X)2} (X = Cl, Br, and I). Chem. Lett. 2013, 42, 204–208. [Google Scholar]
- Tsubomura, T.; Kimura, K.; Nishikawa, M.; Tsukuda, T. Structures and photophysical properties of copper(I) complexes bearing diphenylphenanthroline and bis(diphenylphosphino)alkane: The effect of phenyl groups on the phenanthroline ligand. Dalton Trans. 2015, 44, 7554–7562. [Google Scholar] [CrossRef] [PubMed]
- Kakizoe, D.; Nishikawa, N.; Fuji, Y.; Tsubomura, T. Photophysicle properties of three coordinated copper(I) complexes bearing 1,10-phenanthroline and a monodentate phosphine ligand. Dalton Trans. 2017, 46, 14804–14811. [Google Scholar] [CrossRef] [PubMed]
- Wallesch, M.; Volz, D.; Zink, D.M.; Schepers, U.; Nieger, M.; Baumann, T.; Braese, S. Bright Coppertunities: Multinuclear CuI Complexes with N–P Ligands and Their Applications. Chem. Eur. J. 2014, 20, 6578–6590. [Google Scholar] [CrossRef]
- Giereth, R.; Mengele, A.K.; Frey, W.; Kloss, M.; Stuffen, A.; Karnahl, M.; Tschierlei, S. Copper(I) phosphinooxalozine complexes: Impact of the ligand substitution and steric demand on the electrochemical and photophysical properties. Chem. Eur. J. 2020, 26, 2675–2684. [Google Scholar] [CrossRef]
- Gunaratne, T.; Rodgers, M.A.J.; Felder, D.; Nierengarten, J.-F.; Accorsi, G.; Armaroli, N. Ultrafast dynamics of Cu(I)-phenanthrolines in dichloromethane. Chem. Commun. 2003, 3010–3011. [Google Scholar] [CrossRef]
- Samia, A.C.S.; Cody, J.; Fahrni, C.J.; Burda, C. The effects of ligand constraints on the metal-to-ligand charge transfer relaxation dynamics in copper (I)-phenanthroline complexes: A comparative study by femtosecond time-resolved spectroscopy. J. Phys. Chem. B 2000, 108, 563–569. [Google Scholar] [CrossRef]
- Cody, J.; Dennisson, J.; Gilmore, J.; VanDerveer, D.G.; Henary, M.M.; Gabrielli, A.; Sherrill, C.D.; Zhang, Y.; Pan, C.-P.; Burda, C.; et al. X-ray structures, photophysical characterization, and computational analysis of geometrically constrained copper(I)-phenanthroline complexes. Inorg. Chem. 2003, 42, 4918–4929. [Google Scholar] [CrossRef]
- Kohler, L.; Hayes, D.; Hong, J.Y.; Carter, T.J.; Shelby, M.L.; Fransted, K.A.; Chen, L.X.; Mulfort, K.L. Synthesis, structure, ultrafast kinetics, and light-induced dynamics of CuHETPHEN chromophores. Dalton Trans. 2016, 45, 9871–9883. [Google Scholar] [CrossRef]
- Kelley, M.S.; Shelby, M.L.; Mara, M.W.; Haldrup, K.; Hayes, D.; Hadt, R.G.; Zhang, X.; Stickrath, A.B.; Ruppert, R.; Sauvage, J.-P.; et al. Ultrafast dynamics of two copper bisphenanthroline complexes measured by X-ray transient absorption spectroscopy. J. Phys. B-Atom. Mol. Opt. Phys. 2017, 50, 154006. [Google Scholar] [CrossRef] [Green Version]
- Iwamura, M.; Takeuchi, S.; Tahara, T. Substituent effect on the photoinduced structural change of Cu(I) complexes observed by femtosecond emission spectroscopy. Phys. Chem. Chem. Phys. 2014, 16, 4143–4154. [Google Scholar] [CrossRef]
- Fransted, K.A.; Jackson, N.E.; Zong, R.; Mara, M.W.; Huang, J.; Harpham, M.R.; Shelby, M.L.; Thununel, R.P.; Chen, L.X. Ultrafast Structural Dynamics of Cu(I)-Bicinchoninic Acid and Their Implications for Solar Energy Applications. J. Physical Chem. A 2014, 118, 10497–10506. [Google Scholar] [CrossRef]
- Shaw, G.B.; Grant, C.D.; Shirota, H.; Castner, E.W.; Meyer, G.J.; Chen, L.X. Ultrafast structural rearrangements in the MLCT excited state for copper(I) bis-phenanthrolines in solution. J. Am. Chem. Soc. 2007, 129, 2147–2160. [Google Scholar] [CrossRef] [Green Version]
- Iwamura, M.; Takeuchi, S.; Tahara, T. Real-time observation of the photoinduced structural change of bis(2,9-dimethyl-1,10-phenanthroline)copper(I) by femtosecond fluorescence spectroscopy: A realistic potential curve of the Jahn-Teller distortion. J. Am. Chem. Soc. 2007, 129, 5248–5256. [Google Scholar] [CrossRef] [PubMed]
- Lockard, J.V.; Kabehie, S.; Zink, J.I.; Smolentsev, G.; Soldatov, A.; Chen, L.X. Influence of Ligand Substitution on Excited State Structural Dynamics in Cu(I) Bisphenanthroline Complexes. J. Phys. Chem. B 2010, 114, 14521–14527. [Google Scholar] [CrossRef] [PubMed]
- Iwamura, M.; Takeuchi, S.; Tahara, T. Ultrafast Excited-State Dynamics of Copper(I) Complexes. Acc. Chem. Res. 2015, 48, 782–791. [Google Scholar] [CrossRef]
- Hua, L.; Iwamura, M.; Takeuchi, S.; Tahara, T. The substituent effect on the MLCT excited state dynamics of Cu(I) complexes studied by femtosecond time-resolved absorption and observation of coherent nuclear wavepacket motion. Phys. Chem. Chem. Phys. 2015, 17, 2067–2077. [Google Scholar] [CrossRef]
- Gothard, N.A.; Mara, M.W.; Huang, J.; Szarko, J.M.; Rolczynski, B.; Lockard, J.V.; Chen, L.X. Strong Steric Hindrance Effect on Excited State Structural Dynamics of Cu(I) Diimine Complexes. J. Phys. Chem. A 2012, 116, 1984–1992. [Google Scholar] [CrossRef]
- Papanikolaou, P.A.; Tkachenko, N.V. Probing the excited state dynamics of a new family of Cu(I)-complexes with an enhanced light absorption capacity: Excitation-wavelength dependent population of states through branching. Phys. Chem. Chem. Phys. 2013, 15, 13128–13136. [Google Scholar] [CrossRef] [PubMed]
- Grupe, M.; Bappler, F.; Theiss, M.; Busch, J.M.; Dietrich, F.; Volz, D.; Gerhards, M.; Bräse, S.; Diller, R. Real-time observation of molecular flattening and intersystem crossing in [(DPEPhos)Cu(I)(PyrTet)] via ultrafast UV/Vis- and mid-IR spectroscopy on solution and solid samples. Phys. Chem. Chem. Phys. 2020, 22, 14187–14200. [Google Scholar] [CrossRef] [PubMed]
- Iwamura, M.; Watanabe, H.; Ishii, K.; Takeuchi, S.; Tahara, T. Coherent Nuclear Dynamics in Ultrafast Photoinduced Structural Change of Bis(diimine)copper(I) Complex. J. Am. Chem. Soc. 2011, 133, 7728–7736. [Google Scholar] [CrossRef] [PubMed]
- Capano, G.; Penfold, T.J.; Chergui, M.; Tavernelli, I. Photophysics of a copper phenanthroline elucidated by trajectory and wavepacket-based quantum dynamics: A synergetic approach. Phys. Chem. Chem. Phys. 2017, 19, 19590–19600. [Google Scholar] [CrossRef] [Green Version]
- Capano, G.; Chergui, M.; Rothlisberger, U.; Tavernelli, I.; Penfold, T.J. A Quantum Dynamics Study of the Ultrafast Relaxation in a Prototypical Cu(I)–Phenanthroline. J. Phys. Chem. A 2014, 118, 9861–9869. [Google Scholar] [CrossRef]
- Zgierski, M.Z. Cu(I)-2,9-dimethyl-1,10-phenanthroline: Density functional study of the structure, vibrational force-field, and excited electronic states. J. Chem. Phys. 2003, 118, 4045. [Google Scholar] [CrossRef]
- Stoïanov, A.; Gourlaouen, C.; Vela, S.; Daniel, C. Luminescent Dinuclear Copper(I) Complexes as Potential Thermally Activated Delayed Fluorescence (TADF) Emitters: A Theoretical Study. J. Phys. Chem. A 2018, 122, 1413–1421. [Google Scholar] [CrossRef]
- Levi, G.; Biasin, E.; Dohn, A.O.; Jonsson, H. On the interplay of solvent and conformational effects in simulated excited-state dynamics of a copper phenanthroline photosensitizer. Phys. Chem. Chem. Phys. 2020, 22, 748–757. [Google Scholar] [CrossRef]
- Du, L.; Lan, Z. Ultrafast structural flattening motion in photoinduced excited state dynamics of bis(diamine)copper(I) complex. Phys. Cem. Chem. Phys. 2016, 18, 7641–7650. [Google Scholar] [CrossRef]
- Capano, G.; Rothlisberger, U.; Tavernelli, I.; Penfold, T.J. Theoretical Rationalization of the Emission Properties of Photophysical Cu(I)-phenanthroline Complexes. J. Phys. Chem. A. 2015, 119, 7026–7037. [Google Scholar] [CrossRef]
- Guda, A.; Windisch, J.; Probst, B.; van Bokhoven, J.A.; Alberto, R.; Nachtegaal, M.; Chen, L.X.; Smolentsev, G. Excited-State Structure of Copper Phenanthroline-Based Photosensitizers. Phys. Chem. Chem. Phys. 2021, 23, 26729–26736. [Google Scholar] [CrossRef] [PubMed]
- Kalsani, V.; Schmittel, M.; Listorti, A.; Accorsi, G.; Armaroli, N. Novel Phenanthroline Ligands and Their Kinetically Locked Copper(I) Complexes with Unexpected Photophysical Properties. Inorg. Chem. 2006, 45, 2061–2067. [Google Scholar] [CrossRef] [PubMed]
- Green, O.; Gandhi, B.A.; Burstyn, J.N. Photophysical Characteristics and Reactivity of Bis(2,9-di-tert-butyl-1,10-phenanthroline) Copper(I). Inorg. Chem. 2009, 48, 5704–5714. [Google Scholar] [CrossRef]
- Garakyaraghi, S.; Koutnik, P.; Castellano, F.N. Photoinduced Structural Distortions and Singlet-Triplet Intersystem Crossing in Cu(I) MLCT Excited States Monitored by Optically Gated Fluorescence Spectroscopy. Phys. Chem. Chem. Phys. 2017, 19, 16662–16668. [Google Scholar] [CrossRef] [PubMed]
- Garakyaraghi, S.; McCusker, C.E.; Khan, S.; Koutnik, P.; Bui, A.T.; Castellano, F.N. Enhancing the Visible-Light Absorption and Excited-State Properties of Cu(I) MLCT Excited States. Inorg. Chem. 2018, 57, 2296–2307. [Google Scholar] [CrossRef]
- Brandl, T.; Kerzig, C.; Le Pleux, L.; Prescimone, A.; Wenger, O.S.; Mayor, M. Improved Photostability of a CuI Complex by Macrocyclization of the Phenanthroline Ligands. Chem. Eur. J. 2020, 26, 3119–3128. [Google Scholar] [CrossRef] [PubMed]
- Livshits, M.Y.; Reeves, B.J.; DeWeerd, J.; Strauss, S.H.; Boltalina, O.V.; Rack, J.J. Trifluoromethylated Phenanthroline Ligands Reduce Excited-State Distortion in Homoleptic Copper(I) Complexes. Inorg. Chem. 2020, 59, 2781–2790. [Google Scholar] [CrossRef] [PubMed]
- Rosko, M.C.; Wells, K.A.; Hauke, C.E.; Castellano, F.N. Next Generation Cuprous Phenanthroline MLCT Photosensitizer Featuring Cyclohexyl Substituents. Inorg. Chem. 2021, 60, 8394–8403. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilisation of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ADF, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: https://www.scm.com/doc/ADF/index.html (accessed on 1 June 2019).
- Van Lenthe, E.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1−118. J. Comp. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef] [PubMed]
- Van Lenthe, E.; van Leeuwen, R.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. Int. J. Quant. Chem. 1996, 57, 281–293. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A. Conductor-like Screening Model for Real Solvents: A New Approach to the Quantitative Calculation of Solvation Phenomena. J. Phys. Chem. 1995, 99, 2224–2235. [Google Scholar] [CrossRef]
- Wang, F.; Ziegler, T. A simplified relativistic time-dependent density-functional theory formalism for the calculations of excitation, energies including spin-orbit coupling effect. J. Chem. Phys. 2005, 123, 154102–154112. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ziegler, T.; van Lenthe, E.; van Gisbergen, S.; Baerends, E.J. The calculation of excitation energies based on the relativistic two-component zeroth-order regular approximation and time-dependent density-functional with full use of symmetry. J. Chem. Phys. 2005, 122, 204103–204112. [Google Scholar] [CrossRef]
- Peach, M.J.G.; Tozer, D.J. Overcoming Low Orbital Overlap and Triplet Instability Problems in TDDFT. J. Phys. Chem. A 2012, 116, 9783–9789. [Google Scholar] [CrossRef]
- Plasser, F. TheoDORE: A toolbox for a detailed and automated analysis of electronic excited state computations. J. Chem. Phys. 2020, 152, 084108. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.Z.; Xu, J.Q.; Yang, G.Y.; Bu, W.M.; Xing, Y.H.; Li, D.M.; Liu, S.Q.; Ye, L.; Fan, Y.G. Structure of [Cu(phen)2]2[{Cu(phen)}2Mo8O26].H2O. Pol. J. Chem. 1999, 73, 1909. [Google Scholar]
- Avdeeva, V.V.; Dziova, A.E.; Polyakova, I.N.; Malinina, E.A.; Goeva, L.V.; Kuznetsov, N.T. Copper(I), copper(II), and heterovalent copper(I,II) complexes with 1,10-phenanthroline and the closo-decaborate anion. Inorg. Chim. Acta 2015, 430, 74–81. [Google Scholar] [CrossRef]
- Rheingold, A.L.; Department of Chemistry, University of California, La Jolla, CA, USA; Maatta, E.; Department of Chemistry, Kansas State University, Manhattan, KS, USA. CSD Communication, 2015.
- Huang, Z.; Hartwig, J.F. Copper(I) Enolate complexes in α-Arylation Reactions: Synthesis, Reactivity and Mechanism. Angew. Chem. Int. Ed. 2012, 51, 1028–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; Hou, C.; Li, H.; Weng, Z. Decarboxylative Trifluoromethylating Reagent [Cu(O2CCF3)(phen)] and Difluorocarbene Precursor [Cu(phen)2][O2CCF2Cl]. Chem. Eur. J. 2016, 22, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- Hillhouse, G.; Searle Chemistry Laboratory, Department of Chemistry, The University of Chicago, Chicago, IL, USA; Rheingold, A.L.; Department of Chemistry, University of Delaware, Newark, DE, USA; Allen, M.B. CSD Communication, 1996.
- Huang, Y.; Ajitha, M.J.; Huang, K.-W.; Zhang, Z.; Weng, Z. A class of effective decarboxylative perfluoroalkylating reagents: [(phen)2Cu](O2CRF). Dalton Trans. 2016, 45, 8468–8474. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.-H.; Lu, Z.-L.; Xu, J.-Q.; Bie, H.-Y.; Lu, J.; Zhang, X. Syntheses, characterization and optical properties of some copper(i) halides with 1,10-phenanthroline ligand. New J. Chem. 2004, 28, 940–945. [Google Scholar] [CrossRef]
- Hamalainen, R.; Department of Inorganic Chemistry, University of Helsinki, Helsinki, Finland; Ahlgren, M.; Department of Inorganic Chemistry, University of Helsinki, Helsinki, Finland; Turpeinen, U.; Department of Inorganic Chemistry, University of Helsinki, Helsinki, Finland; Raikas, T.; Department of Inorganic Chemistry, University of Helsinki, Helsinki, Finland. Crystal Structure Communications, 1979.
- Dessy, G.; Laboratorio di Teoria e Struttura Elettronica e Comportamento Spettrochimico dei Composti di Coordinazione del CNR, Monterotondo Stazione, Italy; Fares, V.; Laboratorio di Teoria e Struttura Elettronica e Comportamento Spettrochimico dei Composti di Coordinazione del CNR, Monterotondo Stazione, Italy. Crystal Structure Communications, 1979.
- Royappa, A.T.; Royappa, A.D.; Moral, R.F.; Rheingold, A.L.; Papoular, R.J.; Blum, D.M.; Duong, T.Q.; Stepherson, J.R.; Vu, O.D.; Chen, B.; et al. Copper(I) oxalate complexes: Synthesis, structures and surprises. Polyhedron 2016, 119, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.-L.; Gembicky, M.; Messerschmidt, M.; Dominiak, P.M.; Coppens, P. Effect of the Environment on Molecular Properties: Synthesis, Structure, and Photoluminescence of Cu(I) Bis(2,9-dimethyl-1,10-phenanthroline) Nanoclusters in Eight Different Supramolecular Frameworks. Inorg. Chem. 2006, 45, 9281–9289. [Google Scholar] [CrossRef]
- Kovalevsky, A.Y.; Gembicky, M.; Novozhilova, I.V.; Coppens, P. Solid-State Structure Dependence of the Molecular Distortion and Spectroscopic Properties of the Cu(I) Bis(2,9-dimethyl-1,10-phenanthroline) Ion. Inorg. Chem. 2003, 42, 8794–8802. [Google Scholar] [CrossRef] [PubMed]
- Blake, A.J.; Hill, S.J.; Hubberstey, P.; Li, W.-S. Copper(I) chelated by 2,9-dimethyl-1,10-phenanthroline and bridged by 4,4′-bipyridine or trans-1,2-bis(pyridin-4-yl)ethene to give discrete dinuclear and polymeric cations. J. Chem. Soc. Dalton Trans. 1998, 909–916. [Google Scholar] [CrossRef]
- Gandhi, B.A.; Green, O.; Burstyn, J.N. Facile oxidation-based of sterically encumbered four-coordinate bis(2,9-di-ter-butyl-1,10-phenanthroline)copper(I) and related three-coordinate copper(I) complexes. Inorg. Chem. 2007, 46, 3816–3825. [Google Scholar] [CrossRef] [PubMed]
- Asano, M.S.; Tomiduka, K.; Sekizawa, K.; Yamashita, K.; Sugiura, K. Temperature-dependent Emission of Copper(I) Phenanthroline Complexes with Bulky Substituents: Estimation of an Energy Gap between the Singlet and Triplet MLCT States. Chem. Lett. 2010, 39, 376–378. [Google Scholar] [CrossRef]
- Czerwieniec, R.; Yersin, H. Diversity of Copper(I) Complexes Showing Thermally Activated Delayed Fluorescence: Basis Photophysical Analysis. Inorg. Chem. 2015, 54, 4322–4327. [Google Scholar] [CrossRef] [PubMed]
- Farias, G.; Salla, C.A.M.; Heying, R.S.; Bortoluzzi, A.J.; Curcio, S.F.; Cazati, T.; dos Santos, P.L.; Monkman, A.P.; de Souza, B.; Bechtlod, I.H. Reducing lifetime in Cu(I) Complexes with Thermally Activated Delayed Fluorescence and Phosphorescence Promoted by Chalcogenolate-diimine ligands. J. Mater. Chem. C 2020, 8, 14595–14604. [Google Scholar] [CrossRef]
- Greenough, S.E.; Horbury, M.D.; Thompson, J.O.F.; Roberts, G.M.; Karsili, T.N.V.; Marchetti, B.; Townsend, D.; Stavros, V.G. Solvent induced conformer specific photochemistry of guaiacol. Phys. Chem. Chem. Phys. 2014, 16, 16187–16195. [Google Scholar] [CrossRef]
- Miller, M.T.; Gantzel, P.K.; Karpishin, T.B. Structures of the Copper(I) and Copper(II) Complexes of 2,9-Diphenyl-1,10-phenanthroline: Implications for Excited-State Structural Distortion. Inorg. Chem. 1998, 37, 2285–2290. [Google Scholar] [CrossRef]
- Gimeno, L.; Blart, E.; Rebilly, J.-N.; Coupeau, M.; Allain, M.; Roisnel, T.; Quarré de Verneuil, A.; Gourlaouen, C.; Daniel, C.; Pellegrin, Y. Non-symmetrical sterically challenged phenanthroline ligands and their homoleptic copper(I) complexes with improved excited state properties. Chem. Eur. J. 2020, 26, 11887–11899. [Google Scholar] [CrossRef] [PubMed]
- Heuft, M.A.; Fallis, A.G. Template-directed synthesis of helical phenanthroline cyclophanes. Angew. Chem. Int. Ed. 2002, 41, 4520–4523. [Google Scholar] [CrossRef]
- Dietrich-Buchecker, C.O.; Guilhem, J.; Khemiss, A.K.; Kintzinger, J.-P.; Pascard, C.; Sauvage, J.-P. Molecular Structure of a I3I-Catenate: Curling Up of the Interlocked System by Interaction between the Two Copper Complex Subunits. Angew. Chem. Int. Ed. Engl. 1987, 26, 661–663. [Google Scholar] [CrossRef]
- Gimeno, L.; Phelan, B.T.; Sprague-Klein, E.A.; Roisnel, T.; Blart, E.; Gourlaouen, C.; Chen, L.X.; Pellegrin, Y. Bulky and Stable Copper(I)-Phenanthroline Complex: Impact of Steric Strain and Symmetry on the Excited-State Properties. Inorg. Chem. 2022, 61, 7296–7307. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | 1H | 1Me | 1tBu (δ) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Eabs | λabs | f | Eabs | λabs | f | Eabs | λabs | f | |
| S1 | 2.370 | 523 | 7.91 × 10−13 | 2.361 | 525 | 1.17 × 10−13 | 2.419 | 513 | 1.30 × 10−3 |
| S2 | 2.427 | 511 | 5.49 × 10−11 | 2.421 | 512 | 2.71 × 10−11 | 2.465 | 503 | 9.46 × 10−5 |
| S3 | 2.536 | 489 | 3.35 × 10−1 | 2.531 | 490 | 3.26 × 10−1 | 2.534 | 489 | 1.69 × 10−1 |
| S4 | 2.699 | 459 | 8.94 × 10−4 | 2.775 | 447 | 2.20 × 10−3 | 2.778 | 446 | 7.14 × 10−3 |
| S5 | 2.699 | 459 | 8.77 × 10−4 | 2.790 | 444 | 2.31 × 10−3 | 2.810 | 441 | 2.34 × 10−3 |
| S6 | 2.789 | 445 | 4.48 × 10−2 | 2.873 | 431 | 5.25 × 10−2 | 2.864 | 433 | 3.91 × 10−3 |
| S7 | 2.800 | 443 | 4.24 × 10−2 | 2.873 | 431 | 5.25 × 10−2 | 2.895 | 428 | 3.24 × 10−2 |
| S8 | 3.031 | 409 | 2.21 × 10−6 | 3.029 | 409 | 1.54 × 10−6 | 2.912 | 426 | 1.40 × 10−2 |
| S9 | 3.101 | 400 | 1.19 × 10−3 | 3.101 | 400 | 1.16 × 10−3 | 2.915 | 425 | 2.67 × 10−3 |
| S10 | 3.101 | 400 | 1.20 × 10−3 | 3.121 | 397 | 1.16 × 10−3 | 2.948 | 421 | 6.70 × 10−3 |
| S11 | 3.265 | 380 | 2.96 × 10−5 | 3.287 | 377 | 6.67 × 10−5 | 3.157 | 393 | 9.42 × 10−6 |
| S12 | 3.276 | 378 | 2.68 × 10−5 | 3.298 | 376 | 6.74 × 10−5 | 3.170 | 391 | 1.82 × 10−3 |
| S13 | 3.379 | 367 | 1.27 × 10−4 | 3.401 | 365 | 1.26 × 10−4 | 3.212 | 386 | 6.36 × 10−4 |
| S14 | 3.379 | 367 | 1.26 × 10−4 | 3.401 | 365 | 1.37 × 10−4 | 3.213 | 386 | 6.01 × 10−4 |
| S15 | 3.454 | 359 | 1.51 × 10−14 | 3.526 | 352 | 7.98 × 10−13 | 3.329 | 372 | 3.68 × 10−4 |
| S16 | 3.462 | 358 | 3.21 × 10−12 | 3.554 | 349 | 6.72 × 10−13 | 3.342 | 371 | 7.34 × 10−4 |
| S17 | 3.628 | 342 | 4.70 × 10−2 | 3.722 | 333 | 5.69 × 10−2 | 3.581 | 346 | 2.71 × 10−2 |
| S18 | 3.680 | 337 | 9.90 × 10−7 | 3.793 | 327 | 1.12 × 10−6 | 3.624 | 342 | 4.93 × 10−4 |
| S19 | 3.753 | 330 | 4.65 × 10−9 | 3.829 | 324 | 3.72 × 10−11 | 3.625 | 342 | 1.04 × 10−3 |
| S20 | 3.771 | 329 | 2.53 × 10−14 | 3.857 | 321 | 3.58 × 10−13 | 3.642 | 340 | 4.41 × 10−5 |
| S21 | 3.812 | 325 | 1.14 × 10−12 | 3.895 | 318 | 2.63 × 10−13 | 3.841 | 323 | 2.03 × 10−3 |
| S22 | 3.818 | 325 | 1.38 × 10−9 | 3.895 | 318 | 1.03 × 10−10 | 3.861 | 321 | 1.26 × 10−5 |
| S23 | 3.875 | 320 | 4.99 × 10−2 | 3.929 | 316 | 4.04 × 10−2 | 3.917 | 317 | 3.12 × 10−2 |
| S24 | 4.012 | 309 | 3.19 × 10−7 | 3.958 | 313 | 1.53 × 10−7 | 3.966 | 313 | 3.84 × 10−3 |
| S25 | 4.079 | 304 | 1.48 × 10−2 | 4.027 | 308 | 2.27 × 10−2 | 4.009 | 309 | 3.50 × 10−2 |
| State | 2 | 3 | ||||
|---|---|---|---|---|---|---|
| Eabs | λabs | f | Eabs | λabs | f | |
| S1 | 2.381 | 521 | 1.63 × 10−6 | 2.448 | 506 | 1.69 × 10−4 |
| S2 | 2.434 | 509 | 3.08 × 10−6 | 2.659 | 466 | 6.74 × 10−2 |
| S3 | 2.515 | 493 | 1.65 × 10−1 | 2.669 | 465 | 1.32 × 10−1 |
| S4 | 2.730 | 454 | 2.20 × 10−4 | 2.848 | 435 | 6.53 × 10−4 |
| S5 | 2.767 | 448 | 9.72 × 10−5 | 2.968 | 418 | 9.83 × 10−3 |
| S6 | 2.782 | 446 | 2.33 × 10−2 | 2.990 | 415 | 2.31 × 10−2 |
| S7 | 2.856 | 434 | 6.03 × 10−3 | 3.013 | 411 | 7.92 × 10−3 |
| S8 | 2.877 | 431 | 3.02 × 10−2 | 3.029 | 409 | 2.31 × 10−2 |
| S9 | 2.886 | 430 | 2.08 × 10−4 | 3.125 | 397 | 1.21 × 10−3 |
| S10 | 2.994 | 414 | 2.01 × 10−3 | 3.230 | 384 | 1.92 × 10−3 |
| S11 | 3.093 | 401 | 1.99 × 10−4 | 3.297 | 376 | 3.07 × 10−3 |
| S12 | 3.116 | 398 | 1.25 × 10−5 | 3.320 | 373 | 1.98 × 10−5 |
| S13 | 3.174 | 391 | 1.66 × 10−3 | 3.439 | 360 | 8.82 × 10−4 |
| S14 | 3.212 | 386 | 1.03 × 10−3 | 3.469 | 357 | 3.24 × 10−3 |
| S15 | 3.299 | 376 | 2.67 × 10−6 | 3.501 | 354 | 1.62 × 10−3 |
| S16 | 3.338 | 371 | 1.75 × 10−7 | 3.605 | 344 | 1.83 × 10−4 |
| S17 | 3.515 | 353 | 1.08 × 10−6 | 3.728 | 333 | 6.97 × 10−4 |
| S18 | 3.544 | 350 | 2.26 × 10−2 | 3.759 | 330 | 2.34 × 10−2 |
| S19 | 3.551 | 349 | 2.97 × 10−7 | 3.826 | 324 | 2.27 × 10−5 |
| S20 | 3.618 | 343 | 2.38 × 10−4 | 3.844 | 323 | 2.48 × 10−3 |
| S21 | 3.705 | 335 | 6.04 × 10−7 | 3.904 | 318 | 1.04 × 10−4 |
| S22 | 3.879 | 320 | 3.74 × 10−2 | 3.936 | 315 | 5.64 × 10−3 |
| S23 | 3.892 | 319 | 8.98 × 10−6 | 3.962 | 313 | 3.21 × 10−4 |
| S24 | 3.930 | 315 | 7.24 × 10−3 | 3.992 | 311 | 1.78 × 10−2 |
| S25 | 3.993 | 310 | 1.31 × 10−2 | 4.029 | 308 | 5.14 × 10−2 |
| State | 1H | 1Me | 1tBu | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Eabs | f | Mixing | Eabs | f | Mixing | Eabs | f | Mixing | |
| E1 | 2.241 | 4.16 × 10−6 | T1 83% T4 16% | 2.233 | 6.36 × 10−6 | T1 81% T4 18% | 2.262 | 3.35 × 10−6 | T1 85% T4 9% T2 4% |
| E2 | 2.241 | 4.26 × 10−6 | T1 83% T4 16% | 2.233 | 6.31 × 10−6 | T1 81% T4 18% | 2.263 | 2.38 × 10−5 | T1 85% T4 9% T2 4% |
| E3 | 2.248 | 3.26 × 10−6 | T2 61% T3 38% | 2.235 | 4.81 × 10−6 | T2 68% T3 31% | 2.269 | 3.09 × 10−3 | T1 95% S3 2% |
| E4 | 2.248 | 3.29 × 10−6 | T2 61% T3 38% | 2.235 | 4.79 × 10−6 | T2 68% T3 31% | 2.290 | 1.30 × 10−5 | T2 65% T3 28% |
| E5 | 2.251 | 5.76 × 10−3 | T1 97% S3 2% | 2.243 | 5.29 × 10−3 | T1 98% S3 2% | 2.291 | 4.80 × 10−5 | T2 65% T3 29% T1 3% |
| E6 | 2.265 | 1.56 × 10−8 | T2 89% S1 10% | 2.249 | 1.29 × 10−9 | T2 91% S1 9% | 2.310 | 2.11 × 10−5 | T2 98% |
| E7 | 2.292 | 3.43 × 10−7 | T3 99% | 2.286 | 2.29 × 10−9 | T3 99% | 2.331 | 1.17 × 10−3 | T3 82% S1 16% |
| E8 | 2.303 | 1.35 × 10−11 | T4 90% S2 9% | 2.289 | 1.09 × 10−11 | T4 92% S2 8% | 2.352 | 1.25 × 10−5 | T4 86% S2 12% |
| E9 | 2.322 | 6.05 × 10−7 | T3 61% T2 38% | 2.311 | 5.98 × 10−7 | T3 68% T2 31% | 2.368 | 1.09 × 10−5 | T3 55% T2 27% T4 13% T1 3% |
| E10 | 2.322 | 6.37 × 10−7 | T3 61% T2 38% | 2.311 | 6.40 × 10−7 | T3 68% T2 31% | 2.368 | 1.13 × 10−5 | T3 58% T2 27% T4 5% T1 3% |
| E11 | 2.329 | 1.81 × 10−6 | T4 82% T1 16% | 2.315 | 2.18 × 10−6 | T4 80% T1 18% | 2.380 | 9.55 × 10−6 | T4 77% T3 11% T1 8% T2 2% |
| E12 | 2.329 | 1.84 × 10−6 | T4 82% T1 16% | 2.315 | 2.24 × 10−6 | T4 80% T1 18% | 2.381 | 1.57 × 10−5 | T4 75% T3 13% T1 7% T2 3% |
| E13 | 2.372 | 5.45 × 10−8 | S1 89% T2 10% | 2.361 | 3.09 × 10−11 | S1 90% T2 9% | 2.422 | 1.18 × 10−3 | S1 81% T3 15% T14 1% |
| E14 | 2.428 | 5.05 × 10−11 | S2 90% T4 9% | 2.421 | 6.16 × 10−11 | S2 91% T4 7% | 2.465 | 8.10 × 10−5 | S2 85% T4 12% T13 1% |
| E15 | 2.529 | 3.23 × 10−1 | S3 97% T1 2% | 2.524 | 3.15 × 10−1 | S3 97% T1 2% | 2.523 | 1.60 × 10−1 | S3 95% T1 2% |
| E16 | 2.617 | 5.87 × 10−6 | T5 71% T7 26% | 2.659 | 5.21 × 10−9 | T5 49% T6 49% | 2.718 | 6.82 × 10−6 | T5 72% T7 15% T6 9% |
| E17 | 2.617 | 1.06 × 10−4 | T5 71% T7 26% | 2.659 | 1.41 × 10−11 | T5 49% T6 49% | 2.718 | 8.35 × 10−6 | T5 72% T7 16% T6 9% |
| E18 | 2.624 | 5.84 × 10−4 | T5 80% S4 11% T6 4% S5 4% | 2.679 | 4.66 × 10−4 | T6 81% S4 10% T5 8% | 2.728 | 1.46 × 10−3 | T5 90% S7 4% S5 3% |
| E19 | 2.637 | 1.43 × 10−7 | T6 70% T8 25% T7 3% | 2.680 | 4.43 × 10−4 | T5 83% T6 10% S5 8% | 2.736 | 9.80 × 10−6 | T6 68% T8 20% T5 6% T7 4% |
| E20 | 2.637 | 1.46 × 10−4 | T6 70% T8 25% T7 3% | 2.700 | 1.40 × 10−11 | T7 35% T8 35% T6 14% T5 14% | 2.736 | 2.76 × 10−4 | T6 68% T8 20% T7 5% T5 5% |
| State | 2 | 3 | ||||
|---|---|---|---|---|---|---|
| Eabs | f | Mixing | Eabs | f | Mixing | |
| E1 | 2.223 | 1.26 × 10−6 | T1 56% T2 38% T3 3% | 2.290 | 2.03 × 10−6 | T1 92% T2 5% |
| E2 | 2.223 | 2.24 × 10−5 | T1 56% T2 38% T3 3% | 2.290 | 1.07 × 10−5 | T1 92% T2 6% |
| E3 | 2.243 | 1.26 × 10−7 | T1 92% S1 6% | 2.297 | 8.58 × 10−4 | T1 98% |
| E4 | 2.256 | 3.29 × 10−3 | T2 97% S3 2% | 2.393 | 8.39 × 10−5 | T2 47% S1 41% T2 10% |
| E5 | 2.270 | 2.80 × 10−6 | T3 60% T2 27% T4 6% T1 6% | 2.427 | 2.14 × 10−6 | T2 82% T4 6% T1 4% T3 4% |
| E6 | 2.270 | 1.25 × 10−5 | T3 59% T2 28% T4 6% T1 6% | 2.428 | 4.79 × 10−5 | T2 80% T3 6% T4 5% T1 5% |
| E7 | 2.283 | 2.17 × 10−4 | T3 98% | 2.458 | 2.19 × 10−3 | T3 44% S1 26% T2 26% |
| E8 | 2.297 | 1.90 × 10−6 | T1 35% T2 31% T3 29% T12 1% | 2.461 | 2.37 × 10−5 | T3 71% T4 16% T2 9% |
| E9 | 2.298 | 4.42 × 10−6 | T1 36% T2 32% T3 29% T12 1% | 2.462 | 4.17 × 10−5 | T3 71% T4 16% T2 10% |
| E10 | 2.363 | 1.25 × 10−6 | S1 50% T4 33% S2 13% T1 2% | 2.477 | 1.66 × 10−3 | T3 50% S1 31% T2 15% S2 1% |
| E11 | 2.390 | 1.41 × 10−6 | S1 41% T4 29% S2 24% T1 4% | 2.514 | 2.32 × 10−3 | T4 96% S2 1% |
| E12 | 2.412 | 1.48 × 10−6 | T4 91% T3 6% T14 2% | 2.539 | 9.12 × 10−7 | T4 75% T3 20% |
| E13 | 2.413 | 2.54 × 10−5 | T4 91% T3 6% S13 1% | 2.540 | 1.16 × 10−5 | T4 75% T3 21% |
| E14 | 2.447 | 1.91 × 10−6 | S2 60% T4 36% T14 2% | 2.653 | 6.59 × 10−2 | S2 90% S3 4% |
| E15 | 2.503 | 1.56 × 10−1 | S3 95% T2 2% | 2.662 | 1.22 × 10−1 | S3 91% S2 4% |
| E16 | 2.666 | 1.56 × 10−4 | T5 74% T7 15% T6 6% T8 4% | 2.764 | 1.29 × 10−4 | T5 87% T6 11% |
| E17 | 2.666 | 2.11 × 10−8 | T5 73% T7 15% T6 6% T8 4% | 2.764 | 7.16 × 10−6 | T5 87% T6 11% |
| E18 | 2.671 | 5.13 × 10−4 | T5 88% S5 6% T6 3% S8 2% | 2.771 | 6.64 × 10−4 | T5 96% S6 3% |
| E19 | 2.681 | 9.91 × 10−6 | T6 57% T7 22% T5 15% T8 5% | 2.813 | 4.31 × 10−4 | S4 66% T6 33% |
| E20 | 2.681 | 1.34 × 10−8 | T6 57% T7 22% T5 15% T8 5% | 2.875 | 1.62 × 10−4 | T6 86% T5 12% |
| 1H | S1 | S2 | S3 | S4 | T1 | T2 | T3 | T4 |
|---|---|---|---|---|---|---|---|---|
| T1 | 0.0 | 0.0 | 307.4 | 3.0 | 0.0 | 0.0 | 0.0 | 390.4 |
| T2 | 275.7 | 0.0 | 0.0 | 17.3 | 0.0 | 0.0 | 428.6 | 0.0 |
| T3 | 0.0 | 0.0 | 0.0 | 11.2 | 0.0 | 428.6 | 0.0 | 0.0 |
| T4 | 0.0 | 300.0 | 0.0 | 9.6 | 390.4 | 0.0 | 0.0 | 0.0 |
| 1tBu | S1 | S2 | S3 | S4 | T1 | T2 | T3 | T4 |
| T1 | 114.9 | 0.5 | 298.4 | 21.3 | 0.0 | 107.1 | 1.1 | 423.7 |
| T2 | 1.3 | 57.9 | 0.8 | 171.1 | 107.1 | 0.0 | 441.5 | 2.2 |
| T3 | 289.2 | 3.1 | 118.5 | 58.9 | 1.1 | 441.5 | 0.0 | 88.3 |
| T4 | 0.8 | 322.4 | 4.1 | 25.1 | 423.7 | 2.2 | 88.3 | 0.0 |
| 2 | S1 | S2 | S3 | S4 | T1 | T2 | T3 | T4 |
| T1 | 282.2 | 139.9 | 0.2 | 7.0 | 0.0 | 392.7 | 201.9 | 0.4 |
| T2 | 0.3 | 0.4 | 302.1 | 5.6 | 392.7 | 0.0 | 0.5 | 183.7 |
| T3 | 0.1 | 302.1 | 76.5 | 9.9 | 201.9 | 0.5 | 0.0 | 399.3 |
| T4 | 129.8 | 5.6 | 0.5 | 22.4 | 0.4 | 183.7 | 399.3 | 0.0 |
| 3 | S1 | S2 | S3 | S4 | T1 | T2 | T3 | T4 |
| T1 | 7.9 | 159.0 | 221.6 | 20.5 | 0.0 | 425.3 | 176.5 | 21.4 |
| T2 | 316.8 | 65.3 | 48.9 | 23.0 | 425.3 | 0.0 | 24.8 | 206.9 |
| T3 | 97.4 | 248.0 | 210.6 | 5.4 | 176.5 | 24.8 | 0.0 | 411.0 |
| T4 | 8.6 | 149.2 | 124.8 | 10.2 | 21.4 | 206.9 | 411.0 | 0.0 |
| State | 1H | 1Me | 1tBu (δ) | |
|---|---|---|---|---|
| S1 | Edef | 0.820 | 0.442 | 0.253 |
| Eem | 0.707 | 1.569 | 1.813 | |
| Estab | 1.527 | 2.011 | 2.066 | |
| λem | 1753 | 790 | 684 | |
| fosc | 2.25 × 10−5 | 1.77 × 10−2 | 2.54 × 10−4 | |
| T1 | Edef | 0.610 | 0.443 | 0.307 |
| Eem | 0.704 | 1.246 | 1.656 | |
| Estab | 1.314 | 1.689 | 1.964 | |
| λem | 1761 | 995 | 749 | |
| ΔEST | 0.464 | 0.411 | 0.143 | |
| SOC S1–T1 | 25.5 | 54.2 | 20.1 | |
| fosc | 2.28 × 10−9 | 2.01 × 10−9 | 6.85 × 10−6 | |
| 7.01 × 10−9 | 5.38 × 10−8 | 2.77 × 10−5 | ||
| 2.34 × 10−6 | 1.17 × 10−4 | 1.77 × 10−4 |
| State | Conformer | α | β | γ | δ | ε | ζ |
|---|---|---|---|---|---|---|---|
| GS Symmetry | D2d | D2d | C1 | C2 | C1 | C2v | |
| S1 | S1 Symmetry | C2 | C2 | C1 | C1 | C1 | C2 |
| Edef | 0.542 | 0.497 | 0.279 | 0.286 | 0.376 | 0.249 | |
| Eem | 1.944 | 1.779 | 1.870 | 1.813 | 1.801 | 1.884 | |
| Estab | 2.486 | 2.275 | 2.149 | 2.099 | 2.177 | 2.133 | |
| ΔE | 0.387 | 0.176 | 0.050 | 0.000 | 0.078 | 0.033 | |
| λem | 638 | 697 | 663 | 684 | 688 | 658 | |
| fosc | 1.06 × 10−5 | 1.57 × 10−6 | 1.53 × 10−5 | 2.54 × 10−4 | 2.53 × 10−5 | 1.59 × 10−8 | |
| T1 | T1 Symmetry | C2 | C2 | C1 | C1 | C1 | C2 |
| Edef | 0.585 | 0.513 | 0.295 | 0.341 | 0.397 | 0.270 | |
| Eem | 1.745 | 1.633 | 1.715 | 1.656 | 1.648 | 1.726 | |
| Estab | 2.331 | 2.146 | 2.011 | 1.997 | 2.046 | 1.997 | |
| ΔE | 0.334 | 0.149 | 0.014 | 0.000 | 0.049 | 0.000 | |
| λem | 710 | 759 | 723 | 749 | 752 | 718 | |
| ΔEST | 0.188 | 0.133 | 0.144 | 0.143 | 0.138 | 0.140 | |
| SOC S1–T1 | 73.0 | 6.0 | 3.6 | 20.1 | 8.3 | 0.0 | |
| 3.04 × 10−6 | 2.10 × 10−9 | 7.05 × 10−7 | 6.85 × 10−6 | 4.25 × 10−6 | 1.09 × 10−13 | ||
| fosc | 8.97 × 10−6 | 6.86 × 10−6 | 4.69 × 10−5 | 2.77 × 10−5 | 1.42 × 10−5 | 6.25 × 10−6 | |
| 3.04 × 10−4 | 3.01 × 10−4 | 2.81 × 10−4 | 1.77 × 10−4 | 2.46 × 10−4 | 2.88 × 10−4 |
| State | 2 | 2 | 3 | 3 | |
|---|---|---|---|---|---|
| S1 | Edef | 0.227 | 0.241 | 0.257 | 0.343 |
| Eem | 1.783 | 1.797 | 1.881 | 1.893 | |
| Estab | 2.009 | 2.037 | 2.137 | 2.235 | |
| λem | 695 | 690 | 659 | 655 | |
| fosc | 0.00 | 0.00 | 5.43 × 10−5 | 5.73 × 10−6 | |
| T1 | Edef | 0.243 | 0.26 | 0.277 | 0.365 |
| Eem | 1.634 | 1.643 | 1.717 | 1.733 | |
| Estab | 1.877 | 1.903 | 1.995 | 2.098 | |
| λem | 759 | 754 | 722 | 715 | |
| ΔEST | 0.135 | 0.138 | 0.146 | 0.143 | |
| SOC S1–T1 | 0.0 | 0.0 | 7.7 | 2.9 | |
| fosc | 9.96 × 10−8 | 1.04 × 10−7 | 2.37 × 10−7 | 1.28 × 10−8 | |
| 4.81 × 10−6 | 1.07 × 10−5 | 5.68 × 10−6 | 3.87 × 10−6 | ||
| 3.03 × 10−4 | 1.39 × 10−4 | 3.10 × 10−4 | 1.51 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gourlaouen, C.; Daniel, C. Towards Optimized Photoluminescent Copper(I) Phenanthroline-Functionalized Complexes: Control of the Photophysics by Symmetry-Breaking and Spin–Orbit Coupling. Materials 2022, 15, 5222. https://doi.org/10.3390/ma15155222
Gourlaouen C, Daniel C. Towards Optimized Photoluminescent Copper(I) Phenanthroline-Functionalized Complexes: Control of the Photophysics by Symmetry-Breaking and Spin–Orbit Coupling. Materials. 2022; 15(15):5222. https://doi.org/10.3390/ma15155222
Chicago/Turabian StyleGourlaouen, Christophe, and Chantal Daniel. 2022. "Towards Optimized Photoluminescent Copper(I) Phenanthroline-Functionalized Complexes: Control of the Photophysics by Symmetry-Breaking and Spin–Orbit Coupling" Materials 15, no. 15: 5222. https://doi.org/10.3390/ma15155222
APA StyleGourlaouen, C., & Daniel, C. (2022). Towards Optimized Photoluminescent Copper(I) Phenanthroline-Functionalized Complexes: Control of the Photophysics by Symmetry-Breaking and Spin–Orbit Coupling. Materials, 15(15), 5222. https://doi.org/10.3390/ma15155222