
Structural Defects, Mechanical Behaviors, and Properties of Two-Dimensional Materials



Abstract
1. Introduction
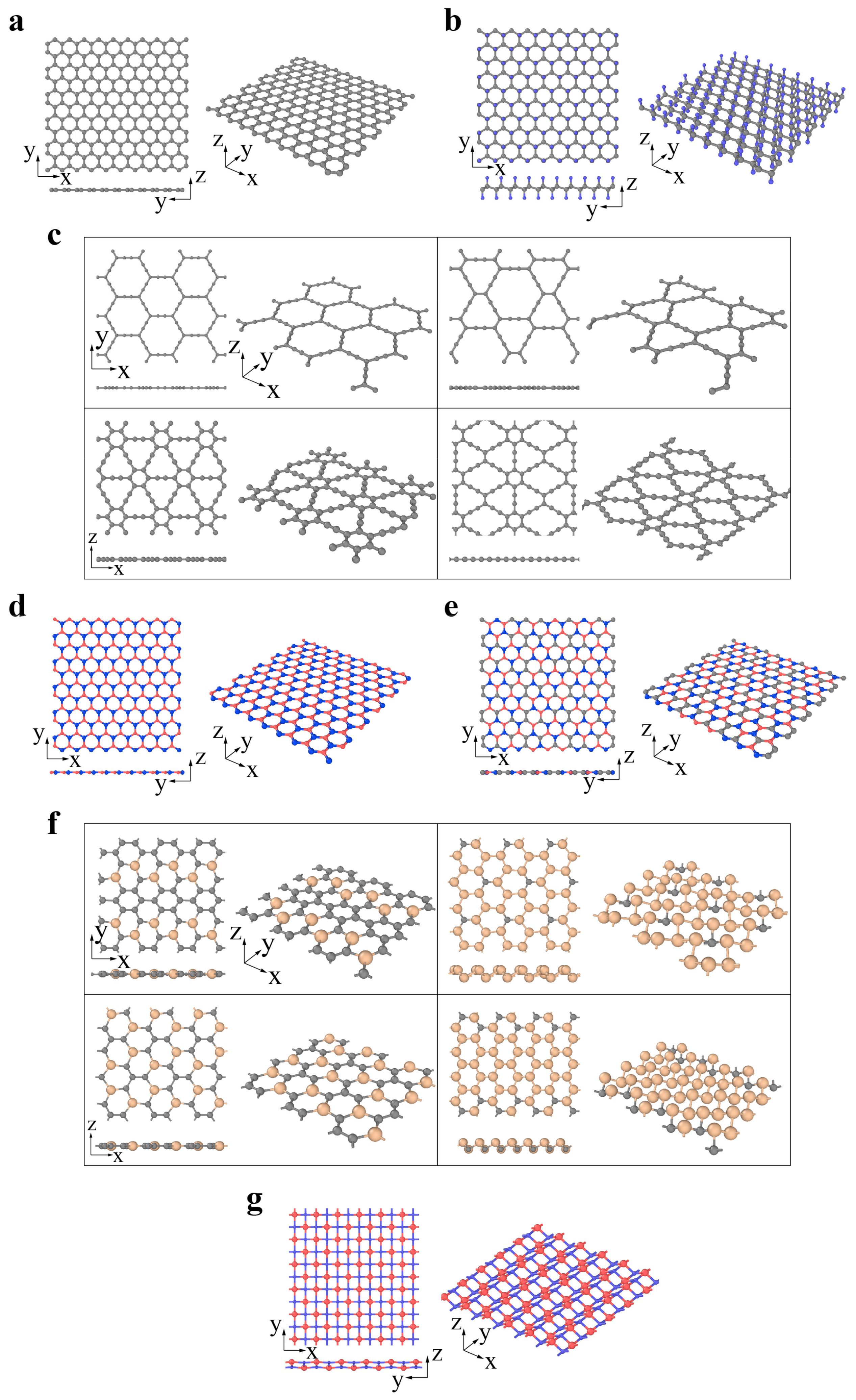
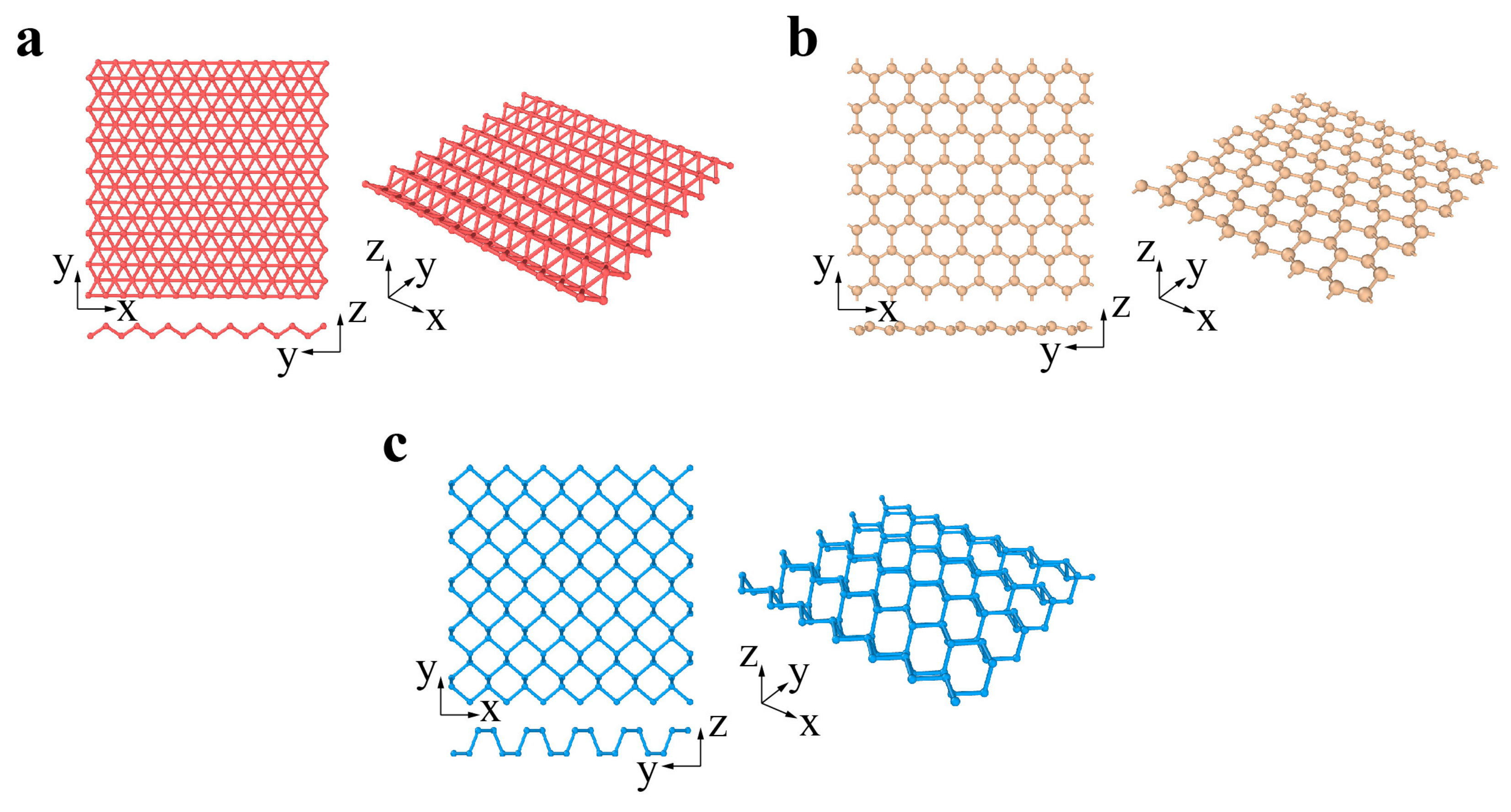
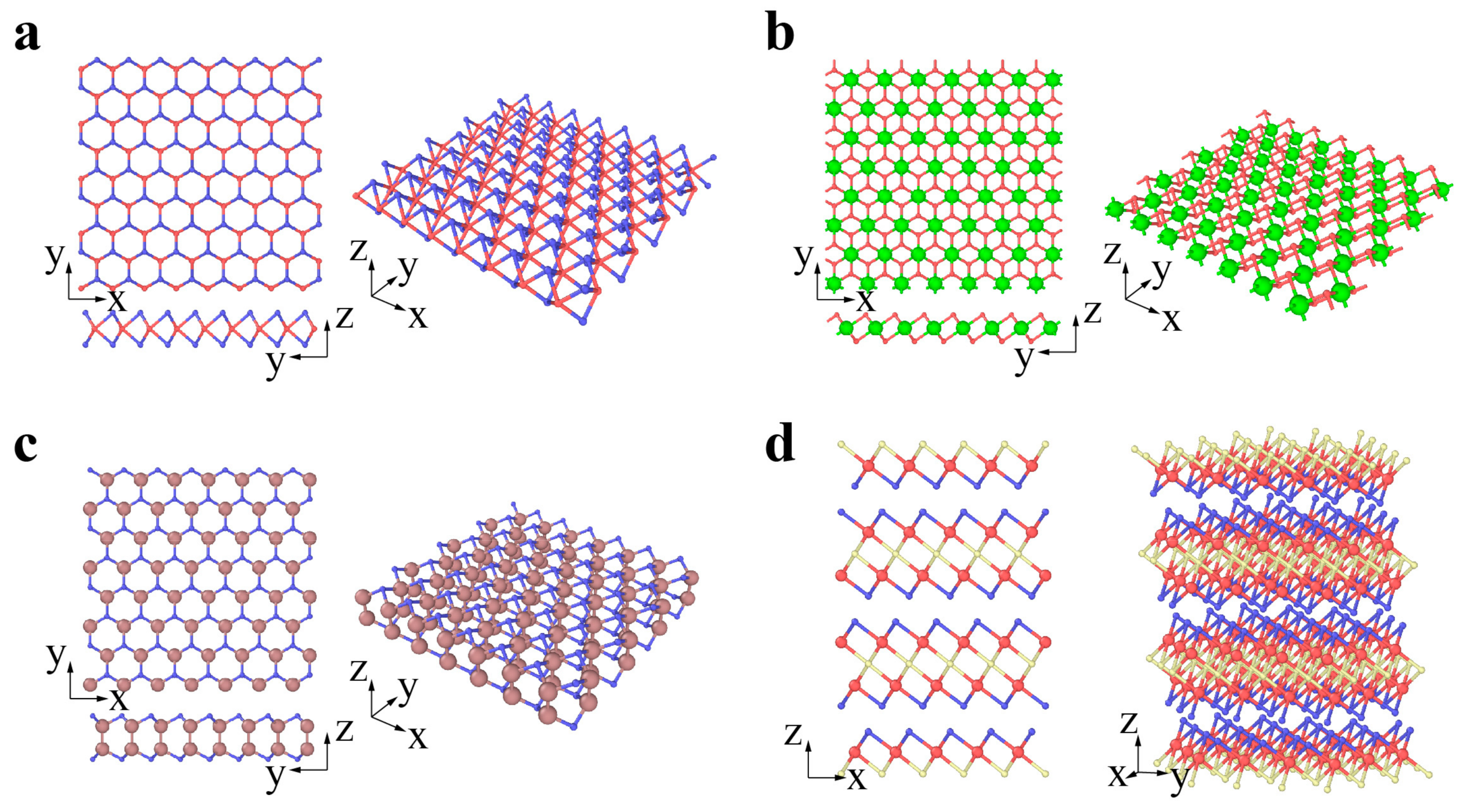
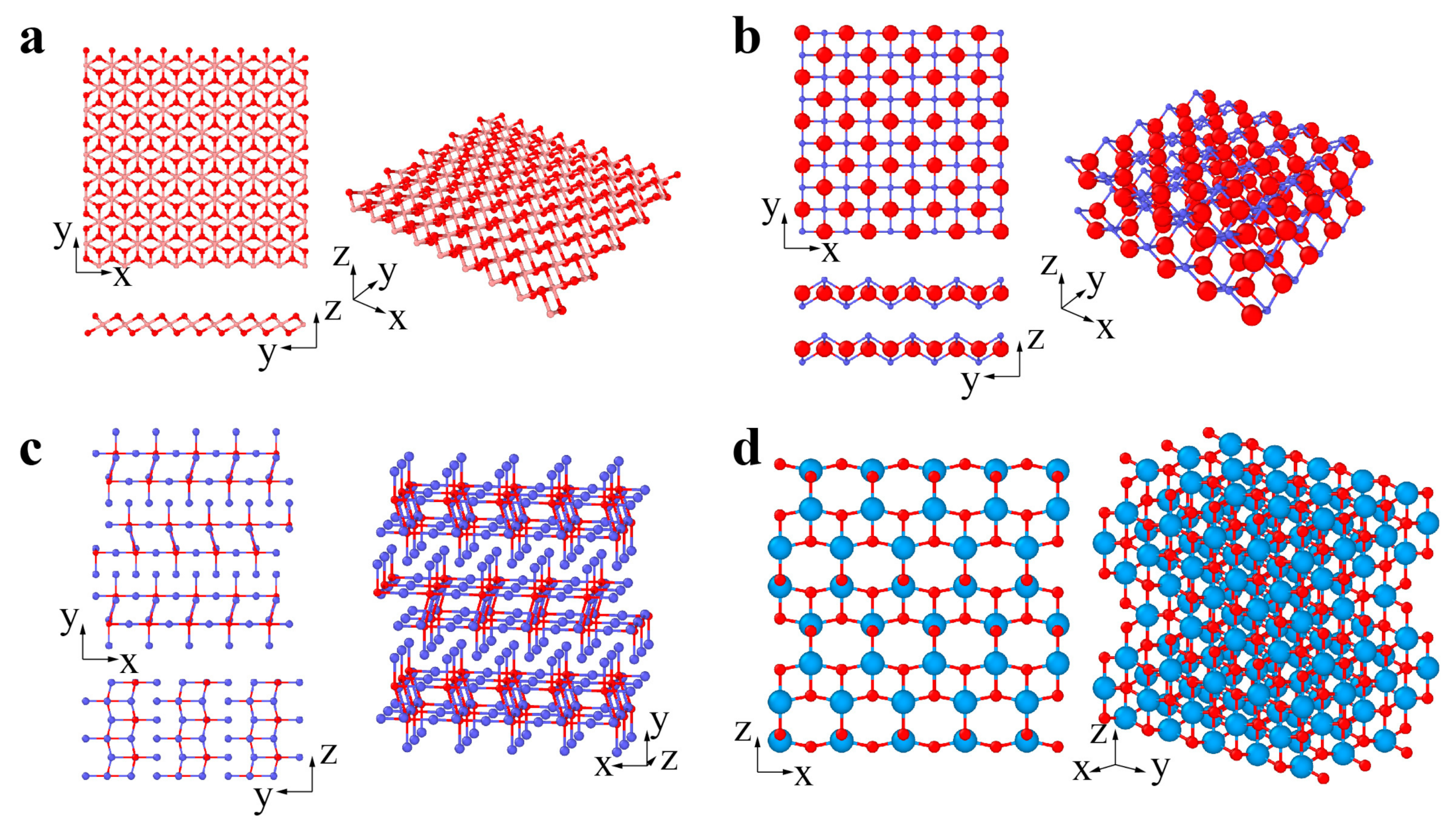
2. Classification and Atomic Structures

3. Structural Defects in Various 2D Materials
3.1. Defects in Graphene and h-BN
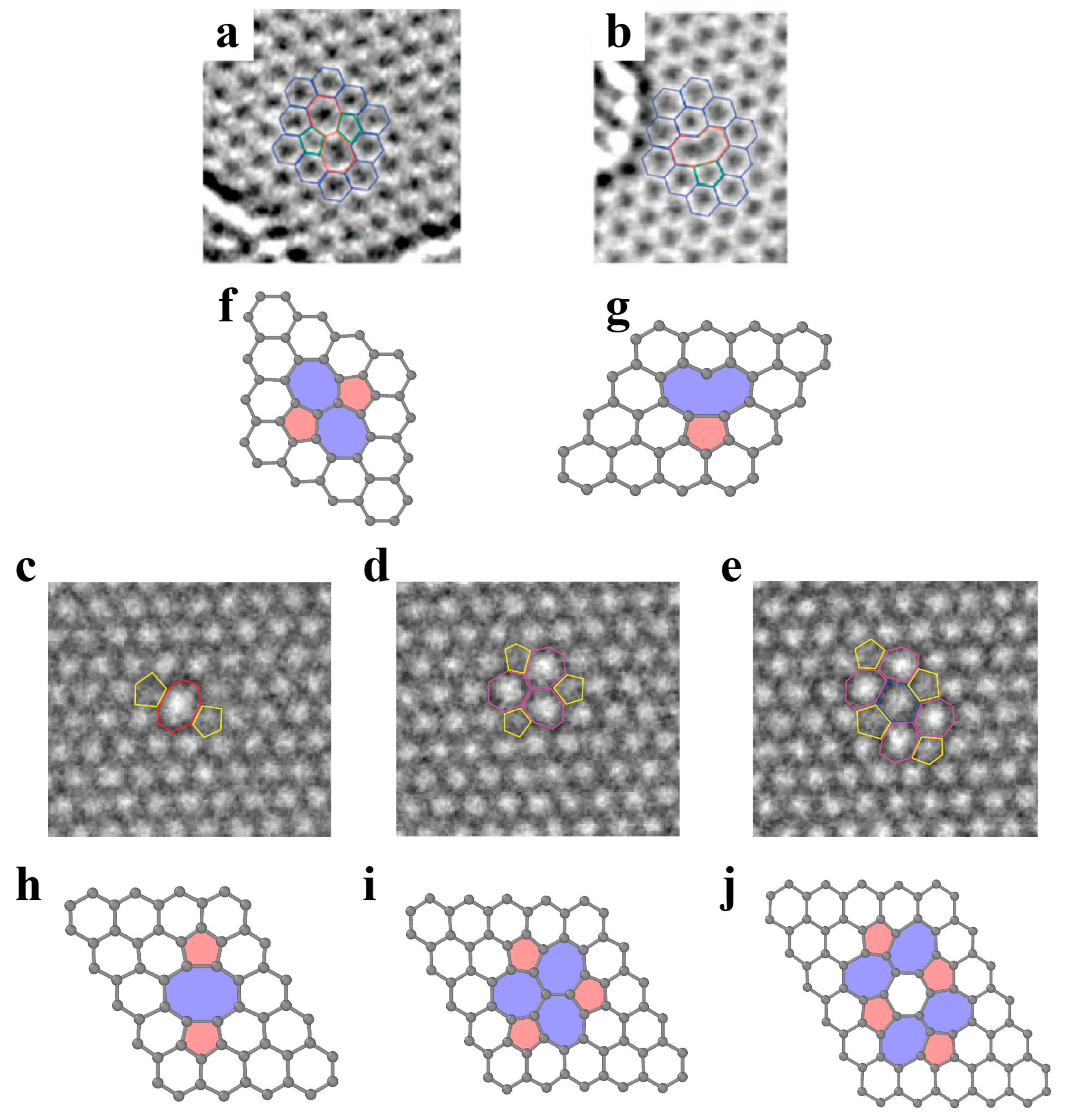
3.1.1. Point Defects


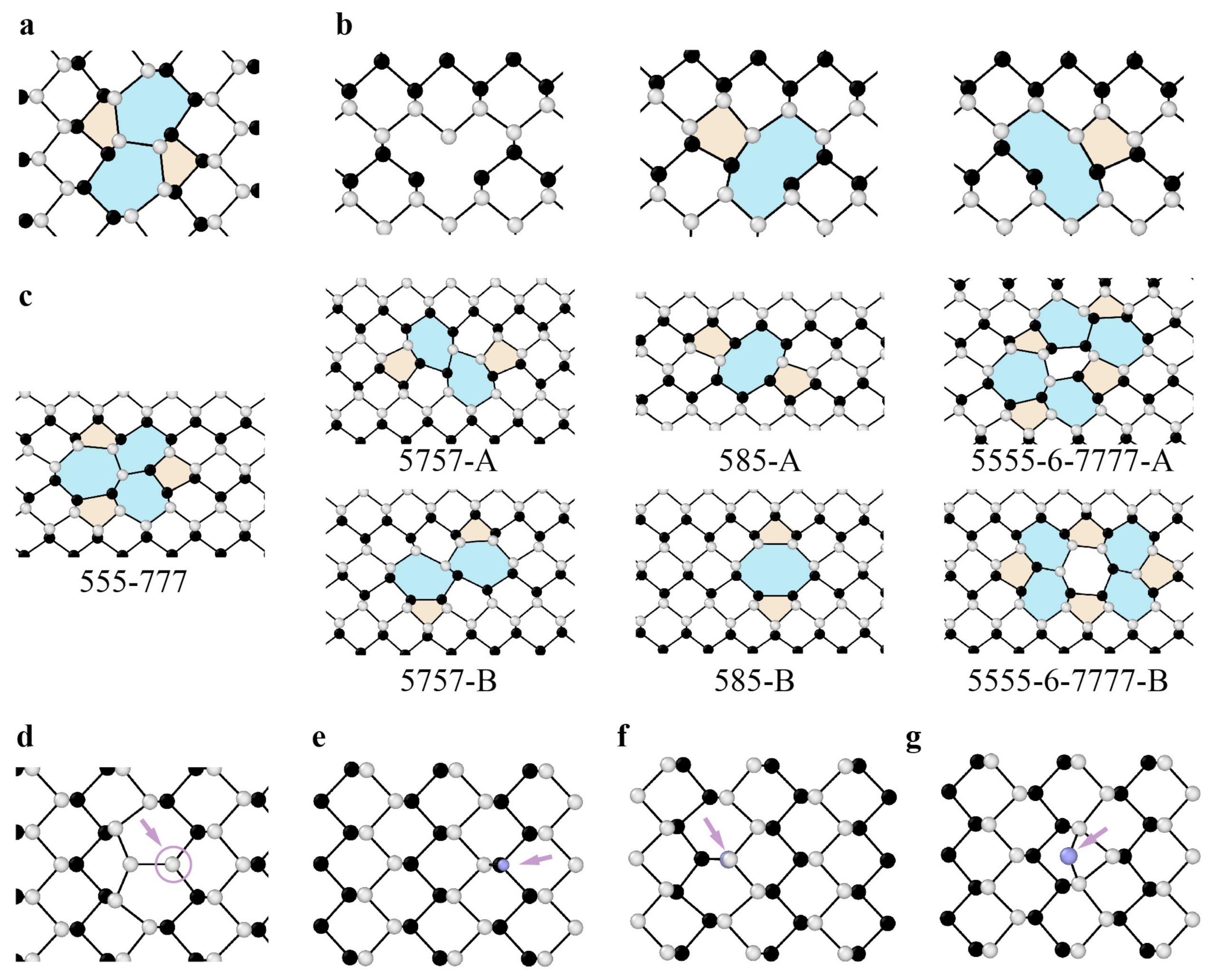
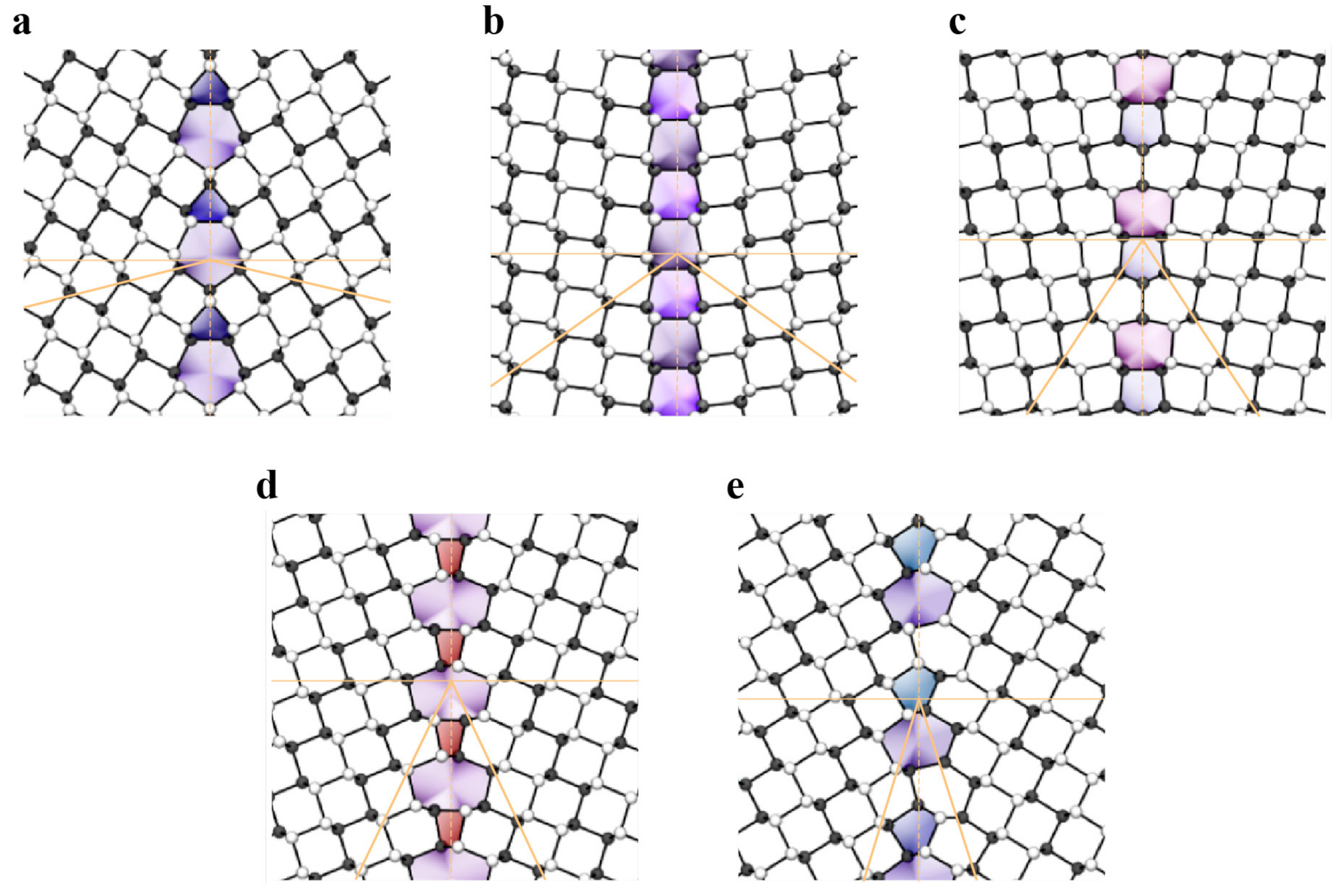
3.1.2. Dislocations and Grain Boundaries

3.2. Defects in MoS2
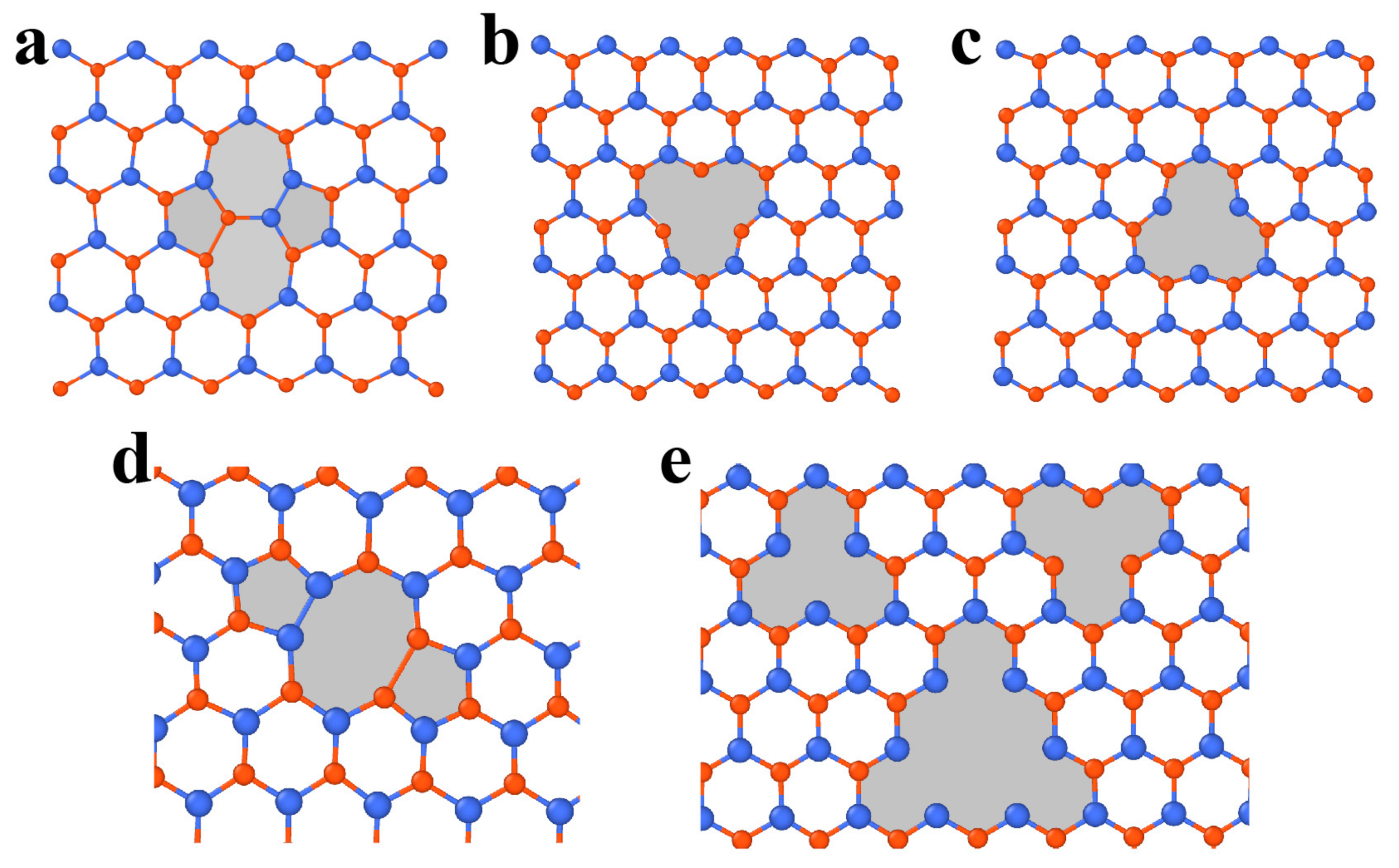
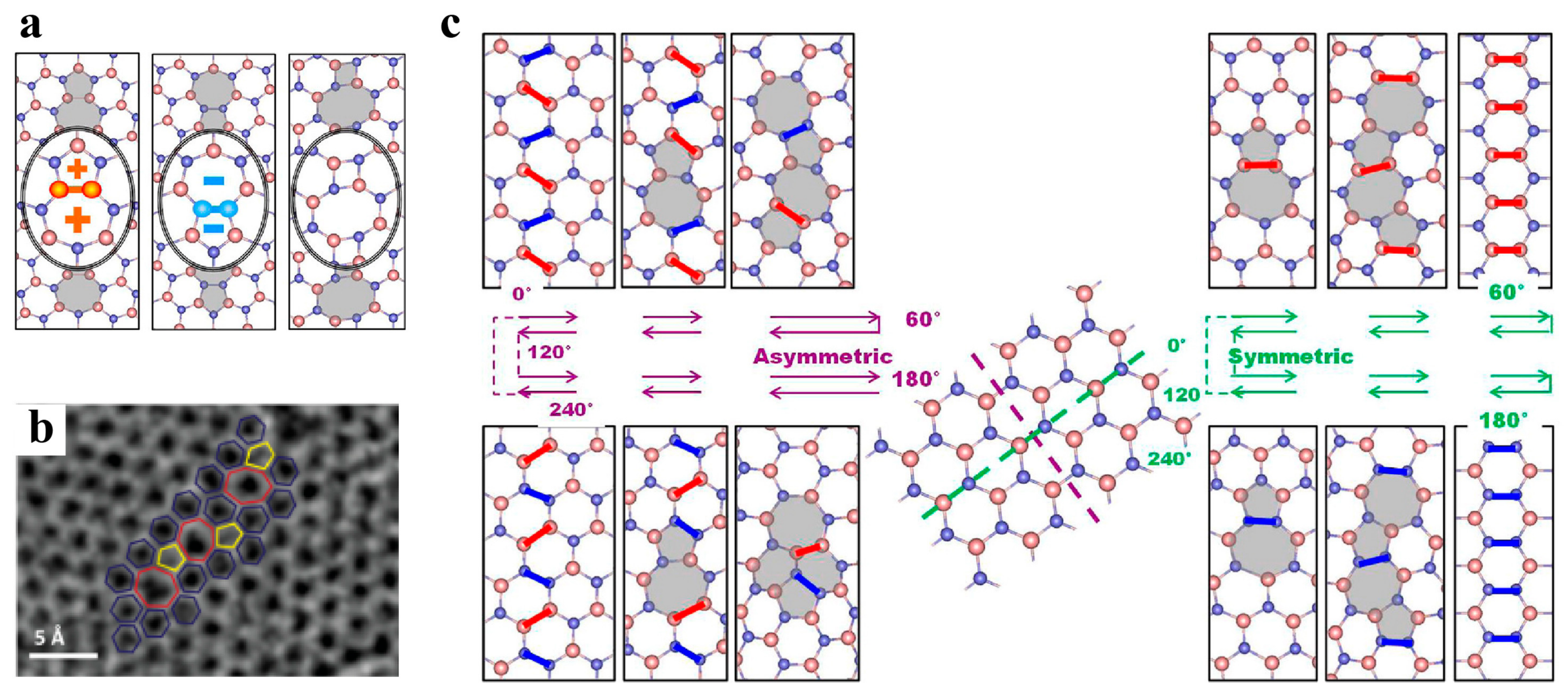
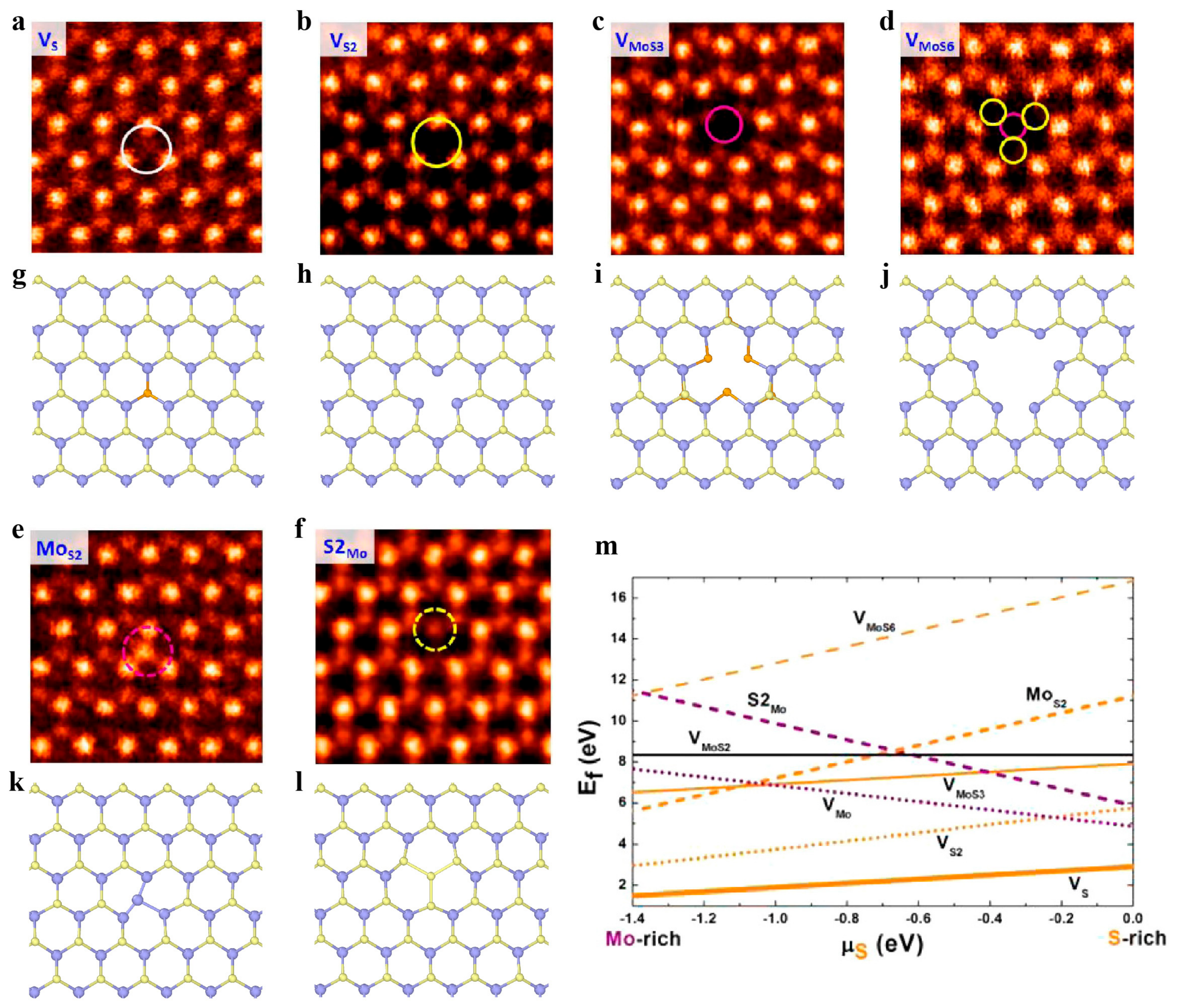
3.2.1. Point Defects
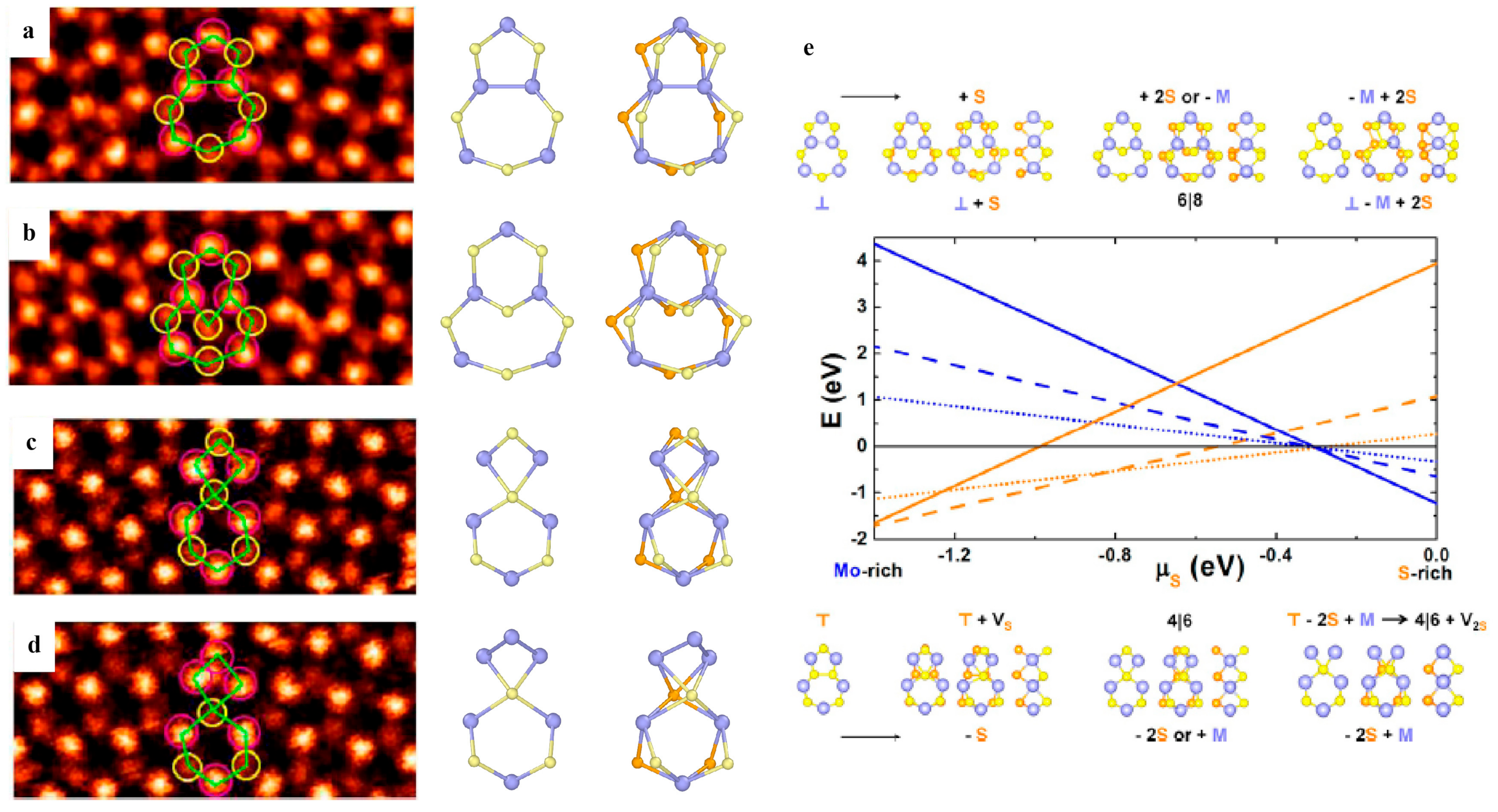
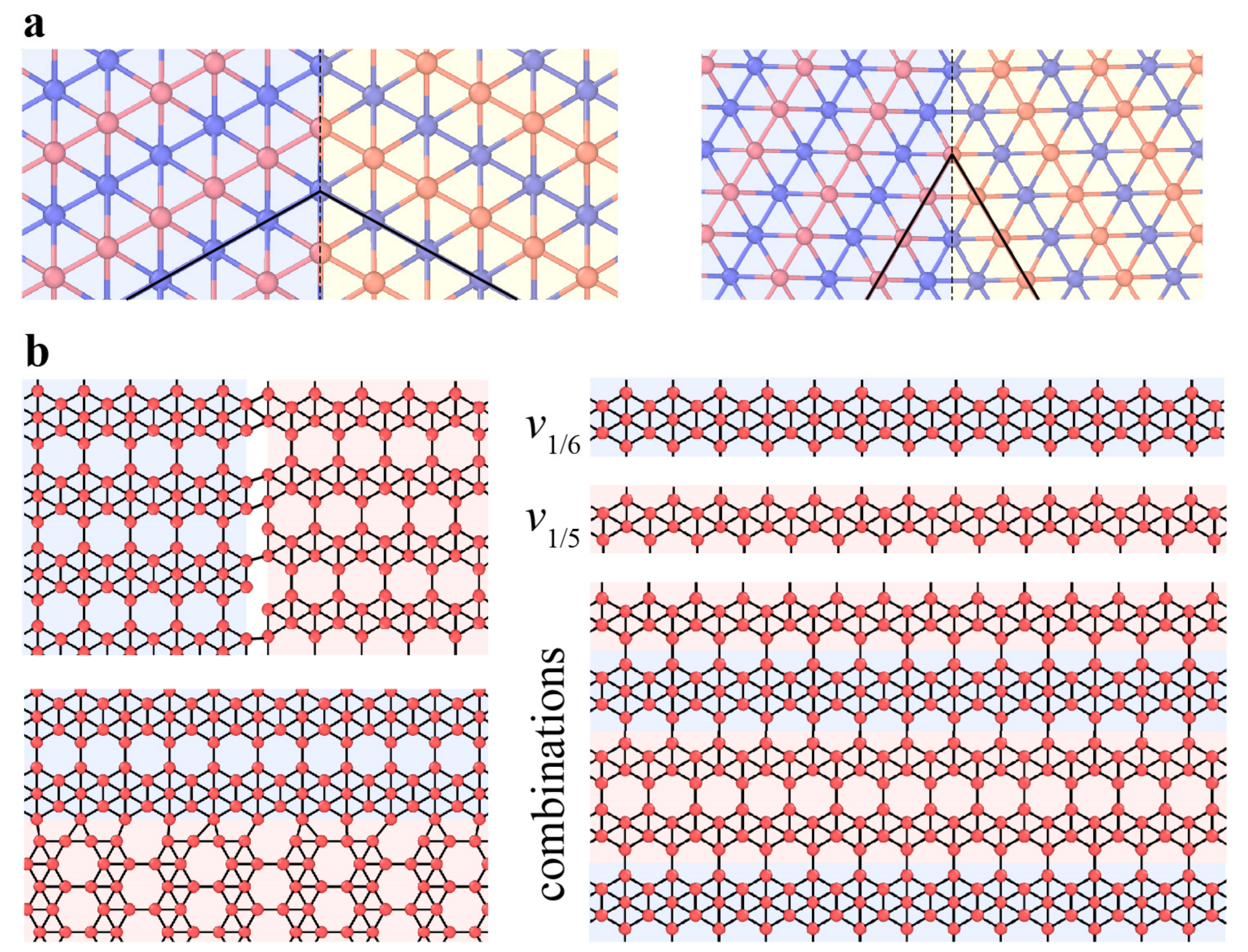
3.2.2. Dislocations and Grain Boundaries
3.3. Defects in Black Phosphorus and Borophene
3.3.1. Point Defects
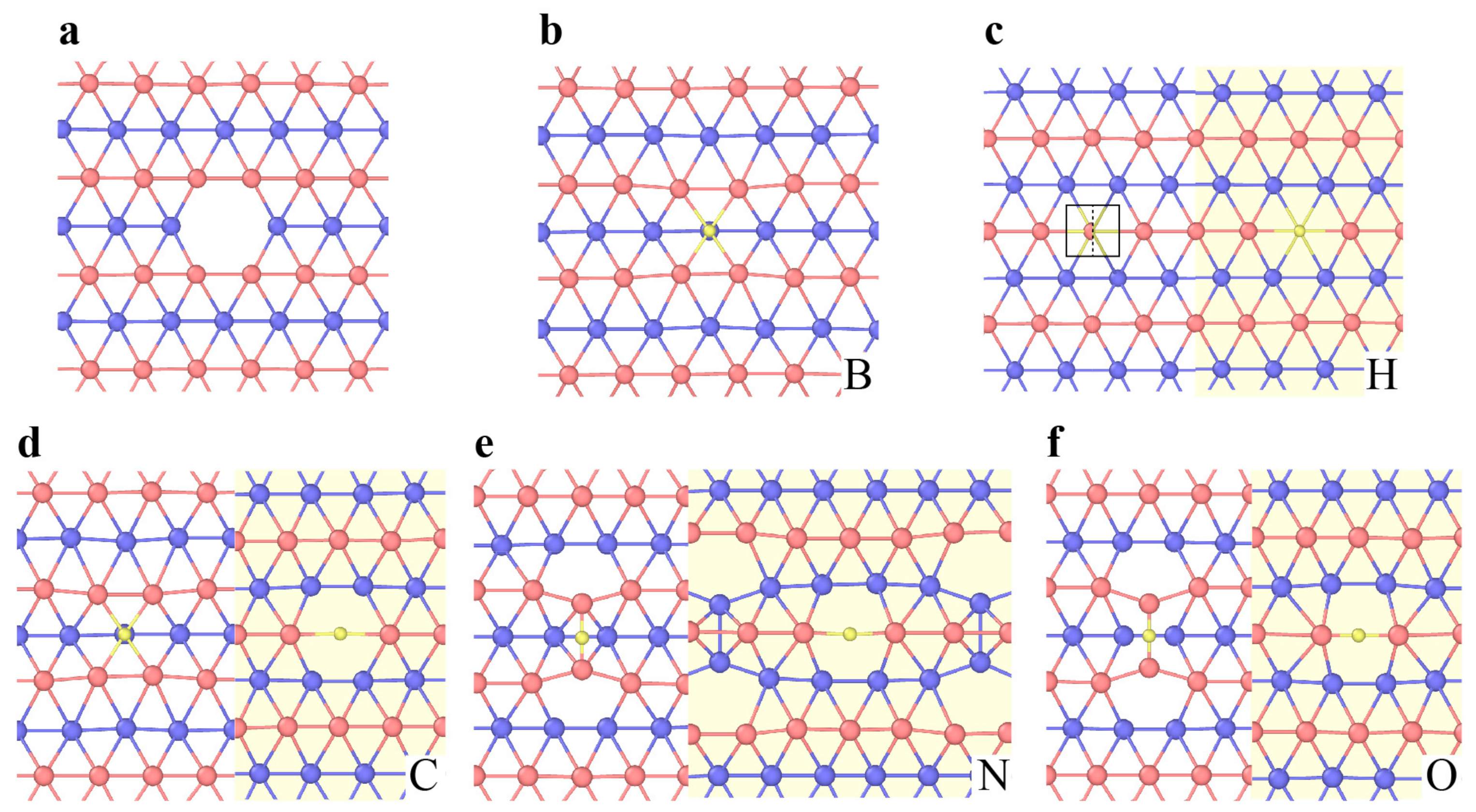
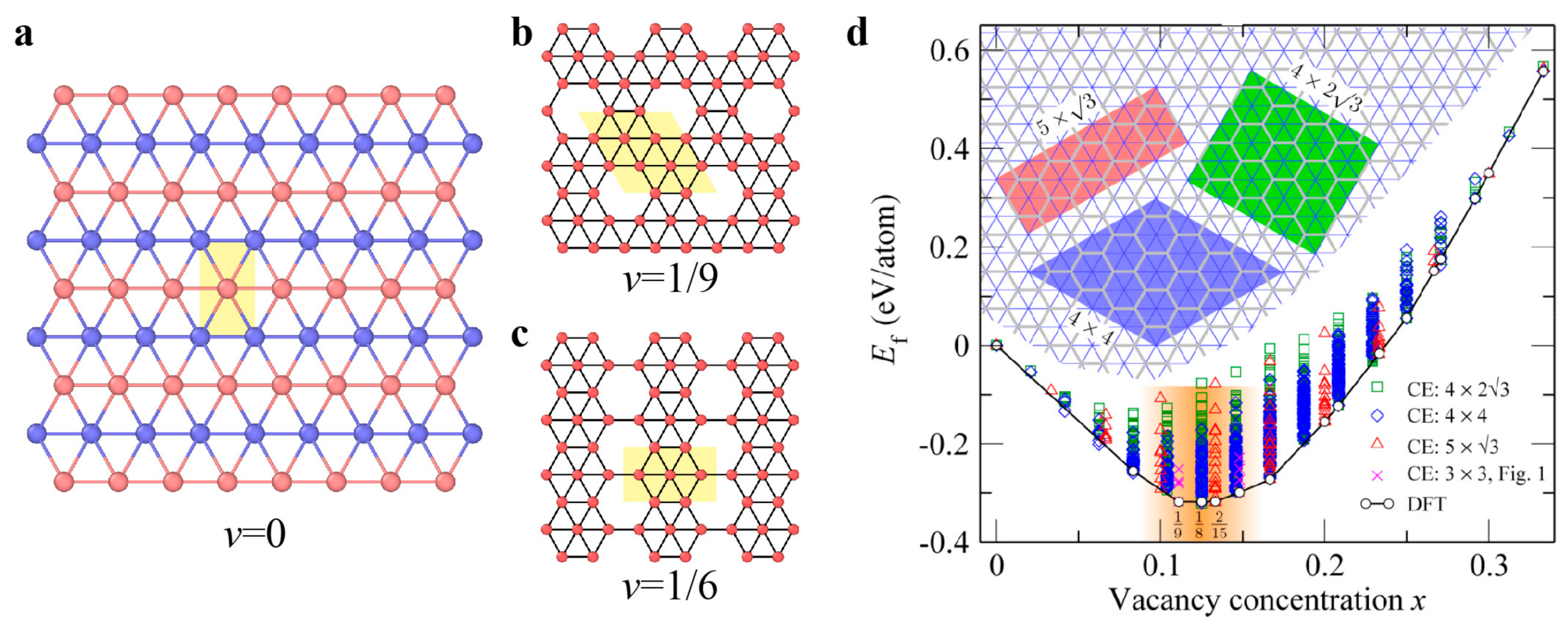
3.3.2. Dislocations and Grain Boundaries

4. Mechanical Behaviors and Properties of 2D Materials
4.1. Modulus and Strength
4.1.1. Experimental Measurements via Nanoindentation in Atomic Force Microscopy
4.1.2. First-Principle Calculations and MD Simulations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | Modulus | Poisson’s Ratio | Strength | Method | Ref. |
|---|---|---|---|---|---|
| graphene | 1.0 ± 0.1 TPa | - | 130 ± 10 GPa | AFM | [8] |
| 1050 GPa | 0.186 | 121 (zz) GPa | DFT | [139] | |
| 110 (ac) GPa | |||||
| h-BN | 223–503 N/m (2–5 layer) | - | 8.8–15.7 N/m (2–5 layer) | AFM | [131] |
| 292.1 N/m | - | 71.7 N/m | Molecular mechanics | [131] | |
| 780 ± 20 (zz) GPa | - | 102 (zz) GPa | DFT | [144] | |
| 773 ± 40 (ac) GPa | 88 (ac) GPa | ||||
| MoS2 | 197.9 ± 4.3 (zz) GPa | 0.21 (in-plane) 0.27 (out-of-plane) | 24.7 (zz) GPa | DFT | [146] |
| 200.3 ± 3.7 (ac) GPa | 25.1 (ac) GPa | ||||
| 129/118 N/m | 0.29/0.31 | - | DFT | [133] | |
| 270 ± 100 GPa | - | 22 ± 4 GPa | AFM | [134] | |
| 330 ± 70 GPa (5–25 layers) | - | - | AFM | [130] | |
| phosphorene | 166 (zz) GPa | 0.62 (zz) | 18 (zz) GPa | DFT | [142] |
| 44 (ac) GPa | 0.17 (ac) | 8 (ac) GPa | |||
| 58.6 ± 11.7 (zz) GPa | - | 4.79 ± 1.43 (zz) GPa | AFM | [135] | |
| 27.2 ± 4.1 (ac) GPa (14~28 nm) | 2.31 ± 0.71 (ac) GPa | ||||
| borophene | 166 (zz) N/m | - | 12.98 (zz) N/m | DFT | [145] |
| 389 (ac) N/m | 20.26 (ac) N/m | ||||
| borophene | 163 (zz) N/m | 0 (zz) –0.23 (ac) | 12.39 (zz) N/m | DFT | [140] |
| 399 (ac) N/m | 21.09 (ac) N/m | ||||
| v1/12 | 161 (zz) N/m | 0.08 (zz) 0.09 (ac) | - | DFT | [140] |
| 208 (ac) N/m | - | ||||
| v1/9 | 212 (zz) N/m | 0.14 (zz) 0.14 (ac) | 18.77 (zz) N/m | DFT | [140] |
| 212 (ac) N/m | 14.38 (ac) N/m | ||||
| v1/8 | 222 (zz) N/m | 0.18 (zz) 0.17 (ac) | 16.87 (zz) N/m | DFT | [140] |
| 216 (ac) N/m | 15.50 (ac) N/m | ||||
| v1/6 | 210 (zz) N/m | 0.17 (zz) 0.15 (ac) | 15.50 (zz) N/m | DFT | [140] |
| 189 (ac) N/m | 16.61 (ac) N/m | ||||
| v1/5 | 208 (zz) N/m | 0.12 (zz) 0.11 (ac) | - | DFT | [140] |
| 196 (ac) N/m | - | ||||
| g-Si | 71.2 N/m | 0.401 | 6.0 (zz) N/m | DFT | [147] |
| 6.3 (ac) N/m | |||||
| b-Si | 63.8 N/m | 0.325 | 5.9 (zz) N/m | DFT | [147] |
| 6.0 (ac) N/m | |||||
| hexagonal silica | 130.5 (zz) N/m | ~0.5 | 38.3 (zz) N/m | DFT | [143] |
| 136.3 (ac) N/m | 35.3 (ac) N/m | ||||
| haeckelite silica | 84.3 (out-of-plane a2) N/m | - | 29.4 (out-of-plane a2) N/m | DFT | [143] |
| 114.8 (in-plane a2) N/m | 27.6 (in-plane a2) N/m |
4.1.3. Theoretical Modeling
4.1.4. Influence of Defects (Grain Boundary and Vacancy) on Strength and Modulus
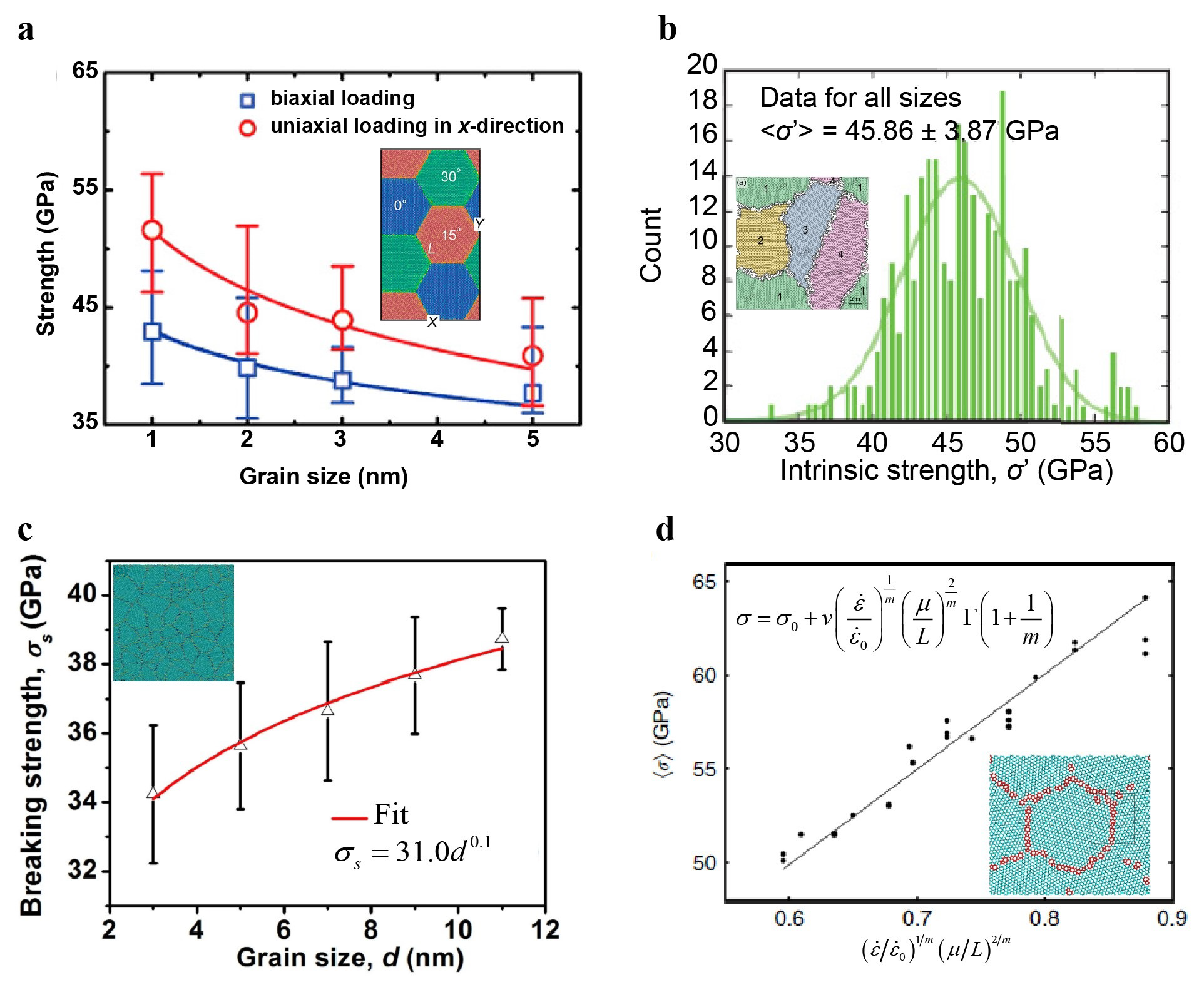
4.1.5. Grain Size Effect on the Strength of Polycrystalline Graphene
4.2. Fracture Behaviors
4.2.1. Model I Fracture of 2D Materials
4.2.2. Toughening Mechanisms

4.2.3. Other Fracture Modes
4.2.4. Fatigue Failure
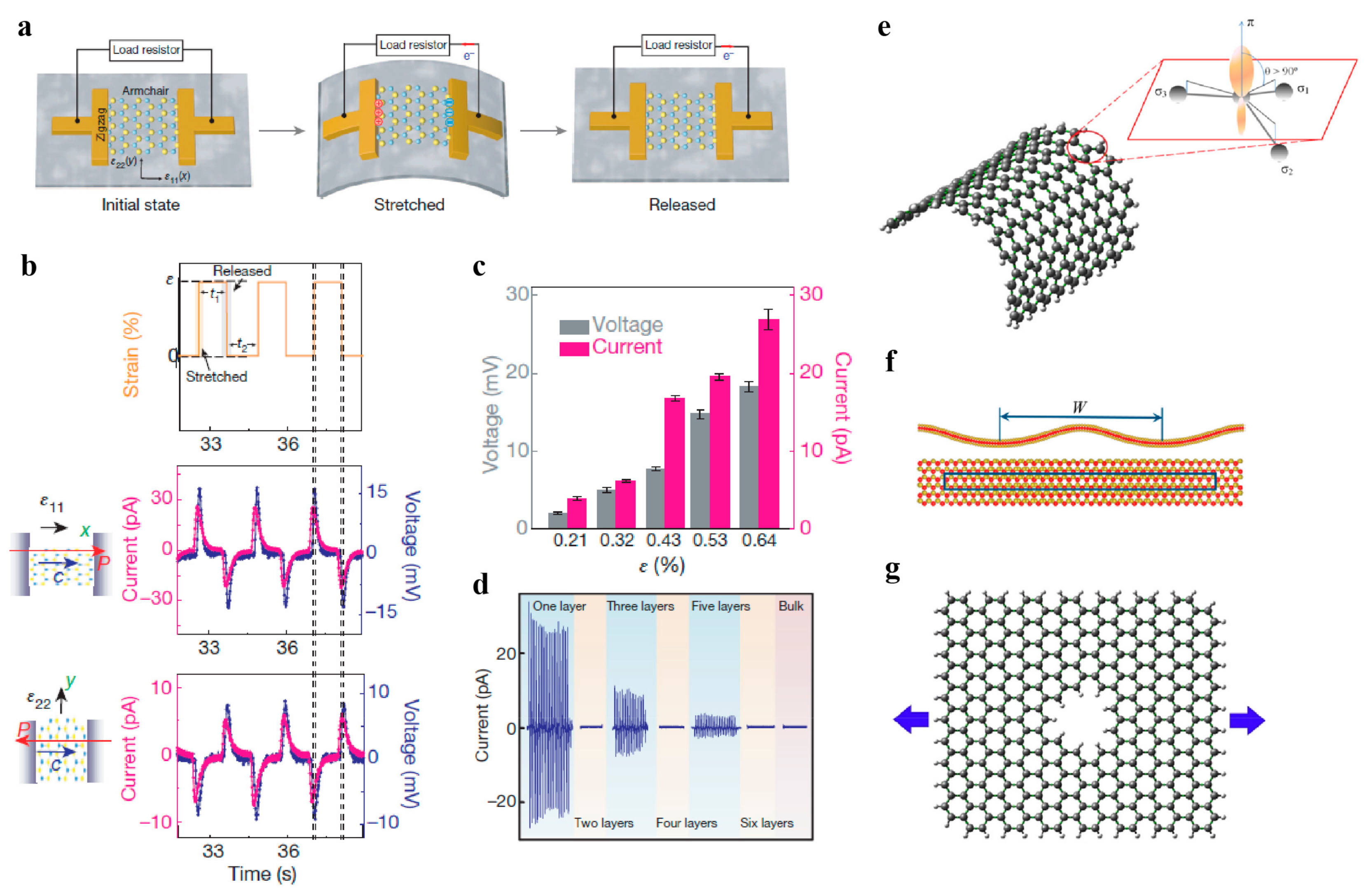
4.3. Piezoelectricity and Flexoelectricity
| Material | e11 (pC/m) | d11 (pm/V) | Material | e11 (pC/m) | d11 (pm/V) |
|---|---|---|---|---|---|
| 2H-CrS2 | 543 | 6.15 | 2H-TaSe2 | 250 | 3.94 |
| 2H-CrSe2 | 575 | 8.25 | 2H-TaTe2 | 207 | 4.72 |
| 2H-CrTe2 | 654 | 13.45 | BeO | 132 | 1.39 |
| 2H-MoS2 | 362 | 3.65 | MgO | 230 | 6.63 |
| 290(Exp) [220] | - | CaO | 155 | 8.47 | |
| 2H-MoSe2 | 383 | 4.55 | ZnO | 266 | 8.65 |
| 2H-MoTe2 | 467 | 7.39 | CdO | 333 | 21.7 |
| 2H-WS2 | 243 | 2.12 | BN | 139 | 0.61 |
| 2H-WSe2 | 257 | 2.64 | BP | 240 | 2.18 |
| 2H-WTe2 | 323 | 4.39 | BAs | 204 | 2.19 |
| 2H-NbS2 | 211 | 3.12 | BSb | 206 | 3.06 |
| 2H-NbSe2 | 222 | 3.87 | AlN | 223 | 2.75 |
| 2H-NbTe2 | 184 | 4.45 | GaN | 148 | 2 |
| 2H-TaS2 | 267 | 3.44 | InN | 224 | 5.5 |
| GaS [209] | 134 | 2.06 | - | - | - |
| GaSe [209] | 147 | 2.30 | - | - | - |
| InSe [209] | 57 | 1.46 | - | - | - |
| Atom(s) | e31 (pC/m) | d31 (pm/V) | Atom(s) | e31 (pC/m) | d31 (pm/V) |
|---|---|---|---|---|---|
| Li | 55 | 0.15 | F | −26 | 0.0018 |
| K | 52 | 0.23 | H, F | −31 | 0.034 |
| H | 20 | 0.11 | F, Li | 30 | 0.3 |
| Material | e11/e31 | d11/d31 | Material | e11/e31 | d11/d31 |
|---|---|---|---|---|---|
| PbO a (p) | 280 | 73.1 | GaAs b | 49/8.2 | 1.5/0.125 |
| AlP a (p) | 3.5 | 0.09 | GaSb a | 33.2/0.8 | 1.42/0.016 |
| AlAs a | 12.7/40.1 | 0.38/0.568 | InP b | 0.5/25.1 | 0.02/0.390 |
| AlSb a | 19.9/18.6 | 0.79/0.351 | InAs b | 1.7/12.6 | 0.08/0.248 |
| GaP b | 52.6/25.9 | 1.29/0.310 | InSb a | 17.9/2.3 | 1.15/0.058 |

4.4. Friction and Interlayer Shear
4.4.1. Sliding Friction on Surfaces
4.4.2. Interlayer Shear/Sliding
4.4.3. Friction Modulation
4.5. Van der Waals Interaction between 2D Materials and Substrate
4.5.1. Tunable Band Gap of 2D Materials on a Substrate
4.5.2. Van der Waals Interface of Heterostructures
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef]
- Akinwande, D.; Brennan, C.J.; Bunch, J.S.; Egberts, P.; Felts, J.R.; Gao, H.; Huang, R.; Kim, J.-S.; Li, T.; Li, Y.; et al. A review on mechanics and mechanical properties of 2D materials—Graphene and beyond. Extreme Mech. Lett. 2017, 13, 42–77. [Google Scholar] [CrossRef]
- Butler, S.Z.; Hollen, S.M.; Cao, L.; Cui, Y.; Gupta, J.A.; Gutiérrez, H.R.; Heinz, T.F.; Hong, S.S.; Huang, J.; Ismach, A.F.; et al. Progress, challenges, and opportunities in two-dimensional materials beyond graphene. ACS Nano 2013, 7, 2898–2926. [Google Scholar] [CrossRef]
- Wang, Q.H.; Kalantar-Zadeh, K.; Kis, A.; Coleman, J.N.; Strano, M.S. Electronics and optoelectronics of two-dimensional transition metal dichalcogenides. Nat. Nanotechnol. 2012, 7, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Wang, K.; Zhu, H. Recent developments in graphene-based membranes: Structure, mass-transport mechanism and potential applications. Adv. Mater. 2016, 28, 2287–2310. [Google Scholar] [CrossRef] [PubMed]
- Mas-Balleste, R.; Gomez-Navarro, C.; Gomez-Herrero, J.; Zamora, F. 2D materials: To graphene and beyond. Nanoscale 2011, 3, 20–30. [Google Scholar] [CrossRef]
- Wei, Y.; Yang, R. Nanomechanics of graphene. Natl. Sci. Rev. 2019, 6, 324–348. [Google Scholar] [CrossRef]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, K.I.; Sikes, K.J.; Jiang, Z.; Klima, M.; Fudenberg, G.; Hone, J.; Kim, P.; Stormer, H.L. Ultrahigh electron mobility in suspended graphene. Solid State Commun. 2008, 146, 351–355. [Google Scholar] [CrossRef]
- Balandin, A.A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior thermal conductivity of single-layer graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.R.; Blake, P.; Grigorenko, A.N.; Novoselov, K.S.; Booth, T.J.; Stauber, T.; Peres, N.M.; Geim, A.K. Fine structure constant defines visual transparency of graphene. Science 2008, 320, 1308. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, Y.; Tian, C.; Girit, C.; Zettl, A.; Crommie, M.; Shen, Y.R. Gate-variable optical transitions in graphene. Science 2008, 320, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.M.; Jenkins, K.A.; Valdes-Garcia, A.; Small, J.P.; Farmer, D.B.; Avouris, P. Operation of graphene transistors at gigahertz frequencies. Nano Lett. 2009, 9, 422–426. [Google Scholar] [CrossRef]
- Wu, Y.; Lin, Y.M.; Bol, A.A.; Jenkins, K.A.; Xia, F.; Farmer, D.B.; Zhu, Y.; Avouris, P. High-frequency, scaled graphene transistors on diamond-like carbon. Nature 2011, 472, 74–78. [Google Scholar] [CrossRef]
- Lin, Y.M.; Dimitrakopoulos, C.; Jenkins, K.A.; Farmer, D.B.; Chiu, H.Y.; Grill, A.; Avouris, P. 100-GHz transistors from wafer-scale epitaxial graphene. Science 2010, 327, 662. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Zhao, Y.; Jang, H.; Lee, S.Y.; Kim, J.M.; Kim, K.S.; Ahn, J.H.; Kim, P.; Choi, J.Y.; Hong, B.H. Large-scale pattern growth of graphene films for stretchable transparent electrodes. Nature 2009, 457, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Park, Y.J.; Chen, X.; Das, T.; Kim, M.S.; Ahn, J.H. Graphene-based flexible and stretchable electronics. Adv. Mater. 2016, 28, 4184–4202. [Google Scholar] [CrossRef]
- Furchi, M.; Urich, A.; Pospischil, A.; Lilley, G.; Unterrainer, K.; Detz, H.; Klang, P.; Andrews, A.M.; Schrenk, W.; Strasser, G.; et al. Microcavity-integrated graphene photodetector. Nano Lett. 2012, 12, 2773–2777. [Google Scholar] [CrossRef]
- Xia, F.; Mueller, T.; Lin, Y.M.; Valdes-Garcia, A.; Avouris, P. Ultrafast graphene photodetector. Nat. Nanotechnol. 2009, 4, 839–843. [Google Scholar] [CrossRef]
- Gan, X.; Shiue, R.-J.; Gao, Y.; Meric, I.; Heinz, T.F.; Shepard, K.; Hone, J.; Assefa, S.; Englund, D. Chip-integrated ultrafast graphene photodetector with high responsivity. Nat. Photonics 2013, 7, 883–887. [Google Scholar] [CrossRef]
- Mueller, T.; Xia, F.; Avouris, P. Graphene photodetectors for high-speed optical communications. Nat. Photonics 2010, 4, 297–301. [Google Scholar] [CrossRef]
- Stankovich, S.; Dikin, D.A.; Dommett, G.H.; Kohlhaas, K.M.; Zimney, E.J.; Stach, E.A.; Piner, R.D.; Nguyen, S.T.; Ruoff, R.S. Graphene-based composite materials. Nature 2006, 442, 282–286. [Google Scholar] [CrossRef]
- El-Kady, M.F.; Strong, V.; Dubin, S.; Kaner, R.B. Laser scribing of high-performance and flexible graphene-based electrochemical capacitors. Science 2012, 335, 1326–1330. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.L.; Srivastava, A.; Gowda, S.R.; Gullapalli, H.; Dubey, M.; Ajayan, P.M. Synthesis of nitrogen-doped graphene films for lithium battery application. ACS Nano 2010, 4, 6337–6342. [Google Scholar] [CrossRef]
- Kim, H.; Park, K.Y.; Hong, J.; Kang, K. All-graphene-battery: Bridging the gap between supercapacitors and lithium ion batteries. Sci. Rep. 2014, 4, 5278. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.J.; Jun, D.H.; Yang, J.H.; Zhou, Z.; Yang, S.S.; Cheng, M.M.-C. Carbon dioxide gas sensor using a graphene sheet. Sens. Actuators B 2011, 157, 310–313. [Google Scholar] [CrossRef]
- Fowler, J.D.; Allen, M.J.; Tung, V.C.; Yang, Y.; Kaner, R.B.; Weiller, B.H. Practical chemical sensors from chemically derived graphene. ACS Nano 2009, 3, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Perkins, F.K.; Snow, E.S.; Wei, Z.; Sheehan, P.E. Reduced graphene oxide molecular sensors. Nano Lett. 2008, 8, 3137–3140. [Google Scholar] [CrossRef]
- Schneider, G.F.; Kowalczyk, S.W.; Calado, V.E.; Pandraud, G.; Zandbergen, H.W.; Vandersypen, L.M.; Dekker, C. DNA translocation through graphene nanopores. Nano Lett. 2010, 10, 3163–3167. [Google Scholar] [CrossRef]
- Heerema, S.J.; Dekker, C. Graphene nanodevices for DNA sequencing. Nat. Nanotechnol. 2016, 11, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Traversi, F.; Raillon, C.; Benameur, S.M.; Liu, K.; Khlybov, S.; Tosun, M.; Krasnozhon, D.; Kis, A.; Radenovic, A. Detecting the translocation of DNA through a nanopore using graphene nanoribbons. Nat. Nanotechnol. 2013, 8, 939–945. [Google Scholar] [CrossRef]
- Liu, J.; Cui, L.; Losic, D. Graphene and graphene oxide as new nanocarriers for drug delivery applications. Acta Biomater. 2013, 9, 9243–9257. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Zhang, S.; Zhang, G.; Sun, X.; Lee, S.T.; Liu, Z. Graphene in mice: Ultrahigh in vivo tumor uptake and efficient photothermal therapy. Nano Lett. 2010, 10, 3318–3323. [Google Scholar] [CrossRef]
- Sun, X.; Liu, Z.; Welsher, K.; Robinson, J.T.; Goodwin, A.; Zaric, S.; Dai, H. Nano-graphene oxide for cellular imaging and drug delivery. Nano Res. 2008, 1, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Mounet, N.; Gibertini, M.; Schwaller, P.; Campi, D.; Merkys, A.; Marrazzo, A.; Sohier, T.; Castelli, I.E.; Cepellotti, A.; Pizzi, G.; et al. Two-dimensional materials from high-throughput computational exfoliation of experimentally known compounds. Nat. Nanotechnol. 2018, 13, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Joensen, P.; Frindt, R.F.; Morrison, S.R. Single-layer MoS2. Mater. Res. Bull. 1986, 21, 457–461. [Google Scholar] [CrossRef]
- Paffett, M.T.; Simonson, R.J.; Papin, P.; Paine, R.T. Borazine adsorption and decomposition at Pt(111) and Ru(001) surfaces. Surf. Sci. 1990, 232, 286–296. [Google Scholar] [CrossRef]
- Nagashima, A.; Tejima, N.; Gamou, Y.; Kawai, T.; Oshima, C. Electronic dispersion relations of monolayer hexagonal boron nitride formed on the Ni(111) surface. Phys. Rev. B 1995, 51, 4606–4613. [Google Scholar] [CrossRef]
- Liu, H.; Neal, A.T.; Zhu, Z.; Luo, Z.; Xu, X.; Tomanek, D.; Ye, P.D. Phosphorene: An unexplored 2D semiconductor with a high hole mobility. ACS Nano 2014, 8, 4033–4041. [Google Scholar] [CrossRef]
- Feng, B.; Zhang, J.; Zhong, Q.; Li, W.; Li, S.; Li, H.; Cheng, P.; Meng, S.; Chen, L.; Wu, K. Experimental realization of two-dimensional boron sheets. Nat. Chem. 2016, 8, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Mannix, A.J.; Zhou, X.-F.; Kiraly, B.; Wood, J.D.; Alducin, D.; Myers, B.D.; Liu, X.; Fisher, B.L.; Santiago, U.; Guest, J.R.; et al. Synthesis of borophenes: Anisotropic, two-dimensional boron polymorphs. Science 2015, 350, 1513–1516. [Google Scholar] [CrossRef] [PubMed]
- Vogt, P.; De Padova, P.; Quaresima, C.; Avila, J.; Frantzeskakis, E.; Asensio, M.C.; Resta, A.; Ealet, B.; Le Lay, G. Silicene: Compelling experimental evidence for graphenelike two-dimensional silicon. Phys. Rev. Lett. 2012, 108, 155501. [Google Scholar] [CrossRef]
- Feng, B.; Ding, Z.; Meng, S.; Yao, Y.; He, X.; Cheng, P.; Chen, L.; Wu, K. Evidence of silicene in honeycomb structures of silicon on Ag(111). Nano Lett. 2012, 12, 3507–3511. [Google Scholar] [CrossRef]
- Dávila, M.E.; Xian, L.; Cahangirov, S.; Rubio, A.; Le Lay, G. Germanene: A novel two-dimensional germanium allotrope akin to graphene and silicene. New J. Phys. 2014, 16, 095002. [Google Scholar]
- Zhu, F.-F.; Chen, W.-J.; Xu, Y.; Gao, C.-L.; Guan, D.-D.; Liu, C.-H.; Qian, D.; Zhang, S.-C.; Jia, J.-F. Epitaxial growth of two-dimensional stanene. Nat. Mater. 2015, 14, 1020. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Song, X.; Liu, J.; Yan, Z.; Huo, C.; Zhang, S.; Su, M.; Liao, L.; Wang, W.; Ni, Z.; et al. Two-dimensional antimonene single crystals grown by van der Waals epitaxy. Nat. Commun. 2016, 7, 13352. [Google Scholar] [CrossRef] [PubMed]
- Nagao, T.; Sadowski, J.T.; Saito, M.; Yaginuma, S.; Fujikawa, Y.; Kogure, T.; Ohno, T.; Hasegawa, Y.; Hasegawa, S.; Sakurai, T. Nanofilm allotrope and phase transformation of ultrathin Bi film on Si(111)-7x7. Phys. Rev. Lett. 2004, 93, 105501. [Google Scholar] [CrossRef]
- Reis, F.; Li, G.; Dudy, L.; Bauernfeind, M.; Glass, S.; Hanke, W.; Thomale, R.; Schafer, J.; Claessen, R. Bismuthene on a SiC substrate: A candidate for a high-temperature quantum spin Hall material. Science 2017, 357, 287–290. [Google Scholar] [CrossRef]
- Zhu, Z.; Cai, X.; Yi, S.; Chen, J.; Dai, Y.; Niu, C.; Guo, Z.; Xie, M.; Liu, F.; Cho, J.H.; et al. Multivalency-driven formation of Te-based monolayer materials: A combined first-principles and experimental study. Phys. Rev. Lett. 2017, 119, 106101. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; McGaughey, A.J. Strongly anisotropic in-plane thermal transport in single-layer black phosphorene. Sci. Rep. 2015, 5, 8501. [Google Scholar] [CrossRef]
- Watanabe, K.; Taniguchi, T.; Kanda, H. Direct-bandgap properties and evidence for ultraviolet lasing of hexagonal boron nitride single crystal. Nat. Mater. 2004, 3, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single-layer MoS2 transistors. Nat. Nanotechnol. 2011, 6, 147–150. [Google Scholar] [CrossRef]
- Lopez-Sanchez, O.; Lembke, D.; Kayci, M.; Radenovic, A.; Kis, A. Ultrasensitive photodetectors based on monolayer MoS2. Nat. Nanotechnol. 2013, 8, 497–501. [Google Scholar] [CrossRef]
- Yin, Z.; Li, H.; Li, H.; Jiang, L.; Shi, Y.; Sun, Y.; Lu, G.; Zhang, Q.; Chen, X.; Zhang, H. Single-layer MoS2 phototransistors. ACS Nano 2012, 6, 74–80. [Google Scholar] [CrossRef]
- Fang, H.; Chuang, S.; Chang, T.C.; Takei, K.; Takahashi, T.; Javey, A. High-performance single layered WSe2 p-FETs with chemically doped contacts. Nano Lett. 2012, 12, 3788–3792. [Google Scholar] [CrossRef] [PubMed]
- Mannix, A.J.; Zhang, Z.; Guisinger, N.P.; Yakobson, B.I.; Hersam, M.C. Borophene as a prototype for synthetic 2D materials development. Nat. Nanotechnol. 2018, 13, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Adamska, L.; Sadasivam, S.; Foley, J.J.; Darancet, P.; Sharifzadeh, S. First-principles investigation of borophene as a monolayer transparent conductor. J. Phys. Chem. C 2018, 122, 4037–4045. [Google Scholar] [CrossRef]
- Ji, Q.; Li, C.; Wang, J.; Niu, J.; Gong, Y.; Zhang, Z.; Fang, Q.; Zhang, Y.; Shi, J.; Liao, L.; et al. Metallic vanadium disulfide nanosheets as a platform material for multifunctional electrode applications. Nano Lett. 2017, 17, 4908–4916. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, Y.; Zhang, S.-C. Two-dimensional time-reversal-invariant topological superconductivity in a doped quantum spin-Hall insulator. Phys. Rev. B 2014, 90, 054503. [Google Scholar] [CrossRef]
- Albrecht, T.R.; Mizes, H.A.; Nogami, J.; Park, S.-I.; Quate, C.F. Observation of tilt boundaries in graphite by scanning tunneling microscopy and associated multiple tip effects. Appl. Phys. Lett. 1988, 52, 362–364. [Google Scholar] [CrossRef]
- Kim, K.; Lee, Z.; Regan, W.; Kisielowski, C.; Crommie, M.F.; Zettl, A. Grain boundary mapping in polycrystalline graphene. ACS Nano 2011, 5, 2142–2146. [Google Scholar] [CrossRef]
- Haugan, T.; Barnes, P.N.; Wheeler, R.; Meisenkothen, F.; Sumption, M. Addition of nanoparticle dispersions to enhance flux pinning of the YBa2Cu3O7−x superconductor. Nature 2004, 430, 867–870. [Google Scholar] [CrossRef] [PubMed]
- Balog, R.; Jorgensen, B.; Nilsson, L.; Andersen, M.; Rienks, E.; Bianchi, M.; Fanetti, M.; Laegsgaard, E.; Baraldi, A.; Lizzit, S.; et al. Bandgap opening in graphene induced by patterned hydrogen adsorption. Nat. Mater. 2010, 9, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Bagri, A.; Mattevi, C.; Acik, M.; Chabal, Y.J.; Chhowalla, M.; Shenoy, V.B. Structural evolution during the reduction of chemically derived graphene oxide. Nat. Chem. 2010, 2, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Tanabe, Y.; Qiu, H.J.; Sugawara, K.; Heguri, S.; Tu, N.H.; Huynh, K.K.; Fujita, T.; Takahashi, T.; Tanigaki, K.; et al. High-quality three-dimensional nanoporous graphene. Angew. Chem. Int. Ed. Engl. 2014, 53, 4822–4826. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, J.; Lin, Y.; Bozkurt, P.; Oleynik, I.I.; Batzill, M. An extended defect in graphene as a metallic wire. Nat. Nanotechnol. 2010, 5, 326–329. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Pei, Q.X.; Wang, C.M. Mechanical properties of graphynes under tension: A molecular dynamics study. Appl. Phys. Lett. 2012, 101, 081909. [Google Scholar] [CrossRef]
- Malko, D.; Neiss, C.; Vines, F.; Gorling, A. Competition for graphene: Graphynes with direction-dependent Dirac cones. Phys. Rev. Lett. 2012, 108, 086804. [Google Scholar] [CrossRef]
- Li, G.; Li, Y.; Liu, H.; Guo, Y.; Li, Y.; Zhu, D. Architecture of graphdiyne nanoscale films. Chem. Commun. 2010, 46, 3256–3258. [Google Scholar] [CrossRef]
- Li, Y.; Xu, L.; Liu, H.; Li, Y. Graphdiyne and graphyne: From theoretical predictions to practical construction. Chem. Soc. Rev. 2014, 43, 2572–2586. [Google Scholar] [CrossRef]
- Beniwal, S.; Hooper, J.; Miller, D.P.; Costa, P.S.; Chen, G.; Liu, S.Y.; Dowben, P.A.; Sykes, E.C.; Zurek, E.; Enders, A. Graphene-like Boron-Carbon-Nitrogen monolayers. ACS Nano 2017, 11, 2486–2493. [Google Scholar] [CrossRef]
- Shi, Z.; Zhang, Z.; Kutana, A.; Yakobson, B.I. Predicting two-dimensional silicon carbide monolayers. ACS Nano 2015, 9, 9802–9809. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, X.; Yakobson, B.I.; Guo, W. Two-dimensional tetragonal tic monolayer sheet and nanoribbons. J. Am. Chem. Soc. 2012, 134, 19326–19329. [Google Scholar] [CrossRef]
- Wang, T.; Wang, H.; Kou, Z.; Liang, W.; Luo, X.; Verpoort, F.; Zeng, Y.J.; Zhang, H. Xenes as an emerging 2D monoelemental family: Fundamental electrochemistry and energy applications. Adv. Funct. Mater. 2020, 30, 2002885. [Google Scholar]
- Kappera, R.; Voiry, D.; Yalcin, S.E.; Branch, B.; Gupta, G.; Mohite, A.D.; Chhowalla, M. Phase-engineered low-resistance contacts for ultrathin MoS2 transistors. Nat. Mater. 2014, 13, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Dumcenco, D.O.; Huang, Y.S.; Suenaga, K. Atomic mechanism of the semiconducting-to-metallic phase transition in single-layered MoS2. Nat. Nanotechnol. 2014, 9, 391–396. [Google Scholar] [CrossRef]
- Duerloo, K.A.; Li, Y.; Reed, E.J. Structural phase transitions in two-dimensional Mo- and W-dichalcogenide monolayers. Nat. Commun. 2014, 5, 4214. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Zhang, H.; Wang, W.; Colombo, L.; Wallace, R.M.; Cho, K. Band alignment of two-dimensional transition metal dichalcogenides: Application in tunnel field effect transistors. Appl. Phys. Lett. 2013, 103, 053513. [Google Scholar] [CrossRef]
- Zhen, Y.-X.; Yang, M.; Zhang, H.; Fu, G.-S.; Wang, J.-L.; Wang, S.-F.; Wang, R.-N. Ultrahigh power factors in P-type 1T-ZrX2 (X = S, Se) single layers. Sci. Bull. 2017, 62, 1530–1537. [Google Scholar] [CrossRef]
- Yagmurcukardes, M.; Senger, R.T.; Peeters, F.M.; Sahin, H. Mechanical properties of monolayer GaS and GaSe crystals. Phys. Rev. B 2016, 94, 245407. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, C.-X.; Qi, X.-L.; Dai, X.; Fang, Z.; Zhang, S.-C. Topological insulators in Bi2Se3, Bi2Te3 and Sb2Te3 with a single Dirac cone on the surface. Nat. Phys. 2009, 5, 438–442. [Google Scholar] [CrossRef]
- Azadmanjiri, J.; Srivastava, V.K.; Kumar, P.; Wang, J.; Yu, A. Graphene-supported 2D transition metal oxide heterostructures. J. Mater. Chem. A 2018, 6, 13509–13537. [Google Scholar] [CrossRef]
- Kumar, P.; Liu, J.; Ranjan, P.; Hu, Y.; Yamijala, S.S.; Pati, S.K.; Irudayaraj, J.; Cheng, G.J. Alpha lead oxide (α-PbO): A new 2D material with visible light sensitivity. Small 2018, 14, 1703346. [Google Scholar] [CrossRef] [PubMed]
- Haque, F.; Daeneke, T.; Kalantar-Zadeh, K.; Ou, J.Z. Two-dimensional transition metal oxide and chalcogenide-based photocatalysts. Nano-Micro Lett. 2018, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Banhart, F.; Kotakoski, J.; Krasheninnikov, A.V. Structural defects in graphene. ACS Nano 2011, 5, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Ni, B.; Zhang, T.; Li, J.; Li, X.; Gao, H. Topological design of graphene. In Handbook of Graphene; Stauber, T., Ed.; Scrivener: New York, NY, USA, 2019; Volume 2, pp. 1–44. [Google Scholar]
- Li, L.; Reich, S.; Robertson, J. Defect energies of graphite: Density-functional calculations. Phys. Rev. B 2005, 72, 184109. [Google Scholar] [CrossRef]
- Ma, J.; Alfè, D.; Michaelides, A.; Wang, E. Stone-Wales defects in graphene and other planar sp2-bonded materials. Phys. Rev. B 2009, 80, 033407. [Google Scholar] [CrossRef]
- Li, Y.F.; Zhou, Z.; Golberg, D.; Bando, Y.; Schleyer, P.V.; Chen, Z.F. Stone-wales defects in single-walled boron nitride nanotubes: Formation energies, electronic structures, and reactivity. J. Phys. Chem. C 2008, 112, 1365–1370. [Google Scholar] [CrossRef]
- Meyer, J.C.; Kisielowski, C.; Erni, R.; Rossell, M.D.; Crommie, M.F.; Zettl, A. Direct imaging of lattice atoms and topological defects in graphene membranes. Nano Lett. 2008, 8, 3582–3586. [Google Scholar] [CrossRef]
- Krasheninnikov, A.V.; Lehtinen, P.O.; Foster, A.S.; Nieminen, R.M. Bending the rules: Contrasting vacancy energetics and migration in graphite and carbon nanotubes. Chem. Phys. Lett. 2006, 418, 132–136. [Google Scholar] [CrossRef]
- El-Barbary, A.A.; Telling, R.H.; Ewels, C.P.; Heggie, M.I.; Briddon, P.R. Structure and energetics of the vacancy in graphite. Phys. Rev. B 2003, 68, 144107. [Google Scholar] [CrossRef]
- Lusk, M.T.; Carr, L.D. Nanoengineering defect structures on graphene. Phys. Rev. Lett. 2008, 100, 175503. [Google Scholar] [CrossRef]
- Kotakoski, J.; Meyer, J.C.; Kurasch, S.; Santos-Cottin, D.; Kaiser, U.; Krasheninnikov, A.V. Stone-Wales-type transformations in carbon nanostructures driven by electron irradiation. Phys. Rev. B 2011, 83, 245420. [Google Scholar] [CrossRef]
- Kim, Y.; Ihm, J.; Yoon, E.; Lee, G.-D. Dynamics and stability of divacancy defects in graphene. Phys. Rev. B 2011, 84, 075445. [Google Scholar] [CrossRef]
- Hou, Z.; Wang, X.; Ikeda, T.; Terakura, K.; Oshima, M.; Kakimoto, M.-A.; Miyata, S. Interplay between nitrogen dopants and native point defects in graphene. Phys. Rev. B 2012, 85, 165439. [Google Scholar] [CrossRef]
- Si, M.S.; Li, J.Y.; Shi, H.G.; Niu, X.N.; Xue, D.S. Divacancies in graphitic boron nitride sheets. EPL 2009, 86. [Google Scholar] [CrossRef]
- Si, M.S.; Xue, D.S. Magnetic properties of vacancies in a graphitic boron nitride sheet by first-principles pseudopotential calculations. Phys. Rev. B 2007, 75, 193409. [Google Scholar] [CrossRef]
- Zobelli, A.; Ewels, C.P.; Gloter, A.; Seifert, G.; Stephan, O.; Csillag, S.; Colliex, C. Defective structure of BN nanotubes: From single vacancies to dislocation lines. Nano Lett. 2006, 6, 1955–1960. [Google Scholar] [CrossRef]
- Jin, C.; Lin, F.; Suenaga, K.; Iijima, S. Fabrication of a freestanding boron nitride single layer and its defect assignments. Phys. Rev. Lett. 2009, 102, 195505. [Google Scholar] [CrossRef]
- Kolesnikova, A.L.; Rozhkov, M.A.; Hussainova, I.; Orlova, T.S.; Yasnikov, I.S.; Zhigilei, L.V.; Romanov, A.E. Structure and energy of intercrystallite boundaries in graphene. Rev. Adv. Mater. Sci. 2017, 52, 91–98. [Google Scholar]
- Romanov, A.E.; Rozhkov, M.A.; Kolesnikova, A.L. Disclinations in polycrystalline graphene and pseudo-graphenes. Rev. Lett. Mater. 2018, 8, 384–400. [Google Scholar] [CrossRef]
- Yazyev, O.V.; Louie, S.G. Topological defects in graphene: Dislocations and grain boundaries. Phys. Rev. B 2010, 81, 195420. [Google Scholar] [CrossRef]
- Liu, Y.; Zou, X.; Yakobson, B.I. Dislocations and grain boundaries in two-dimensional boron nitride. ACS Nano 2012, 6, 7053–7058. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.Y.; Ruiz-Vargas, C.S.; van der Zande, A.M.; Whitney, W.S.; Levendorf, M.P.; Kevek, J.W.; Garg, S.; Alden, J.S.; Hustedt, C.J.; Zhu, Y.; et al. Grains and grain boundaries in single-layer graphene atomic patchwork quilts. Nature 2011, 469, 389–392. [Google Scholar] [CrossRef]
- Gibb, A.L.; Alem, N.; Chen, J.-H.; Erickson, K.J.; Ciston, J.; Gautam, A.; Linck, M.; Zettl, A. Atomic resolution imaging of grain boundary defects in monolayer chemical vapor deposition-grown hexagonal boron nitride. J. Am. Chem. Soc. 2013, 135, 6758–6761. [Google Scholar] [CrossRef]
- Zhou, W.; Zou, X.; Najmaei, S.; Liu, Z.; Shi, Y.; Kong, J.; Lou, J.; Ajayan, P.M.; Yakobson, B.I.; Idrobo, J.-C. Intrinsic structural defects in monolayer molybdenum disulfide. Nano Lett. 2013, 13, 2615–2622. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Liu, Y.; Yakobson, B.I. Predicting dislocations and grain boundaries in two-dimensional metal-disulfides from the first principles. Nano Lett. 2013, 13, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Najmaei, S.; Liu, Z.; Zhou, W.; Zou, X.; Shi, G.; Lei, S.; Yakobson, B.I.; Idrobo, J.-C.; Ajayan, P.M.; Lou, J. Vapour phase growth and grain boundary structure of molybdenum disulphide atomic layers. Nat. Mater. 2013, 12, 754. [Google Scholar] [CrossRef] [PubMed]
- Azizi, A.; Zou, X.; Ercius, P.; Zhang, Z.; Elías, A.L.; Perea-López, N.; Stone, G.; Terrones, M.; Yakobson, B.I.; Alem, N. Dislocation motion and grain boundary migration in two-dimensional tungsten disulphide. Nat. Commun. 2014, 5, 4867. [Google Scholar] [CrossRef]
- Van der Zande, A.M.; Huang, P.Y.; Chenet, D.A.; Berkelbach, T.C.; You, Y.; Lee, G.-H.; Heinz, T.F.; Reichman, D.R.; Muller, D.A.; Hone, J.C. Grains and grain boundaries in highly crystalline monolayer molybdenum disulphide. Nat. Mater. 2013, 12, 554. [Google Scholar] [CrossRef]
- Lu, W.; Nan, H.; Hong, J.; Chen, Y.; Zhu, C.; Liang, Z.; Ma, X.; Ni, Z.; Jin, C.; Zhang, Z. Plasma-assisted fabrication of monolayer phosphorene and its Raman characterization. Nano Res. 2014, 7, 853–859. [Google Scholar] [CrossRef]
- Li, L.; Yu, Y.; Ye, G.J.; Ge, Q.; Ou, X.; Wu, H.; Feng, D.; Chen, X.H.; Zhang, Y. Black phosphorus field-effect transistors. Nat. Nanotechnol. 2014, 9, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Yakobson, B.I. An open canvas-2D materials with defects, disorder, and functionality. Acc. Chem. Res. 2015, 48, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Ke, Q.; Zhang, G.; Yakobson, B.I.; Zhang, Y.-W. Highly itinerant atomic vacancies in phosphorene. J. Am. Chem. Soc. 2016, 138, 10199–10206. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, F.; Zhang, Z.; Penev, E.S.; Yakobson, B.I. Two-dimensional mono-elemental semiconductor with electronically inactive defects: The case of phosphorus. Nano Lett. 2014, 14, 6782–6786. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Yang, J. Defects in phosphorene. J. Phys. Chem. C 2015, 119, 20474–20480. [Google Scholar] [CrossRef]
- Ziletti, A.; Carvalho, A.; Campbell, D.K.; Coker, D.F.; Castro Neto, A.H. Oxygen defects in phosphorene. Phys. Rev. Lett. 2015, 114, 046801. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Slough, W.J.; Pandey, R.; Karna, S.P. Degradation of phosphorene in air: Understanding at atomic level. 2D Mater. 2016, 3, 025011. [Google Scholar] [CrossRef]
- Wang, V.; Geng, W.T. Lattice defects and the mechanical anisotropy of borophene. J. Phys. Chem. C 2017, 121, 10224–10232. [Google Scholar] [CrossRef]
- Penev, E.S.; Bhowmick, S.; Sadrzadeh, A.; Yakobson, B.I. Polymorphism of two-dimensional boron. Nano Lett. 2012, 12, 2441–2445. [Google Scholar] [CrossRef]
- Zhang, Z.; Penev, E.S.; Yakobson, B.I. Two-dimensional materials: Polyphony in B flat. Nat. Chem. 2016, 8, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, Y.; Gao, G.; Yakobson, B.I. Two-dimensional boron monolayers mediated by metal substrates. Angew. Chem. Int. Ed. Engl. 2015, 54, 13022–13026. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Mannix, A.J.; Hu, Z.; Kiraly, B.; Guisinger, N.P.; Hersam, M.C.; Yakobson, B.I. Substrate-induced nanoscale undulations of borophene on silver. Nano Lett. 2016, 16, 6622–6627. [Google Scholar] [CrossRef]
- Xu, S.; Zhao, Y.; Liao, J.; Yang, X.; Xu, H. The nucleation and growth of borophene on the Ag(111) surface. Nano Res. 2016, 9, 2616–2622. [Google Scholar] [CrossRef]
- Penev, E.S.; Artyukhov, V.I.; Ding, F.; Yakobson, B.I. Unfolding the fullerene: Nanotubes, graphene and poly-elemental varieties by simulations. Adv. Mater. 2012, 24, 4956–4976. [Google Scholar] [CrossRef] [PubMed]
- Kulish, V.V. Surface reactivity and vacancy defects in single-layer borophene polymorphs. Phys. Chem. Chem. Phys. 2017, 19, 11273–11281. [Google Scholar] [CrossRef]
- Guo, Y.; Zhou, S.; Zhang, J.; Bai, Y.; Zhao, J. Atomic structures and electronic properties of phosphorene grain boundaries. 2D Mater. 2016, 3, 025008. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Wang, L.; Yakobson, B.I.; Hersam, M.C. Intermixing and periodic self-assembly of borophene line defects. Nat. Mater. 2018, 17, 783–788. [Google Scholar] [CrossRef]
- Castellanos-Gomez, A.; Poot, M.; Steele, G.A.; van der Zant, H.S.; Agrait, N.; Rubio-Bollinger, G. Elastic properties of freely suspended MoS2 nanosheets. Adv. Mater. 2012, 24, 772–775. [Google Scholar] [CrossRef]
- Song, L.; Ci, L.; Lu, H.; Sorokin, P.B.; Jin, C.; Ni, J.; Kvashnin, A.G.; Kvashnin, D.G.; Lou, J.; Yakobson, B.I.; et al. Large scale growth and characterization of atomic hexagonal boron nitride layers. Nano Lett. 2010, 10, 3209–3215. [Google Scholar] [CrossRef]
- Liu, K.; Yan, Q.; Chen, M.; Fan, W.; Sun, Y.; Suh, J.; Fu, D.; Lee, S.; Zhou, J.; Tongay, S.; et al. Elastic properties of chemical-vapor-deposited monolayer MoS2, WS2, and their bilayer heterostructures. Nano Lett. 2014, 14, 5097–5103. [Google Scholar] [CrossRef] [PubMed]
- Cooper, R.C.; Lee, C.; Marianetti, C.A.; Wei, X.; Hone, J.; Kysar, J.W. Nonlinear elastic behavior of two-dimensional molybdenum disulfide. Phys. Rev. B 2013, 87, 035423. [Google Scholar] [CrossRef]
- Bertolazzi, S.; Brivio, J.; Kis, A. Stretching and breaking of ultrathin MoS2. ACS Nano 2011, 5, 9703–9709. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Shen, W.; Wu, S.; Liu, L.; Feng, Z.; Wang, C.; Hu, C.; Yao, P.; Zhang, H.; Pang, W.; et al. Mechanical and electrical anisotropy of few-layer black phosphorus. ACS Nano 2015, 9, 11362–11370. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Medrano Sandonas, L.; Zhang, T.; Gall, M.; Dianat, A.; Gutierrez, R.; Muhle, U.; Gluch, J.; Jordan, R.; Cuniberti, G.; et al. In-situ stretching patterned graphene nanoribbons in the transmission electron microscope. Sci. Rep. 2017, 7, 211. [Google Scholar] [CrossRef]
- Yang, Y.; Li, X.; Wen, M.; Hacopian, E.; Chen, W.; Gong, Y.; Zhang, J.; Li, B.; Zhou, W.; Ajayan, P.M.; et al. Brittle fracture of 2D MoSe2. Adv. Mater. 2017, 29, 1604201. [Google Scholar] [CrossRef]
- Li, X.; Gao, H. Atomistic modelling of deformation and failure mechanisms in nanostructured materials. Natl. Sci. Rev. 2015, 2, 133–136. [Google Scholar] [CrossRef]
- Liu, F.; Ming, P.; Li, J. Ab initio calculation of ideal strength and phonon instability of graphene under tension. Phys. Rev. B 2007, 76, 064120. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, Y.; Penev, E.S.; Yakobson, B.I. Elasticity, flexibility, and ideal strength of borophenes. Adv. Funct. Mater. 2017, 27, 1605059. [Google Scholar] [CrossRef]
- Wang, B.; Wu, J.; Gu, X.; Yin, H.; Wei, Y.; Yang, R.; Dresselhaus, M. Stable planar single-layer hexagonal silicene under tensile strain and its anomalous Poisson’s ratio. Appl. Phys. Lett. 2014, 104, 081902. [Google Scholar] [CrossRef]
- Wei, Q.; Peng, X. Superior mechanical flexibility of phosphorene and few-layer black phosphorus. Appl. Phys. Lett. 2014, 104, 251915. [Google Scholar] [CrossRef]
- Gao, E.; Xie, B.; Xu, Z. Two-dimensional silica: Structural, mechanical properties, and strain-induced band gap tuning. J. Appl. Phys. 2016, 119, 014301. [Google Scholar] [CrossRef]
- Wang, B.; Wei, Y.; Yang, R.; Dresselhaus, M. Mechanics and mechanically tunable band gap in single-layer hexagonal boron-nitride. Mater. Res. Lett. 2013, 1, 200–206. [Google Scholar]
- Wang, H.; Li, Q.; Gao, Y.; Miao, F.; Zhou, X.-F.; Wan, X.G. Strain effects on borophene: Ideal strength, negative Possion’s ratio and phonon instability. New J. Phys. 2016, 18, 073016. [Google Scholar] [CrossRef]
- Li, T. Ideal strength and phonon instability in single-layer MoS2. Phys. Rev. B 2012, 85, 235407. [Google Scholar] [CrossRef]
- Peng, Q.; Wen, X.; De, S. Mechanical stabilities of silicene. RSC Adv. 2013, 3, 13772–13781. [Google Scholar] [CrossRef]
- Brenner, D.W.; Shenderova, O.A.; Harrison, J.A.; Stuart, S.J.; Ni, B.; Sinnott, S.B. A second-generation reactive empirical bond order (REBO) potential energy expression for hydrocarbons. J. Phys.: Condens. Matter 2002, 14, 783–802. [Google Scholar] [CrossRef]
- Stuart, S.J.; Tutein, A.B.; Harrison, J.A. A reactive potential for hydrocarbons with intermolecular interactions. J. Chem. Phys. 2000, 112, 6472–6486. [Google Scholar] [CrossRef]
- O’Connor, T.C.; Andzelm, J.; Robbins, M.O. AIREBO-M: A reactive model for hydrocarbons at extreme pressures. J. Chem. Phys. 2015, 142, 024903. [Google Scholar]
- Tersoff, J. Empirical interatomic potential for carbon, with applications to amorphous carbon. Phys. Rev. Lett. 1988, 61, 2879. [Google Scholar] [CrossRef]
- Lindsay, L.; Broido, D.A.; Mingo, N. Flexural phonons and thermal transport in graphene. Phys. Rev. B 2010, 82, 115427. [Google Scholar] [CrossRef]
- van Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: A reactive force field for hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef]
- Kınacı, A.; Haskins, J.B.; Sevik, C.; Çağın, T. Thermal conductivity of BN-C nanostructures. Phys. Rev. B 2012, 86, 115410. [Google Scholar]
- Jiang, J.-W.; Park, H.S.; Rabczuk, T. Molecular dynamics simulations of single-layer molybdenum disulphide (MoS2): Stillinger-Weber parametrization, mechanical properties, and thermal conductivity. J. Appl. Phys. 2013, 114, 064307. [Google Scholar] [CrossRef]
- Chang, T.; Gao, H. Size-dependent elastic properties of a single-walled carbon nanotube via a molecular mechanics model. J. Mech. Phys. Solids 2003, 51, 1059–1074. [Google Scholar] [CrossRef]
- Qu, J.; Zhang, H.; Li, J.; Zhao, S.; Chang, T. Structure-dependent mechanical properties of extended beta-graphyne. Carbon 2017, 120, 350–357. [Google Scholar] [CrossRef]
- Hou, J.; Yin, Z.; Zhang, Y.; Chang, T. An analytical molecular mechanics model for elastic properties of graphyne-n. J. Appl. Mech. 2015, 82, 094501. [Google Scholar] [CrossRef]
- Hou, J.; Yin, Z.; Zhang, Y.; Chang, T.-C. Structure dependent elastic properties of supergraphene. Acta Mech. Sin. 2016, 32, 684–689. [Google Scholar] [CrossRef]
- Jiang, L.; Guo, W. A molecular mechanics study on size-dependent elastic properties of single-walled boron nitride nanotubes. J. Mech. Phys. Solids 2011, 59, 1204–1213. [Google Scholar] [CrossRef]
- Xiao, J.R.; Staniszewskia, J.; Gillespie, J.W., Jr. Tensile behaviors of graphene sheets and carbon nanotubes with multiple Stone-Wales defects. Mater. Sci. Eng. A 2010, 527, 715–723. [Google Scholar] [CrossRef]
- Li, C.Y.; Chou, T.S. A structural mechanics approach for the analysis of carbon nanotubes. Int. J. Solids Struct. 2003, 40, 2487–2499. [Google Scholar] [CrossRef]
- Xiao, J.R.; Gama, B.A.; Gillespie, J.W., Jr. An analytical molecular structural mechanics model for the mechanical properties of carbon nanotubes. Int. J. Solids Struct. 2005, 42, 3075–3092. [Google Scholar] [CrossRef]
- Ghaderi, S.H.; Hajiesmaili, E. Molecular structural mechanics applied to coiled carbon nanotubes. Comput. Mater. Sci. 2012, 55, 344–349. [Google Scholar] [CrossRef]
- Eberhardt, O.; Wallmersperger, T. Advanced molecular structural mechanics model for carbon nanotubes incorporating the 2nd generation REBO potential. Int. J. Eng. Sci. 2019, 144, 103137. [Google Scholar] [CrossRef]
- Firouz-Abadi, R.D.; Moshrefzadeh-Sany, H.; Mohammadkhani, H.; Sarmadi, M. A modified molecular structural mechanics model for the buckling analysis of single layer graphene sheet. Solid State Commun. 2016, 225, 12–16. [Google Scholar] [CrossRef]
- López-Polín, G.; Gómez-Navarro, C.; Parente, V.; Guinea, F.; Katsnelson, M.I.; Pérez-Murano, F.; Gómez-Herrero, J. Increasing the elastic modulus of graphene by controlled defect creation. Nat. Phys. 2014, 11, 26–31. [Google Scholar] [CrossRef]
- Zandiatashbar, A.; Lee, G.-H.; An, S.J.; Lee, S.; Mathew, N.; Terrones, M.; Hayashi, T.; Picu, C.R.; Hone, J.; Koratkar, N. Effect of defects on the intrinsic strength and stiffness of graphene. Nat. Commun. 2014, 5, 3186. [Google Scholar] [CrossRef] [PubMed]
- Hao, F.; Fang, D.; Xu, Z. Mechanical and thermal transport properties of graphene with defects. Appl. Phys. Lett. 2011, 99, 041901. [Google Scholar] [CrossRef]
- Wang, M.C.; Yan, C.; Ma, L.; Hu, N.; Chen, M.W. Effect of defects on fracture strength of graphene sheets. Comput. Mater. Sci. 2012, 54, 236–239. [Google Scholar] [CrossRef]
- Dewapriya, M.A.N.; Rajapakse, R.K.N.D. Molecular dynamics simulations and continuum modeling of temperature and strain rate dependent fracture strength of graphene with vacancy defects. J. Appl. Mech. 2014, 81, 081010. [Google Scholar] [CrossRef]
- Lee, G.-H.; Cooper, R.C.; An, S.J.; Lee, S.; van der Zande, A.; Petrone, N.; Hammerberg, A.G.; Lee, C.; Crawford, B.; Oliver, W.; et al. High-strength chemical-vapor–deposited graphene and grain boundaries. Science 2013, 340, 1073–1076. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Vargas, C.S.; Zhuang, H.L.; Huang, P.Y.; van der Zande, A.M.; Garg, S.; McEuen, P.L.; Muller, D.A.; Hennig, R.G.; Park, J. Softened elastic response and unzipping in chemical vapor deposition graphene membranes. Nano Lett. 2011, 11, 2259–2263. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wu, J.; Yin, H.; Shi, X.; Yang, R.; Dresselhaus, M. The nature of strength enhancement and weakening by pentagon–heptagon defects in graphene. Nat. Mater. 2012, 11, 759. [Google Scholar] [CrossRef] [PubMed]
- Grantab, R.; Shenoy, V.B.; Ruoff, R.S. Anomalous strength characteristics of tilt grain boundaries in graphene. Science 2010, 330, 946–948. [Google Scholar] [CrossRef]
- Rasool, H.I.; Ophus, C.; Klug, W.S.; Zettl, A.; Gimzewski, J.K. Measurement of the intrinsic strength of crystalline and polycrystalline graphene. Nat. Commun. 2013, 4, 2811. [Google Scholar] [CrossRef]
- Song, Z.; Artyukhov, V.I.; Yakobson, B.I.; Xu, Z. Pseudo Hall–Petch strength reduction in polycrystalline graphene. Nano Lett. 2013, 13, 1829–1833. [Google Scholar] [CrossRef] [PubMed]
- Shekhawat, A.; Ritchie, R.O. Toughness and strength of nanocrystalline graphene. Nat. Commun. 2016, 7, 10546. [Google Scholar] [CrossRef]
- Kotakoski, J.; Meyer, J.C. Mechanical properties of polycrystalline graphene based on a realistic atomistic model. Phys. Rev. B 2012, 85, 195447. [Google Scholar] [CrossRef]
- Sha, Z.D.; Quek, S.S.; Pei, Q.X.; Liu, Z.S.; Wang, T.J.; Shenoy, V.B.; Zhang, Y.W. Inverse pseudo Hall-Petch relation in polycrystalline graphene. Sci. Rep. 2014, 4, 5991. [Google Scholar] [CrossRef]
- Zhang, T.; Li, X.; Gao, H. Fracture of graphene: A review. Int. J. Fracture 2015, 196, 1–31. [Google Scholar] [CrossRef]
- Zhang, P.; Ma, L.; Fan, F.; Zeng, Z.; Peng, C.; Loya, P.E.; Liu, Z.; Gong, Y.; Zhang, J.; Zhang, X.; et al. Fracture toughness of graphene. Nat. Commun. 2014, 5, 3782. [Google Scholar] [CrossRef]
- Khare, R.; Mielke, S.L.; Paci, J.T.; Zhang, S.; Ballarini, R.; Schatz, G.C.; Belytschko, T. Coupled quantum mechanical/molecular mechanical modeling of the fracture of defective carbon nanotubes and graphene sheets. Phys. Rev. B 2007, 75, 075412. [Google Scholar] [CrossRef]
- Terdalkar, S.S.; Huang, S.; Yuan, H.; Rencis, J.J.; Zhu, T.; Zhang, S. Nanoscale fracture in graphene. Chem. Phys. Lett. 2010, 494, 218–222. [Google Scholar] [CrossRef]
- Yin, H.; Qi, H.J.; Fan, F.; Zhu, T.; Wang, B.; Wei, Y. Griffith criterion for brittle fracture in graphene. Nano Lett. 2015, 15, 1918–1924. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, X.; Lee, J.D. An atomistic methodology of energy release rate for graphene at nanoscale. J. Appl. Phys. 2014, 115, 114314. [Google Scholar] [CrossRef]
- Kim, K.; Artyukhov, V.I.; Regan, W.; Liu, Y.; Crommie, M.F.; Yakobson, B.I.; Zettl, A. Ripping graphene: Preferred directions. Nano Lett. 2012, 12, 293–297. [Google Scholar] [CrossRef]
- Jung, G.; Qin, Z.; Buehler, M.J. Molecular mechanics of polycrystalline graphene with enhanced fracture toughness. Extreme Mech. Lett. 2015, 2, 52–59. [Google Scholar] [CrossRef]
- Zhang, T.; Li, X.; Kadkhodaei, S.; Gao, H. Flaw insensitive fracture in nanocrystalline graphene. Nano Lett. 2012, 12, 4605–4610. [Google Scholar] [CrossRef]
- Gao, H.; Chen, S. Flaw tolerance in a thin strip under tension. J. Appl. Mech. 2005, 72, 732–737. [Google Scholar] [CrossRef]
- Wang, X.; Tabarraei, A.; Spearot, D.E. Fracture mechanics of monolayer molybdenum disulfide. Nanotechnology 2015, 26, 175703. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Hong, J.; Pidaparti, R.; Wang, X. Fracture patterns and the energy release rate of phosphorene. Nanoscale 2016, 8, 5728–5736. [Google Scholar] [CrossRef]
- Zhang, T.; Li, X.; Gao, H. Defects controlled wrinkling and topological design in graphene. J. Mech. Phys. Solids 2014, 67, 2–13. [Google Scholar] [CrossRef]
- Zhang, T.; Li, X.; Gao, H. Designing graphene structures with controlled distributions of topological defects: A case study of toughness enhancement in graphene ruga. Extreme Mech. Lett. 2014, 1, 3–8. [Google Scholar] [CrossRef]
- Meng, F.; Chen, C.; Song, J. Dislocation shielding of a nanocrack in graphene: Atomistic simulations and continuum modeling. J. Phys. Chem. Lett. 2015, 6, 4038–4042. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Gao, H. Toughening graphene with topological defects: A perspective. J. Appl. Mech. 2015, 82, 051001. [Google Scholar] [CrossRef]
- Qi, Z.; Campbell, D.K.; Park, H.S. Atomistic simulations of tension-induced large deformation and stretchability in graphene kirigami. Phys. Rev. B 2014, 90, 245437. [Google Scholar] [CrossRef]
- Bahamon, D.A.; Qi, Z.; Park, H.S.; Pereira, V.M.; Campbell, D.K. Graphene kirigami as a platform for stretchable and tunable quantum dot arrays. Phys. Rev. B 2016, 93, 235408. [Google Scholar] [CrossRef]
- Hanakata, P.Z.; Qi, Z.; Campbell, D.K.; Park, H.S. Highly stretchable MoS2 kirigami. Nanoscale 2016, 8, 458–463. [Google Scholar] [CrossRef]
- Butler, K.T.; Davies, D.W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nature 2018, 559, 547–555. [Google Scholar] [CrossRef]
- Carrasquilla, J.; Melko, R.G. Machine learning phases of matter. Nat. Phys. 2017, 13, 431–434. [Google Scholar] [CrossRef]
- Ramprasad, R.; Batra, R.; Pilania, G.; Mannodi-Kanakkithodi, A.; Kim, C. Machine learning in materials informatics: Recent applications and prospects. Comput. Mater. 2017, 54. [Google Scholar] [CrossRef]
- Hanakata, P.Z.; Cubuk, E.D.; Campbell, D.K.; Park, H.S. Accelerated search and design of stretchable graphene kirigami using machine learning. Phys. Rev. Lett. 2018, 121, 255304. [Google Scholar] [CrossRef]
- Blees, M.K.; Barnard, A.W.; Rose, P.A.; Roberts, S.P.; McGill, K.L.; Huang, P.Y.; Ruyack, A.R.; Kevek, J.W.; Kobrin, B.; Muller, D.A.; et al. Graphene kirigami. Nature 2015, 524, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.W. Crack tip shielding by micro-cracking in brittle solids. Acta Metall. 1987, 33, 1605–1619. [Google Scholar] [CrossRef]
- Hacopian, E.F.; Yang, Y.; Ni, B.; Li, Y.; Li, X.; Chen, Q.; Guo, H.; Tour, J.M.; Gao, H.; Lou, J. Toughening graphene by integrating carbon nanotubes. ACS Nano 2018, 12, 7901–7910. [Google Scholar] [CrossRef]
- Min, K.; Aluru, N.R. Mechanical properties of graphene under shear deformation. Appl. Phys. Lett. 2011, 98, 013113. [Google Scholar] [CrossRef]
- Wang, C.; Liu, Y.; Lan, L.; Tan, H. Graphene wrinkling: Formation, evolution and collapse. Nanoscale 2013, 5, 4454–4461. [Google Scholar] [CrossRef]
- Wang, C.G.; Lan, L.; Liu, Y.P.; Tan, H.F. Defect-guided wrinkling in graphene. Comput. Mater. Sci. 2013, 77, 250–253. [Google Scholar] [CrossRef]
- Annett, J.; Cross, G.L. Self-assembly of graphene ribbons by spontaneous self-tearing and peeling from a substrate. Nature 2016, 535, 271–275. [Google Scholar] [CrossRef]
- Moura, M.J.; Marder, M. Tearing of free-standing graphene. Phys. Rev. E 2013, 88, 032405. [Google Scholar] [CrossRef]
- Kawai, T.; Okada, S.; Miyamoto, Y.; Hiura, H. Self-redirection of tearing edges in graphene: Tight-binding molecular dynamics simulations. Phys. Rev. B 2009, 80, 033401. [Google Scholar] [CrossRef]
- Sen, D.; Novoselov, K.S.; Reis, P.M.; Buehler, M.J. Tearing graphene sheets from adhesive substrates produces tapered nanoribbons. Small 2010, 6, 1108–1116. [Google Scholar] [CrossRef]
- Ye, S.; Cai, Y.; Liu, X.; Yao, X.; Luo, S.-N. Crack propagation in graphene monolayer under tear loading. Phys. Chem. Chem. Phys. 2019, 21, 2659–2664. [Google Scholar] [CrossRef]
- Cui, T.; Mukherjee, S.; Sudeep, P.M.; Colas, G.; Najafi, F.; Tam, J.; Ajayan, P.M.; Singh, C.V.; Sun, Y.; Filleter, T. Fatigue of graphene. Nat. Mater. 2020, 19, 405–411. [Google Scholar] [CrossRef]
- Cui, T.; Yip, K.; Hassan, A.; Wang, G.; Liu, X.; Sun, Y.; Filleter, T. Graphene fatigue through van der Waals interactions. Sci. Adv. 2020, 6, eabb1335. [Google Scholar] [CrossRef]
- Kim, S.K.; Bhatia, R.; Kim, T.-H.; Seol, D.; Kim, J.H.; Kim, H.; Seung, W.; Kim, Y.; Lee, Y.H.; Kim, S.-W. Directional dependent piezoelectric effect in CVD grown monolayer MoS2 for flexible piezoelectric nanogenerators. Nano Energy 2016, 22, 483–489. [Google Scholar] [CrossRef]
- Wu, W.; Wang, L.; Li, Y.; Zhang, F.; Lin, L.; Niu, S.; Chenet, D.; Zhang, X.; Hao, Y.; Heinz, T.F.; et al. Piezoelectricity of single-atomic-layer MoS2 for energy conversion and piezotronics. Nature 2014, 514, 470–474. [Google Scholar] [CrossRef]
- Qi, J.; Lan, Y.W.; Stieg, A.Z.; Chen, J.H.; Zhong, Y.L.; Li, L.J.; Chen, C.D.; Zhang, Y.; Wang, K.L. Piezoelectric effect in chemical vapour deposition-grown atomic-monolayer triangular molybdenum disulfide piezotronics. Nat. Commun. 2015, 6, 7430. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, Y.; Xiao, J.; Liu, M.; Xiong, S.; Wong, Z.J.; Ye, Z.; Ye, Y.; Yin, X.; Zhang, X. Observation of piezoelectricity in free-standing monolayer MoS2. Nat. Nanotechnol. 2014, 10, 151. [Google Scholar] [CrossRef]
- Blonsky, M.N.; Zhuang, H.L.; Singh, A.K.; Hennig, R.G. Ab initio prediction of piezoelectricity in two-dimensional materials. ACS Nano 2015, 9, 9885–9891. [Google Scholar] [CrossRef]
- Li, W.; Li, J. Piezoelectricity in two-dimensional group-III monochalcogenides. Nano Res. 2015, 8, 3796–3802. [Google Scholar] [CrossRef]
- Gomes, L.C.; Carvalho, A.; Castro Neto, A.H. Enhanced piezoelectricity and modified dielectric screening of two-dimensional group-IV monochalcogenides. Phys. Rev. B 2015, 92, 214103. [Google Scholar] [CrossRef]
- Duerloo, K.-A.N.; Ong, M.T.; Reed, E.J. Intrinsic piezoelectricity in two-dimensional materials. J. Phys. Chem. Lett. 2012, 3, 2871–2876. [Google Scholar] [CrossRef]
- Elias, D.C.; Nair, R.R.; Mohiuddin, T.M.; Morozov, S.V.; Blake, P.; Halsall, M.P.; Ferrari, A.C.; Boukhvalov, D.W.; Katsnelson, M.I.; Geim, A.K.; et al. Control of graphene’s properties by reversible hydrogenation: Evidence for graphane. Science 2009, 323, 610–613. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Burgess, J.S.; Junkermeier, C.E.; Badescu, S.C.; Reinecke, T.L.; Perkins, F.K.; Zalalutdniov, M.K.; Baldwin, J.W.; Culbertson, J.C.; Sheehan, P.E.; et al. Properties of fluorinated graphene films. Nano Lett. 2010, 10, 3001–3005. [Google Scholar] [CrossRef] [PubMed]
- Ong, M.T.; Reed, E.J. Engineered piezoelectricity in graphene. ACS Nano 2012, 6, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.D.; Mao, S.; Yeh, Y.W.; Purohit, P.K.; McAlpine, M.C. Nanoscale flexoelectricity. Adv. Mater. 2013, 25, 946–974. [Google Scholar] [CrossRef] [PubMed]
- Ahmadpoor, F.; Sharma, P. Flexoelectricity in two-dimensional crystalline and biological membranes. Nanoscale 2015, 7, 16555–16570. [Google Scholar] [CrossRef] [PubMed]
- Dumitrica, T.; Landis, C.M.; Yakobson, B.I. Curvature-induced polarization in carbon nanoshells. Chem. Phys. Lett. 2002, 360, 182–188. [Google Scholar] [CrossRef]
- Kalinin, S.V.; Meunier, V. Electronic flexoelectricity in low-dimensional systems. Phys. Rev. B 2008, 77, 033403. [Google Scholar] [CrossRef]
- Kvashnin, A.G.; Sorokin, P.B.; Yakobson, B.I. Flexoelectricity in carbon nanostructures: Nanotubes, fullerenes, and nanocones. J. Phys. Chem. Lett. 2015, 6, 2740–2744. [Google Scholar] [CrossRef]
- Naumov, I.; Bratkovsky, A.M.; Ranjan, V. Unusual flexoelectric effect in two-dimensional noncentrosymmetric sp2-bonded crystals. Phys. Rev. Lett. 2009, 102, 217601. [Google Scholar] [CrossRef]
- Duerloo, K.A.; Reed, E.J. Flexural electromechanical coupling: A nanoscale emergent property of boron nitride bilayers. Nano Lett. 2013, 13, 1681–1686. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Guo, Y.; Zhang, Z.; Guo, W. Flexoelectricity in monolayer transition metal dichalcogenides. J. Phys. Chem. Lett. 2018, 9, 6841–6846. [Google Scholar] [CrossRef] [PubMed]
- Chandratre, S.; Sharma, P. Coaxing graphene to be piezoelectric. Appl. Phys. Lett. 2012, 100, 023114. [Google Scholar] [CrossRef]
- Kundalwal, S.I.; Meguid, S.A.; Weng, G.J. Strain gradient polarization in graphene. Carbon 2017, 117, 462–472. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, J.; Liang, J.; Jaroniec, M.; Qiao, S.Z. Graphitic carbon nitride materials: Controllable synthesis and applications in fuel cells and photocatalysis. Energy Environ. Sci. 2012, 5, 6717–6731. [Google Scholar] [CrossRef]
- Zelisko, M.; Hanlumyuang, Y.; Yang, S.; Liu, Y.; Lei, C.; Li, J.; Ajayan, P.M.; Sharma, P. Anomalous piezoelectricity in two-dimensional graphene nitride nanosheets. Nat. Commun. 2014, 5, 4284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Ma, T.; Erdemir, A.; Li, Q. Tribology of two-dimensional materials: From mechanisms to modulating strategies. Mater. Today 2019, 26, 67–86. [Google Scholar] [CrossRef]
- Filleter, T.; McChesney, J.L.; Bostwick, A.; Rotenberg, E.; Emtsev, K.V.; Seyller, T.; Horn, K.; Bennewitz, R. Friction and dissipation in epitaxial graphene films. Phys. Rev. Lett. 2009, 102, 086102. [Google Scholar] [CrossRef]
- Lee, C.; Li, Q.; Kalb, W.; Liu, X.Z.; Berger, H.; Carpick, R.W.; Hone, J. Frictional characteristics of atomically thin sheets. Science 2010, 328, 76–80. [Google Scholar] [CrossRef]
- Deng, Z.; Smolyanitsky, A.; Li, Q.; Feng, X.Q.; Cannara, R.J. Adhesion-dependent negative friction coefficient on chemically modified graphite at the nanoscale. Nat. Mater. 2012, 11, 1032–1037. [Google Scholar] [CrossRef]
- Sun, X.-Y.; Qi, Y.-Z.; Ouyang, W.; Feng, X.-Q.; Li, Q. Energy corrugation in atomic-scale friction on graphite revisited by molecular dynamics simulations. Acta Mech. Sin. 2015, 32, 604–610. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, J.; Grey, F.; Liu, J.Z.; Liu, Y.; Wang, Y.; Yang, Y.; Cheng, Y.; Zheng, Q. Observation of microscale superlubricity in graphite. Phys. Rev. Lett. 2012, 108, 205503. [Google Scholar] [CrossRef]
- Dienwiebel, M.; Verhoeven, G.S.; Pradeep, N.; Frenken, J.W.; Heimberg, J.A.; Zandbergen, H.W. Superlubricity of graphite. Phys. Rev. Lett. 2004, 92, 126101. [Google Scholar] [CrossRef]
- Feng, X.; Kwon, S.; Park, J.Y.; Salmeron, M. Superlubric sliding of graphene nanoflakes on graphene. ACS Nano 2013, 7, 1718–1724. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Guo, W.; Chen, C. Modifying atomic-scale friction between two graphene sheets: A molecular-force-field study. Phys. Rev. B 2007, 76, 155429. [Google Scholar] [CrossRef]
- Zhang, H.; Guo, Z.; Gao, H.; Chang, T. Stiffness-dependent interlayer friction of graphene. Carbon 2015, 94, 60–66. [Google Scholar] [CrossRef]
- Wang, G.; Dai, Z.; Wang, Y.; Tan, P.; Liu, L.; Xu, Z.; Wei, Y.; Huang, R.; Zhang, Z. Measuring interlayer shear stress in bilayer graphene. Phys. Rev. Lett. 2017, 119, 036101. [Google Scholar] [CrossRef]
- Hopster, J.; Kozubek, R.; Ban-d’Etat, B.; Guillous, S.; Lebius, H.; Schleberger, M. Damage in graphene due to electronic excitation induced by highly charged ions. 2D Mater. 2014, 1, 011011. [Google Scholar] [CrossRef]
- Sun, X.-Y.; Wu, R.; Xia, R.; Chu, X.-H.; Xu, Y.-J. Effects of Stone-Wales and vacancy defects in atomic-scale friction on defective graphite. Appl. Phys. Lett. 2014, 104, 183109. [Google Scholar] [CrossRef]
- Gong, P.; Ye, Z.; Yuan, L.; Egberts, P. Evaluation of wetting transparency and surface energy of pristine and aged graphene through nanoscale friction. Carbon 2018, 132, 749–759. [Google Scholar] [CrossRef]
- Vilhena, J.G.; Pimentel, C.; Pedraz, P.; Luo, F.; Serena, P.A.; Pina, C.M.; Gnecco, E.; Perez, R. Atomic-scale sliding friction on graphene in water. ACS Nano 2016, 10, 4288–4293. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Peng, Y.; Lang, H. A novel approach to decrease friction of graphene. Carbon 2017, 118, 233–240. [Google Scholar] [CrossRef]
- Spear, J.C.; Custer, J.P.; Batteas, J.D. The influence of nanoscale roughness and substrate chemistry on the frictional properties of single and few layer graphene. Nanoscale 2015, 7, 10021–10029. [Google Scholar] [CrossRef]
- Quereda, J.; Castellanos-Gomez, A.; Agraït, N.; Rubio-Bollinger, G. Single-layer MoS2 roughness and sliding friction quenching by interaction with atomically flat substrates. Appl. Phys. Lett. 2014, 105, 053111. [Google Scholar] [CrossRef]
- Ni, Z.H.; Yu, T.; Lu, Y.H.; Wang, Y.Y.; Feng, Y.P.; Shen, Z.X. Uniaxial strain on graphene: Raman spectroscopy study and band-gap opening. ACS Nano 2008, 2, 2301–2305. [Google Scholar] [CrossRef]
- Gui, G.; Li, J.; Zhong, J. Band structure engineering of graphene by strain: First-principles calculations. Phys. Rev. B 2008, 78, 075435. [Google Scholar] [CrossRef]
- Johari, P.; Shenoy, V.B. Tuning the electronic properties of semiconducting transition metal dichalcogenides by applying mechanical strains. ACS Nano 2012, 6, 5449–5456. [Google Scholar] [CrossRef]
- Scalise, E.; Houssa, M.; Pourtois, G.; Afanas’ev, V.; Stesmans, A. Strain-induced semiconductor to metal transition in the two-dimensional honeycomb structure of MoS2. Nano Res. 2011, 5, 43–48. [Google Scholar] [CrossRef]
- Lu, P.; Wu, X.; Guo, W.; Zeng, X.C. Strain-dependent electronic and magnetic properties of MoS2 monolayer, bilayer, nanoribbons and nanotubes. Phys. Chem. Chem. Phys. 2012, 14, 13035–13040. [Google Scholar] [CrossRef]
- Dai, Z.; Liu, L.; Zhang, Z. Strain engineering of 2D materials: Issues and opportunities at the interface. Adv. Mater. 2019, 31, 1805417. [Google Scholar] [CrossRef]
- Wang, Y.; Cong, C.; Qiu, C.; Yu, T. Raman spectroscopy study of lattice vibration and crystallographic orientation of monolayer MoS2 under uniaxial strain. Small 2013, 9, 2857–2861. [Google Scholar] [CrossRef]
- Geim, A.K.; Grigorieva, I.V. Van der Waals heterostructures. Nature 2013, 499, 419–425. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Mishchenko, A.; Carvalho, A.; Castro Neto, A.H. 2D materials and van der Waals heterostructures. Science 2016, 353, aac9439. [Google Scholar] [CrossRef]
- Dean, C.R.; Young, A.F.; Meric, I.; Lee, C.; Wang, L.; Sorgenfrei, S.; Watanabe, K.; Taniguchi, T.; Kim, P.; Shepard, K.L.; et al. Boron nitride substrates for high-quality graphene electronics. Nat. Nanotechnol. 2010, 5, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, L.A.; Geim, A.K.; Zhukov, A.A.; Jalil, R.; Morozov, S.V.; Novoselov, K.S.; Grigorieva, I.V.; Hill, E.H.; Cheianov, V.V.; Fal’ko, V.I.; et al. Tunable metal–insulator transition in double-layer graphene heterostructures. Nat. Phys. 2011, 7, 958–961. [Google Scholar] [CrossRef]
- Britnell, L.; Ribeiro, R.M.; Eckmann, A.; Jalil, R.; Belle, B.D.; Mishchenko, A.; Kim, Y.J.; Gorbachev, R.V.; Georgiou, T.; Morozov, S.V.; et al. Strong light-matter interactions in heterostructures of atomically thin films. Science 2013, 340, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Britnell, L.; Gorbachev, R.V.; Jalil, R.; Belle, B.D.; Schedin, F.; Mishchenko, A.; Georgiou, T.; Katsnelson, M.I.; Eaves, L.; Morozov, S.V.; et al. Field-effect tunneling transistor based on vertical graphene heterostructures. Science 2012, 335, 947–950. [Google Scholar] [CrossRef] [PubMed]
- Yankowitz, M.; Xue, J.; Cormode, D.; Sanchez-Yamagishi, J.D.; Watanabe, K.; Taniguchi, T.; Jarillo-Herrero, P.; Jacquod, P.; LeRoy, B.J. Emergence of superlattice Dirac points in graphene on hexagonal boron nitride. Nat. Phys. 2012, 8, 382–386. [Google Scholar] [CrossRef]
- Kang, J.; Li, J.; Li, S.S.; Xia, J.B.; Wang, L.W. Electronic structural Moire pattern effects on MoS2/MoSe2 2D heterostructures. Nano Lett. 2013, 13, 5485–5490. [Google Scholar] [CrossRef]
- Zhang, C.; Chuu, C.-P.; Ren, X.; Li, M.-Y.; Li, L.-J.; Jin, C.; Chou, M.-Y.; Shih, C.-K. Interlayer couplings, Moiré patterns, and 2D electronic superlattices in MoS2/WSe2 hetero-bilayers. Sci. Adv. 2017, 3, 1601459. [Google Scholar] [CrossRef] [PubMed]
- Haigh, S.J.; Gholinia, A.; Jalil, R.; Romani, S.; Britnell, L.; Elias, D.C.; Novoselov, K.S.; Ponomarenko, L.A.; Geim, A.K.; Gorbachev, R. Cross-sectional imaging of individual layers and buried interfaces of graphene-based heterostructures and superlattices. Nat. Mater. 2012, 11, 764–767. [Google Scholar] [CrossRef]
- Kretinin, A.V.; Cao, Y.; Tu, J.S.; Yu, G.L.; Jalil, R.; Novoselov, K.S.; Haigh, S.J.; Gholinia, A.; Mishchenko, A.; Lozada, M.; et al. Electronic properties of graphene encapsulated with different two-dimensional atomic crystals. Nano Lett. 2014, 14, 3270–3276. [Google Scholar] [CrossRef]
- Cao, G.; Gao, H. Mechanical properties characterization of two-dimensional materials via nanoindentation experiments. Prog. Mater. Sci. 2019, 103, 558–595. [Google Scholar] [CrossRef]
- Han, J.; Pugno, N.M.; Ryu, S. Nanoindentation cannot accurately predict the tensile strength of graphene or other 2D materials. Nanoscale 2015, 7, 15672–15679. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.W.; Allen, C.S.; Wu, Y.A.; He, K.; Olivier, J.; Neethling, J.; Kirkland, A.I.; Warner, J.H. Spatial control of defect creation in graphene at the nanoscale. Nat. Commun. 2012, 3, 1144. [Google Scholar] [CrossRef]
- Ma, T.; Liu, Z.; Wen, J.; Gao, Y.; Ren, X.; Chen, H.; Jin, C.; Ma, X.L.; Xu, N.; Cheng, H.M.; et al. Tailoring the thermal and electrical transport properties of graphene films by grain size engineering. Nat. Commun. 2017, 8, 14486. [Google Scholar] [CrossRef]
- Shi, Y.; Zhou, W.; Lu, A.Y.; Fang, W.; Lee, Y.H.; Hsu, A.L.; Kim, S.M.; Kim, K.K.; Yang, H.Y.; Li, L.J.; et al. Van der Waals epitaxy of MoS2 layers using graphene as growth templates. Nano Lett. 2012, 12, 2784–2791. [Google Scholar] [CrossRef] [PubMed]
- Miwa, J.A.; Dendzik, M.; Gronborg, S.S.; Bianchi, M.; Lauritsen, J.V.; Hofmann, P.; Ulstrup, S. Van der Waals epitaxy of two-dimensional MoS2-graphene heterostructures in ultrahigh vacuum. ACS Nano 2015, 9, 6502–6510. [Google Scholar] [CrossRef]
- Cao, Y.; Fatemi, V.; Demir, A.; Fang, S.; Tomarken, S.L.; Luo, J.Y.; Sanchez-Yamagishi, J.D.; Watanabe, K.; Taniguchi, T.; Kaxiras, E.; et al. Correlated insulator behaviour at half-filling in magic-angle graphene superlattices. Nature 2018, 556, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Fatemi, V.; Fang, S.; Watanabe, K.; Taniguchi, T.; Kaxiras, E.; Jarillo-Herrero, P. Unconventional superconductivity in magic-angle graphene superlattices. Nature 2018, 556, 43–50. [Google Scholar] [CrossRef] [PubMed]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, Z.; Zhong, L.; Wang, H.; Li, X. Structural Defects, Mechanical Behaviors, and Properties of Two-Dimensional Materials. Materials 2021, 14, 1192. https://doi.org/10.3390/ma14051192
Xiong Z, Zhong L, Wang H, Li X. Structural Defects, Mechanical Behaviors, and Properties of Two-Dimensional Materials. Materials. 2021; 14(5):1192. https://doi.org/10.3390/ma14051192
Chicago/Turabian StyleXiong, Zixin, Lei Zhong, Haotian Wang, and Xiaoyan Li. 2021. "Structural Defects, Mechanical Behaviors, and Properties of Two-Dimensional Materials" Materials 14, no. 5: 1192. https://doi.org/10.3390/ma14051192
APA StyleXiong, Z., Zhong, L., Wang, H., & Li, X. (2021). Structural Defects, Mechanical Behaviors, and Properties of Two-Dimensional Materials. Materials, 14(5), 1192. https://doi.org/10.3390/ma14051192