
Single-Component Organic Solar Cells Based on Intramolecular Charge Transfer Photoabsorption

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Materials and Methods
3. Results
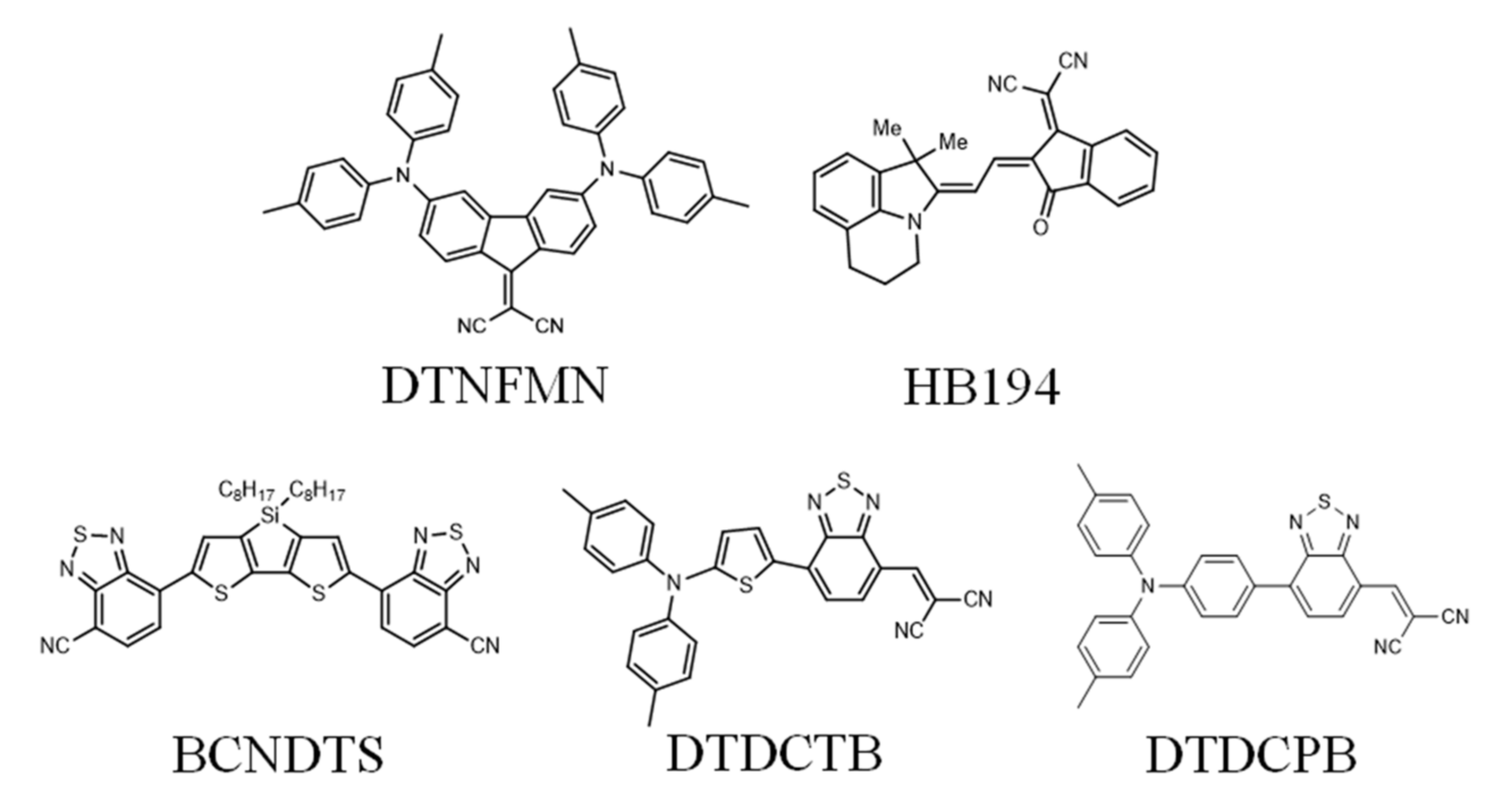
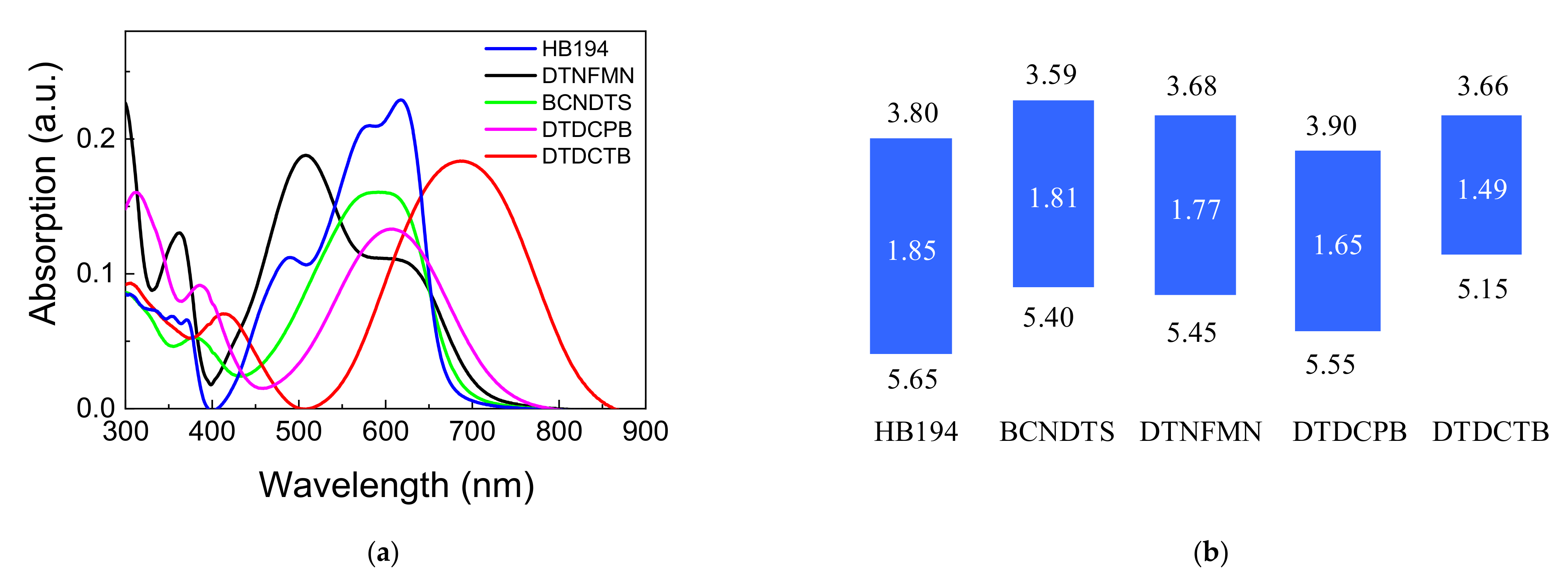
3.1. Film Characterization
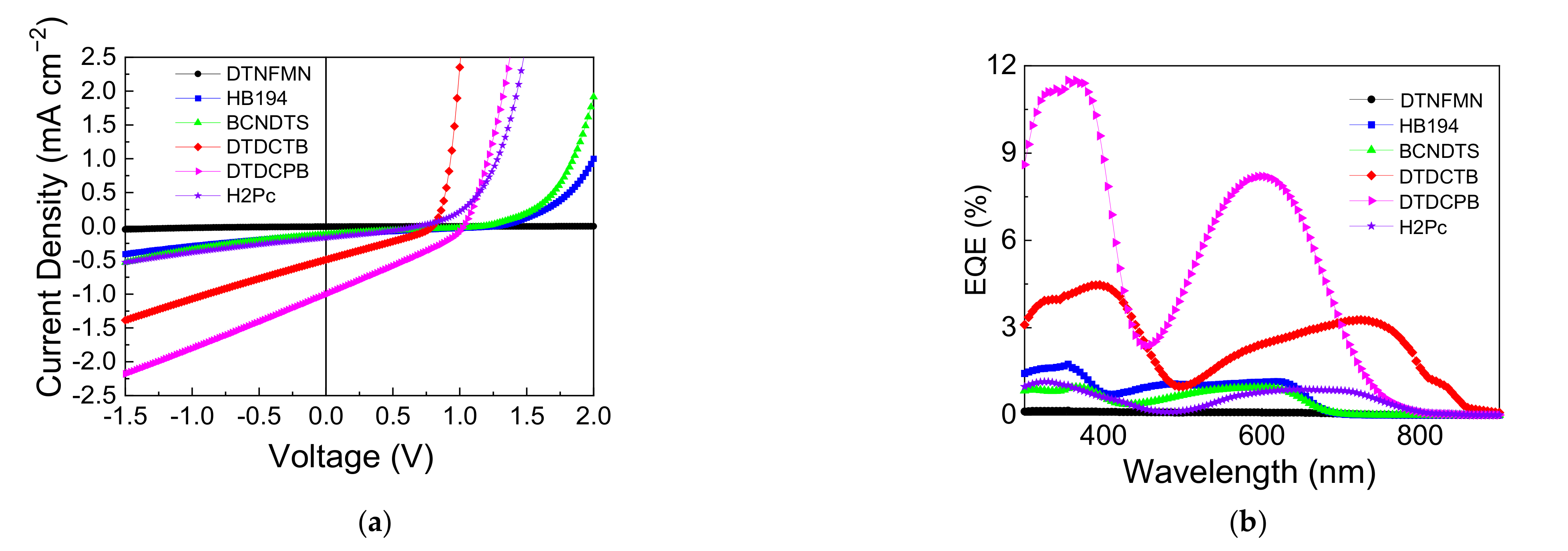
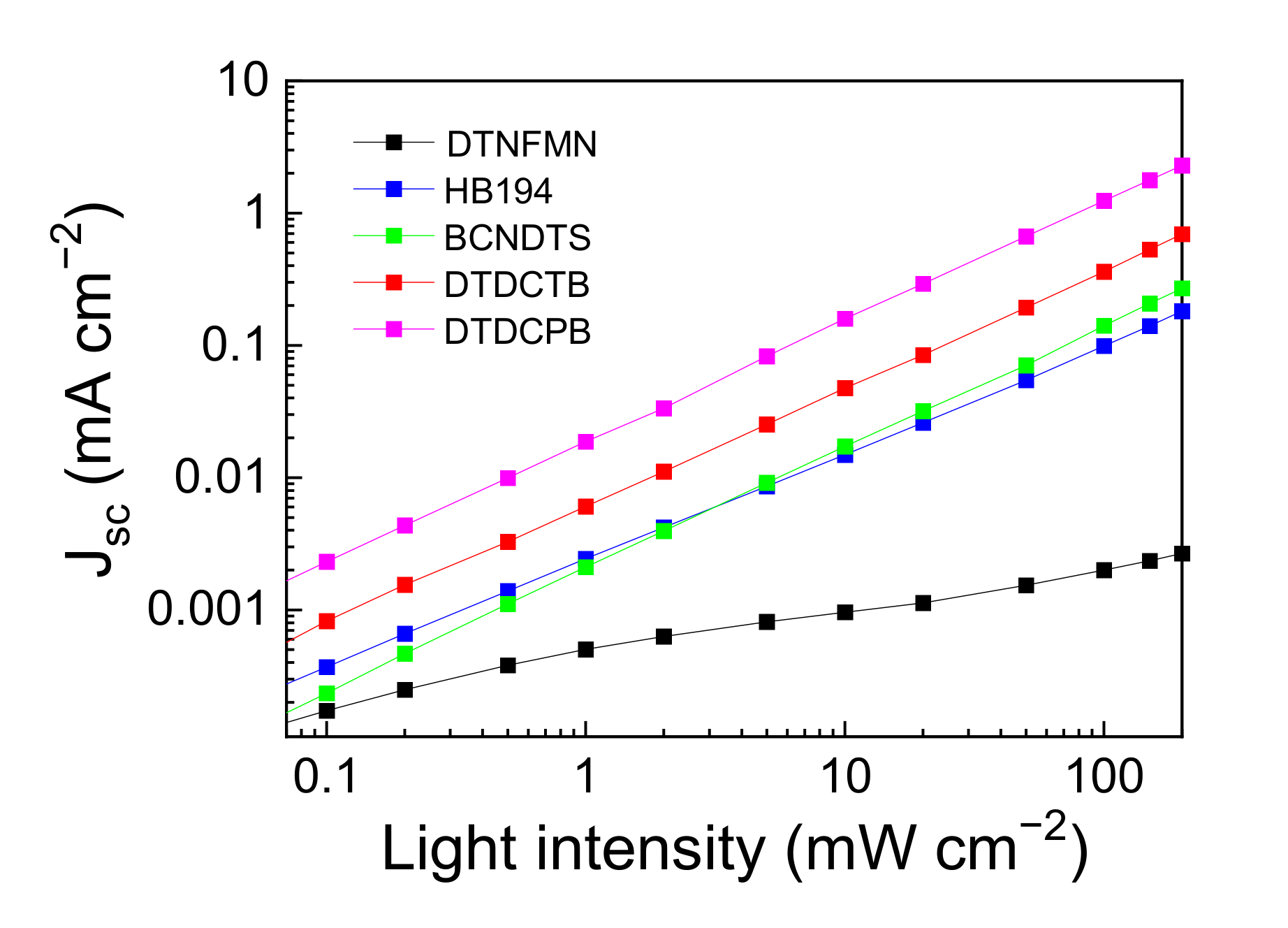
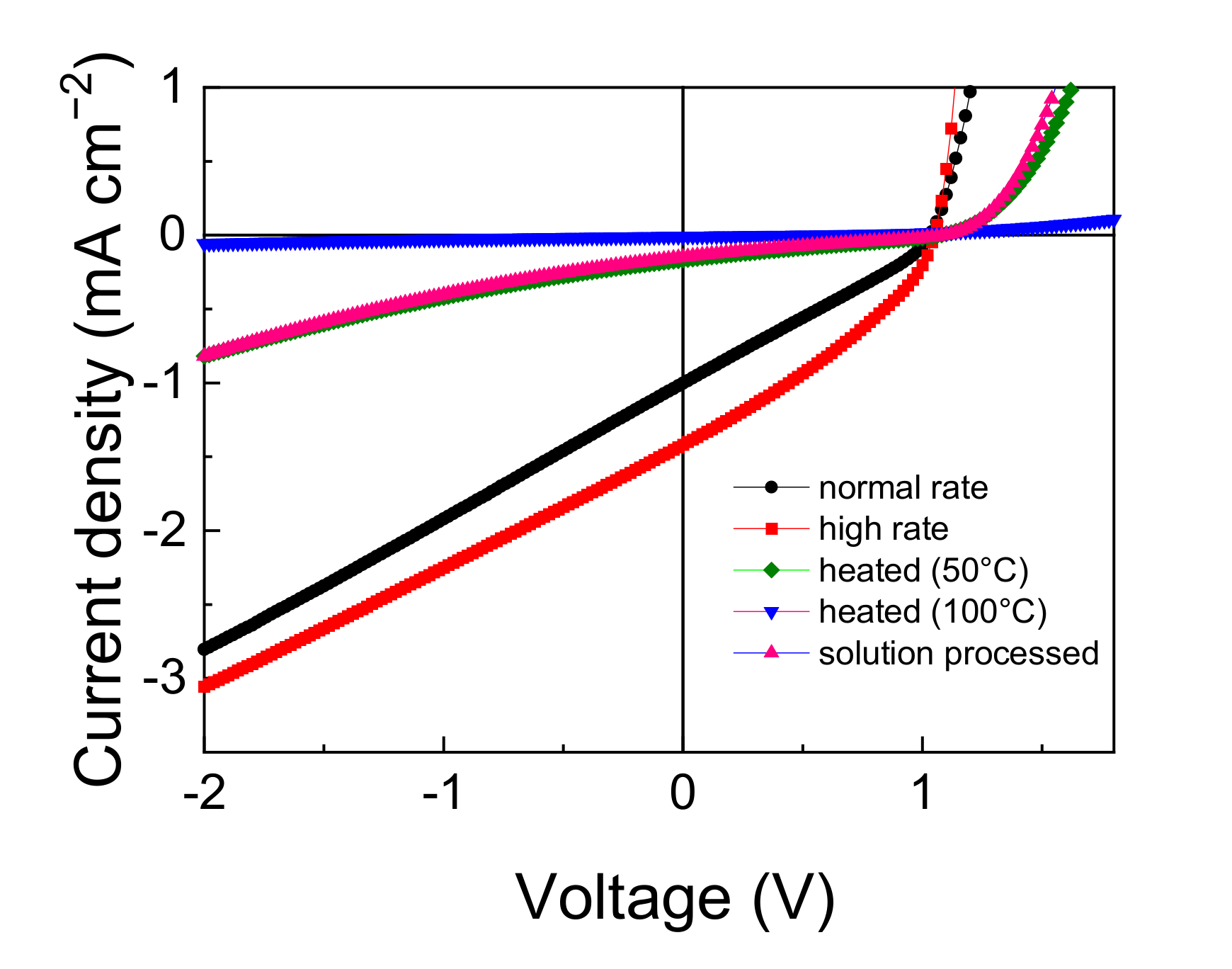
3.2. Device Performance Comparison
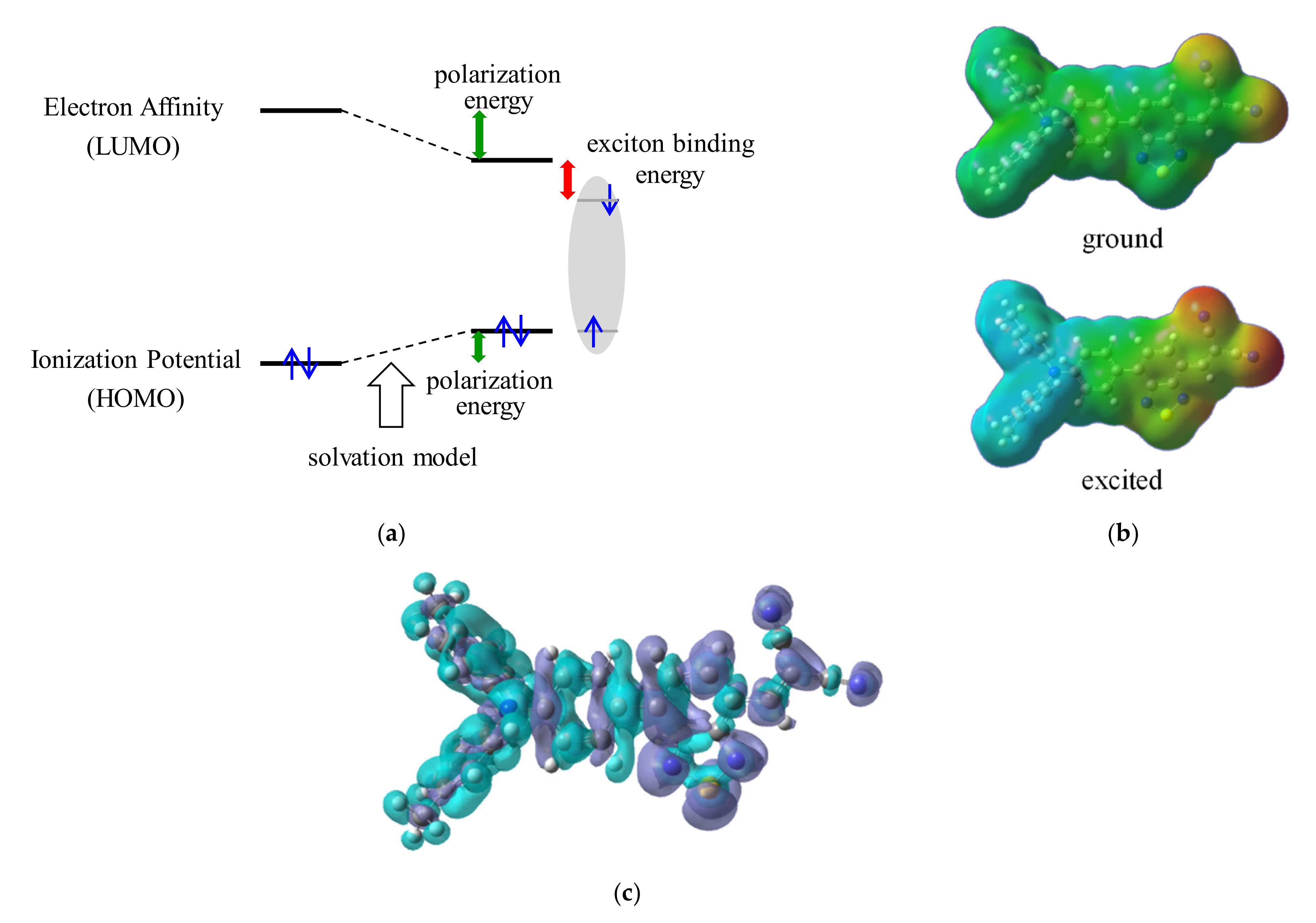
3.3. Exciton Binding Energy (EBE)
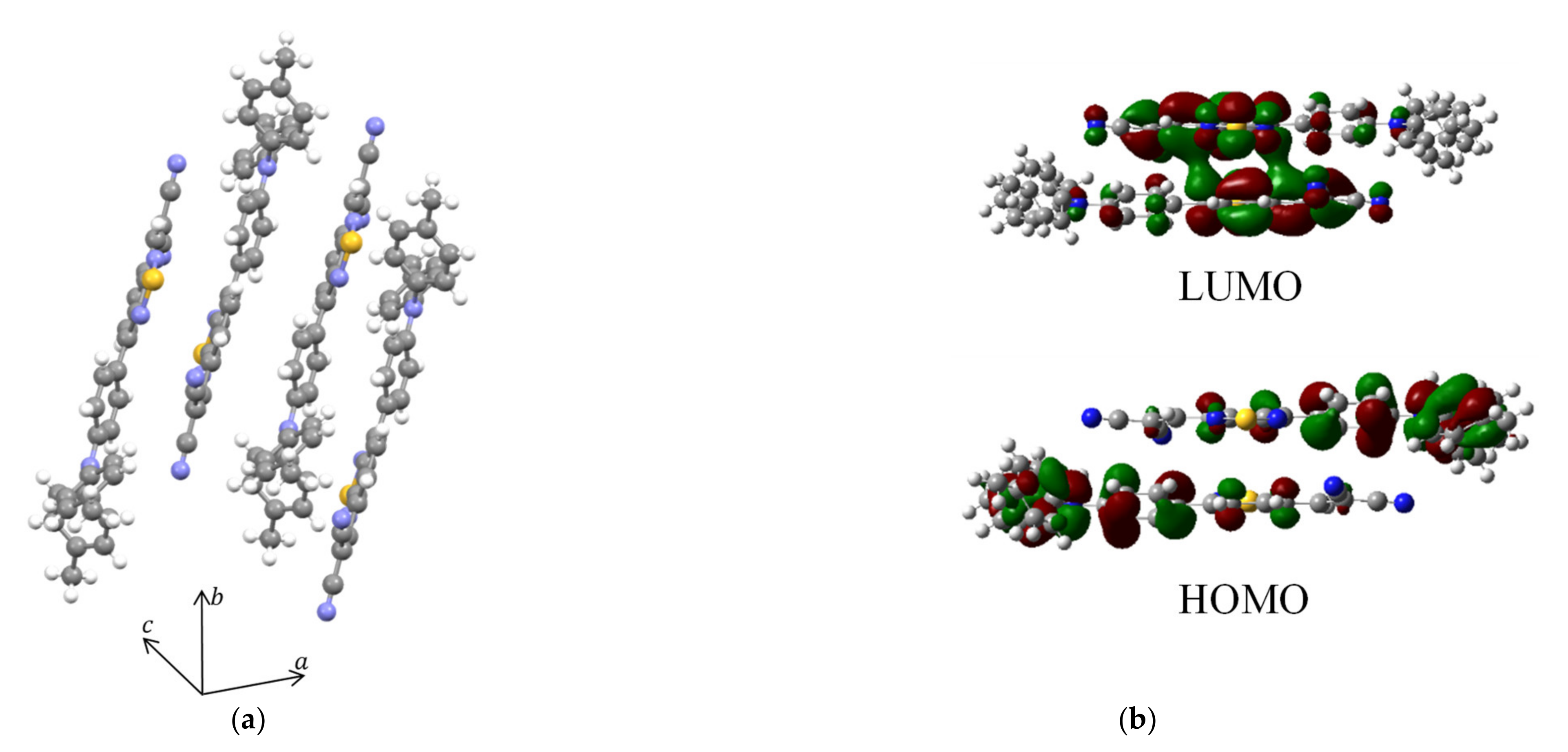
3.4. Effects of Film Structure in the DTDCPB Device
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, H.; Yao, H.; Hou, J.; Zhu, J.; Zhang, J.; Li, W.; Yu, R.; Gao, B.; Zhang, S.; Hou, J. Over 14% Efficiency in Organic Solar Cells Enabled by Chlorinated Nonfullerene Small-Molecule Acceptors. Adv. Mater. 2018, 30, 1800613. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, K.; Tamai, Y.; Ohkita, H.; Osaka, I.; Takimiya, K. High-efficiency polymer solar cells with small photon energy loss. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Du, X.; Wang, J.; Duan, C.; Tang, X.; Heumueller, T.; Liu, G.; Li, Y.; Wang, Z.; Wang, J.; et al. Efficient organic solar cells with extremely high open-circuit voltages and low voltage losses by suppressing nonradiative recombination losses. Adv. Energy Mater. 2018, 8, 1801699. [Google Scholar] [CrossRef]
- Linderl, T.; Zechel, T.; Brendel, M.; Moseguí González, D.; Müller-Buschbaum, P.; Pflaum, J.; Brütting, W. Energy losses in small-molecule organic photovoltaics. Adv. Energy Mater. 2017, 7, 1700237. [Google Scholar] [CrossRef]
- Sariciftci, N.S. Primary Photoexcitations in Conjugated Polymers: Molecular Exciton Versus Semiconductor Band Model; World Scientific: Singapore, 1998; ISBN 978-981-02-2880-4. [Google Scholar]
- Vandewal, K.; Albrecht, S.; Hoke, E.T.; Graham, K.R.; Widmer, J.; Douglas, J.D.; Schubert, M.; Mateker, W.R.; Bloking, J.T.; Burkhard, G.F.; et al. Efficient charge generation by relaxed charge-transfer states at organic interfaces. Nat. Mater. 2014, 13, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Imahori, H.; Guldi, D.M.; Tamaki, K.; Yoshida, Y.; Luo, C.; Sakata, Y.; Fukuzumi, S. Charge separation in a novel artificial photosynthetic reaction center lives 380 Ms. J. Am. Chem. Soc. 2001, 123, 6617–6628. [Google Scholar] [CrossRef]
- Ohkubo, K.; Fukuzumi, S. Rational design and functions of electron donor–acceptor dyads with much longer charge-separated lifetimes than natural photosynthetic reaction centers. BCSJ 2009, 82, 303–315. [Google Scholar] [CrossRef]
- Yamamoto, M.; Föhlinger, J.; Petersson, J.; Hammarström, L.; Imahori, H. A ruthenium complex–porphyrin–fullerene-linked molecular pentad as an integrative photosynthetic model. Angew. Chem. Int. Ed. 2017, 56, 3329–3333. [Google Scholar] [CrossRef]
- Miyanishi, S.; Zhang, Y.; Tajima, K.; Hashimoto, K. Fullerene attached all-semiconducting diblock copolymers for stable single-component polymer solar cells. Chem. Commun. 2010, 46, 6723–6725. [Google Scholar] [CrossRef]
- Lai, W.; Li, C.; Zhang, J.; Yang, F.; Colberts, F.J.M.; Guo, B.; Wang, Q.M.; Li, M.; Zhang, A.; Janssen, R.A.J.; et al. Diketopyrrolopyrrole-based conjugated polymers with perylene bisimide side chains for single-component organic solar cells. Chem. Mater. 2017, 29, 7073–7077. [Google Scholar] [CrossRef]
- Yu, P.; Feng, G.; Li, J.; Li, C.; Xu, Y.; Xiao, C.; Li, W. A selenophene substituted double-cable conjugated polymer enables efficient single-component organic solar cells. J. Mater. Chem. C 2020, 8, 2790–2797. [Google Scholar] [CrossRef]
- Armin, A.; Stoltzfus, D.M.; Donaghey, J.E.; Clulow, A.J.; Nagiri, R.C.R.; Burn, P.L.; Gentle, I.R.; Meredith, P. Engineering dielectric constants in organic semiconductors. J. Mater. Chem. C 2017, 5, 3736–3747. [Google Scholar] [CrossRef]
- Lee, J.H.; Park, C.G.; Kim, A.; Kim, H.J.; Kim, Y.; Park, S.; Cho, M.J.; Choi, D.H. High-performance polymer solar cell with single active material of fully conjugated block copolymer composed of wide-band gap donor and narrow-band gap acceptor blocks. ACS Appl. Mater. Interfaces 2018, 10, 18974–18983. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Lin, L.-Y.; Lu, C.-W.; Lin, F.; Huang, Z.-Y.; Lin, H.-W.; Wang, P.-H.; Liu, Y.-H.; Wong, K.-T.; Wen, J.; et al. Vacuum-deposited small-molecule organic solar cells with high power conversion efficiencies by judicious molecular design and device optimization. J. Am. Chem. Soc. 2012, 134, 13616–13623. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-Y.; Chen, Y.-H.; Huang, Z.-Y.; Lin, H.-W.; Chou, S.-H.; Lin, F.; Chen, C.-W.; Liu, Y.-H.; Wong, K.-T. A low-energy-gap organic dye for high-performance small-molecule organic solar cells. J. Am. Chem. Soc. 2011, 133, 15822–15825. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, V.; Kronenberg, N.M.; Lenze, M.R.; Graf, S.M.; Hertel, D.; Meerholz, K.; Bürckstümmer, H.; Tulyakova, E.V.; Würthner, F. Simple, highly efficient vacuum-processed bulk heterojunction solar cells based on merocyanine dyes. Adv. Energy Mater. 2011, 1, 888–893. [Google Scholar] [CrossRef]
- Chi, L.-C.; Chen, H.-F.; Hung, W.-Y.; Hsu, Y.-H.; Feng, P.-C.; Chou, S.-H.; Liu, Y.-H.; Wong, K.-T. Donor-acceptor small molecule with coplanar and rigid π-bridge for efficient organic solar cells. Sol. Energy Mater. Sol. Cells 2013, 109, 33–39. [Google Scholar] [CrossRef]
- Lin, L.-Y.; Lu, C.-W.; Huang, W.-C.; Chen, Y.-H.; Lin, H.-W.; Wong, K.-T. New A-A-D-A-A-type electron donors for small molecule organic solar cells. Org. Lett. 2011, 13, 4962–4965. [Google Scholar] [CrossRef]
- Liang, Y.; Xu, Z.; Xia, J.; Tsai, S.-T.; Wu, Y.; Li, G.; Ray, C.; Yu, L. For the bright future—Bulk heterojunction polymer solar cells with power conversion efficiency of 7.4%. Adv. Mater. 2010, 22, E135–E138. [Google Scholar] [CrossRef]
- Nayak, P.K.; Periasamy, N. Calculation of ionization potential of amorphous organic thin-films using solvation model and DFT. Org. Electron. 2009, 10, 532–535. [Google Scholar] [CrossRef]
- Nayak, P.K.; Periasamy, N. Calculation of electron affinity, ionization potential, transport gap, optical band gap and exciton binding energy of organic solids using ‘solvation’ model and DFT. Org. Electron. 2009, 10, 1396–1400. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Scharber, M.C.; Sariciftci, N.S. Efficiency of bulk-heterojunction organic solar cells. Prog. Polym. Sci. 2013, 38, 1929–1940. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | JSC | VOC | FF | PCE | Photon Energy Loss |
|---|---|---|---|---|---|
| (mA/cm2) | (V) | (%) | (%) | (V) | |
| DTNFMN | 0.00 | 1.05 | 15.5 | 0.00 | 0.72 |
| HB194 | 0.12 | 1.25 | 22.6 | 0.04 | 0.60 |
| BCNDTS | 0.16 | 1.22 | 23.7 | 0.05 | 0.59 |
| DTDCTB | 0.50 | 0.79 | 28.8 | 0.11 | 0.70 |
| DTDCPB | 1.00 | 1.03 | 29.1 | 0.30 | 0.69 |
| H2Pc | 0.16 | 0.67 | 31.0 | 0.03 | 0.94 |
| Compounds | IP (eV) | EA (eV) | Eg (eV) | Ex (eV) | EBE (eV) | Δr (ang) | S |
|---|---|---|---|---|---|---|---|
| DTDCPB | −5.524 | −3.438 | 2.086 | 2.292 | −0.206 | 6.177 | 0.301 |
| DTDCTB | −5.499 | −3.400 | 2.099 | 1.980 | 0.119 | 4.125 | 0.372 |
| DTNFMN | −5.554 | −2.837 | 2.717 | 2.425 | 0.292 | 2.694 | 0.378 |
| HB194 | −5.901 | −2.800 | 3.101 | 2.561 | 0.540 | 2.505 | 0.307 |
| BCNDTS | −5.746 | −3.562 | 2.184 | 2.216 | −0.032 | 0.201 | 0.476 |
| Device | JSC | VOC | FF | PCE |
|---|---|---|---|---|
| (mA/cm2) | (V) | (%) | (%) | |
| Normal rate | 1.00 | 1.03 | 29.1 | 0.30 |
| High rate | 1.42 | 1.05 | 33.2 | 0.49 |
| Heated at 50 °C | 0.17 | 1.07 | 24.5 | 0.04 |
| Heated at 100 °C | 0.01 | 0.56 | 25.6 | 0.00 |
| Spin-coated | 0.15 | 1.09 | 23.1 | 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakayama, K.-i.; Okura, T.; Okuda, Y.; Matsui, J.; Masuhara, A.; Yoshida, T.; White, M.S.; Yumusak, C.; Stadler, P.; Scharber, M.; et al. Single-Component Organic Solar Cells Based on Intramolecular Charge Transfer Photoabsorption. Materials 2021, 14, 1200. https://doi.org/10.3390/ma14051200
Nakayama K-i, Okura T, Okuda Y, Matsui J, Masuhara A, Yoshida T, White MS, Yumusak C, Stadler P, Scharber M, et al. Single-Component Organic Solar Cells Based on Intramolecular Charge Transfer Photoabsorption. Materials. 2021; 14(5):1200. https://doi.org/10.3390/ma14051200
Chicago/Turabian StyleNakayama, Ken-ichi, Tatsuya Okura, Yuki Okuda, Jun Matsui, Akito Masuhara, Tsukasa Yoshida, Matthew Schuette White, Cigdem Yumusak, Phillip Stadler, Markus Scharber, and et al. 2021. "Single-Component Organic Solar Cells Based on Intramolecular Charge Transfer Photoabsorption" Materials 14, no. 5: 1200. https://doi.org/10.3390/ma14051200
APA StyleNakayama, K.-i., Okura, T., Okuda, Y., Matsui, J., Masuhara, A., Yoshida, T., White, M. S., Yumusak, C., Stadler, P., Scharber, M., & Sariciftci, N. S. (2021). Single-Component Organic Solar Cells Based on Intramolecular Charge Transfer Photoabsorption. Materials, 14(5), 1200. https://doi.org/10.3390/ma14051200