
Electrochemical and Spectroelectrochemical Studies on the Reactivity of Perimidine–Carbazole–Thiophene Monomers towards the Formation of Multidimensional Macromolecules versus Stable π-Dimeric States

 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
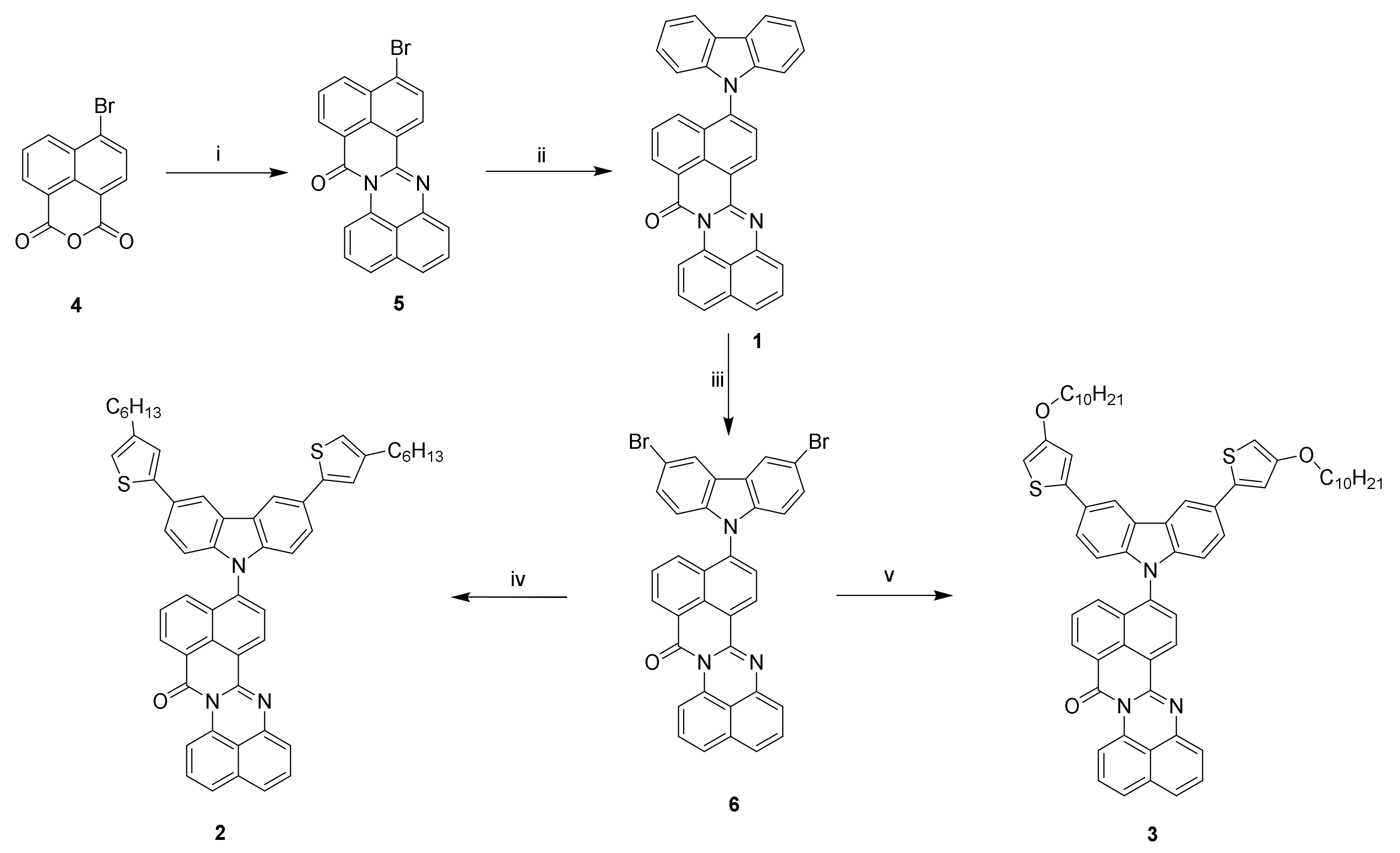
2.2. Synthesis of Monomers
2.3. Characterization Methods
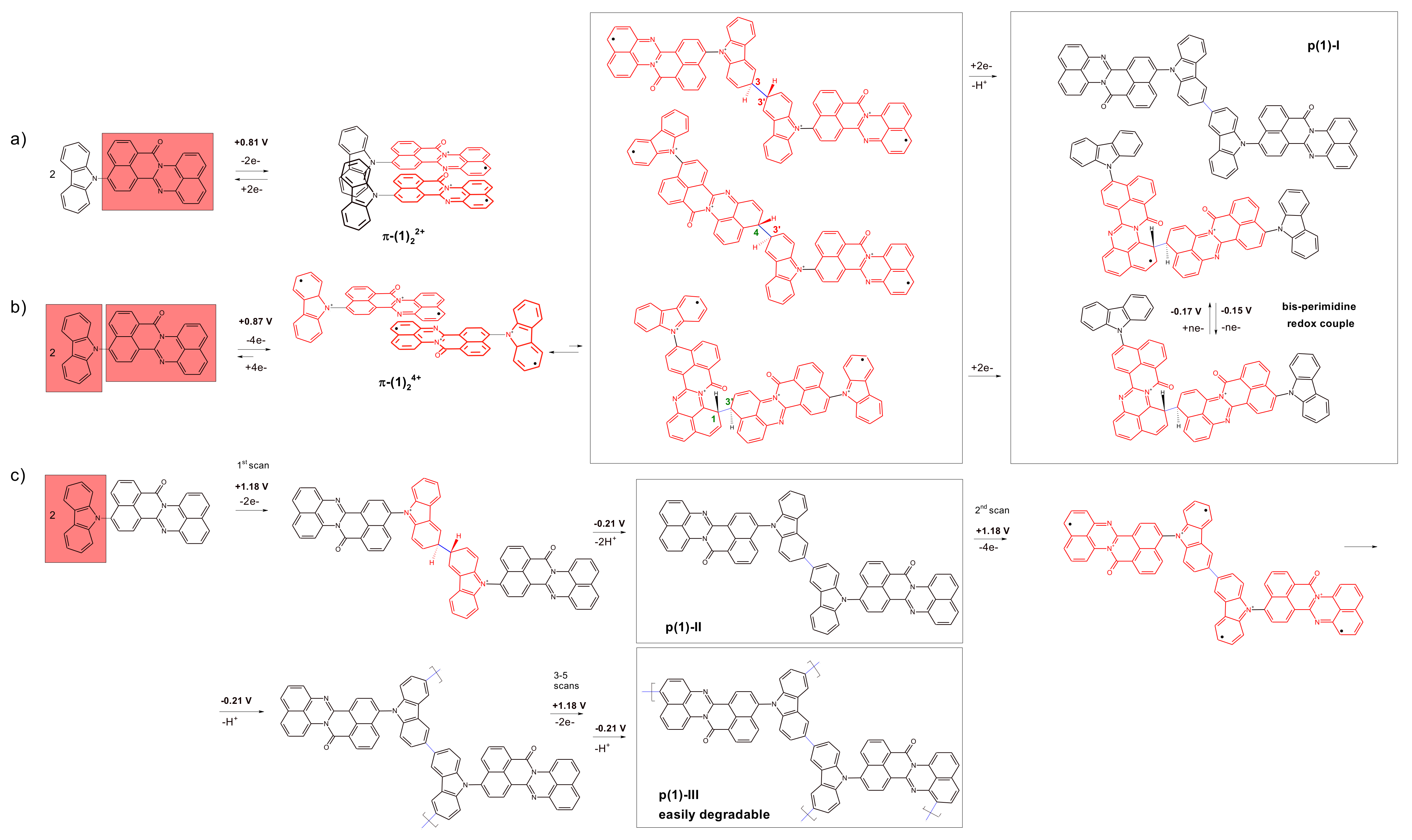
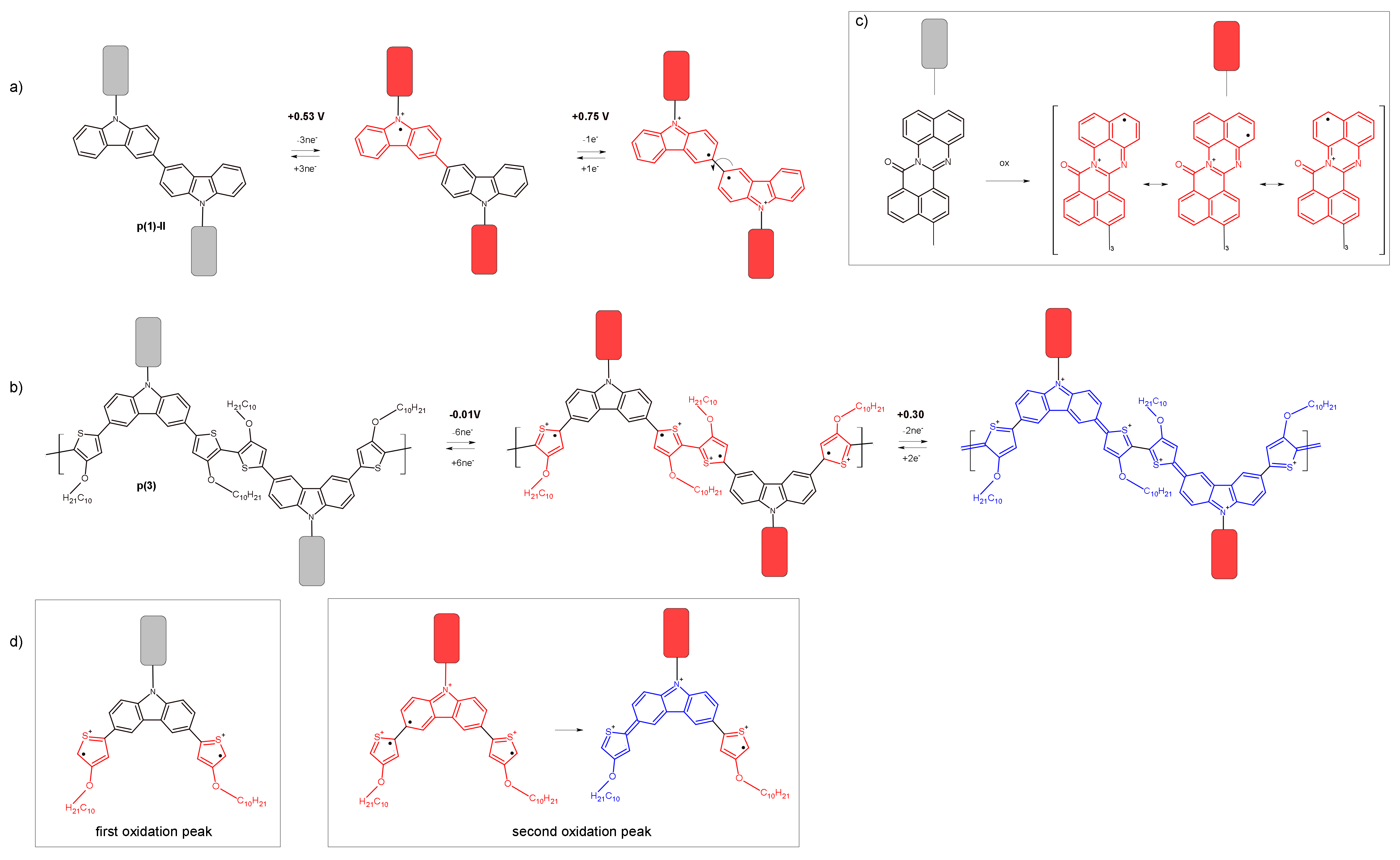
3. Results and Discussion
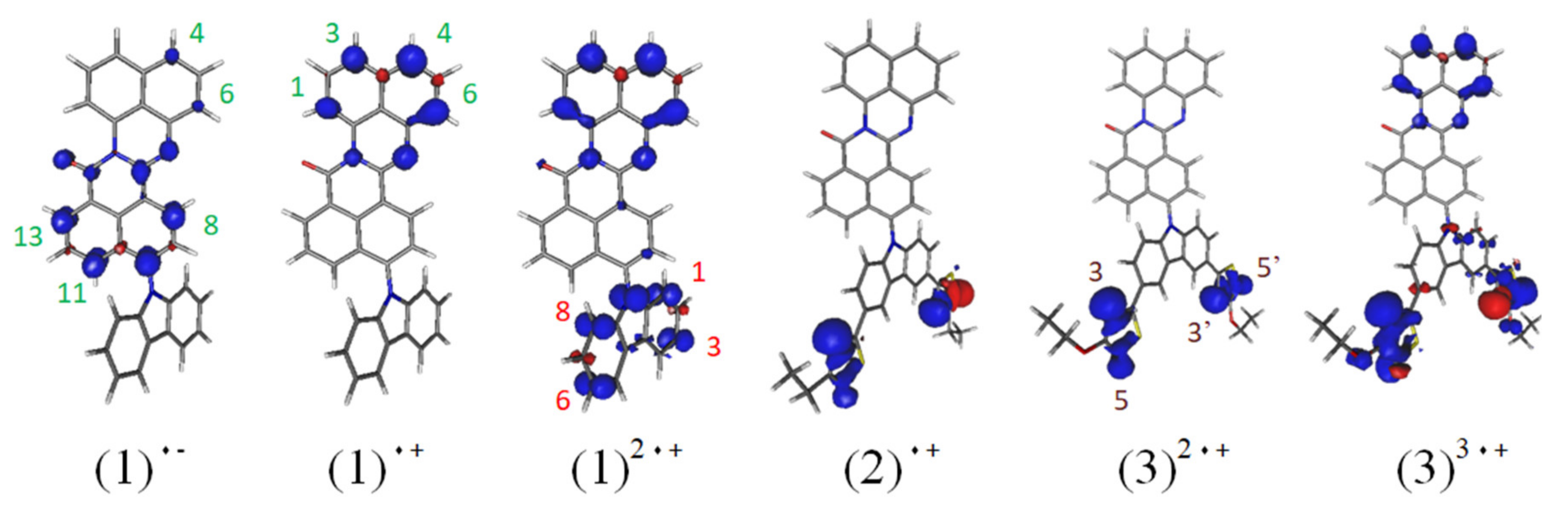
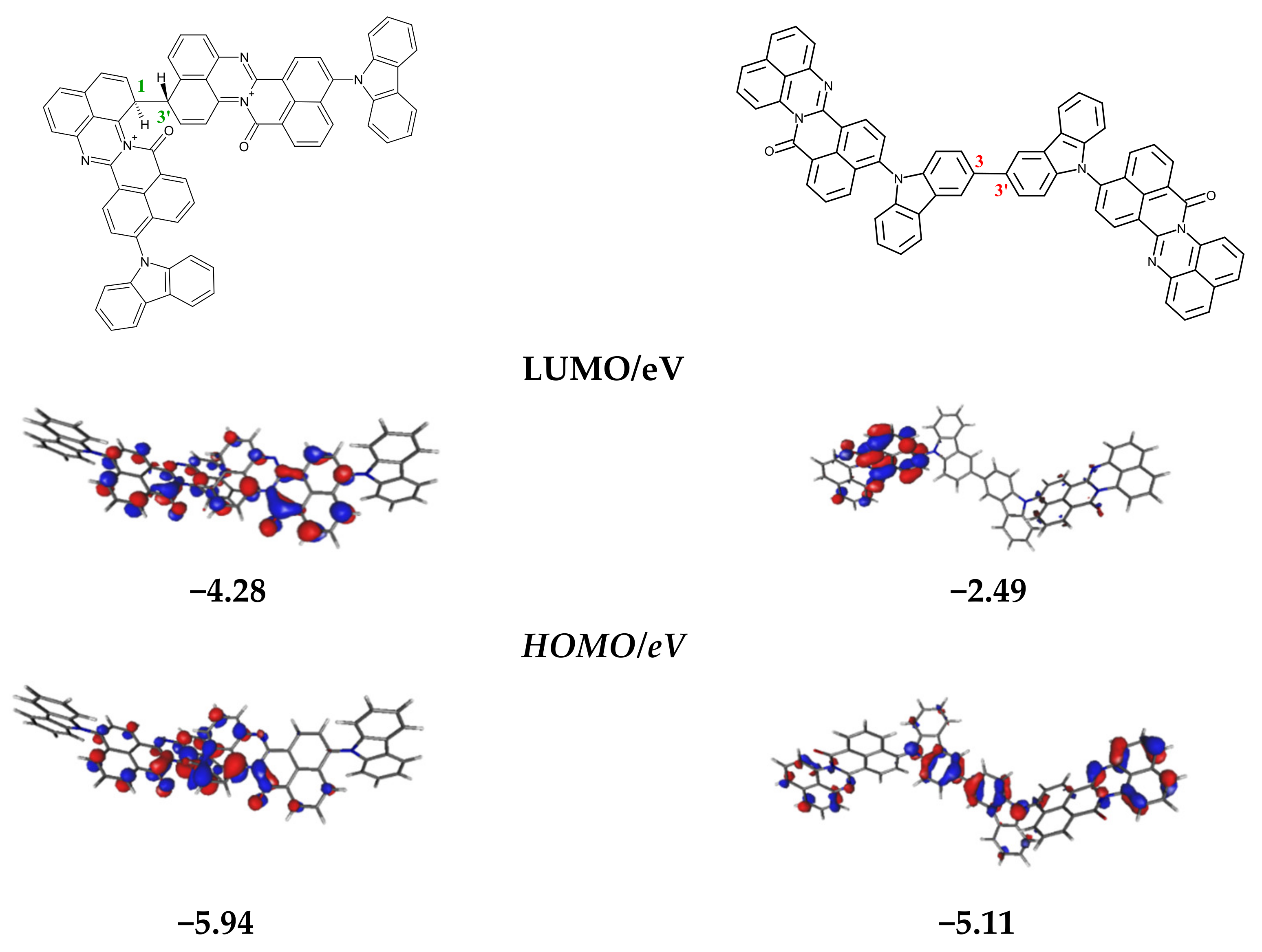
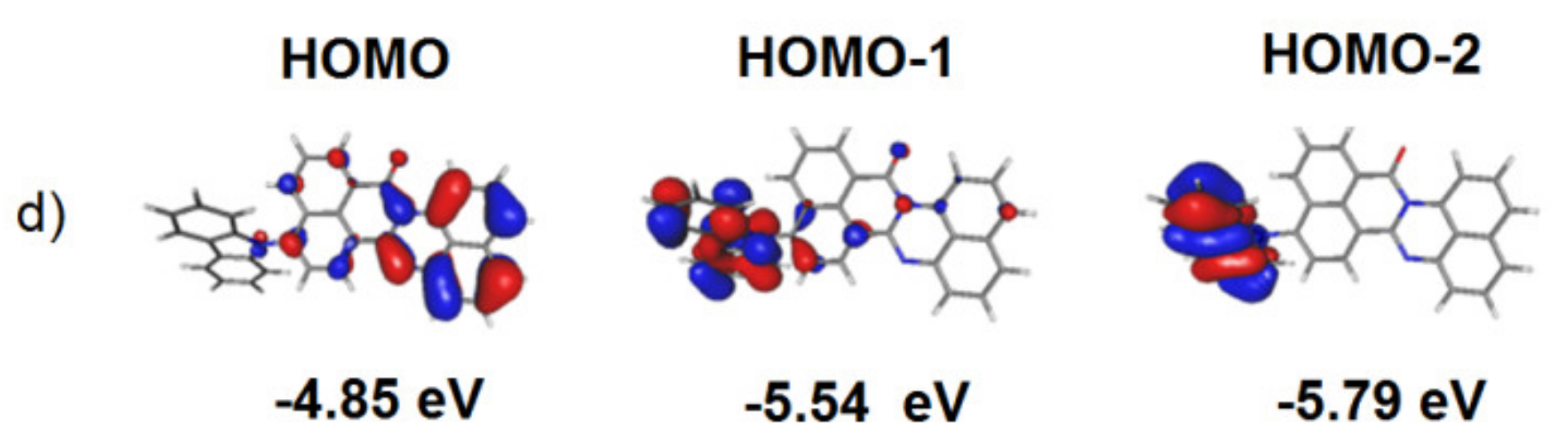
3.1. Quantum Chemical Calculations
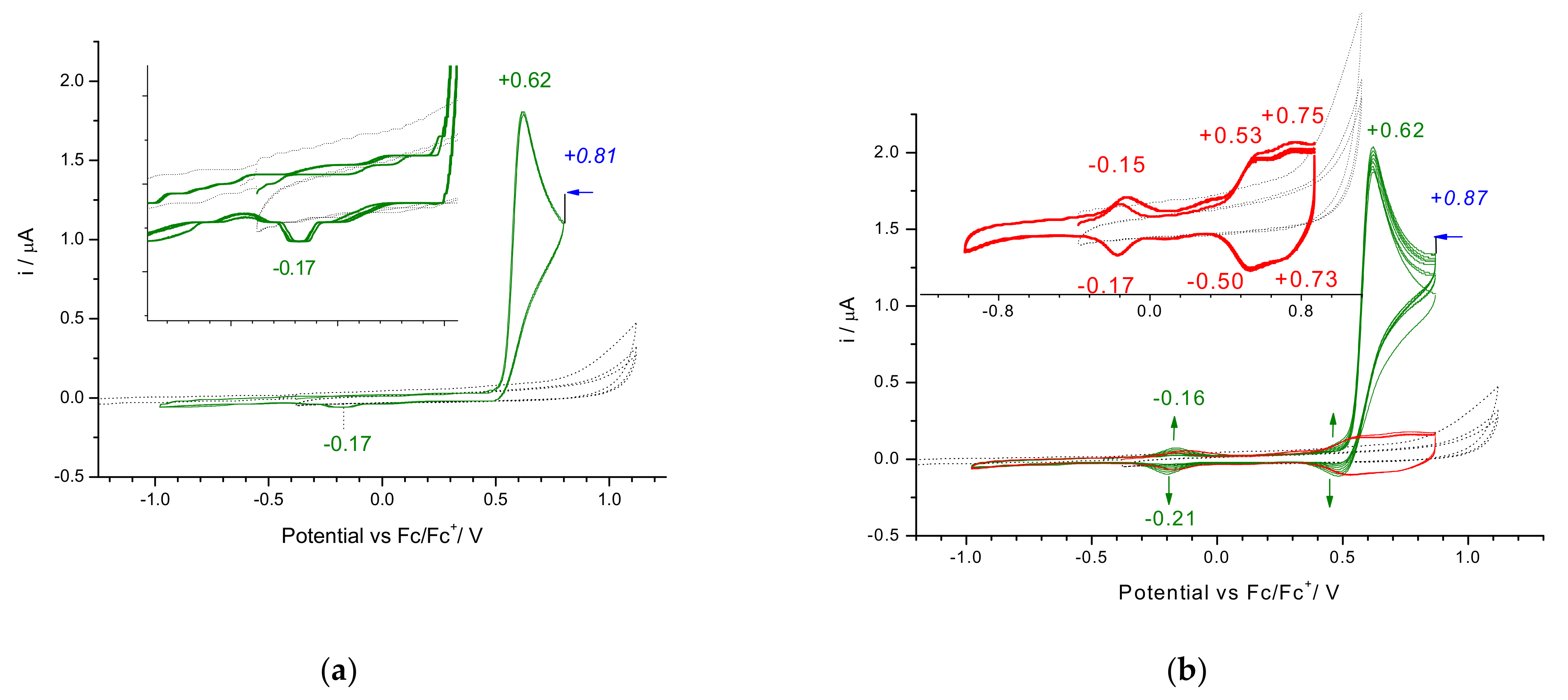
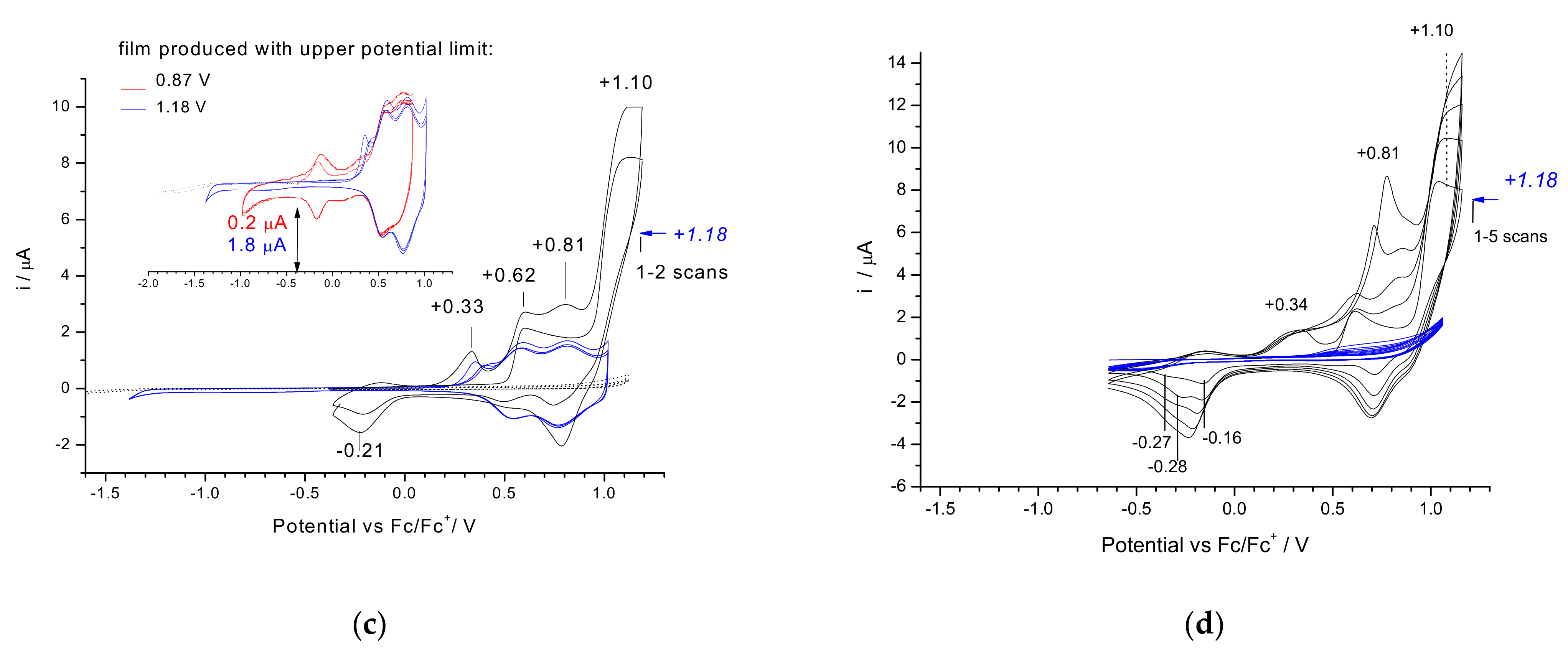
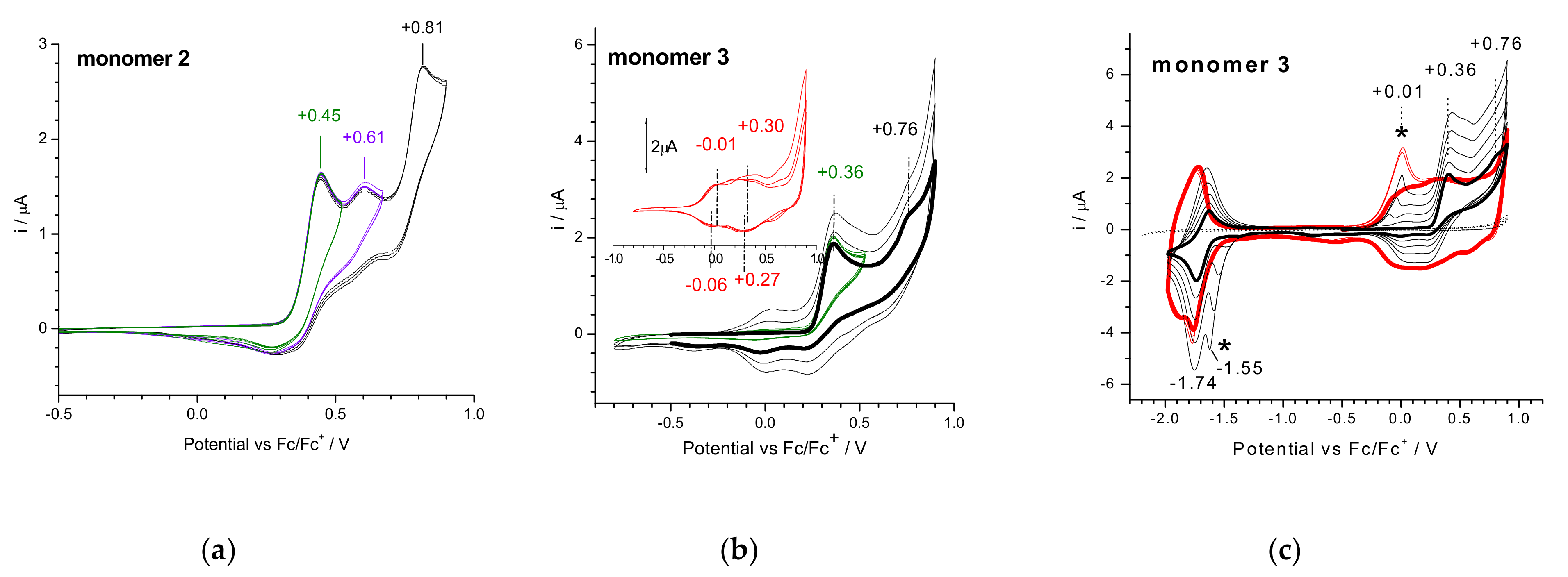
3.2. Electrochemical Reduction and Oxidation of Monomers 1–3
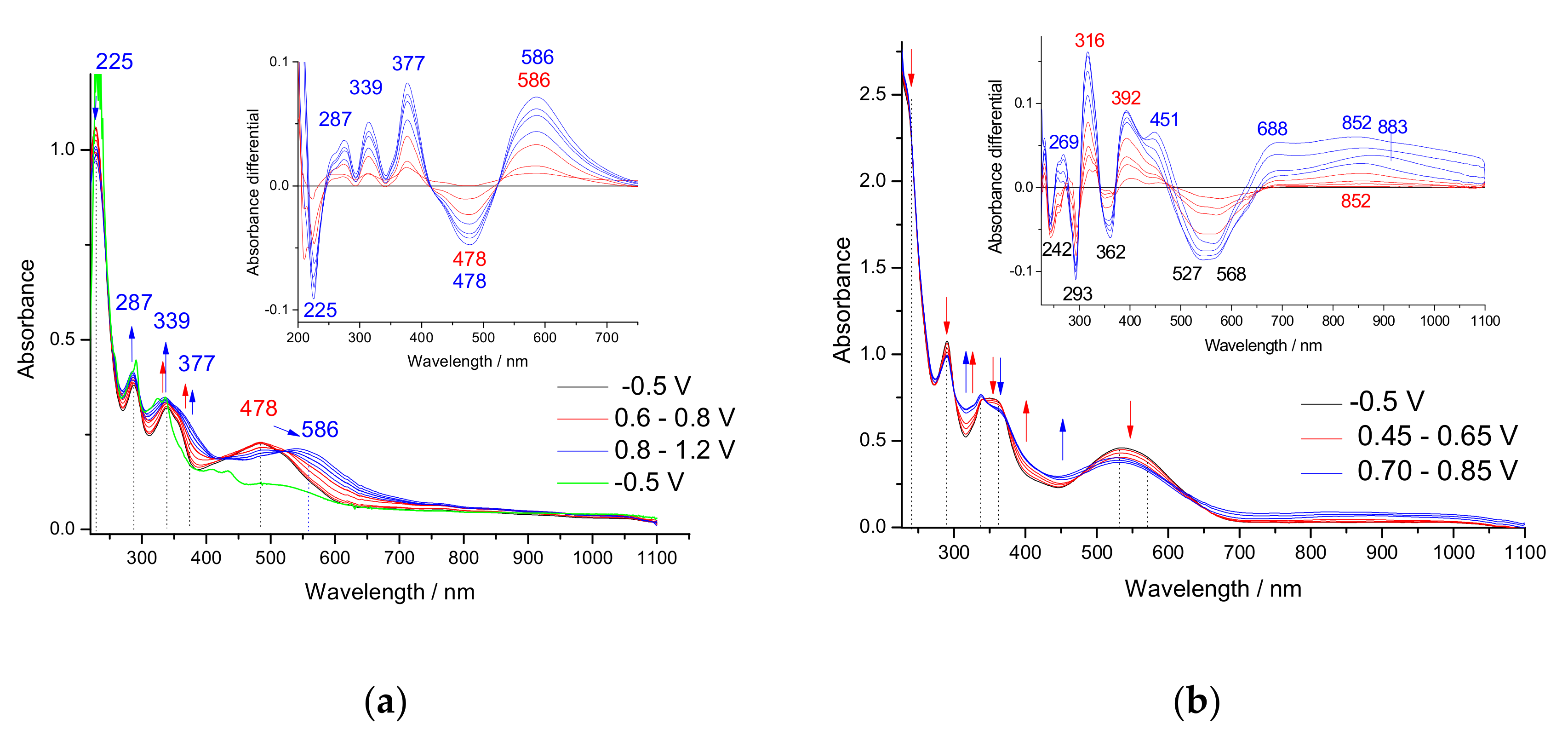
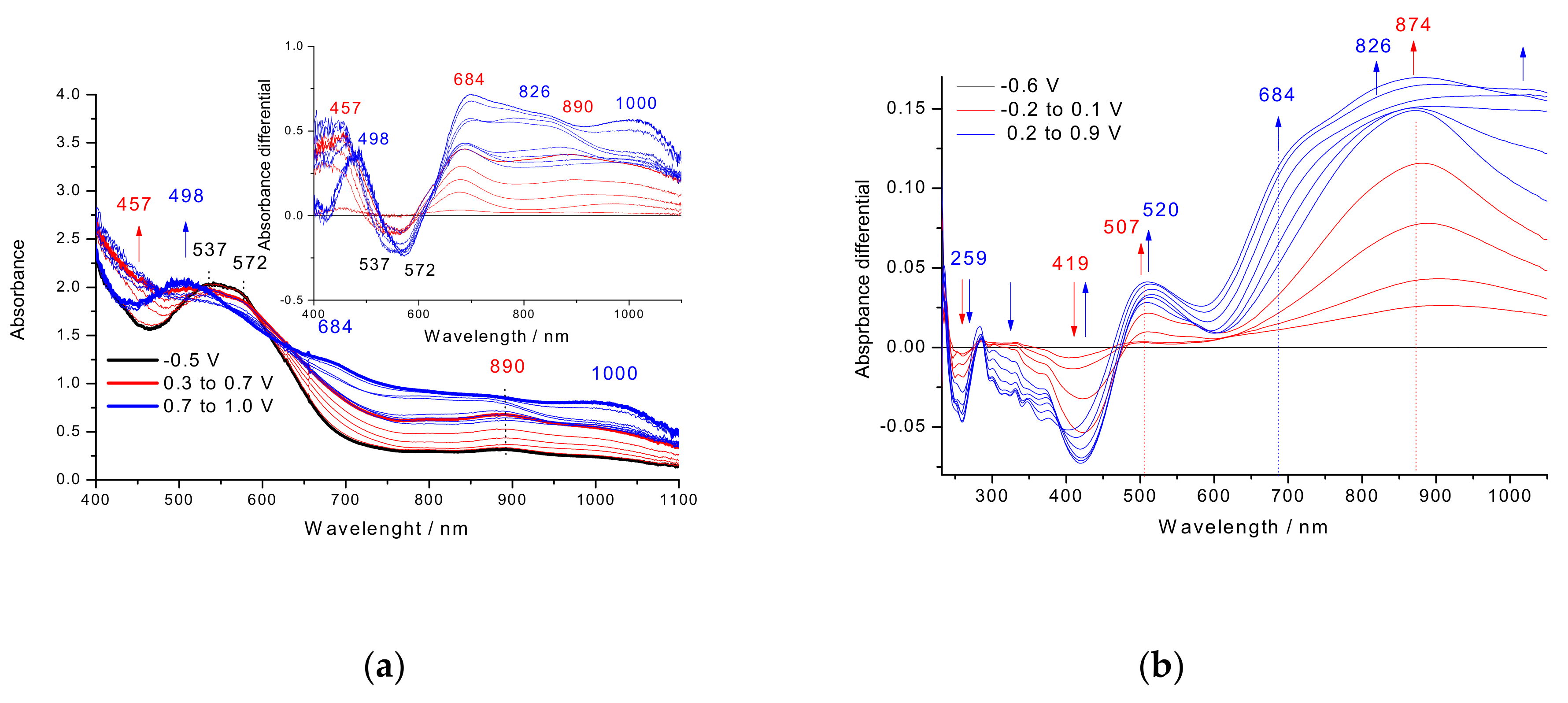
3.3. In Situ UV-Vis-NIR Study of Polarization of 1–3 Monomers, Products p(1) and p(3)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Perepichka, D.F.; Rosei, F. CHEMISTRY: Extending Polymer Conjugation into the Second Dimension. Science 2009, 323, 216–217. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Dai, L. Conducting Polymers for Flexible Supercapacitors. Macromol. Chem. Phys. 2019, 220, 1800355. [Google Scholar] [CrossRef]
- Cavallari, M.R.; Pastrana, L.M.; Sosa, C.D.F.; Marquina, A.M.R.; Izquierdo, J.E.E.; Fonseca, F.J.; De Amorim, C.A.; Paterno, L.G.; Kymissis, I. Organic Thin-Film Transistors as Gas Sensors: A Review. Materials 2020, 14, 3. [Google Scholar] [CrossRef] [PubMed]
- Stadler, P. Isotropic metallic transport in conducting polymers. Synth. Met. 2019, 254, 106–113. [Google Scholar] [CrossRef]
- Li, X.; Cai, S.; Sun, B.; Yang, C.; Zhang, J.; Liu, Y. Chemically Robust Covalent Organic Frameworks: Progress and Perspective. Matter 2020, 3, 1507–1540. [Google Scholar] [CrossRef]
- Brophy, J.; Summerfield, K.; Yin, J.; Kephart, J.; Stecher, J.; Adams, J.; Yanase, T.; Brant, J.; Li-Oakey, K.; Hoberg, J.; et al. The Influence of Disorder in the Synthesis, Characterization and Applications of a Modifiable Two-Dimensional Covalent Organic Framework. Materials 2020, 14, 71. [Google Scholar] [CrossRef]
- Li, H.; Yu, C.; Zhou, Y.; Tuo, H.; Zhong, W. Biligand metal-organic coordination polymer to prepare high N-doped content and structure controllable porous carbon with high-electrochemical performance. Electrochim. Acta 2019, 308, 263–276. [Google Scholar] [CrossRef]
- Evtugyn, G.; Belyakova, S.; Porfireva, A.; Hianik, T. Electrochemical Aptasensors Based on Hybrid Metal-Organic Frameworks. Sensors 2020, 20, 6963. [Google Scholar] [CrossRef]
- Clemons, T.D.; Stupp, S.I. Design of materials with supramolecular polymers. Prog. Polym. Sci. 2020, 111, 101310. [Google Scholar] [CrossRef]
- Ikeda, T.; Haino, T. Supramolecular polymeric assemblies of π-conjugated molecules possessing phenylisoxazoles. Polymer 2017, 128, 243–256. [Google Scholar] [CrossRef]
- Cataldo, S.; Pignataro, B. Polymeric Thin Films for Organic Electronics: Properties and Adaptive Structures. Materials 2013, 6, 1159–1190. [Google Scholar] [CrossRef]
- Gospodinova, N.; Tomšík, E. Hydrogen-bonding versus π–π stacking in the design of organic semiconductors: From dyes to oligomers. Prog. Polym. Sci. 2015, 43, 33–47. [Google Scholar] [CrossRef]
- Liu, B.; Yang, T.; Mu, X.; Mai, Z.; Li, H.; Wang, Y.; Zhou, G. Smart Supramolecular Self-Assembled Nanosystem: Stimulus-Responsive Hydrogen-Bonded Liquid Crystals. Nanomaterials 2021, 11, 448. [Google Scholar] [CrossRef]
- Yang, J.; Chen, S.; Xu, J.; Zhang, Q.; Liu, H.; Liu, Z.; Yuan, M. A Review on Improving the Quality of Perovskite Films in Perovskite Solar Cells via the Weak Forces Induced by Additives. Appl. Sci. 2019, 9, 4393. [Google Scholar] [CrossRef]
- Zhuang, W.-R.; Wang, Y.; Cui, P.-F.; Xing, L.; Lee, J.; Kim, D.; Jiang, H.-L.; Oh, Y.-K. Applications of π-π stacking interactions in the design of drug-delivery systems. J. Control. Release 2019, 294, 311–326. [Google Scholar] [CrossRef]
- Jiang, Q.; Zhang, Q.; Wu, X.; Wu, L.; Lin, J.-H. Exploring the Interfacial Phase and π–π Stacking in Aligned Carbon Nanotube/Polyimide Nanocomposites. Nanomaterials 2020, 10, 1158. [Google Scholar] [CrossRef]
- Yang, X.; Yuan, D.; Hou, J.; Sedgwick, A.C.; Xu, S.; James, T.D.; Wang, L. Organic/inorganic supramolecular nano-systems based on host/guest interactions. Coord. Chem. Rev. 2021, 428, 213609. [Google Scholar] [CrossRef]
- Buntkowsky, G.; Vogel, M. Small Molecules, Non-Covalent Interactions, and Confinement. Molecules 2020, 25, 3311. [Google Scholar] [CrossRef]
- Inaba, S.; Vohra, V. Fabrication Processes to Generate Concentration Gradients in Polymer Solar Cell Active Layers. Materials 2017, 10, 518. [Google Scholar] [CrossRef]
- Appel, G.; Schmeißer, D.; Bauer, J.; Bauer, M.; Egelhaaf, H.J.; Oelkrug, D. The formation of oligomers in the electrolyte upon polymerization of pyrrole. Synth. Met. 1999, 99, 69–77. [Google Scholar] [CrossRef]
- Kaynak, A.; Zolfagharian, A.; Featherby, T.; Bodaghi, M.; Mahmud, M.A.P.; Kouzani, A.Z. Electrothermal Modeling and Analysis of Polypyrrole-Coated Wearable E-Textiles. Materials 2021, 14, 550. [Google Scholar] [CrossRef]
- Ferjani, H.; Chebbi, H.; Fettouhi, M. One-Dimensional Organic–Inorganic Material (C6H9N2)2BiCl5: From Synthesis to Structural, Spectroscopic, and Electronic Characterizations. Int. J. Mol. Sci. 2021, 22, 2030. [Google Scholar] [CrossRef]
- Li, D.-Y.; Xie, H.; Yao, X.-Q.; Ma, H.-C.; Lei, Z.-Q.; Liu, J.-C. A luminescent coordination polymer based on a π-conjugated ligand: Syntheses, structure and luminescent property. J. Mol. Struct. 2017, 1134, 171–173. [Google Scholar] [CrossRef]
- Holliday, S.; Li, Y.; Luscombe, C.K. Recent advances in high performance donor-acceptor polymers for organic photovoltaics. Prog. Polym. Sci. 2017, 70, 34–51. [Google Scholar] [CrossRef]
- Xu, B.; Saianand, G.; Roy, V.A.L.; Qiao, Q.; Reza, K.M.; Kang, S.-W. Employing PCBTDPP as an Efficient Donor Polymer for High Performance Ternary Polymer Solar Cells. Polymers 2019, 11, 1423. [Google Scholar] [CrossRef]
- Belaish, I.; Davidov, D.; Selig, H.; McLean, M.R.; Dalton, L.R. Transport properties of heat treated and doped leader type polymers BBB and BBL. Synth. Met. 1991, 42, 1601–1605. [Google Scholar] [CrossRef]
- Kim, F.S.; Park, C.H.; Na, Y.; Jenekhe, S.A. Effects of ladder structure on the electronic properties and field-effect transistor performance of Poly(benzobisimidazobenzophenanthroline). Org. Electron. 2019, 69, 301–307. [Google Scholar] [CrossRef]
- Chen, S.; Liu, F.; Wang, C.; Shen, J.; Wu, Y. Wu Simple Route to Synthesize Fully Conjugated Ladder Isomer Copolymers with Carbazole Units. Polymers 2019, 11, 1619. [Google Scholar] [CrossRef] [PubMed]
- Vetrichelvan, M.; Valiyaveettil, S. Intramolecular Hydrogen-Bond-Assisted Planarization of Asymmetrically Functionalized Alternating Phenylene-Pyridinylene Copolymers. Chem. Eur. J. 2005, 11, 5889–5898. [Google Scholar] [CrossRef] [PubMed]
- Osaka, I.; Takimiya, K. Backbone orientation in semiconducting polymers. Polymer 2015, 59, A1–A15. [Google Scholar] [CrossRef]
- Ha, J.-W.; Park, J.B.; Park, H.J.; Hwang, D.-H. Novel Conjugated Polymers Containing 3-(2-Octyldodecyl)thieno[3,2-b]thiophene as a π-Bridge for Organic Photovoltaic Applications. Polymers 2020, 12, 2121. [Google Scholar] [CrossRef]
- Ligon-Auer, S.C.; Schwentenwein, M.; Gorsche, C.; Stampfl, J.; Liska, R. Toughening of photo-curable polymer networks: A review. Polym. Chem. 2016, 7, 257–286. [Google Scholar] [CrossRef]
- Zembrzuska, D.; Kalecki, J.; Cieplak, M.; Lisowski, W.; Borowicz, P.; Noworyta, K.; Sharma, P.S. Electrochemically initiated co-polymerization of monomers of different oxidation potentials for molecular imprinting of electroactive analyte. Sens. Actuators B Chem. 2019, 298, 126884. [Google Scholar] [CrossRef]
- Schlotthauer, T.; Friebe, C.; Schwenke, A.M.; Jäger, M.; Schubert, U.S. Mild electropolymerization and monitoring of continuous film formation for photoredox-active Ru metallopolymers. J. Mater. Chem. C 2017, 5, 2636–2648. [Google Scholar] [CrossRef]
- Fulghum, T.; Karim, S.M.A.; Baba, A.; Taranekar, P.; Nakai, T.; Masuda, T.; Advincula, R.C. Conjugated Poly(phenylacetylene) Films Cross-Linked with Electropolymerized Polycarbazole Precursors. Macromolecules 2006, 39, 1467–1473. [Google Scholar] [CrossRef]
- Qu, J.; Shiotsuki, M.; Kobayashi, N.; Sanda, F.; Masuda, T. Synthesis and properties of carbazole-based hyperbranched conjugated polymers. Polymer 2007, 48, 6481–6490. [Google Scholar] [CrossRef]
- Kocaeren, A.A. Synthesis and Electrochromic Performance of a Novel Polymer Based on an Oxidative Polymer Derived from Carbazole and Thiophene. J. Polym. Res. 2016, 23, 66. [Google Scholar] [CrossRef]
- Piron, F.; Leriche, P.; Grosu, I.; Roncali, J. Electropolymerizable 3Dπ-conjugated architectures with ethylenedioxythiophene (EDOT) end-groups as precursors of electroactive conjugated networks. J. Mater. Chem. 2010, 20, 10260–10268. [Google Scholar] [CrossRef][Green Version]
- Hsiao, S.-H.; Wang, H.-M. Electrochemically fabricated electrochromic films from 4-(N-carbazolyl)triphenylamine and its dimethoxy derivative. RSC Adv. 2016, 6, 43470–43479. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, Y.; Gu, C.; Ma, Y. Electropolymerized Conjugated Microporous Poly(zinc-porphyrin) Films as Potential Electrode Materials in Supercapacitors. Adv. Energy Mater. 2015, 5, 1402175. [Google Scholar] [CrossRef]
- Mothika, V.S.; Räupke, A.; Brinkmann, K.O.; Riedl, T.; Brunklaus, G.; Scherf, U. Nanometer-Thick Conjugated Microporous Polymer Films for Selective and Sensitive Vapor-Phase TNT Detection. ACS Appl. Nano Mater. 2018, 1, 6483–6492. [Google Scholar] [CrossRef]
- Tao, L.; Cinquanta, E.; Chiappe, D.; Grazianetti, C.; Fanciulli, M.; Dubey, M.; Molle, A.; Akinwande, D. Silicene field-effect transistors operating at room temperature. Nat. Nanotechnol. 2015, 10, 227–231. [Google Scholar] [CrossRef]
- Jiang, L.; Dong, H.; Meng, Q.; Li, H.; He, M.; Wei, Z.; He, Y.; Hu, W. Millimeter-Sized Molecular Monolayer Two-Dimensional Crystals. Adv. Mater. 2011, 23, 2059–2063. [Google Scholar] [CrossRef]
- Chevrier, M.; Kesters, J.; Blayo, C.; Richeter, S.; Van Der Lee, A.; Coulembier, O.; Surin, M.; Mehdi, A.; Lazzaroni, R.; Evans, R.C.; et al. Regioregular Polythiophene-Porphyrin Supramolecular Copolymers for Optoelectronic Applications. Macromol. Chem. Phys. 2016, 217, 445–458. [Google Scholar] [CrossRef]
- Ghosh, T.; Panicker, J.; Nair, V. Self-Assembled Organic Materials for Photovoltaic Application. Polymers 2017, 9, 112. [Google Scholar] [CrossRef]
- González-Rodríguez, D.; Schenning, A.P.H.J. Hydrogen-bonded Supramolecular π-Functional Materials. Chem. Mater. 2011, 23, 310–325. [Google Scholar] [CrossRef]
- Okamoto, K.; Chithra, P.; Richards, G.; Hill, J.; Ariga, K. Self-Assembly of Optical Molecules with Supramolecular Concepts. Int. J. Mol. Sci. 2009, 10, 1950–1966. [Google Scholar] [CrossRef]
- Voorhaar, L.; Chan, E.W.C.; Baek, P.; Wang, M.; Nelson, A.; Barker, D.; Travas-Sejdic, J. Self-healing polythiophene phenylenes for stretchable electronics. Eur. Polym. J. 2018, 105, 331–338. [Google Scholar] [CrossRef]
- Aksenov, A.V.; Shcherbakov, S.V.; Lobach, I.V.; Aksenova, I.V.; Rubin, M. Pyrimidines as Surrogates for 1,3-Dicarbonyl Compounds in peri Annulation of Perimidines en Route to 1,3-Diazapyrenes. Eur. J. Org. Chem. 2017, 2017, 1666–1673. [Google Scholar] [CrossRef]
- Czichy, M.; Zhylitskaya, H.; Zassowski, P.; Navakouski, M.; Chulkin, P.; Janasik, P.; Lapkowski, M.; Stępień, M. Electrochemical Polymerization of Pyrrole–Perimidine Hybrids: Low-Band-Gap Materials with High n-Doping Activity. J. Phys. Chem. C 2020, 124, 14350–14362. [Google Scholar] [CrossRef]
- Booysen, I.N.; Ebinumoliseh, I.; Sithebe, S.; Akerman, M.P.; Xulu, B. Coordination behaviours of perimidine ligands incorporating fused N-donor heterocyclics towards rhenium(I) and -(V). Polyhedron 2016, 117, 755–760. [Google Scholar] [CrossRef]
- Lee, J.F.; Hsu, S.L.C.; Lee, P.; Chuang, H.Y.; Yang, M.L.; Chen, J.S.; Chou, W.Y. A new intramolecular donor–acceptor polyfluorene copolymer for bulk heterojunction solar cells. Sol. Energy Mater. Sol. Cells 2010, 94, 1166–1172. [Google Scholar] [CrossRef]
- Lee, J.F.; Hsu, S.L.C.; Lee, P.I.; Chuang, H.Y.; Yang, M.L.; Chen, J.S.; Chou, W.Y. Low bandgap carbazole copolymers containing an electron-withdrawing side chain for solar cell applications. Sol. Energy Mater. Sol. Cells 2011, 95, 2795–2804. [Google Scholar] [CrossRef]
- Zotti, G.; Gallazzi, M.C.; Zerbi, G.; Meille, S.V. Conducting polymers from anodic coupling of some regiochemically defined dialkoxy-substituted thiophene oligomers. Synth. Met. 1995, 73, 217–225. [Google Scholar] [CrossRef]
- Pei, J.; Ni, J.; Zhou, X.-H.; Cao, X.-Y.; Lai, Y.-H. Head-to-Tail Regioregular Oligothiophene-Functionalized 9,9‘-Spirobifluorene Derivatives. 1. Synthesis. J. Org. Chem. 2002, 67, 4924–4936. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Comput. Mol. Sci. 2018, 8, 8. [Google Scholar] [CrossRef]
- Allouche, A.-R. Gabedit-A graphical user interface for computational chemistry softwares. J. Comput. Chem. 2011, 32, 174–182. [Google Scholar] [CrossRef]
- Fabian, J. TDDFT-calculations of Vis/NIR absorbing compounds. Dye. Pigment. 2010, 84, 36–53. [Google Scholar] [CrossRef]
- Trasatti, S. The absolute electrode potential: An explanatory note (Recommendations 1986). Pure Appl. Chem. 1986, 58, 955–966. [Google Scholar] [CrossRef]
- Czichy, M.; Motyka, R.; Zassowski, P.; Grabiec, E.; Janasik, P.; Brzeczek-Szafran, A.; Laba, K.; Wolinska-Grabczyk, A.; Lapkowski, M. Effects of solution-phase ordering on the spectroscopic properties and electrooxidative reactivity of isomeric mixtures and isolated isomers of synthesized amidine derivatives. Dye. Pigment. 2020, 178, 108309. [Google Scholar] [CrossRef]
- Kortekaas, L.; Lancia, F.; Steen, J.D.; Browne, W.R. Reversible Charge Trapping in Bis-Carbazole-Diimide Redox Polymers with Complete Luminescence Quenching Enabling Nondestructive Read-Out by Resonance Raman Spectroscopy. J. Phys. Chem. C 2017, 121, 14688–14702. [Google Scholar] [CrossRef]
- Karon, K.; Lapkowski, M. Carbazole electrochemistry: A short review. J. Solid State Electrochem. 2015, 19, 2601–2610. [Google Scholar] [CrossRef]
- Zhang, L.-P.; Chen, B.; Wu, L.-Z.; Tung, C.-H.; Cao, H.; Tanimoto, Y. Photoinduced Intramolecular Electron Transfer and Triplet Energy Transfer in a Steroid-Linked Norbornadiene–Carbazole Dyad. Chem. Eur. J. 2003, 9, 2763–2769. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czichy, M.; Janasik, P.; Wagner, P.; Officer, D.L.; Lapkowski, M. Electrochemical and Spectroelectrochemical Studies on the Reactivity of Perimidine–Carbazole–Thiophene Monomers towards the Formation of Multidimensional Macromolecules versus Stable π-Dimeric States. Materials 2021, 14, 2167. https://doi.org/10.3390/ma14092167
Czichy M, Janasik P, Wagner P, Officer DL, Lapkowski M. Electrochemical and Spectroelectrochemical Studies on the Reactivity of Perimidine–Carbazole–Thiophene Monomers towards the Formation of Multidimensional Macromolecules versus Stable π-Dimeric States. Materials. 2021; 14(9):2167. https://doi.org/10.3390/ma14092167
Chicago/Turabian StyleCzichy, Malgorzata, Patryk Janasik, Pawel Wagner, David L. Officer, and Mieczyslaw Lapkowski. 2021. "Electrochemical and Spectroelectrochemical Studies on the Reactivity of Perimidine–Carbazole–Thiophene Monomers towards the Formation of Multidimensional Macromolecules versus Stable π-Dimeric States" Materials 14, no. 9: 2167. https://doi.org/10.3390/ma14092167
APA StyleCzichy, M., Janasik, P., Wagner, P., Officer, D. L., & Lapkowski, M. (2021). Electrochemical and Spectroelectrochemical Studies on the Reactivity of Perimidine–Carbazole–Thiophene Monomers towards the Formation of Multidimensional Macromolecules versus Stable π-Dimeric States. Materials, 14(9), 2167. https://doi.org/10.3390/ma14092167