
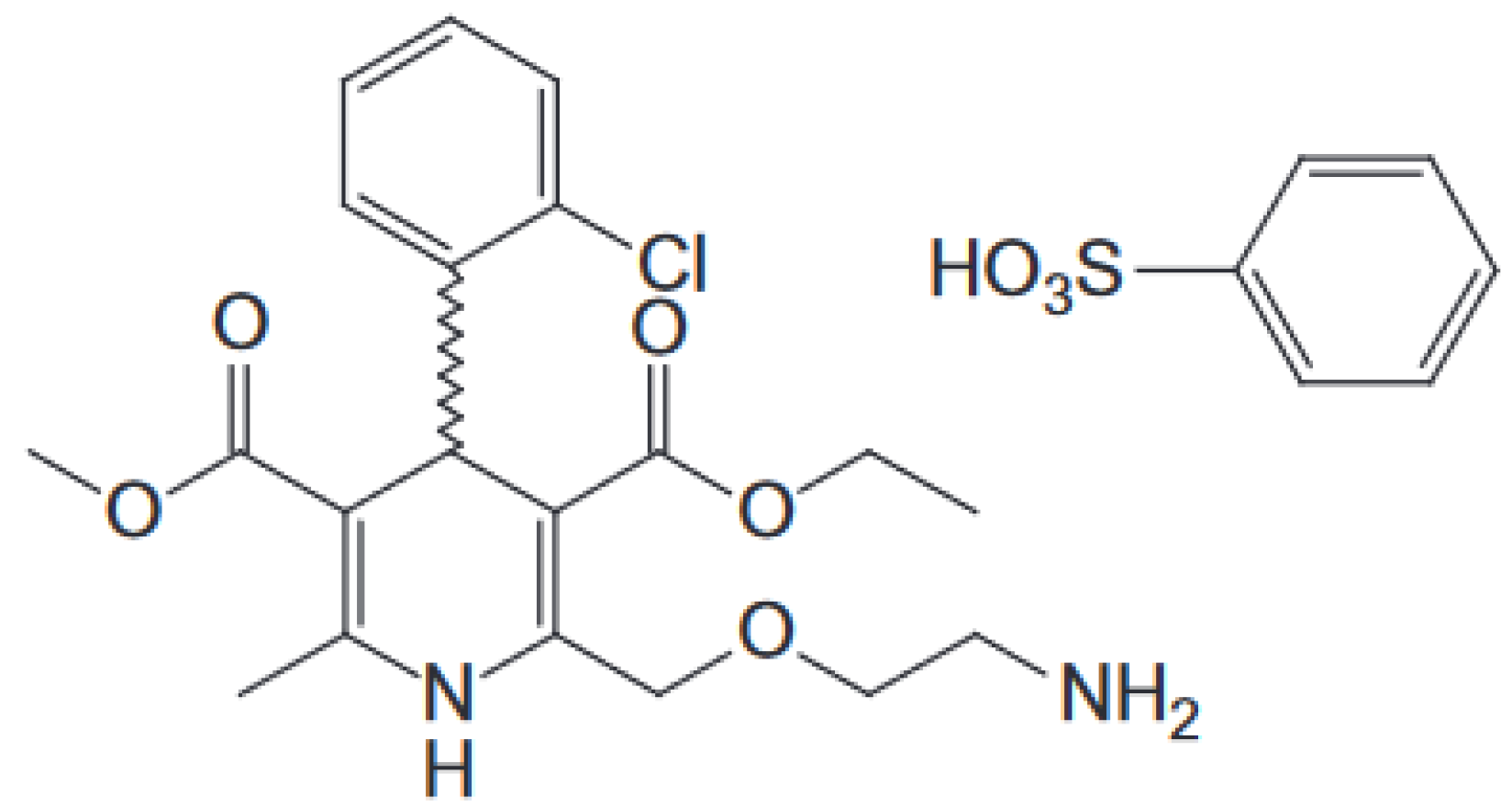
Design and Evaluation of Orally Dispersible Tablets Containing Amlodipine Inclusion Complexes in Hydroxypropyl-β-cyclodextrin and Methyl-β-cyclodextrin


 ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. The Binary System Obtaining Processes
Kneading Product
Preparation of the Inclusion Compounds Using Liquid-State Coprecipitation and Lyophilization Techniques
Preparation of Physical Mixtures
2.2.2. Physicochemical Characterization
2.2.3. Preformulation and Formulation Studies for Orodispersible Tablets Containing AML-HP-β-CD and AML-Me-β-CD Inclusion Complexes
Formulation of Direct Compression Blends
Materials for Direct Compression Preparation
The Pharmacotechnical Properties of the Direct Compression Powders
2.2.4. Development and Manufacturing of Orodispersible Tablets Containing AML-HP-β-CD and AML-Me-β-CD Inclusion Complexes
Formulation of the Orodispersible Tablets Containing AML-HP-β-CD and AML-Me-β-CD Inclusion Complexes
Manufacturing Process of Tablets for Oral Dispersion
2.2.5. Quality Assessment Parameters
In Vitro Disintegration Time
In Vitro Dissolution Studies
3. Results
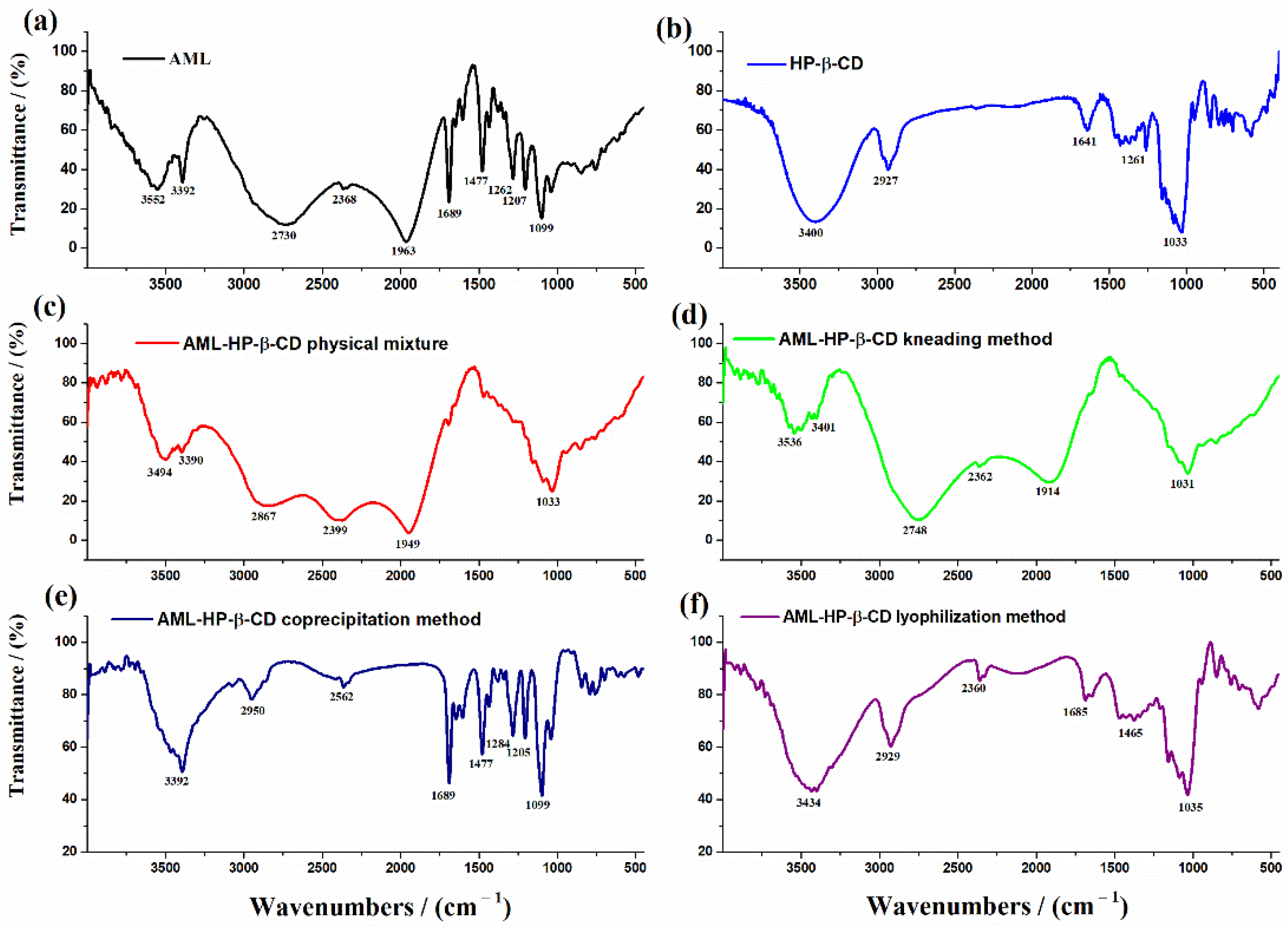
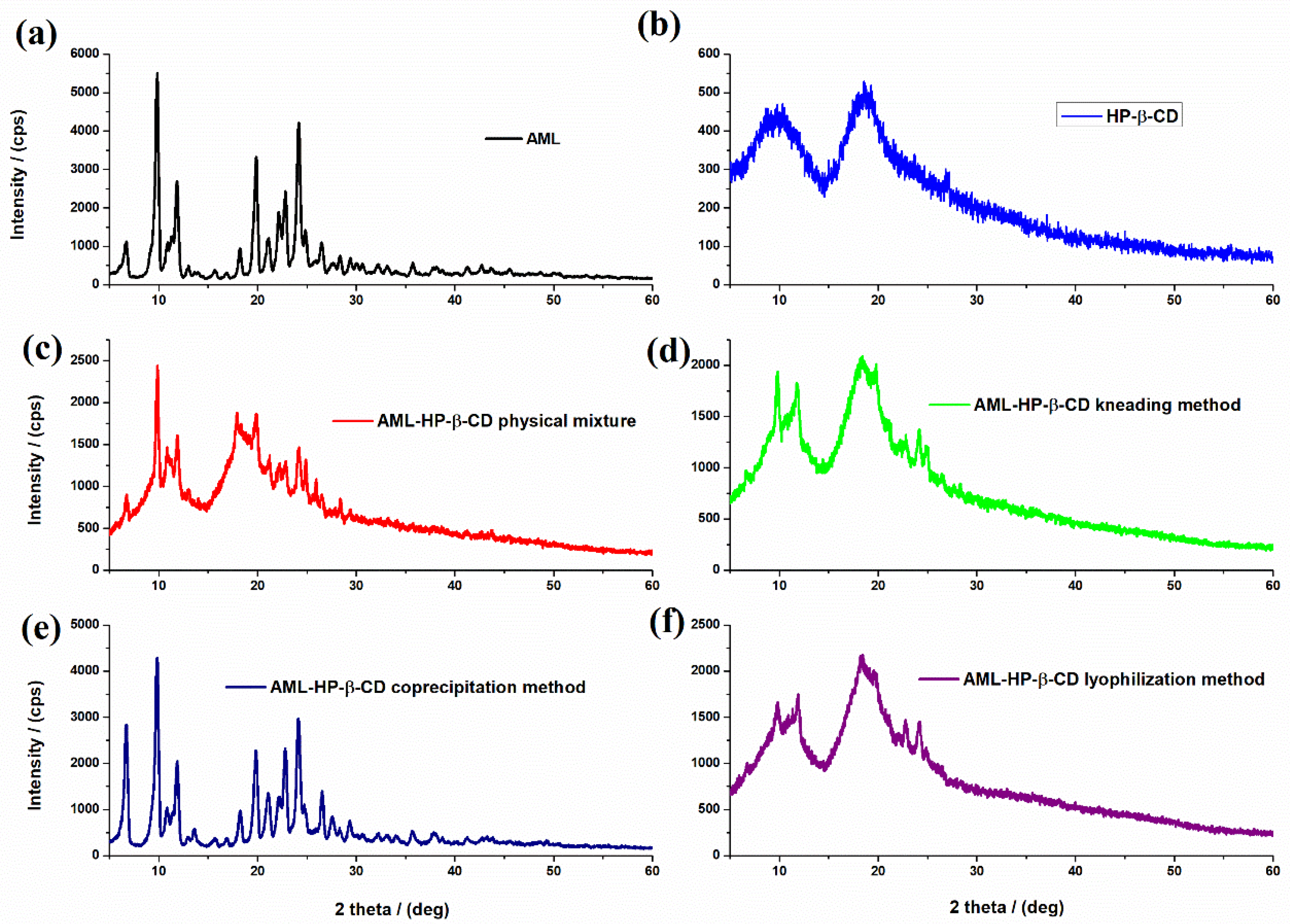
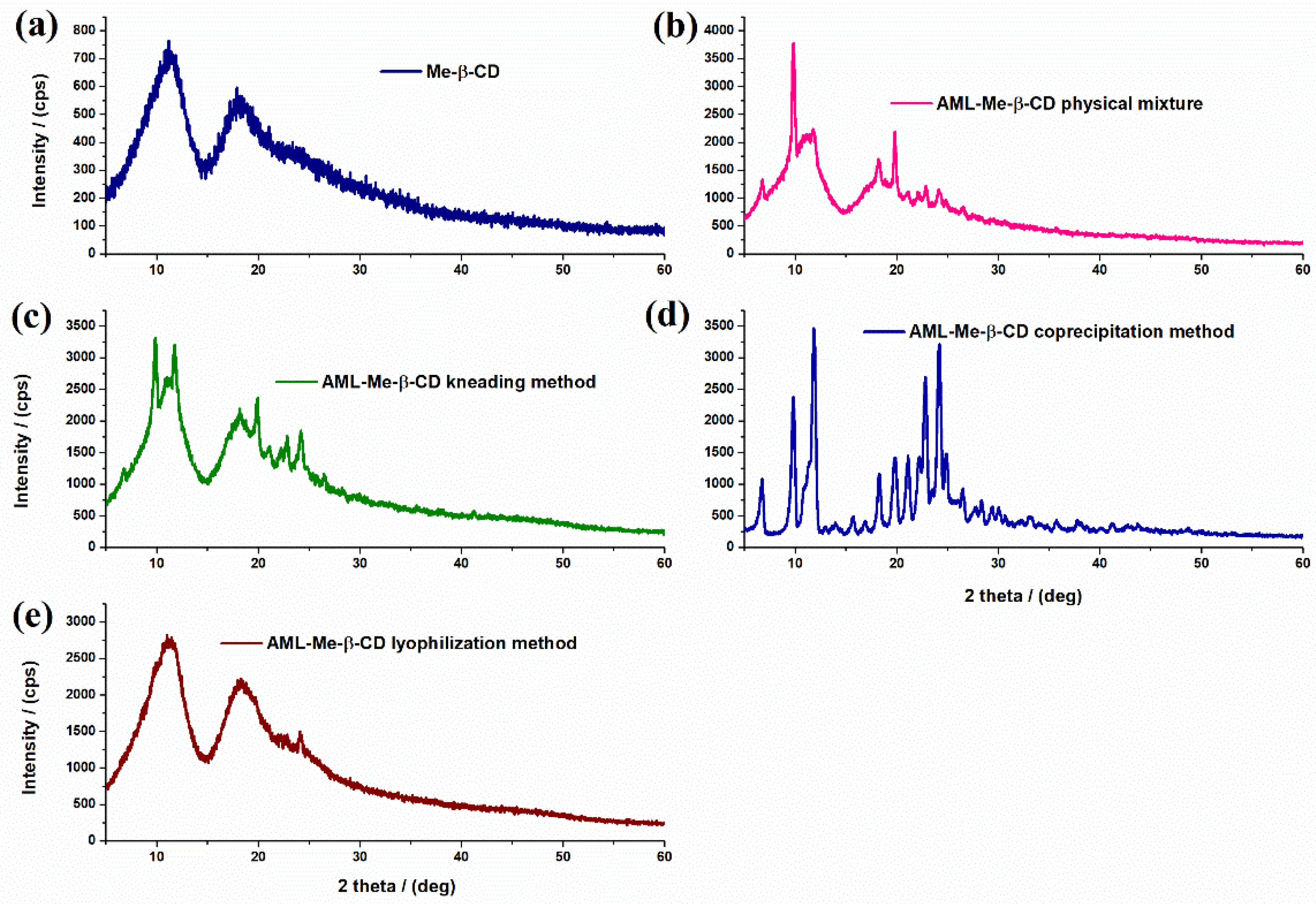
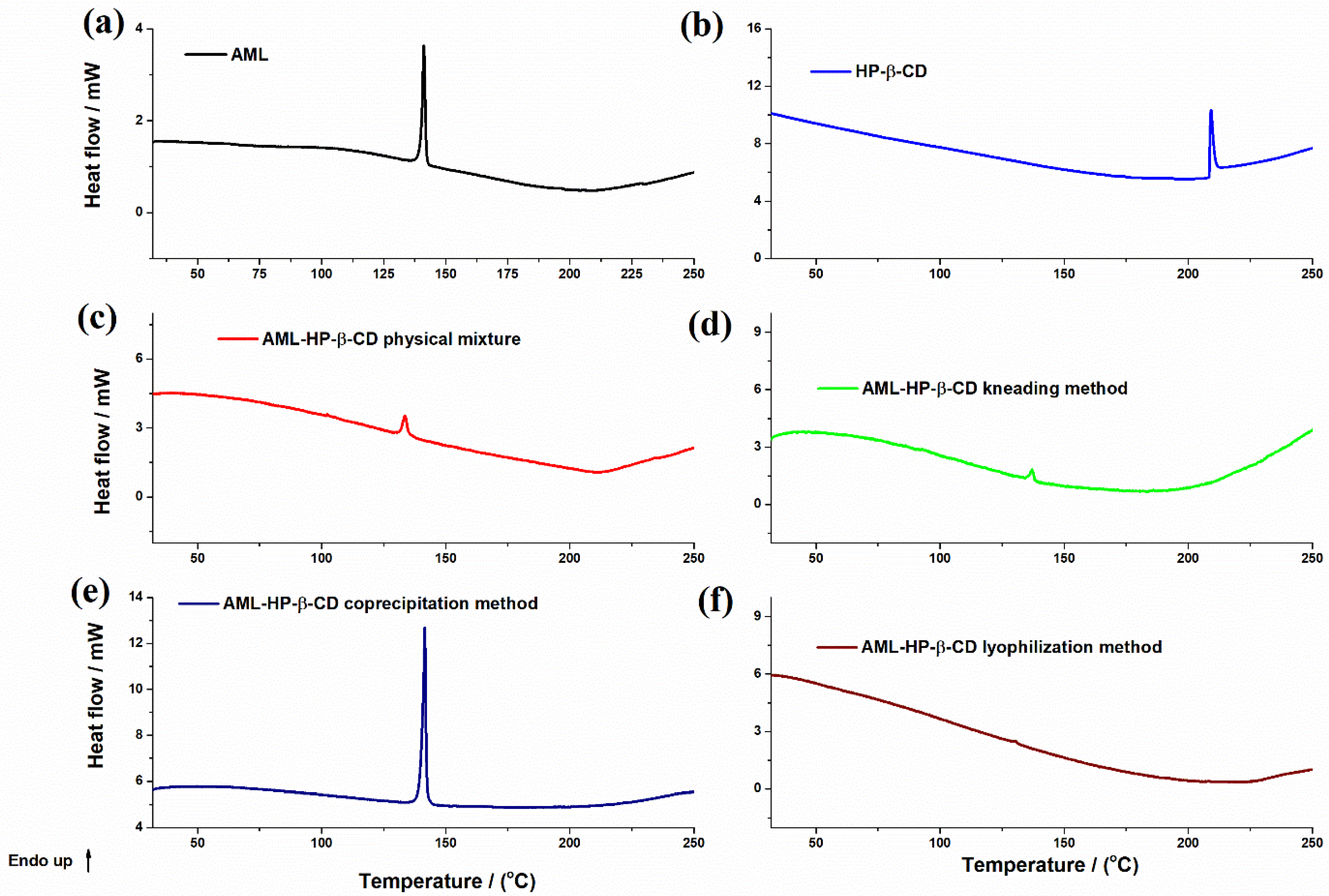
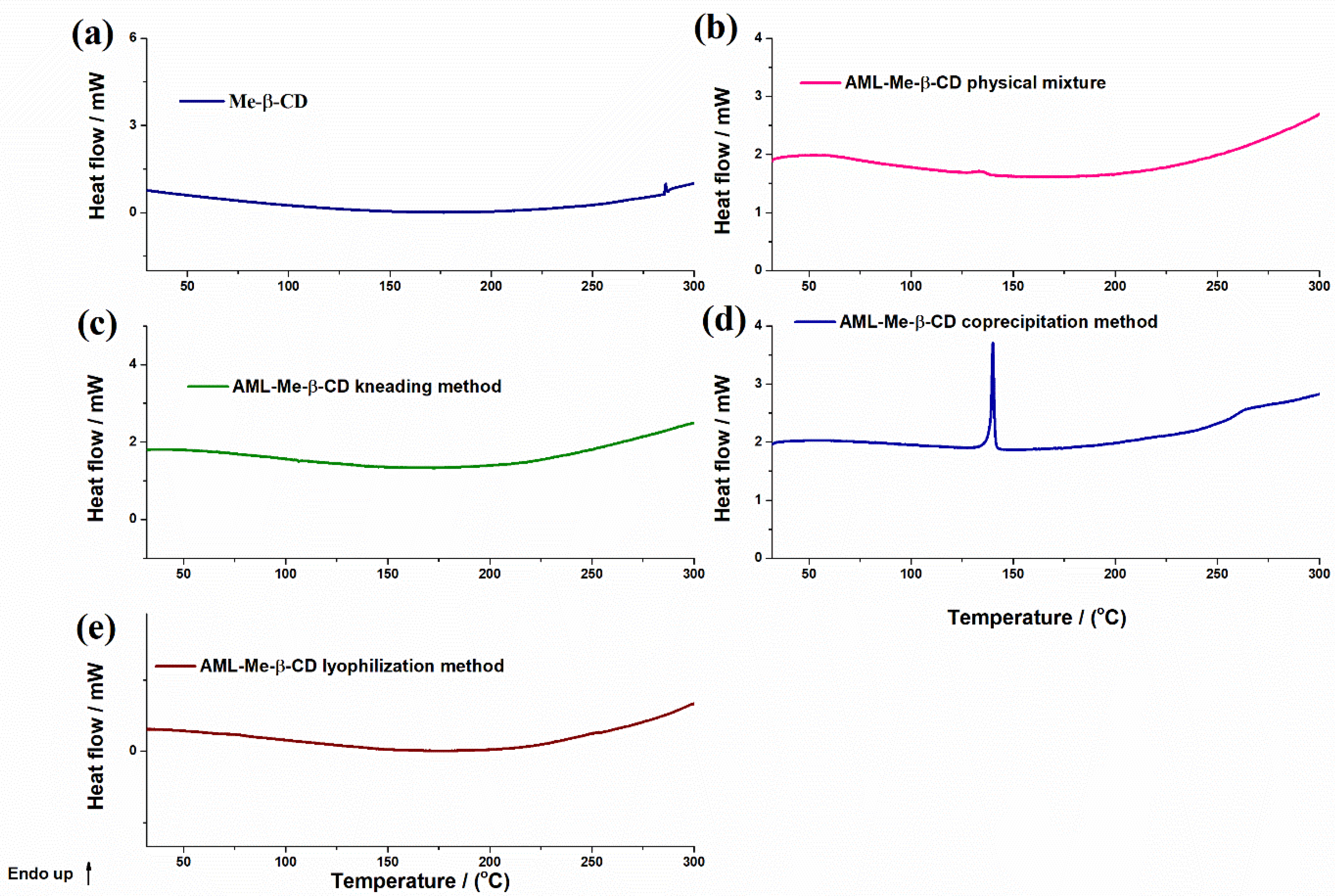
3.1. Binary Systems Characterization
3.2. Characterization of the Inclusion Complexes
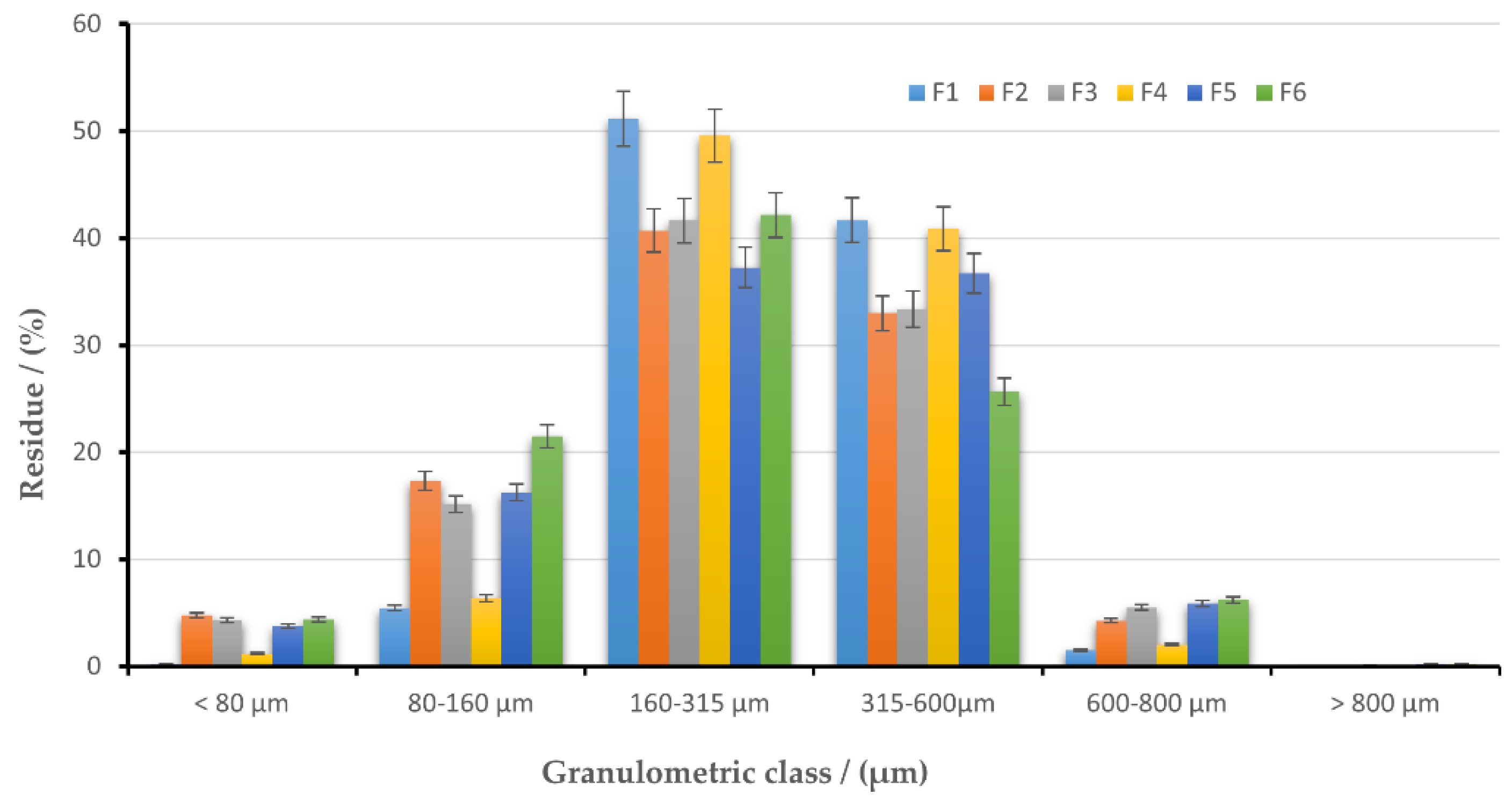
3.3. Preformulation and Formulation Studies for Orodispersible Tablets Containing AML-HP-β-CD and AML-Me-β-CD Inclusion Complexes
Particle Size Distribution
3.4. Quality Assessment of Orally Dispersible Tablets
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Sahoo, S.K.; Padhee, K.; Kochar, P.S.; Sathapathy, A.; Pathak, N. Review on solubility enhancement techniques for hydrophobic drugs. Pharm. Glob. 2011, 3, 1–7. [Google Scholar]
- Yuan, C.; Liu, B.; Liu, H. Characterization of hydroxypropyl-β-cyclodextrins with different substitution patterns via FTIR, GC-MS, and TG-DTA. Carbohydr. Polym. 2015, 118, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Szejtli, J. Introduction and general overview of cyclodextrin chemistry. Chem. Rev. 1998, 98, 1743–1754. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Han, J.; Song, T.; Sung, J.; Kwon, O.; Song, S.; Chung, Y.B. Physicochemical characterization and bioavailability of a novel intestinal metabolite of ginseng saponin complexes with β-cyclodextrin. Int. J. Pharm. 2007, 316, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Ying, W.; Yang, S.; Wu, L. Determination of the inclusion complex between gossypol and β-cyclodextrin. Spectrochim. Acta Part A 2006, 65, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Pfammatter, J.P.; Clericetti-Affolter, C.; Truttmann, A.C.; Busch, K.; Laux-End, R.; Bianchetti, M.G. Amlodipine once-daily in systemic hypertension. Eur. J. Pediatr. 1998, 157, 618–621. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.T.; Smoyer, W.E.; Bunchman, T.E. Treatment of hypertensive children with amlodipine. Am. J. Hypertens. 2000, 13, 1061–1066. [Google Scholar] [CrossRef]
- Somagoni, J.; Reddy, S.; Katakam, V.K.; Koorelli, S.; Manda, S.; Rao, M.Y. Preparation of Inclusion Complexes of Amlodipine Base and its Besylate and Maleate Salts with Hydroxy Propyl b -cyclodextrin—A study on Stereospecific Dissolution. Pharm. Anal. Acta 2011, 2, 1000123. [Google Scholar] [CrossRef] [Green Version]
- Meredith, P.A.; Elliott, H.L. Clinical pharmacokinetics of amlodipine. Clin. Pharmacokinet. 1992, 22, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Laufen, H.; Leitold, M. Enantioselective disposition of oral amlodipine in healthy volunteers. Chirality 1994, 6, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Lauro, M.; Carbone, C.; Auditore, R.; Musumeci, T.; Santagati, N.; Aquino, R.; Puglisi, G. A new inclusion complex of amlodipine besylate and soluble β-cyclodextrin polymer: Preparation, characterization and dissolution profile. J. Incl. Phenom. Macrocycl. Chem. 2013, 76, 19–28. [Google Scholar] [CrossRef]
- Rollinge, J.M.; Burger, A. Physico-chemical characterization of hydrated and anhydrous crystal forms of amlodipine besylate. J. Therm. Anal. Calorim. 2002, 68, 361–372. [Google Scholar]
- Clavijo, G.A.; de Clavijo, I.V.; Weart, C.W. Amlodipine: A new calcium antagonist. Am. J. Hosp. Pharm. 1994, 51, 59–68. [Google Scholar] [CrossRef]
- Dahima, R.; Pachori, A.; Netam, S. Formulation and evaluation of mouth dissolving tablet containing amlodipine besylate solid dispersion. Int. J. ChemTech Res. 2010, 2, 706–715. [Google Scholar]
- Jaya, S.; Amala, V. Formulation and In-vitroEvaluation of Oral Disintegrating Tablets of Amlodipine Besylate. Int. J. Adv. Pharm. 2019, 11, 49–54. [Google Scholar]
- Sabar, M.H. Formulation and in vitro evaluation of fast dissolving film containing amlodipine besylate solid dispersion. Int. J. Pharm. Pharm. Sci. 2013, 5, 419–428. [Google Scholar]
- Kamagoni, K.; Gunnala, P.G.; Somagoni, J. Preparation and enhancement of dissolution rate of amlodipine besylate and valsartan using liquisolid technique. Pharm. Glob. 2013, 4, 1–6. [Google Scholar]
- Tyagi, V.K.; Singh, D.; Pathak, K. Semisolid matrix-filled hard gelatin capsules for rapid dissolution of amlodipine besilate: Development and assessment. J. Adv. PharmTech Res. 2013, 4, 42–49. [Google Scholar]
- Ayorinde, J.O.; Odeniyi, M.A.; Balogun-Agbaje, O. Formulation and Evaluation of Oral Dissolving Films of amlodipine besylate using blends of starches with hydroxypropyl methyl cellulose. Polim. Med. 2016, 46, 45–51. [Google Scholar] [CrossRef]
- Jang, D.-J.; Sim, T.; Oh, E. Formulation and optimization of spray-dried amlodipine solid dispersion for enhanced oral absorption. Drug Dev. Ind. Pharm. 2013, 39, 1133–1141. [Google Scholar] [CrossRef]
- Pare, A.; Yadav, S.K.; Patil, U.K. Formulation and evaluation of effervescent floating tablet of amlodipine besylate. Res. J. Pharm. Technol. 2008, 1, 526–530. [Google Scholar]
- Kapor, A.; Nikolic, V.; Nikolic, L.; Stankovic, M.; Cakic, M.; Stanojevic, L.; Ilic, D. Inclusion complexes of amlodipine besylate and cyclodextrins. Cent. Eur. J. Chem. 2010, 8, 834–841. [Google Scholar] [CrossRef]
- Mielcarek, J.; Czernielewska, A.; Czarczyńska, B. Inclusion complexes of felodipine and amlodipine with methyl-b-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2006, 54, 17–21. [Google Scholar] [CrossRef]
- Ling, W.; Xuehua, J.; Weijuan, X.; Chenrui, L. Complexation of tanshinone IIA with 2-hydroxypropyl-b-cyclodextrin: Effect on aqueous solubility, dissolution rate, and intestinal absorption behavior in rats. Int. J. Pharm. 2007, 341, 58–67. [Google Scholar]
- Bhardwaj, V.; Bansal, M.; Sharma, P.K. Formulation and evaluation of fast dissolving tablets of amlodipine besylate using different super disintegrants and camphor as sublimating agent. Am. Eurasian J. Sci. Res. 2010, 5, 264–269. [Google Scholar]
- Raj, B.S.; Punitha, I.S.R.; Dube, S. Formulation and characterization of fast disintegrating tablets of amlodipine using superdisintegrants. J. Appl. Pharm. Sci. 2012, 2, 118–123. [Google Scholar]
- Sukhavasi, S.; Kishore, V.S. Formulation and evaluation of fast dissolving tablets of amlodipine besylate by using Fenugreek seed mucilage and Ocimum basilicum gum. Int. Curr. Pharm. J. 2012, 1, 243–249. [Google Scholar] [CrossRef]
- Malakzadeh, S.; Alizadeh, N. Spectroscopic study and antioxidant activity of the inclusion complexes of cyclodextrins and amlodipine besylate drug. J. Incl. Phenom. Macrocycl. Chem. 2018, 90, 89–98. [Google Scholar] [CrossRef]
- Gavriloaia, M.R.; Budura, E.A.; Toma, C.C.; Mitu, M.A.; Karampelas, O.; Arama, C.; Lupuleasa, D. In vitro evaluation of diffusion and rheological profiles for dexamethasone inclusion complexes with beta-cyclodextrin or hydroxypropyl beta-cyclodextrin. Farmacia 2012, 60, 895–904. [Google Scholar]
- Sbora, R.; Budura, E.A.; Nitulescu, G.M.; Balaci, T.; Lupuleasa, D. Preparation and characterization of inclusion complexes formed by avobenzone with beta-cyclodextrin, hydroxypropyl-beta-cyclodextrin and hydroxypropyl-alpha-cyclodextrin. Farmacia 2015, 63, 548–555. [Google Scholar]
- El-Kosasy, A.M.; Tawakkol, S.M.; Ayad, M.F.; Sheta, A.I. New methods for amlodipine and valsartan native spectrofluorimetric determination, with factors optimization study. Talanta 2015, 143, 402–413. [Google Scholar] [CrossRef]
- Musuc, A.M.; Anuta, V.; Atkinson, I.; Sarbu, I.; Popa, V.T.; Munteanu, C.; Mircioiu, C.; Ozon, E.A.; Nitulescu, G.M.; Mitu, M.A. Formulation of Chewable Tablets Containing Carbamazepine-β-cyclodextrin Inclusion Complex and F-Melt Disintegration Excipient. The Mathematical Modeling of the Release Kinetics of Carbamazepine. Pharmaceutics 2021, 13, 915. [Google Scholar] [CrossRef]
- Moreton, C. Functionality and performance of excipients in a quality-bydesign world, part VIII: Excipient specifications. Am. Pharm. Rev. 2010, 13, 46–50. [Google Scholar]
- Council of Europe. European Pharmacopoeia, 10th ed.; EDQM, Council of Europe: Strasbourg, France, 2019. [Google Scholar]
- United States Pharmacopeia and the National Formulary (USP 35–NF 30); The United States Pharmacopeial Convention: Rockville, MD, USA, 2012.
- Musuc, A.M.; Anuta, V.; Atkinson, I.; Popa, V.T.; Sarbu, I.; Mircioiu, C.; Abdalrb, G.A.; Mitu, M.A.; Ozon, E.A. Development and Characterization of Orally Disintegrating Tablets Containing a Captopril-Cyclodextrin Complex. Pharmaceutics 2020, 12, 744. [Google Scholar] [CrossRef]
- Szabo, L.; Chiş, V.; Pîrnău, A.; Leopold, N.; Cozar, O.; Orosz, S. Spectroscopic and theoretical study of amlodipine besylate. J. Mol. Struct. 2009, 924–926, 385–392. [Google Scholar] [CrossRef]
- Novac, M.; Musuc, A.M.; Ozon, E.A.; Sarbu, I.; Mitu, M.A.; Rusu, A.; Gheorghe, D.; Petrescu, S.; Atkinson, I.; Lupuliasa, D. Manufacturing and Assessing the New Orally Disintegrating Tablets, Containing Nimodipine-hydroxypropyl-β-cyclodextrin and Nimodipine-methyl-β-cyclodextrin Inclusion Complexes. Molecules 2022, 27, 2012. [Google Scholar] [CrossRef]
- Chelu, M.; Calderon-Moreno, J.; Atkinson, I.; Pandele Cusu, J.; Rusu, A.; Bratan, V.; Aricov, L.; Anastasescu, M.; Seciu-grama, A.-M.; Musuc, A.M. Green synthesis of bioinspired chitosan-ZnO-based polysaccharide gums hydrogels with propolis extract as novel functional natural biomaterials. Int. J. Biol. Macromol. 2022, 211, 410–424. [Google Scholar] [CrossRef]
- Hu, Q.; Fu, X.; Su, Y.; Wang, Y.; Xiaoqin, G.; Wang, S.; Xu, Y.; Yu, C. Enhanced oral bioavailability of koumine by complexation with hydroxypropyl-β-cyclodextrin: Preparation, optimization, ex vivo and in vivo characterization. Drug Deliv. 2021, 28, 2415–2426. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, D.; Lai, J.; Lu, Y.; Yin, Z.; Wu, W. Piroxicam/2-hydroxypropyl-beta-cyclodextrin inclusion complex prepared by a new fluid-bed coating technique. J. Pharm. Sci. 2009, 98, 665–675. [Google Scholar] [CrossRef]
- Mrozek, M.F.; Weaver, M.J. Detection and identification of aqueous saccharides by using surface-enhanced Raman spectroscopy. Anal. Chem. 2002, 74, 4069–4075. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.; Figueiras, A.; Santos, D.; Veiga, F. Preparation and Solid-State Characterization of Inclusion Complexes Formed Between Miconazole and Methyl-β-Cyclodextrin. AAPS PharmSciTech 2008, 9, 1102–1109. [Google Scholar] [CrossRef] [Green Version]
- Naidu, N.B.; Chowdary, K.P.R.; Murthy, K.V.R.; Satyanarayana, V.; Hayman, A.R.; Becket, G. Physicochemical characterization and dissolution properties of meloxicam cyclodextrin binary systems. J. Pharmaceut. Biomed. 2004, 35, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.D.; Ford, L.; Lim, S.; Han, S.; Baumann, J.; Sullivan, H.; Vodak, D.; Igonin, A.; Benameur, H.; Pouton, C.W.; et al. Transformation of Biopharmaceutical Classification System Class I and III Drugs Into Ionic Liquids and Lipophilic Salts for Enhanced Developability Using Lipid Formulations. J. Pharm. Sci. 2018, 107, 203–216. [Google Scholar] [CrossRef] [Green Version]
- Ananchenko, G.; Novakovic, J.; Lewis, J. Amlodipine besylate. Profiles Drug Subst. Excip. Relat. Methodol. 2012, 37, 31–77. [Google Scholar] [CrossRef] [PubMed]
- Marini, A.; Berbenni, V.; Bruni, G.; Mustarelli, P.; Giordano, F.; Villa, M. Thermoanalytical and spectroscopic characterization of betacyclodextrin/ketoprofen inclusion complexes. J. Incl. Phenom. Mol. Recognit. Chem. 1995, 22, 221–234. [Google Scholar] [CrossRef]
- Huang, J.; Kaul, G.; Cai, C.; Chatlapalli, R.; Hernandez-Abad, P.; Ghosh, K.; Nagi, A. Quality by design case study: An integrated multivariate approach to drug product and process development. Int. J. Pharm. 2009, 382, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-S. Study of Flavonoid/Hydroxypropyl-β-Cyclodextrin Inclusion Complexes by UV-Vis, FT-IR, DSC, and X-Ray Diffraction Analysis. Prev. Nutr. Food Sci. 2020, 25, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Figueiras, A.; Ribeiro, L.; Vieira, M.T.; Veiga, F. Preparation and physicochemical characterization of omeprazole:methyl-beta-cyclodextrin inclusion complex in solid state. J. Incl. Phenom. Macrocycl. Chem. 2007, 57, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Shekunov, B.Y.; Chattopadhyay, P.; Tong, H.H.Y.; Chow, A.H.L. Particle Size analysis in Pharmaceutics: Principles, Methods and Applications. Pharm. Res. 2006, 24, 203–227. [Google Scholar] [CrossRef]
- Heinicke, G.; Matthews, F.; Schwartz, J.B. The Effects of Substrate Size, Surface Area, and Density on Coat Thickness of Multi-Particulate Dosage Forms. Pharm. Dev. Technol. 2005, 10, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Balaci, T.; Velescu, B.; Karampelas, O.; Musuc, A.M.; Nitulescu, G.M.; Ozon, E.A.; Nitulescu, G.; Gird, C.E.; Fita, C.; Lupuliasa, D. Physico-Chemical and Pharmaco-Technical Characterization of Inclusion Complexes Formed by Rutoside with beta-Cyclodextrin and Hydroxypropyl-beta-Cyclodextrin Used to Develop Solid Dosage Forms. Processes 2021, 9, 26. [Google Scholar] [CrossRef]
- Swaminathan, V.; Kildsig, D.O. Polydisperse powder mixtures: Effect of particle size and shape on mixture stability. Drug. Dev. Ind. Pharm. 2002, 28, 41–48. [Google Scholar] [CrossRef]
- Mitu, M.A.; Cretu, E.A.; Novac, M.; Karampelas, O.; Nicoara, A.; Nitulescu, G.; Lupuleasa, D. The Flowing Characteristics of Some Composed Powders Containing Inclusion Complexes in Beta-Cyclodextrin. In Proceedings of the 17th Romanian National Congress of Pharmacy, 17th Edition: 21st Century Pharmacy—Between Intelligent Specialization and Social Responsibility 2018, Bucharest, Romania, 26–29 September 2018; Draganescu, D., Arsene, A., Eds.; InFOROmatica S.r.l.: Bologna, Italy, 2018; pp. 129–133. [Google Scholar]
- Mititelu, M.; Moroșan, E.; Nicoară, A.C.; Secăreanu, A.A.; Musuc, A.M.; Atkinson, I.; Pandele Cusu, J.; Nițulescu, G.M.; Ozon, E.A.; Sarbu, I.; et al. Development of Immediate Release Tablets Containing Calcium Lactate Synthetized from Black Sea Mussel Shells. Mar. Drugs 2022, 20, 45. [Google Scholar] [CrossRef]
- Pop, A.L.; Crișan, S.; Bârcă, M.; Ciobanu, A.-M.; Varlas, V.N.; Pop, C.; Pali, M.-A.; Cauni, D.; Ozon, E.A.; Udeanu, D.; et al. Evaluation of Dissolution Profiles of a Newly Developed Solid Oral Immediate-Release Formula Containing Alpha-Lipoic Acid. Processes 2021, 9, 176. [Google Scholar] [CrossRef]
- Mansour, H.M.; Sohn, M.; Al-Ghananeem, A.; DeLuca, P.P. Materials for Pharmaceutical Dosage Forms: Molecular Pharmaceutics and Controlled Release Drug Delivery Aspects. Int. J. Mol. Sci. 2010, 11, 3298–3322. [Google Scholar] [CrossRef] [Green Version]
- Kaerger, J.; Edge, S.; Price, R. Influence of particle size and shape on flowability and compactibility of binary mixtures of paracetamol and microcrystalline cellulose. Eur. J. Pharm. Sci. 2004, 2–3, 173–179. [Google Scholar] [CrossRef]
- Fu, X.; Huck, D.; Makein, L.; Armstrong, B.; Willen, U.; Freeman, T. Effect of particle shape and size on flow properties of lactose powders. Particuology 2012, 10, 203–208. [Google Scholar] [CrossRef]
- Tunuguntla, D.R.; Weinhart, T.; Thornton, A.R. Comparing and contrasting size-based particle segregation models. Comput. Part. Mech. 2017, 4, 387–405. [Google Scholar] [CrossRef] [Green Version]
- Bahar Basim, G.; Khalili, M. Particle size analysis on wide size distribution powders; effect of sampling and characterization technique. Adv. Powder Technol. 2015, 26, 200–207. [Google Scholar] [CrossRef]
- Cartwright, J.J.; Robertson, J.; D’Haene, D.; Burke, M.D.; Hennenkamp, J.R. Twin screw wet granulation: Loss in weight feeding of a poorly flowing active pharmaceutical ingredient. Powder Technol. 2013, 238, 116–121. [Google Scholar] [CrossRef]
- Velikov, V.; Borick, S.; Angell, C.A. The Glass Transition of Water, Based on Hyperquenching Experiments. Science 2001, 294, 2335–2338. [Google Scholar] [CrossRef]
- Charmathy, S.P.; Pinal, R. Moisture-Induced Antiplasticization In Microcrystalline Cellulose Compacts. Tablets Capsul. 2007, 5, 22–33. [Google Scholar]
- Wang, Y.; Li, T.; Muzzio, F.J.; Glasser, B.J. Predicting feeder performance based on material flow properties. Powder Technol. 2017, 308, 135–148. [Google Scholar] [CrossRef] [Green Version]
- Manescu, O.; Lupuleasa, D.; Miron, D.S.; Budura, E.A.; Radulescu, F.S. In vitro drug release from topical antifungal pharmaceutical formulations. Farmacia 2011, 59, 15–23. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients | Formulation/Amount (%) | |||||
|---|---|---|---|---|---|---|
| F1 | F2 | F3 | F4 | F5 | F6 | |
| AML-HP-β-CD | 5.39 | 5.39 | 5.39 | - | - | - |
| AML-Me-β-CD | - | - | - | 6.00 | 6.00 | 6.00 |
| PROSOLV® SMCC HD 90-Silicified microcrystalline cellulose | 91.75 | - | - | 91.14 | - | - |
| F-MELT® | - | 91.75 | - | - | 91.14 | - |
| Ludiflash® | - | - | 91.75 | - | - | 91.14 |
| EXPLOTAB®-Sodium starch glycolate | 1.86 | 1.86 | 1.86 | 1.86 | 1.86 | 1.86 |
| LIGAMED® MF-2-V-Magnesium stearate | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| Ingredients | Quantity mg/Tablet | Role in Formulation | |||
|---|---|---|---|---|---|
| F7 (F1-30 kN) | F8 (F1-60 kN) | F9 (F4-30 kN) | F10 (F4-60 kN) | ||
| Inclusion complex AML-HP-β-CD (1:1) | 18.87 | 18.87 | - | - | Active ingredient |
| Inclusion complex AML-Me-β-CD (1:1) | - | - | 21.02 | 21.02 | Active ingredient |
| PROSOLV® SMCC HD 90-Silicified microcrystalline cellulose | 321.13 | 321.13 | 318.98 | 318.98 | Filler Binder |
| EXPLOTAB®-Sodium starch glycolate | 6.50 | 6.50 | 6.50 | 6.50 | Superdisintegrant |
| LIGAMED® MF-2-V-Magnesium stearate | 3.50 | 3.50 | 3.50 | 3.50 | Lubricant |
| TOTAL | 350.00 | 350.00 | 350.00 | 350.00 | |
| Sample | Process | Tonset/(°C) | Tmax/(°C) | Tend/(°C) | ΔH/(Jg−1) |
|---|---|---|---|---|---|
| AML | Dehydration | 82.1 | 105.9 | 109.7 | 21.7 |
| Melting | 140.2 | 141.2 | 142.3 | 80.3 | |
| HP-β-CD | Melting | 208.6 | 209.2 | 210.9 | 43.2 |
| AML-HP-β-CD physical mixture | Dehydration | 101.9 | 102.2 | 104.1 | 0.9 |
| Melting | 131.7 | 133.6 | 135.1 | 13.3 | |
| Decomposition | 275.4 | 285.2 | 287.1 | 11.8 | |
| AML-HP-β-CD kneading | Dehydration | 116.2 | 118.3 | 120.0 | 7.7 |
| Melting | 136.5 | 137.1 | 138.2 | 6.2 | |
| Decomposition | 291.2 | 291.9 | 292.9 | 39.0 | |
| AML-HP-β-CD coprecipitation | Melting | 140.5 | 141.6 | 142.4 | 78.8 |
| AML-HP-β-CD lyophilization | Melting | 126.3 | 130.5 | 131.3 | 1.3 |
| Decomposition | 265.4 | 266.5 | 268.5 | 9.5 | |
| Me-β-CD | Melting | 209.9 | 210.3 | 211.1 | 0.19 |
| Decomposition | 285.5 | 286.0 | 286.8 | 13.9 | |
| 287.3 | 289.1 | 295.0 | 21.8 | ||
| AML-Me-β-CD physical mixture | Melting | 129.1 | 133.6 | 139.0 | 6.8 |
| AML-Me-β-CD kneading | Dehydration | 66.0 | 66.4 | 66.8 | 0.5 |
| AML-Me-β-CD coprecipitation | Melting | 139.0 | 140.1 | 141.2 | 72.7 |
| Decomposition | 235.6 | 263.9 | 293.6 | 41.7 | |
| AML-Me-β-CD lyophilization | Dehydration | 72.9 | 76.0 | 80.7 | 3.3 |
| Decomposition | 227.9 | 250.4 | 250.4 | 1.1 |
| Parameter | Formulation Code | |||||
|---|---|---|---|---|---|---|
| F1 | F2 | F3 | F4 | F5 | F6 | |
| Moisture content (%) | 3.52 ± 1.31 | 6.29 ± 1.47 | 7.11 ± 2.33 | 4.03 ± 1.57 | 6.83 ± 2.12 | 7.35 ± 1.78 |
| Flow time (s) * | 9.3 ± 1.06 | 12.8 ± 1.17 | 13.1 ± 1.69 | 10.2 ± 1.31 | 16.5 ± 1.95 | 17.2 ± 2.36 |
| Angle of repose (θ degrees) * | 26.42 ± 1.26 | 31.19 ± 2.10 | 30.89 ± 2.85 | 27.37 ± 1.48 | 35.12 ± 2.61 | 36.44 ± 2.94 |
| Flow rate (g/s) * | 6.451 | 4.687 | 4.580 | 5.882 | 3.636 | 3.488 |
| Bulk density (g/mL) | 0.512 | 0.577 | 0.525 | 0.534 | 0.563 | 0.582 |
| Tapped density (g/mL) | 0.585 | 0.682 | 0.679 | 0.621 | 0.681 | 0.723 |
| Carr Index (CI) (%) | 12.48 | 15.4 | 22.68 | 14.01 | 17.33 | 19.50 |
| Hausner’s ratio (HR) | 1.14 | 1.18 | 1.29 | 1.12 | 1.20 | 1.24 |
| Characteristics | Formulation Code | |||
|---|---|---|---|---|
| F7 | F8 | F9 | F10 | |
| Thickness (mm) | 4.02 ± 0.44 | 4.00 ± 1.12 | 4.08 ± 0.78 | 4.05 ± 0.95 |
| Diameter (mm) | 10 ± 0.31 | 10 ± 0.63 | 10 ± 0.11 | 10 ± 0.59 |
| Mass uniformity | 349 ± 1.95 | 347 ± 2.46 | 350 ± 1.78 | 348 ± 3.65 |
| Hardness (N) | 44 ± 3.71 | 57.8 ± 3.55 | 42.2 ± 3.14 | 61.2 ± 4.29 |
| Friability (%) | 0.03 ± 0.09 | 0.05 ± 0.12 | 0.12 ± 0.25 | 0.34 ± 0.33 |
| In vitro disintegration time–in water medium (seconds) | 14 ± 1.24 | 21 ± 2.71 | 13 ± 1.15 | 19 ± 1.97 |
| In vitro disintegration time–in simulated saliva medium (seconds) | 12 ± 3.55 | 16 ± 2.81 | 11 ± 3.22 | 18 ± 4.55 |
| In vitro dissolution rate–in acidic medium, after 30 min (%) | 98.76 ± 1.45 | 96.31 ± 2.08 | 98.11 ± 1.35 | 97.13 ± 1.92 |
| In vitro dissolution rate–in simulated saliva medium, after 30 min (%) | 90.18 ± 1.26 | 88.12 ± 1.84 | 89.84 ± 1.12 | 85.15 ± 2.23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novac, M.; Musuc, A.M.; Ozon, E.A.; Sarbu, I.; Mitu, M.A.; Rusu, A.; Petrescu, S.; Atkinson, I.; Gheorghe, D.; Lupuliasa, D. Design and Evaluation of Orally Dispersible Tablets Containing Amlodipine Inclusion Complexes in Hydroxypropyl-β-cyclodextrin and Methyl-β-cyclodextrin. Materials 2022, 15, 5217. https://doi.org/10.3390/ma15155217
Novac M, Musuc AM, Ozon EA, Sarbu I, Mitu MA, Rusu A, Petrescu S, Atkinson I, Gheorghe D, Lupuliasa D. Design and Evaluation of Orally Dispersible Tablets Containing Amlodipine Inclusion Complexes in Hydroxypropyl-β-cyclodextrin and Methyl-β-cyclodextrin. Materials. 2022; 15(15):5217. https://doi.org/10.3390/ma15155217
Chicago/Turabian StyleNovac, Marian, Adina Magdalena Musuc, Emma Adriana Ozon, Iulian Sarbu, Mirela Adriana Mitu, Adriana Rusu, Simona Petrescu, Irina Atkinson, Daniela Gheorghe, and Dumitru Lupuliasa. 2022. "Design and Evaluation of Orally Dispersible Tablets Containing Amlodipine Inclusion Complexes in Hydroxypropyl-β-cyclodextrin and Methyl-β-cyclodextrin" Materials 15, no. 15: 5217. https://doi.org/10.3390/ma15155217
APA StyleNovac, M., Musuc, A. M., Ozon, E. A., Sarbu, I., Mitu, M. A., Rusu, A., Petrescu, S., Atkinson, I., Gheorghe, D., & Lupuliasa, D. (2022). Design and Evaluation of Orally Dispersible Tablets Containing Amlodipine Inclusion Complexes in Hydroxypropyl-β-cyclodextrin and Methyl-β-cyclodextrin. Materials, 15(15), 5217. https://doi.org/10.3390/ma15155217