
Interatomic Potential for InP


Abstract
:1. Introduction
2. Materials and Methods
2.1. First-Principles Calculations
2.2. Molecular Dynamics Simulations
2.3. The Atomic Bond Order Potential
2.4. Fitting Procedure
3. Results and Discussion
3.1. Parametrization of the BO Potential
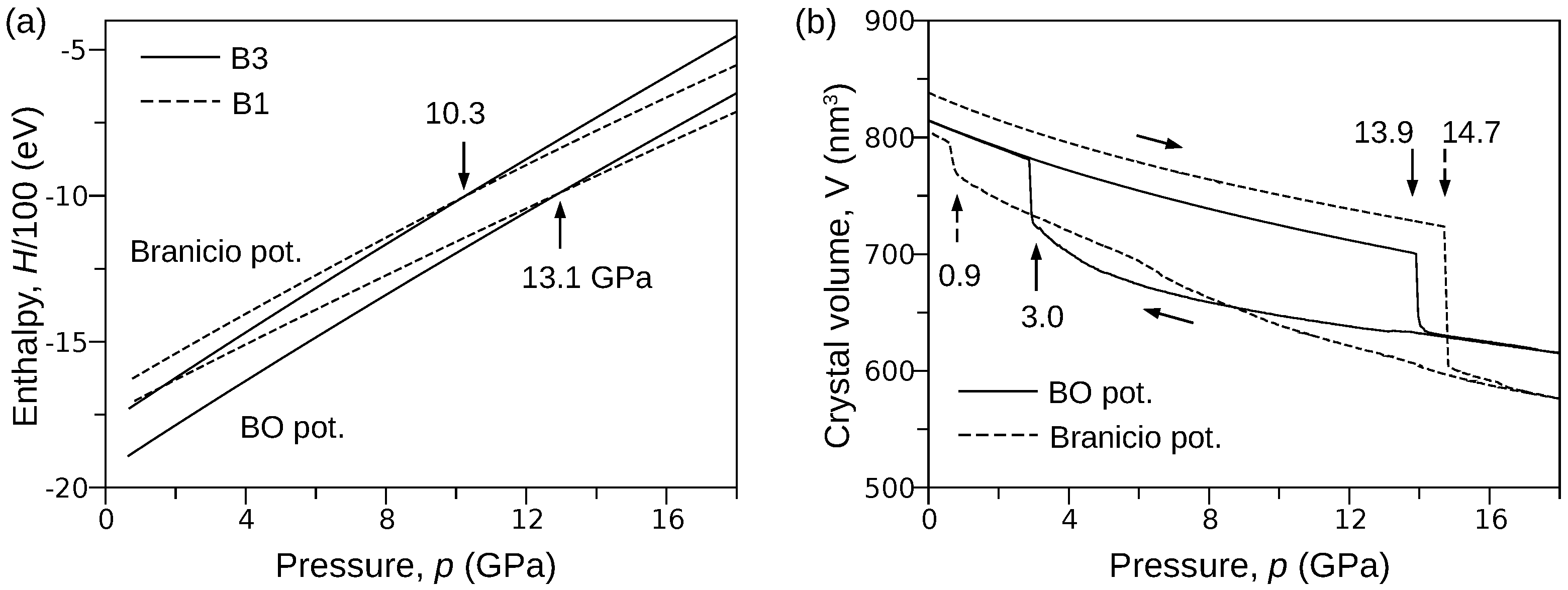
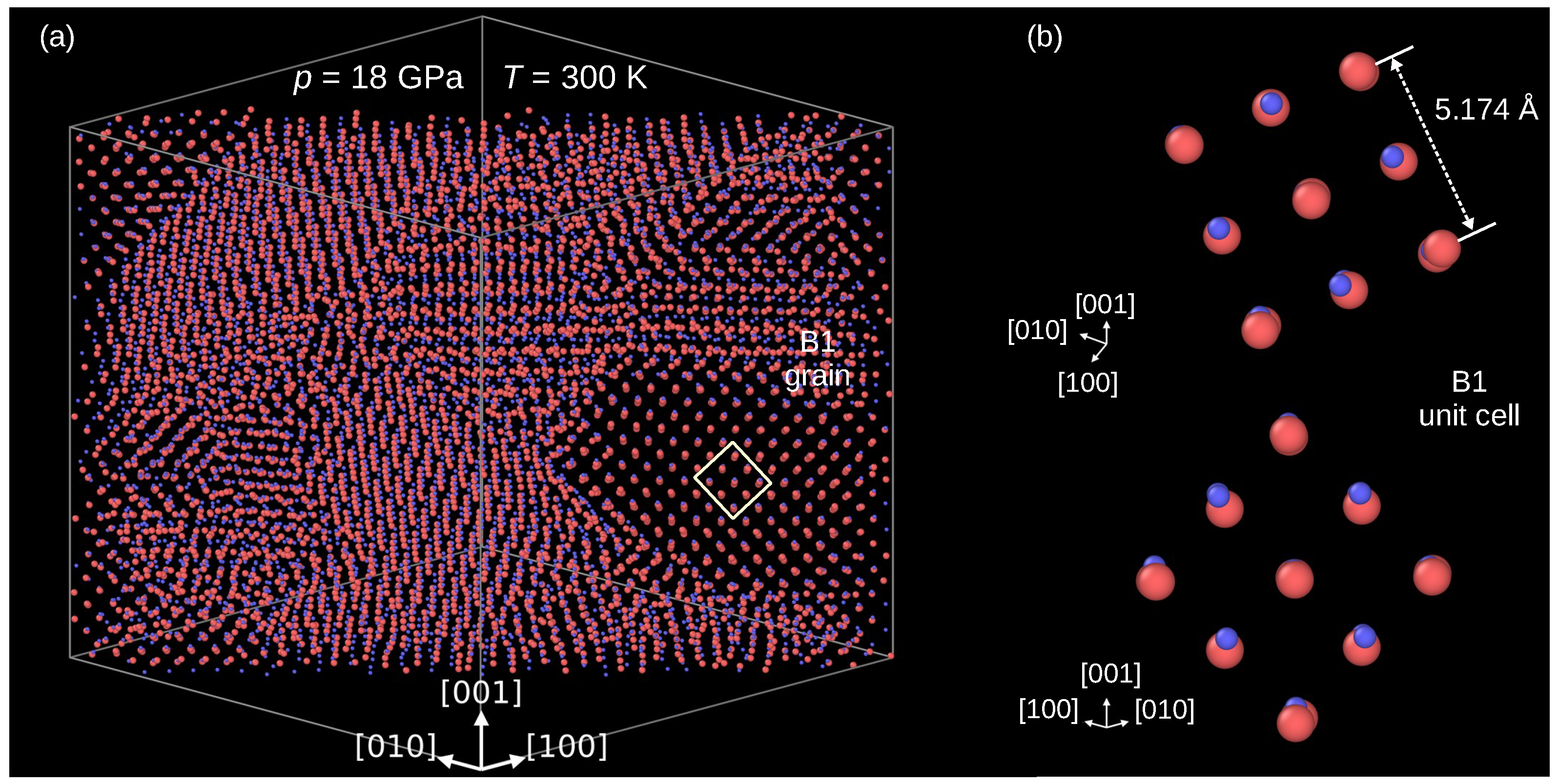
3.2. B3→B1 Phase Transformation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kobayashi, K.W.; Cowles, J.; Tran, L.T.; Gutierrez-Aitken, A.; Block, T.R.; Oki, A.K.; Streit, D.C. 50-MHz-55-GHz multidecade InP-based HBT distributed amplifier. IEEE Microw. Guid. Wave Lett. 1997, 7, 353–355. [Google Scholar] [CrossRef]
- Mukherjee, C.; Deng, M.; Nodjiadjim, V.; Riet, M.; Mismer, C.; Guendouz, D.; Caillaud, C.; Bertin, H.; Vaissiere, N.; Luisier, M.; et al. Towards monolithic indiumm phosphide (InP)-Based electronic photonic technologies for beyond 5G communication systems. Appl. Sci. 2021, 11, 2393. [Google Scholar] [CrossRef]
- Yap, D.; Elliott, K.R.; Brown, Y.K.; Kost, A.R.; Ponti, E.S. High-speed integrated optoelectronic modulation circuit. IEEE Photonics Technol. Lett. 2001, 13, 626–628. [Google Scholar] [CrossRef]
- Green, M.A.; Hishikawa, Y.; Warta, W.; Dunlop, E.D.; Levi, D.H.; Hohl-Ebinger, J.; Ho-Baillie, A.W. Solar cell efficiency tables (version 50). Prog. Photovoltaics 2017, 25, 668–676. [Google Scholar] [CrossRef] [Green Version]
- May, M.M.; Lewerenz, H.J.; Lackner, H.D.; Dimroth, F.; Hannappel, T. Efficient direct solar-to-hydrogen conversion by in situ interface transformation of a tandem structure. Nat. Commun. 2015, 6, 1–7. [Google Scholar] [CrossRef]
- Ramasamy, P.; Kim, N.; Kang, Y.S.; Ramirez, O.; Lee, J.S. Tunable, bright, and narrow-band luminescence from colloidal indium phosphide quantumdots. Chem. Mater. 2017, 29, 6893–6899. [Google Scholar] [CrossRef]
- Ramasamy, P.; Ko, K.J.; Kang, J.W.; Lee, J.S. Two-step “seed-mediated” synthetic approach to colloidal indium phosphide quantum dots withhigh-purity photo- and electroluminescence. Chem. Mater. 2018, 30, 3643–3647. [Google Scholar] [CrossRef]
- Li, C.; Liu, Z.; Chen, J.; Gao, Y.; Li, M.; Zhang, Q. Semiconductor nanowire plasmonic lasers. Nanophotonics 2019, 8, 2091–2110. [Google Scholar] [CrossRef]
- Luo, M.C.; Ren, F.F.; Gagrani, N.; Qiu, K.; Wang, Q.; Yu, L.; Ye, J.; Yan, F.; Zhang, R.; Tan, H.H.; et al. Polarization-independent indium phosphide nanowire photodetectors. Adv. Opt. Mater. 2020, 8, 2000514. [Google Scholar] [CrossRef]
- Chrobak, D.; Chrobak, A.; Nowak, R. Effect of doping on nanoindentation inducedincipient plasticity in InP crystal. AIP Adv. 2019, 9, 125323. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Restrepo, O.D.; Kumar, A.; Windl, W. Native point defects in binary InP semiconductors. J. Mater. Sci. 2012, 47, 7482–7497. [Google Scholar] [CrossRef]
- Malozovsky, Y.; Franklin, L.; Ekuma, E.C.; Zhao, L.; Bagayoko, D. Ab-initio calculations of electronic properties of InP and GaP. Int. J. Mod. Phys. B 2013, 27, 1362013. [Google Scholar] [CrossRef]
- Baida, A.; Ghezali, M. Structural, electronic and optical properties of InP under presure: An ab-initio study. Comput. Condens. Matter 2018, 17, 1–14. [Google Scholar] [CrossRef]
- Kabita, K.; Maibam, J.; Sharma, B.I.; Thapa, R.K.; Brojen Singh, R.K. First principle study on pressure-induced electronic structure and elastic properties of indium phosphide (InP). Indian J. Phys. 2015, 89, 1–7. [Google Scholar] [CrossRef]
- Powell, D.; Migliorato, M.A.; Cullis, A.G. Optimized Tersoff potential parameters for tetrahedrally bonded III-V semiconductors. Phys. Rev. B 2007, 75, 115202. [Google Scholar] [CrossRef]
- Han, P.; Bester, G. Interatomic potentials for the vibrational properties of III-V semiconductor nanostructures. Phys. Rev. B 2011, 83, 174304. [Google Scholar] [CrossRef] [Green Version]
- Stillinger, F.H.; Weber, T.A. Computer-simulation of local order in condensed phases of silicon. Phys. Rev. B 1985, 31, 5262–5271. [Google Scholar] [CrossRef] [Green Version]
- Branicio, P.S.; Rino, J.P.; Shimojo, F. High-pressure phases of InP: An ab initio and molecular-dynamics study. Appl. Phys. Lett. 2006, 88, 161919. [Google Scholar] [CrossRef]
- Branicio, P.S.; Rino, J.P.; Gan, C.K.; Tsuzuki, H. Interaction potential for indium phosphide: A molecular dynamics and first-principles study of the elastic constants, generalized stacking fault and surface energies. J. Phys. Condens. Matter 2009, 21, 1–9. [Google Scholar] [CrossRef]
- Nayir, V.; Tasci, E.S.; Erkoç, Ş. Structural and Thermal Properties of Indium Phosphide Nanoparticles: Molecular Dynamics Simulations. J. Comput. Theor. Nanosci. 2015, 12, 2134–2139. [Google Scholar] [CrossRef]
- Gordiz, K.; Henry, A. Interface conductance modal analysis of lattice matched InGaAs/InP. Applied Physics Letters 2016, 108, 181606. [Google Scholar] [CrossRef]
- Pial, T.H.; Rakib, T.; Mojumder, S.; Motalab, M.; Salam Akanda, M.A. Atomistic investigations on the mechanical properties and fracture mechanisms of indium phosphide nanowires. Phys. Chem. Chem. Phys. 2018, 20, 8647–8657. [Google Scholar] [CrossRef] [PubMed]
- Tersoff, J. Modeling solid-state chemistry: Interatomic potentials for multicomponent systems. Phys. Rev. B 1989, 39, 5566–5568. [Google Scholar] [CrossRef] [PubMed]
- Albe, K.; Nordlund, K.; Averback, R.S. Modeling the metal-semiconductor interaction: Analytical bond-order potential for platinum-carbon. Phys. Rev. B 2002, 65, 195124. [Google Scholar] [CrossRef] [Green Version]
- Albe, K.; Nordlund, K.; Nord, J.; Kuronen, A. Modeling of compound semiconductors: Analytical bond-order potential for Ga, As, and GaAs. Phys. Rev. B 2002, 66, 035205. [Google Scholar] [CrossRef] [Green Version]
- Erhart, P.; Albe, K. Analytical potential for atomistic simulations of silicon, carbon, and silicon carbide. Phys. Rev. B 2005, 71, 035211. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1996, 13, 5188. [Google Scholar] [CrossRef]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in ’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comp. Phys. Comm. 2022, 271, 10817. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO – the Open Visualization Tool. Modelling Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Wang, S.Q.; Ye, H.Q. First-principles study on elastic properties and phase stability of III–V compounds. Phys. Stat. Sol. B 2003, 240, 45–54. [Google Scholar] [CrossRef]
- Stukowski, A.; Fransson, E.; Mock, M.; Erhart, P. Atomicrex - a general purpose tool for the construction of atomic interaction models. Modelling Simul. Mater. Sci. Eng. 2017, 25, 055003. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, M.; Da Silva, J.L.F.; Stampfl, C.; Neugebauer, J.; Scheffler, M. Cohesive properties of group-III nitrides: A comparative study of all-electron and pseudopotential calculations using the generalized gradient approximation. Phys. Rev. B 2002, 65, 245212. [Google Scholar] [CrossRef] [Green Version]
- Råsander, M.; Moram, M.A. On the accuracy of commonly used density functional approximations in determining the elastic constants of insulators and semiconductors. J. Chem. Phys. 2015, 143, 144104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madelung, O. Semiconductors: Data Handbook, 3rd ed.; Springer: Berlin/Heidelberg, Germany, 2003; pp. 139–147. [Google Scholar]
- Pearson, R.G. Cohesive energies of simple ionic and covalent solids. J. Mol. Struct. Theochem. 1992, 260, 11–26. [Google Scholar] [CrossRef]
- Kabita, K.; Indrajit sharma, B.; Brojen Singh, R.K.; Thapa, R.K. Structural properties, phase transition and electronic structure of InP and ZnS compound: Density-functional theory LDA, GGA, mbj-GGA calculations. IOSR J. Appl. Phys. 2018, 10, 44–50. [Google Scholar]
- Hu, C.; Wang, F.; Zheng, Z. Pressure-induced metallic phase transition and elastic properties of indium phosphide III–V semiconductor. J. Mater. Res. 2012, 27, 1105–1111. [Google Scholar] [CrossRef]
- Lin, C.M.; Sheu, H.S.; Tsai, M.H.; Wu, B.R.; Jian, S.R. High pressure induced phase transition in sulfur doped indium phosphide: An angular-dispersive X-ray diffraction and Raman study. Solid State Commun. 2009, 149, 136–141. [Google Scholar] [CrossRef]
- Lin, C.M.; Hsu, I.J.; Lin, S.C.; Chuang, Y.C.; Juang, J.Y. Pressure effect on impurity local vibrational mode and phase transitions in n-type iron-doped indium phosphide. Sci. Rep. 2018, 8, 1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrobak, D.; Trębala, M.; Chrobak, A.; Nowak, R. Origin of nanoscale incipient plasticity in GaAs and InP crystal. Crystals 2019, 9, 651. [Google Scholar] [CrossRef] [Green Version]
- Chrobak, D.; Ziółkowski, G.; Chrobak, A. On incipient plasticity of InP crystal: A molecular dynamics study. Materials 2021, 14, 4157. [Google Scholar] [CrossRef]
- Chiruta, D.; Linares, J.; Dimian, M.; Garcia, Y. Size effect and role of short- and long-range interactions on 1D spin-crossover systems within the framework of an Ising-like model. Eur. J. Inorg. Chem. 2013, 2013, 951–957. [Google Scholar] [CrossRef]
- Catti, M. First-principles study of the orthorhombic mechanism for the B3/B1 high-pressure phase transition of ZnS. Phys. Rev. B 2002, 65, 224115. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DFT | Other | |||||||
|---|---|---|---|---|---|---|---|---|
| LDA | GGA | Data | Branicio | This Work | ||||
| InP B3 | ||||||||
| a | 5.829 | 5.965 | 5.869 [37] | 5.869 | 5.831 | |||
| 7.99 | 6.24 | 6.72 [38] | 6.97 | 7.59 | ||||
| 2.524 | 2.583 | 2.541 | 2.541 | 2.525 | ||||
| 2.00 | 1.56 | 1.68 | 1.74 | 1.90 | ||||
| B | 71.1 | 59.1 | 71.1 [37] | 71.5 | 69.1 | |||
| 100.4 | 86.5 | 101.1 [37] | 101.4 | 94.3 | ||||
| 56.4 | 45.4 | 56.1 [37] | 56.5 | 56.5 | ||||
| 45.3 | 41.1 | 45.6 [37] | 37.7 | 44.1 | ||||
| InP B1 | ||||||||
| a | 5.418 | 5.546 | 5.42 [39] | 5.643 | 5.391 | |||
| 7.64 | 5.75 | 6.57 | 6.85 | |||||
| 2.709 | 2.773 | 2.822 | 2.696 | |||||
| 1.27 | 0.96 | 1.64 | 1.41 | |||||
| B | 89.9 | 73.4 | 88.6 [39] | 49.5 | 90.0 | |||
| 182.5 | 146.9 | 246.6 [40] | 71.5 | 186.2 | ||||
| 43.6 | 36.6 | 48.8 [40] | 38.5 | 41.9 | ||||
| 31.8 | 27.7 | 30.7 [40] | 20.4 | 32.6 | ||||
| In | ||||||||
| dimer | diamond | RS like | FCC | |||||
| LDA | GGA | LDA | GGA | LDA | GGA | LDA | GGA | |
| a | 6.463 | 6.737 | 5.940 | 6.186 | 4.616 | 4.792 | ||
| 2.67 | 2.00 | 2.97 | 2.24 | 3.04 | 2.33 | |||
| 2.930 | 3.092 | 2.799 | 2.917 | 2.970 | 3.093 | 3.264 | 3.389 | |
| 1.64 | 1.42 | 1.34 | 1.00 | 0.99 | 0.75 | 0.51 | 0.39 | |
| B | 32.3 | 21.4 | 45.0 | 30.9 | 50.0 | 35.8 | ||
| P | ||||||||
| a | 5.337 | 5.495 | 4.822 | 4.889 | 3.811 | 3.879 | ||
| 4.00 | 2.90 | 4.55 | 3.33 | 3.82 | 2.59 | |||
| 1.887 | 1.905 | 2.311 | 2.379 | 2.411 | 2.445 | 2.695 | 2.743 | |
| 6.44 | 5.16 | 2.00 | 1.45 | 1.52 | 1.11 | 0.64 | 0.43 | |
| B | 51.4 | 43.2 | 125.9 | 109.1 | 107.8 | 90.2 | ||
| B3 Native Point Defects, | |||
|---|---|---|---|
| Mishra (DFT) | Branicio | This Work | |
| In rich | |||
| , | −1.52, −6.94 | −0.03, −6.94 | −1.66, −5.93 |
| 5.26 | 6.34 | 5.21 | |
| 0.98 | −1.03 | 0.93 | |
| 0.0 | 0.0 | 0 | |
| 4.88 | 16.07 | 4.88 | |
| 1.62 | −0.55 | 1.66 | |
| 4.19 | 8.41 | 5.81 | |
| P rich | |||
| , | −3.96, −4.50 | −8.06, 1.09 | −4.10, −3.49 |
| 2.83 | −1.69 | 2.77 | |
| 3.42 | 7.00 | 3.37 | |
| 4.88 | 16.07 | 4.88 | |
| 0.0 | 0.0 | 0 | |
| 4.06 | 7.48 | 4.10 | |
| 1.76 | 0.37 | 3.37 | |
| S | n | c | d | h | |||||
|---|---|---|---|---|---|---|---|---|---|
| (eV) | (1/Å) | (Å) | (1/Å) | ||||||
| In-P | |||||||||
| 2.039682 | 2.218147 | 1.410176 | 2.500450 | 4.768664 | 0.209249 | 0.686344 | 0.411612 | 0.175824 | 1.712155 |
| In-In | |||||||||
| 0.775299 | 8.540000 | 0.567841 | 2.770000 | 2.286299 | 0.088789 | 1.063382 | 0.486561 | 0.461374 | 1.146285 |
| P-P | |||||||||
| 4.530000 | 3.555819 | 1.804033 | 1.910011 | 0.770884 | 0.450794 | 1.812700 | 0.730891 | 0.473916 | 1.607665 |
| , , Å, Å, Å, Å | |||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrobak, D.; Majtyka-Piłat, A.; Ziółkowski, G.; Chrobak, A. Interatomic Potential for InP. Materials 2022, 15, 4960. https://doi.org/10.3390/ma15144960
Chrobak D, Majtyka-Piłat A, Ziółkowski G, Chrobak A. Interatomic Potential for InP. Materials. 2022; 15(14):4960. https://doi.org/10.3390/ma15144960
Chicago/Turabian StyleChrobak, Dariusz, Anna Majtyka-Piłat, Grzegorz Ziółkowski, and Artur Chrobak. 2022. "Interatomic Potential for InP" Materials 15, no. 14: 4960. https://doi.org/10.3390/ma15144960
APA StyleChrobak, D., Majtyka-Piłat, A., Ziółkowski, G., & Chrobak, A. (2022). Interatomic Potential for InP. Materials, 15(14), 4960. https://doi.org/10.3390/ma15144960