
Anticancer Molecular Mechanism of Protocatechuic Acid Loaded on Folate Coated Functionalized Graphene Oxide Nanocomposite Delivery System in Human Hepatocellular Carcinoma

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Culture Cell Line
2.3. Synthesis of Graphene Oxide (GO)
2.4. Conjugation of Graphene Oxide with Polyethylene Glycol (GOP) Nanocarrier System
2.5. Protocatechuic Acid Loaded on GOP and Coated with Folic Acid (FA)
2.6. Physicochemical Characterization of Nanocomposites
2.7. Determination of Encapsulation Efficacy and Loading Content Using HPLC Analysis
2.8. Protocatechuic Acid In Vitro Drug Release from Nanocomposites
2.9. Lactate Dehydrogenase Assay for Plasma Membrane Integrity Analysis
2.10. Colony Formation Evaluated by Clonogenic Assay
2.11. Apoptosis/Necrosis Cell Death Analysis
2.12. Cell Cycle Arrest Analysis Using Propidium Iodide
2.13. Mitochondrial Membrane Potential Analysis
2.14. Measurement of Cellular Reactive Oxygen Species
2.15. Proteome Profiler Human Apoptotic Antibody Array Detection
2.16. Apoptotic Gene Expression Markers Detection
2.17. Statistical Analysis
3. Results
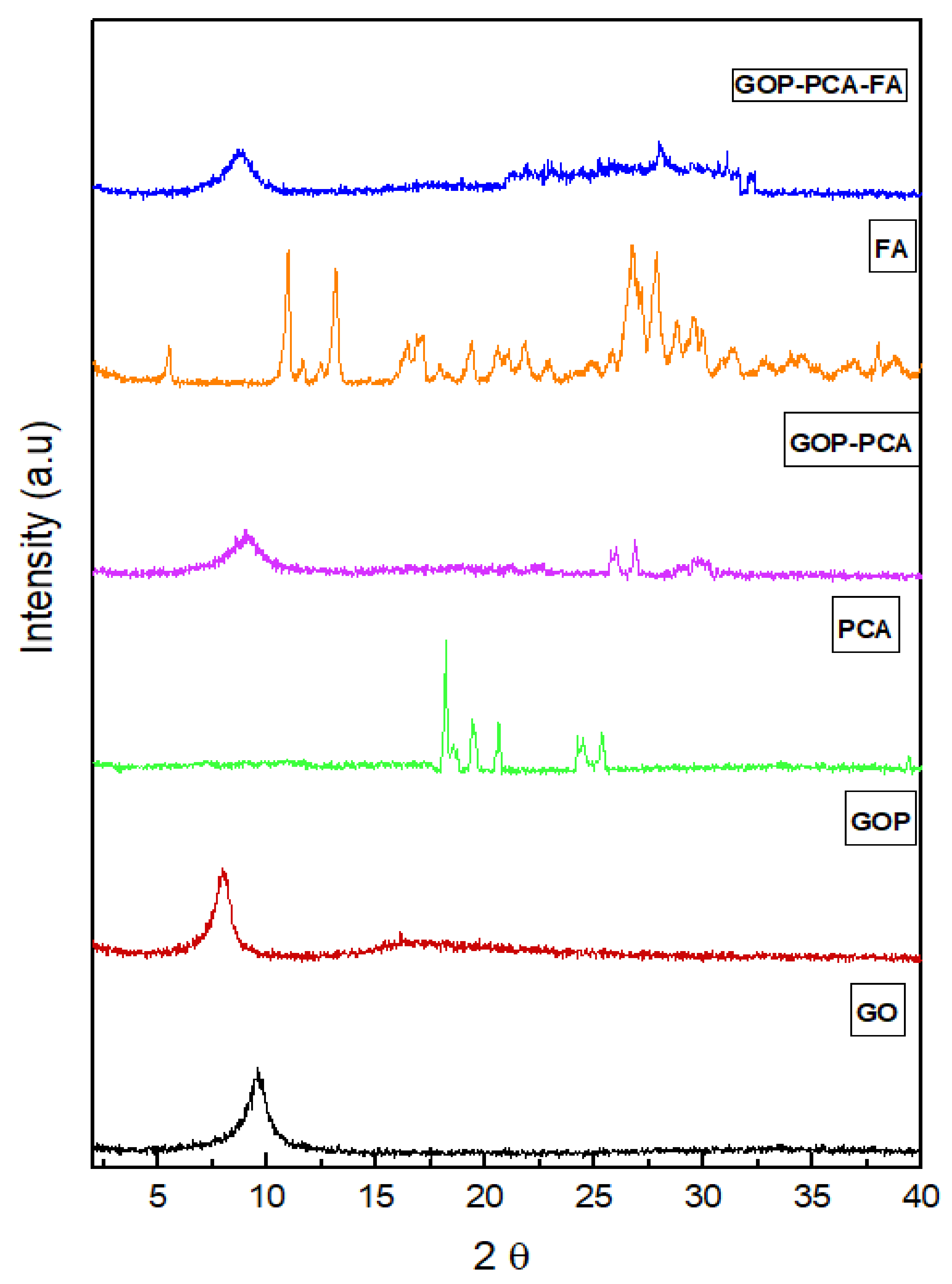
3.1. X-Ray Diffraction Analysis
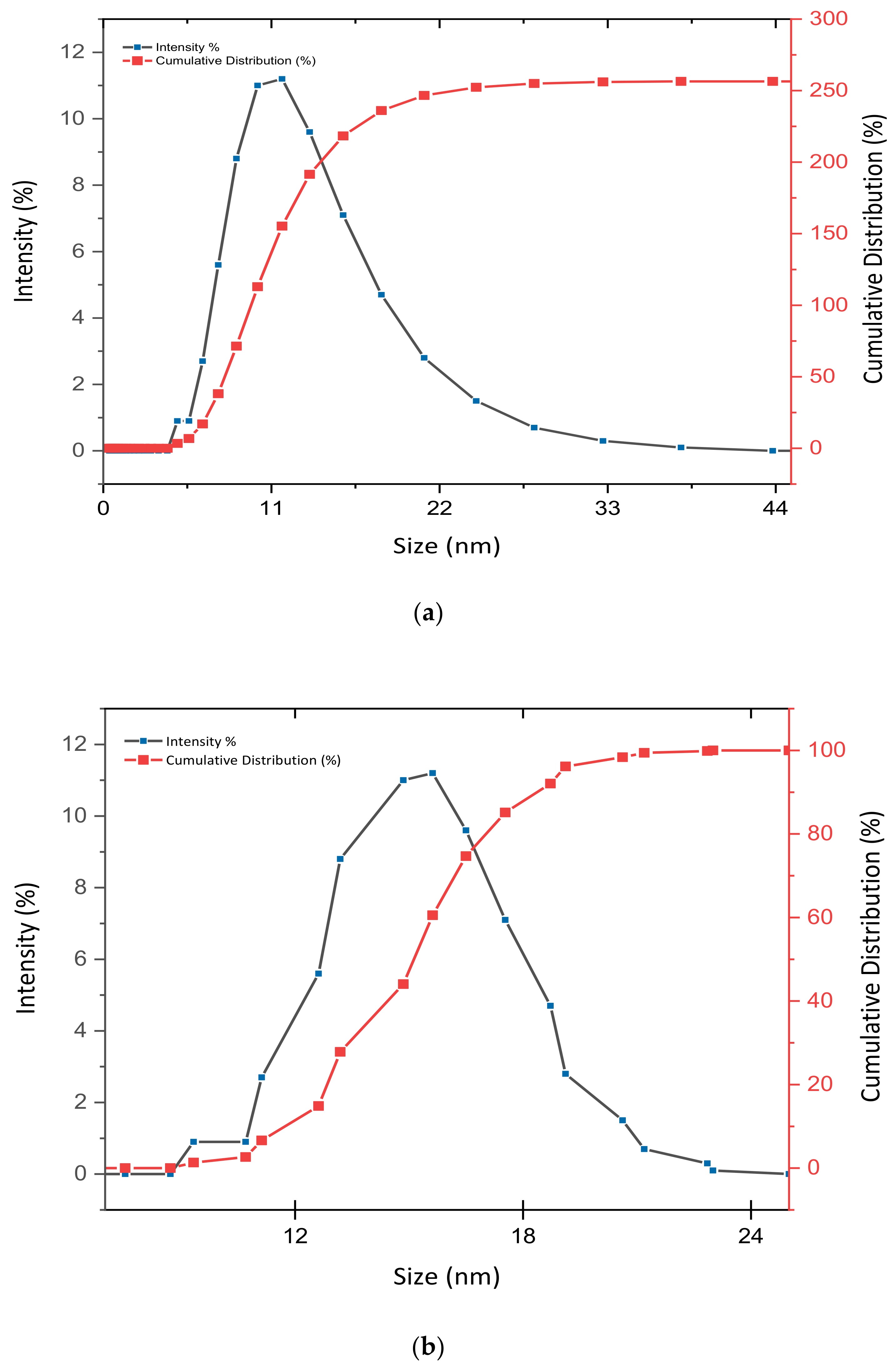
3.2. Determination of Size Distribution and Zeta Potential Measurement of Nanocomposites
3.3. Surface Properties Analysis
3.4. Quantification of Encapsulation Efficacy and Drug Loading Analysis
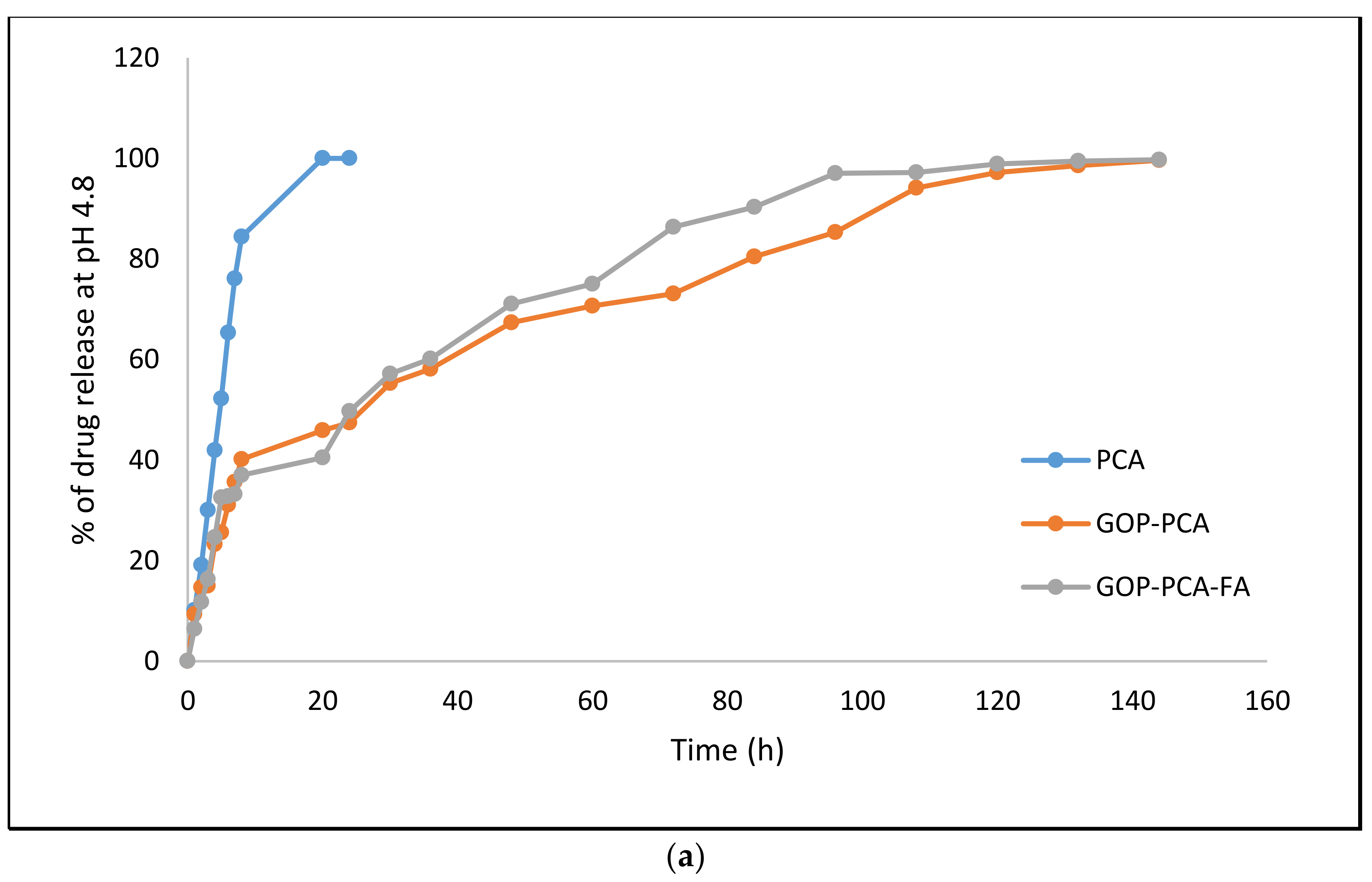
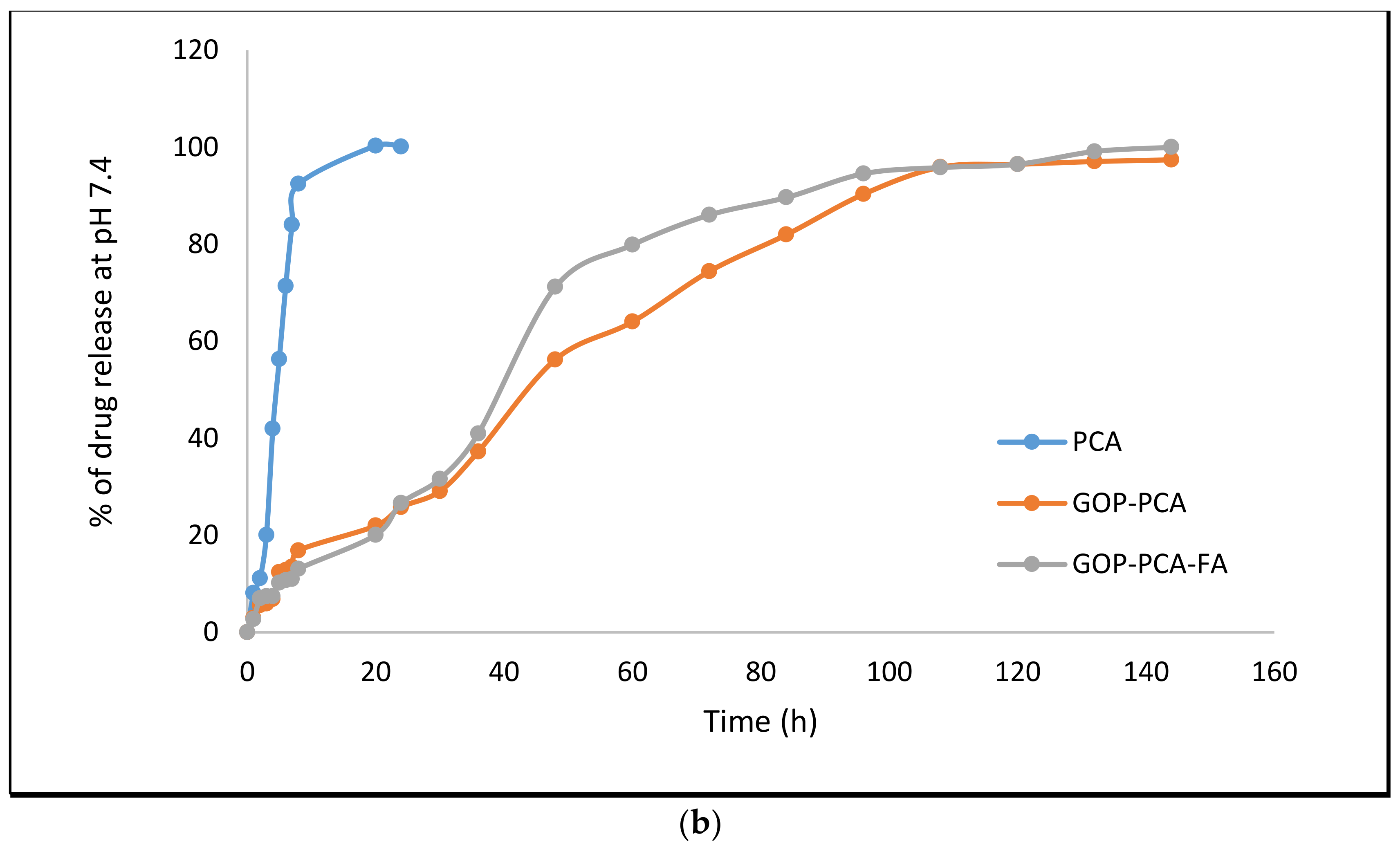
3.5. Protocatechuic Acid in vitro Drug Release Study of Nanocomposite
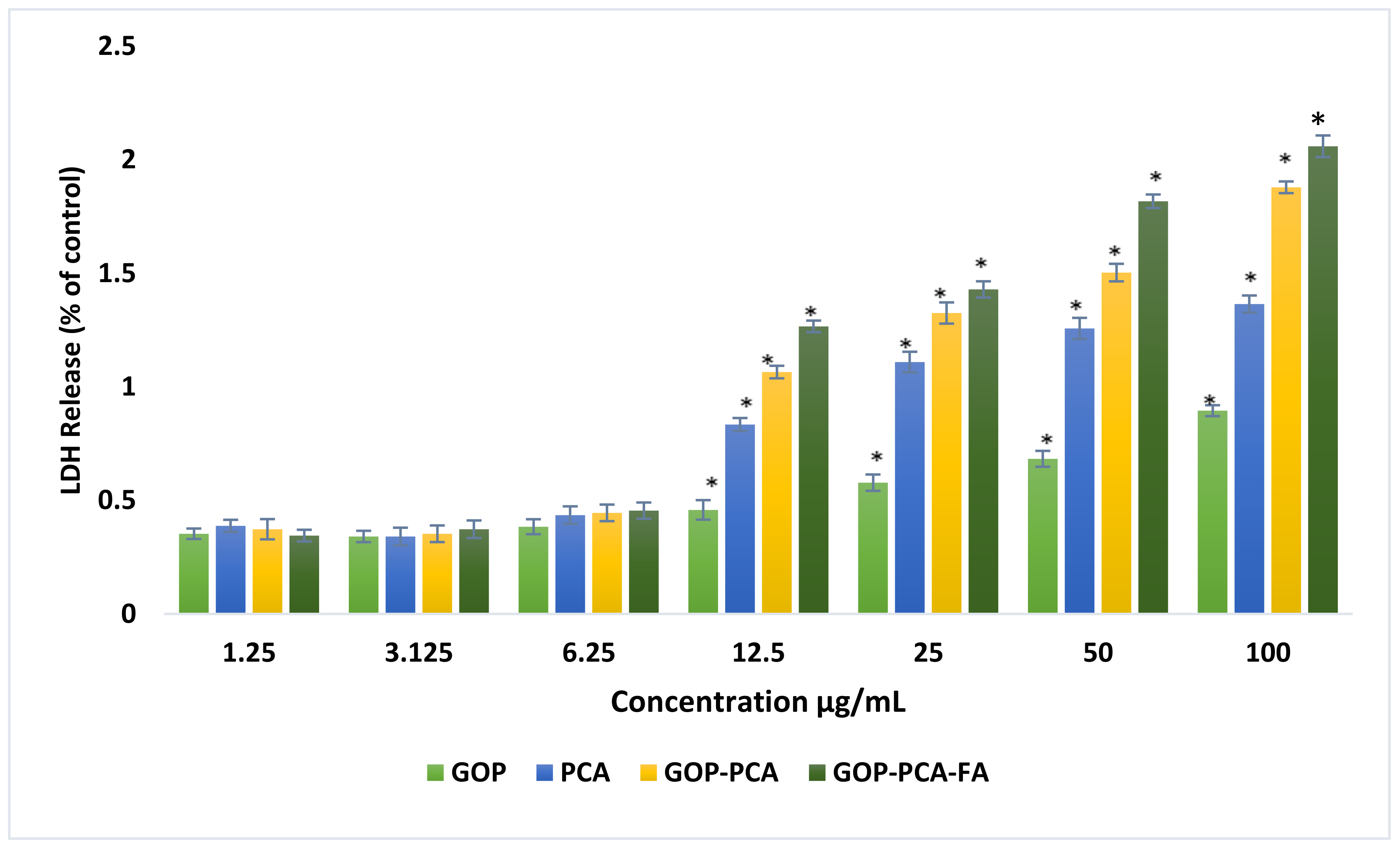
3.6. Lactate Dehydrogenase (LDH) Release from HepG2 Cells Treated with PCA Drug, GOP–PCA, and GOP–PCA–FA Nanocomposites
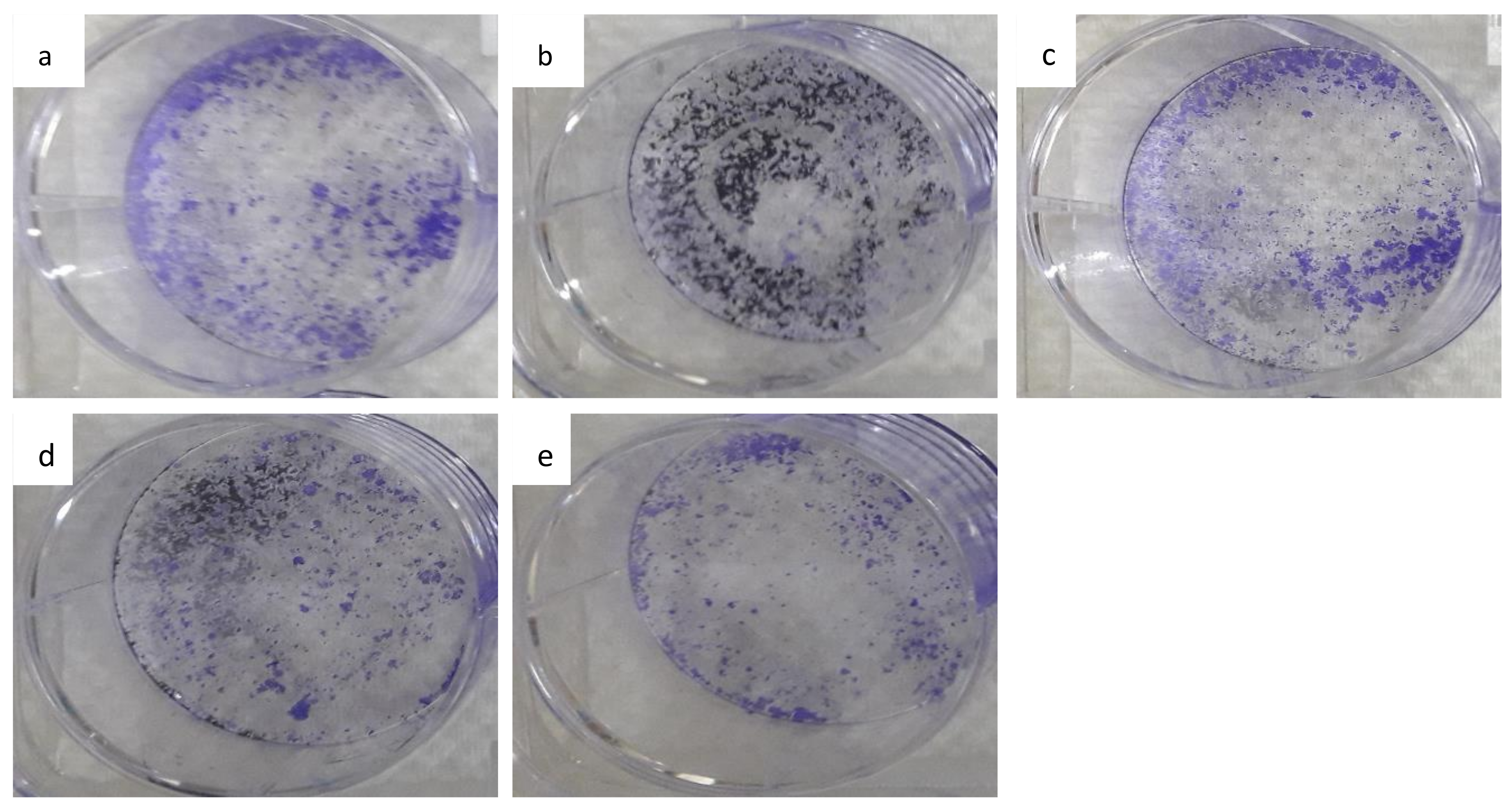
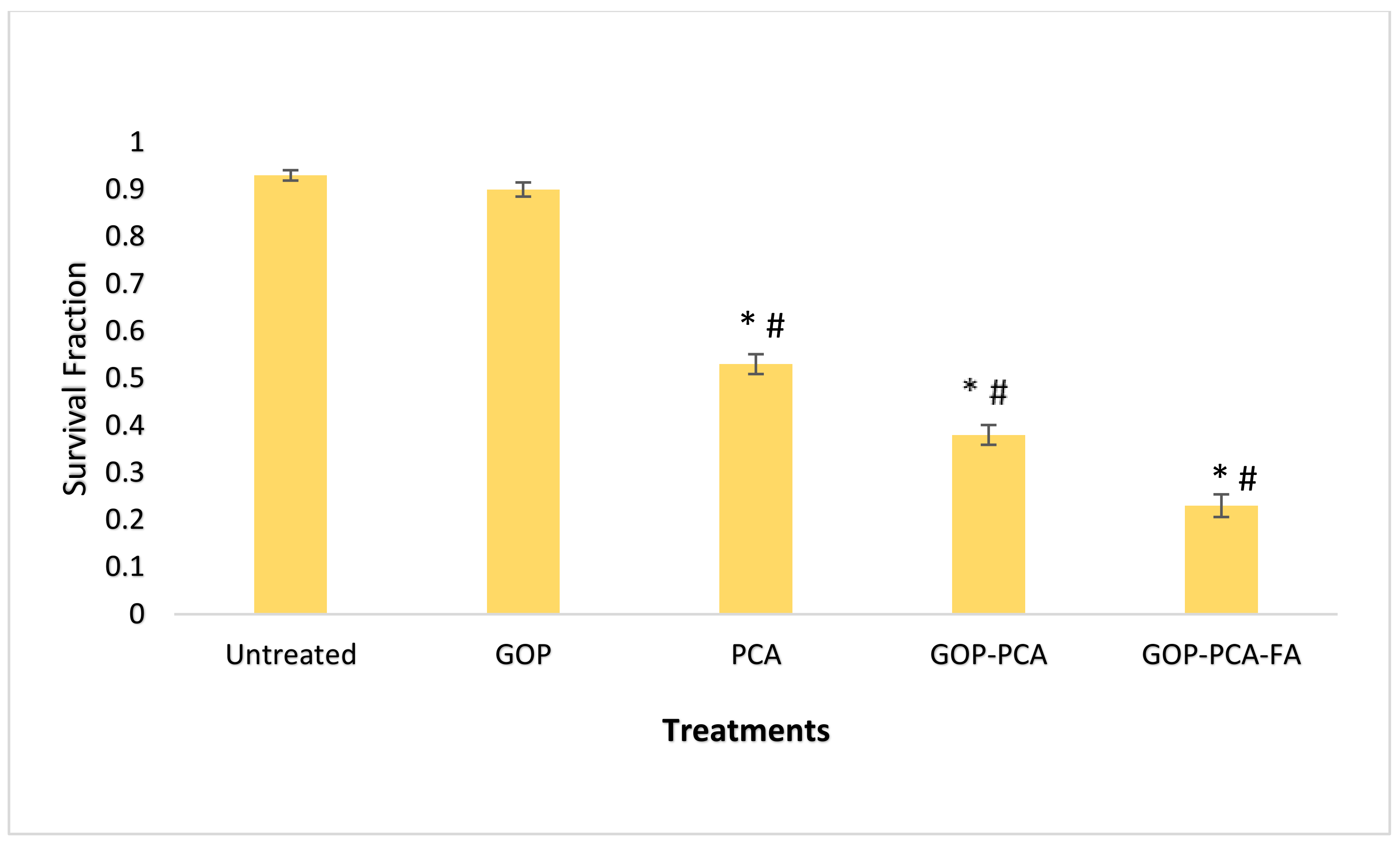
3.7. The Colony-Forming Ability of HepG2 Cells Following Long-Term Exposure to Nanocomposite
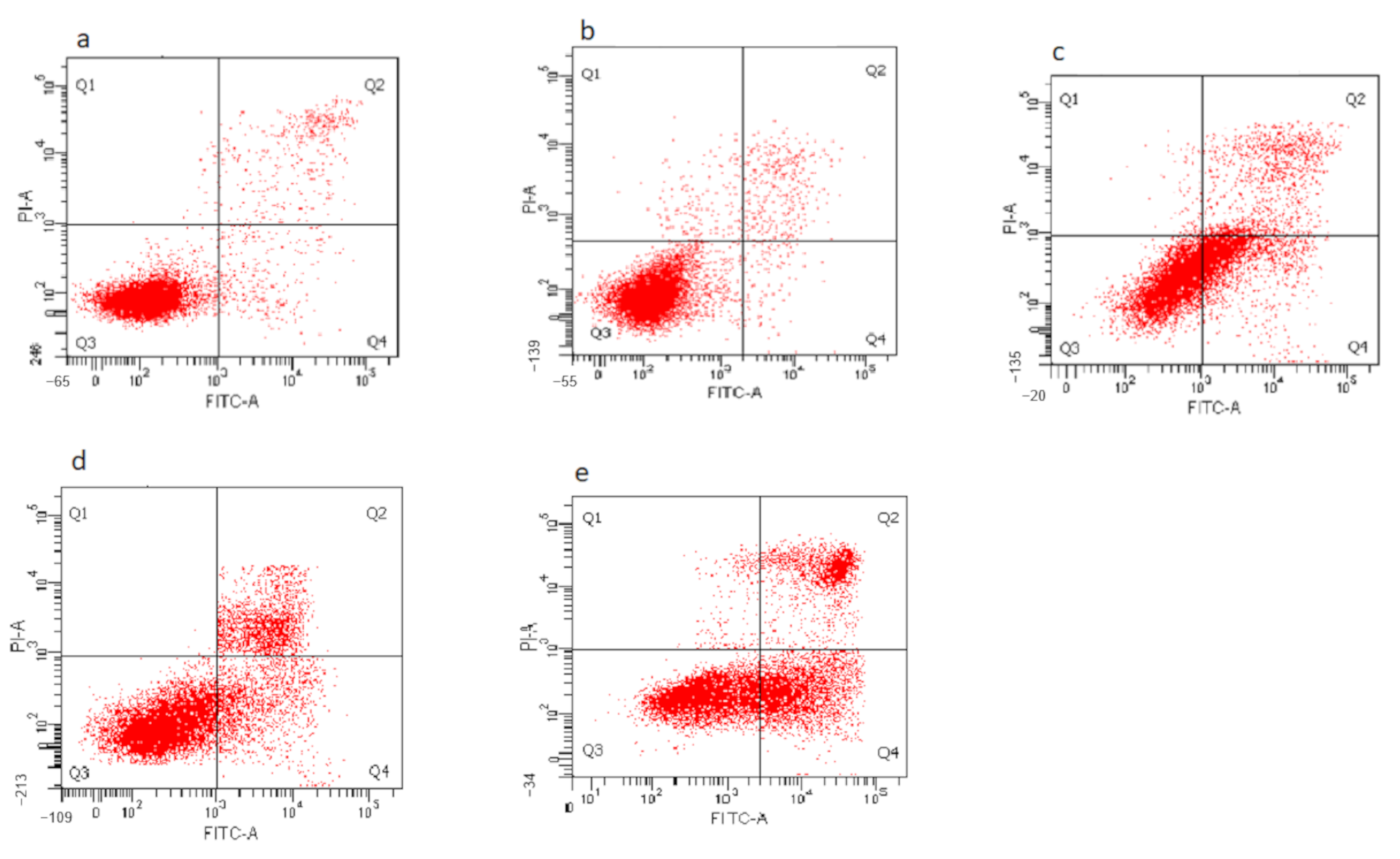
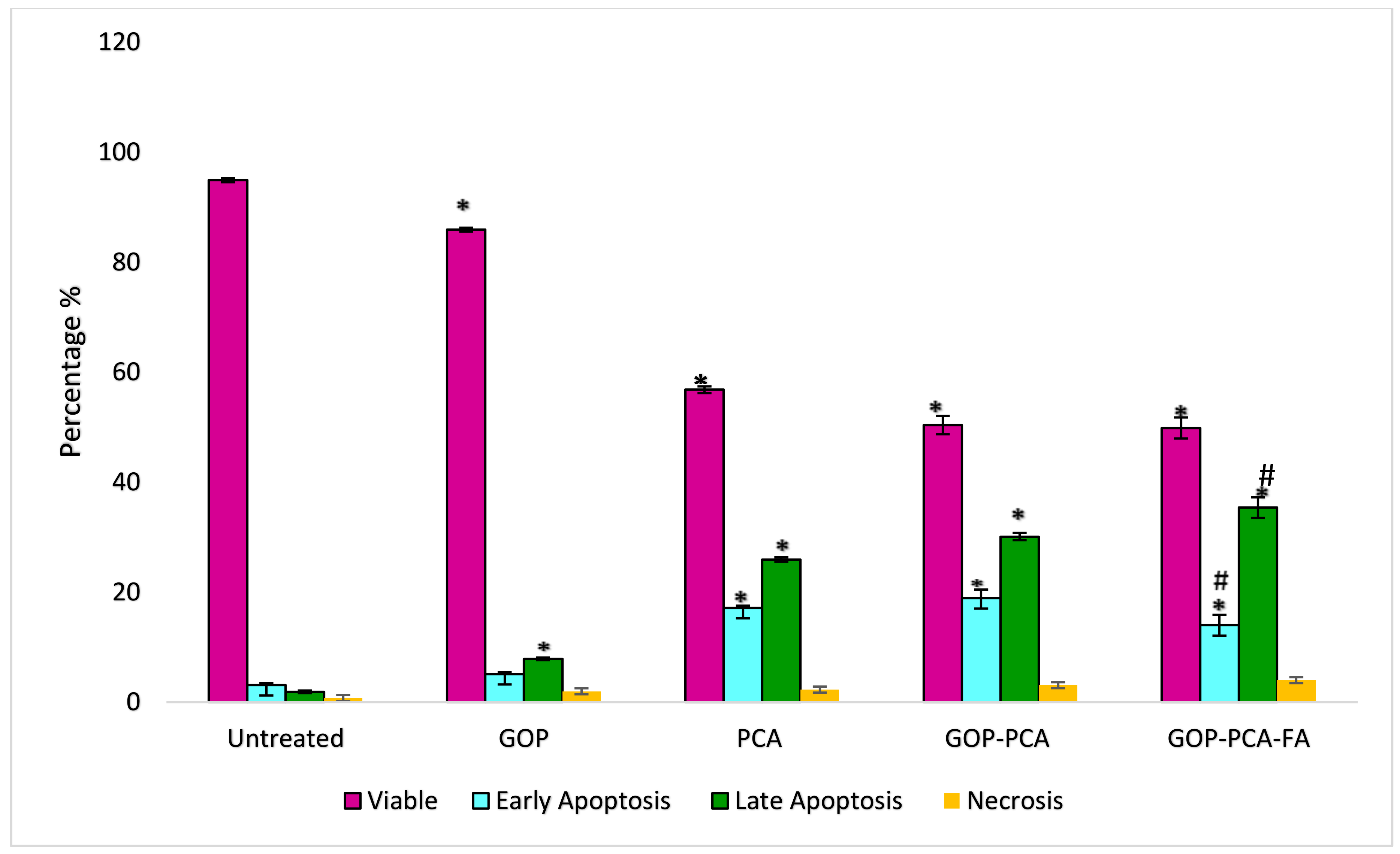
3.8. Determination of Nanocomposite-Induced Apoptosis in HepG2 Cells Using Annexin V FITC/Propidium Iodide (PI) Staining
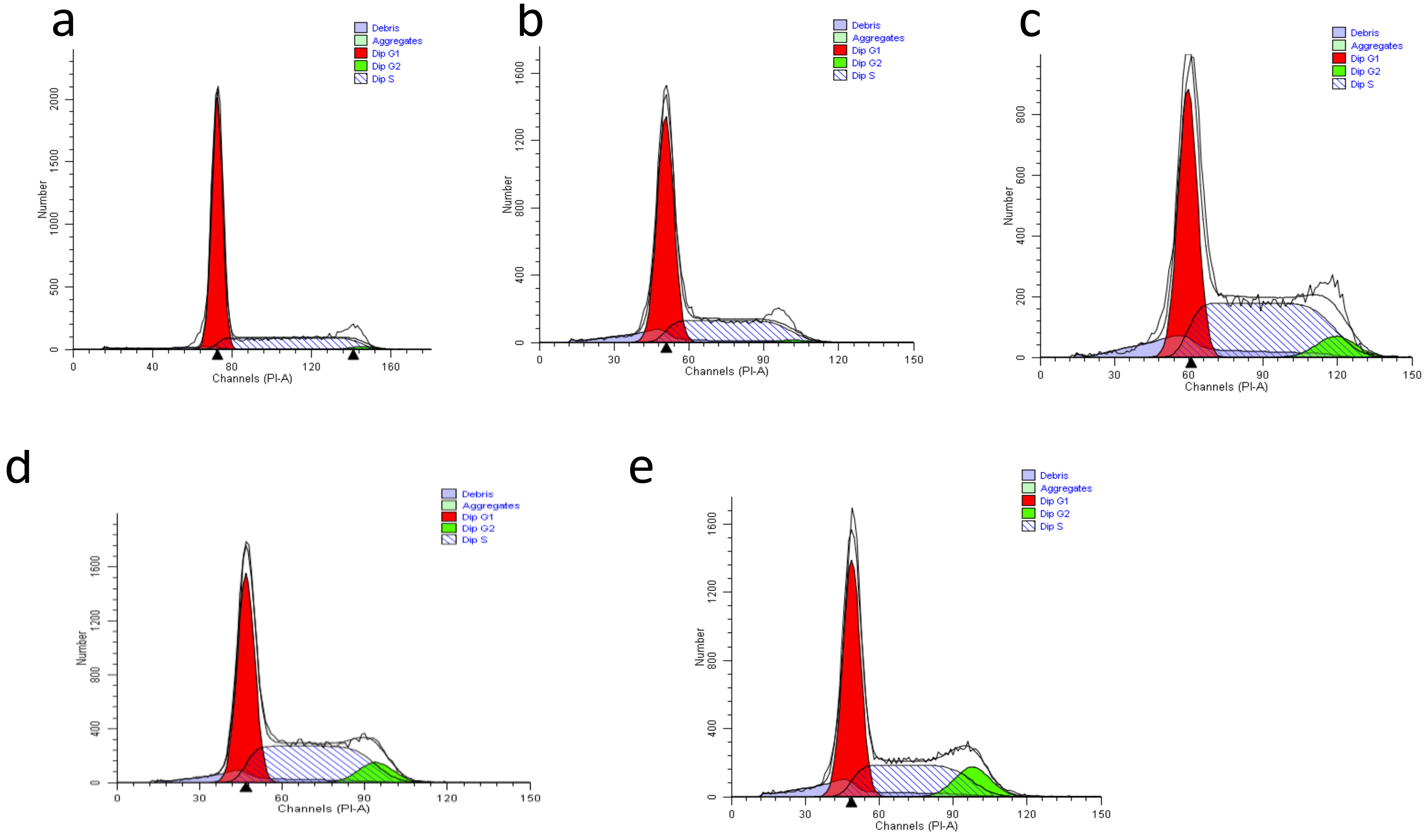
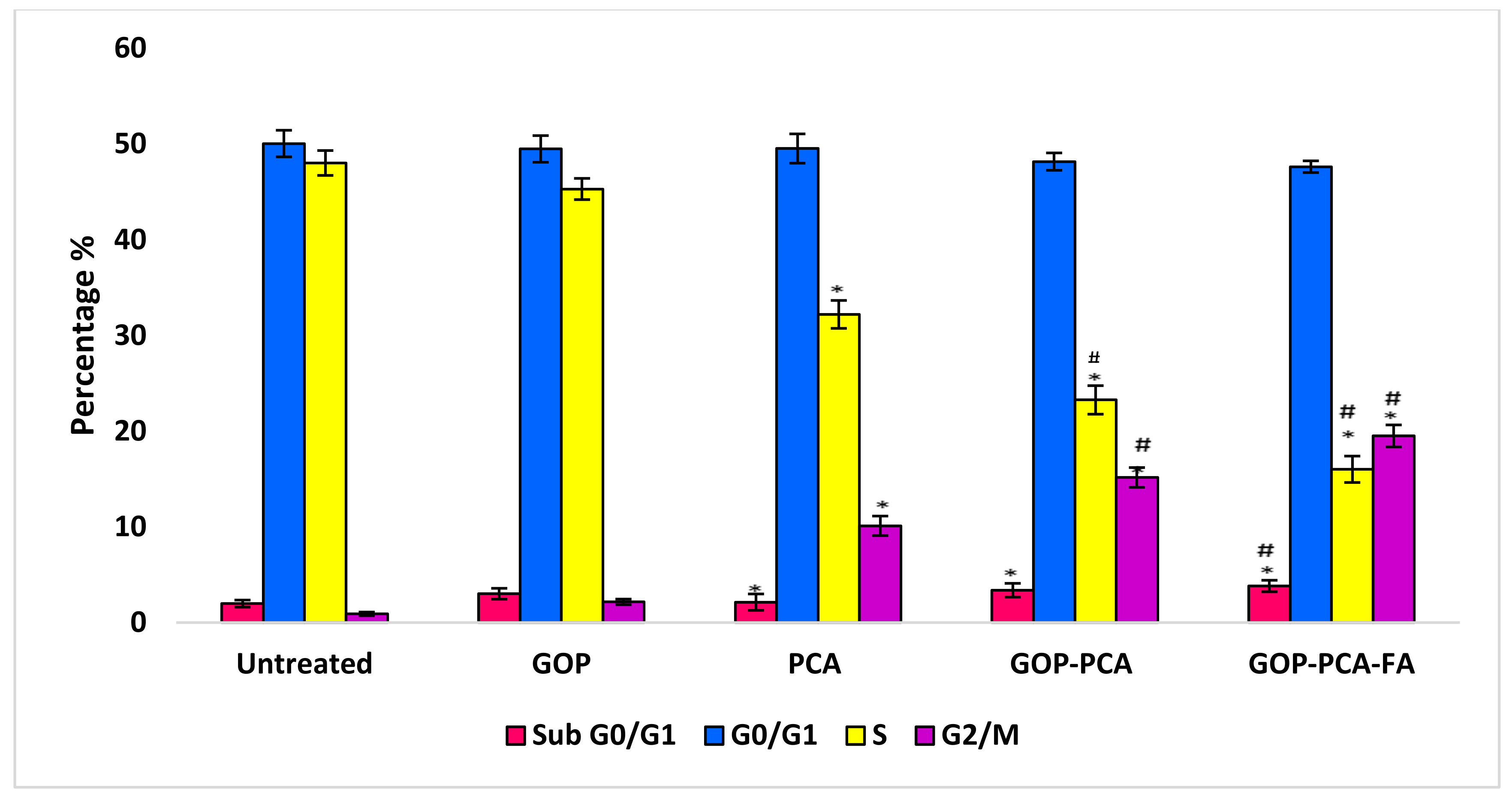
3.9. The Effect of Nanocomposites on Cell Cycle Distribution in HepG2 Cells
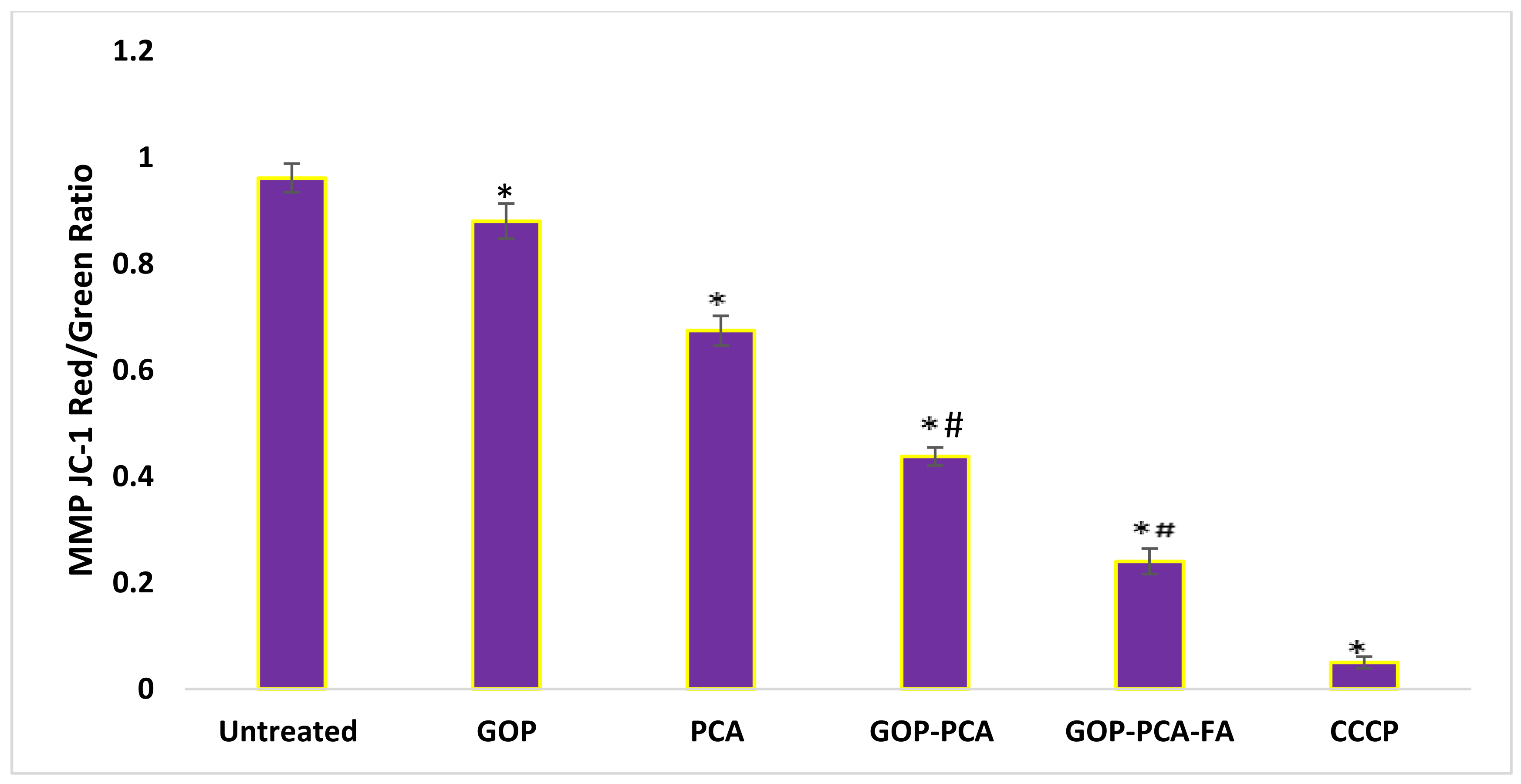
3.10. Nanocomposite Stimulates Mitochondrial Membrane Potential in HepG2 Cells.
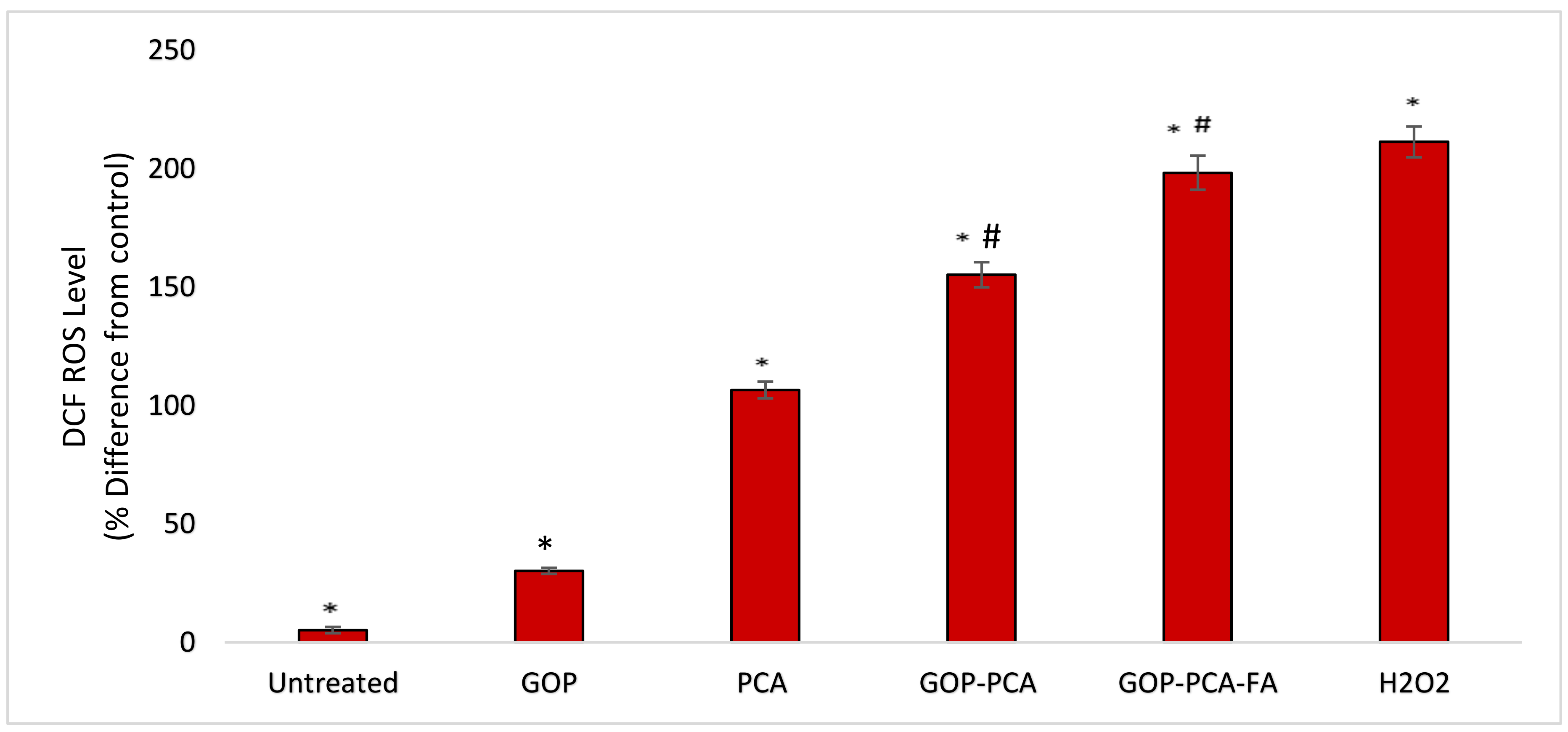
3.11. Nanocomposite Induces Intracellular ROS Generation in HepG2 Cells
3.12. Apoptosis-Related Proteins’ Expression in HepG2 Cells Treated with GOP–PCA–FA Nanocomposite
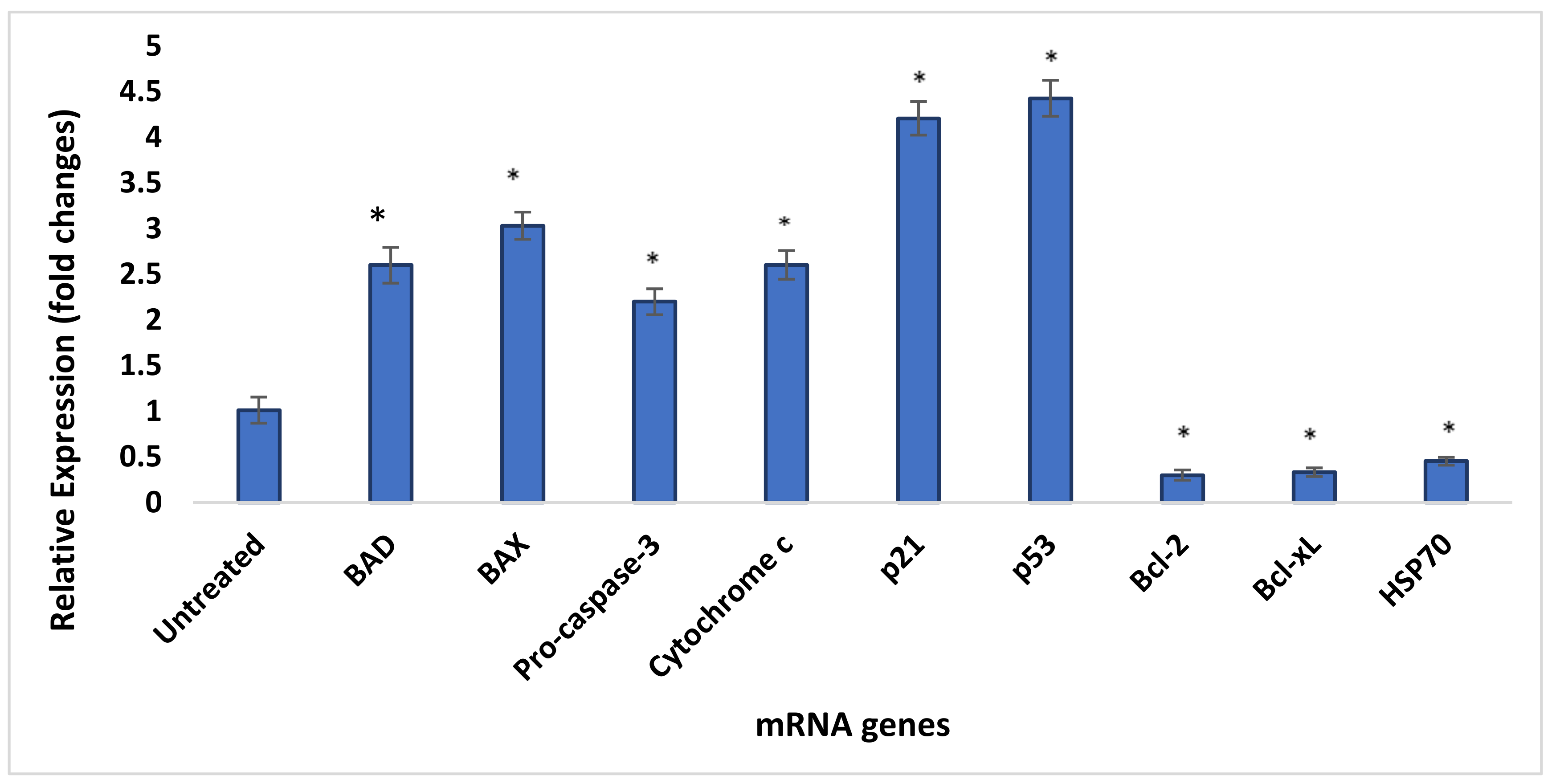
3.13. Quantity Assessment Using qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [PubMed]
- Altekruse, S.F.; McGlynn, K.A.; Reichman, M.E. Hepatocellular Carcinoma Incidence, Mortality, and Survival Trends in the United States From 1975 to 2005. J. Clin. Oncol. 2009, 27, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.-W.; Lin, S.-C.; Su, J.-H.; Chen, Y.K.; Lin, C.-C.; Chan, H.-L. Sinularin induces DNA damage, G2/M phase arrest, and apoptosis in human hepatocellular carcinoma cells. BMC Complement. Altern. Med. 2017, 17, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Latest World Cancer Statistics-GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012-IARC. Available online: https://www.iarc.fr/news-events/latest-world-cancer-statistics-globocan-2012-estimated-cancer-incidence-mortality-and-prevalence-worldwide-in-2012/ (accessed on 12 December 2020).
- Kakkar, S.; Bais, S. A Review on Protocatechuic Acid and Its Pharmacological Potential. ISRN Pharmacol. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Barahuie, F.; Hussein, M.Z.; Gani, S.A.; Fakurazi, S.; Zainal, Z. Synthesis of protocatechuic acid–zinc/aluminium–layered double hydroxide nanocomposite as an anticancer nanodelivery system. J. Solid State Chem. 2015, 221, 21–31. [Google Scholar] [CrossRef]
- Semaming, Y.; Pannengpetch, P.; Chattipakorn, S.C.; Chattipakorn, N. Pharmacological Properties of Protocatechuic Acid and Its Potential Roles as Complementary Medicine. Evidence-Based Complement. Altern. Med. 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Xu, B.; Watkins, R.; Wu, L.; Zhang, C.; Davis, R.M. Natural product-based nanomedicine: Recent advances and issues. Int. J. Nanomedicine. 2015, 10, 6055–6074. [Google Scholar] [CrossRef]
- Joseph, R.R.; Venkatraman, S. Drug delivery to the eye: What benefits do nanocarriers offer? Nanomedicine 2017, 12, 683–702. [Google Scholar] [CrossRef]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Sharma, H.; Mondal, S. Functionalized Graphene Oxide for Chemotherapeutic Drug Delivery and Cancer Treatment: A Promising Material in Nanomedicine. Int. J. Mol. Sci. 2020, 21, 6280. [Google Scholar] [CrossRef]
- Gonçalves, C.; Pereira, P.A.C.; Gama, M. Self-Assembled Hydrogel Nanoparticles for Drug Delivery Applications. Materials 2010, 3, 1420–1460. [Google Scholar] [CrossRef]
- Pattnaik, S.; Swain, K.; Lin, Z. Graphene and graphene-based nanocomposites: Biomedical applications and biosafety. J. Mater. Chem. B 2016, 4, 7813–7831. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Javaid, F.; Chudasama, V. Advances in targeting the folate receptor in the treatment/imaging of cancers. Chem. Sci. 2018, 9, 790–810. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Xu, Y.; Sun, K.; Wang, H.; Zhang, F.; Zhou, Z.; Zhang, J.; Zhang, F.; Caliskan, B.; Qiu, Z.; et al. Identification of a peptide for folate receptor alpha by phage display and its tumor targeting activity in ovary cancer xenograft. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Saifullah, B.; Buskaran, K.; Shaikh, R.B.; Barahuie, F.; Fakurazi, S.; Moklas, M.A.M.; Hussein, M.Z. Graphene Oxide–PEG–Protocatechuic Acid Nanocomposite Formulation with Improved Anticancer Properties. Nanomaterials 2018, 8, 820. [Google Scholar] [CrossRef] [PubMed]
- Buskaran, K.; Hussein, M.Z.; Moklas, M.A.M.; Fakurazi, S. Morphological Changes and Cellular Uptake of Functionalized Graphene Oxide Loaded with Protocatechuic Acid and Folic Acid in Hepatocellular Carcinoma Cancer Cell. Int. J. Mol. Sci. 2020, 21, 5874. [Google Scholar] [CrossRef] [PubMed]
- Barahuie, F.; Hussein, M.Z.; Hussein-Al-Ali, S.H.; Arulselvan, P.; Fakurazi, S.; Zainal, Z. Preparation and controlled-release studies of a protocatechuic acid-magnesium/aluminum-layered double hydroxide nanocomposite. Int. J. Nanomed. 2013, 8, 1975–1987. [Google Scholar] [CrossRef] [PubMed]
- Kazempour, M.; Namazi, H.; Akbarzadeh, A.; Kabiri, R. Synthesis and characterization of PEG-functionalized graphene oxide as an effective pH-sensitive drug carrier. Artif. Cells Nanomed. Biotechnol. 2019, 47, 90–94. [Google Scholar] [CrossRef]
- Colone, M.; Calcabrini, A.; Stringaro, A. Drug Delivery Systems of Natural Products in Oncology. Molecules 2020, 25, 4560. [Google Scholar] [CrossRef]
- Naso, L.G.; Valcarcel, M.; Roura-Ferrer, M.; Kortazar, D.; Salado, C.; Lezama, L.; Rojo, T.; González-Baró, A.C.; Williams, P.A.; Ferrer, E.G. Promising antioxidant and anticancer (human breast cancer) oxidovanadium(IV) complex of chlorogenic acid. Synthesis, characterization and spectroscopic examination on the transport mechanism with bovine serum albumin. J. Inorg. Biochem. 2014, 135, 86–99. [Google Scholar] [CrossRef]
- Fong, Y.T.; Chen, C.-H.; Chen, J.-P. Intratumoral Delivery of Doxorubicin on Folate-Conjugated Graphene Oxide by In-Situ Forming Thermo-Sensitive Hydrogel for Breast Cancer Therapy. Nanomaterials 2017, 7, 388. [Google Scholar] [CrossRef] [PubMed]
- Liechty, W.B.; Peppas, N.A. Expert opinion: Responsive polymer nanoparticles in cancer therapy. Eur. J. Pharm. Biopharm. 2012, 80, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Koirala, N.; Das, D.; Fayazzadeh, E.; Sen, S.; McClain, A.; Puskas, J.E.; Drazba, J.A.; McLennan, G. Folic acid conjugated polymeric drug delivery vehicle for targeted cancer detection in hepatocellular carcinoma. J. Biomed. Mater. Res. Part A 2019, 107, 2522–2535. [Google Scholar] [CrossRef]
- Zwicke, G.L.; Mansoori, G.A.; Jeffery, C. Utilizing the folate receptor for active targeting of cancer nanotherapeutics. Nano Rev. 2012, 3, 18496. [Google Scholar] [CrossRef] [PubMed]
- McGuire, J.J.; Haile, W.H.; Yeh, C.-C. 5-Amino-4-Imidazolecarboxamide Riboside Potentiates Both Transport of Reduced Folates and Antifolates by the Human Reduced Folate Carrier and Their Subsequent Metabolism. Cancer Res. 2006, 66, 3836–3844. [Google Scholar] [CrossRef]
- Gao, B.; Shen, L.; He, K.-W.; Xiao, W.-H. GNRs@SiO2-FA in combination with radiotherapy induces the apoptosis of HepG2 cells by modulating the expression of apoptosis-related proteins. Int. J. Mol. Med. 2015, 36, 1282–1290. [Google Scholar] [CrossRef]
- Saifullah, B.; Buskaran, K.; Baby, R.; Dorniani, D.; Fakurazi, S.; Hussein, M.Z. Dual Drugs Anticancer Nanoformulation using Graphene Oxide-PEG as Nanocarrier for Protocatechuic Acid and Chlorogenic Acid. Pharm. Res. 2019, 36, 1–11. [Google Scholar] [CrossRef]
- Paradee, N.; Sirivat, A. Encapsulation of Folic Acid in Zeolite Y for Controlled Release via Electric Field. Mol. Pharm. 2015, 13, 155–162. [Google Scholar] [CrossRef]
- Din, F.U.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int. J. Nanomed. 2017, 12, 7291–7309. [Google Scholar] [CrossRef]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.D.; Ghandehari, H. Nanoparticle uptake: The phagocyte problem. Nano Today 2015, 10, 487–510. [Google Scholar] [CrossRef]
- Malachowski, T.; Hassel, A. Engineering nanoparticles to overcome immunological barriers for enhanced drug delivery. Eng. Regen. 2020, 1, 35–50. [Google Scholar] [CrossRef]
- Navya, P.; Kaphle, A.; Srinivas, S.P.; Bhargava, S.K.; Rotello, V.M.; Daima, H.K. Current trends and challenges in cancer management and therapy using designer nanomaterials. Nano Converg. 2019, 6, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal. Transduct. Target. Ther. 2018, 3, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Lammel, T.; Boisseaux, P.; Fernández-Cruz, M.-L.; Navas, J.M. Internalization and cytotoxicity of graphene oxide and carboxyl graphene nanoplatelets in the human hepatocellular carcinoma cell line Hep G2. Part. Fibre Toxicol. 2013, 10, 1–21. [Google Scholar] [CrossRef]
- Wang, S.-B.; Ma, Y.-Y.; Chen, X.-Y.; Zhao, Y.-Y.; Mou, X.-Z. Ceramide-Graphene Oxide Nanoparticles Enhance Cytotoxicity and Decrease HCC Xenograft Development: A Novel Approach for Targeted Cancer Therapy. Front. Pharmacol. 2019, 10, 69. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Reutelingsperger, C. Flow cytometry of apoptotic cell death. J. Immunol. Methods 2000, 243, 167–190. [Google Scholar] [CrossRef]
- Wlodkowic, D.; Telford, W.; Skommer, J.; Darzynkiewicz, Z. Apoptosis and Beyond: Cytometry in Studies of Programmed Cell Death. Methods Cell Biol. 2011, 103, 55–98. [Google Scholar] [CrossRef]
- Demchenko, A.P. Beyond annexin V: Fluorescence response of cellular membranes to apoptosis. Cytotechnology 2013, 65, 157–172. [Google Scholar] [CrossRef]
- Tian, J.; Luo, Y.; Huang, L.; Feng, Y.; Ju, H.; Yu, B. Pegylated folate and peptide-decorated graphene oxide nanovehicle for in vivo targeted delivery of anticancer drugs and therapeutic self-monitoring. Biosens. Bioelectron. 2016, 80, 519–524. [Google Scholar] [CrossRef]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nat. Cell Biol. 2001, 411, 342–348. [Google Scholar] [CrossRef]
- Gogineni, V.R.; Nalla, A.K.; Gupta, R.; Dinh, D.H.; Klopfenstein, J.D.; Rao, J.S. Chk2-mediated G2/M cell cycle arrest maintains radiation resistance in malignant meningioma cells. Cancer Lett. 2011, 313, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Solomon, M.; Liu, Y.; Berezin, M.Y.; Achilefu, S. Optical Imaging in Cancer Research: Basic Principles, Tumor Detection, and Therapeutic Monitoring. Med. Princ. Pr. 2011, 20, 397–415. [Google Scholar] [CrossRef] [PubMed]
- Barnum, K.J.; O’Connell, M.J. Cell Cycle Regulation by Checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Dai, X.; Wang, X.; Tan, P.; Gu, L.; Luo, Q.; Zheng, X.; Li, Z.; Zhu, H.; Zhang, H.; et al. A Nanostrategy for Efficient Imaging-Guided Antitumor Therapy through a Stimuli-Responsive Branched Polymeric Prodrug. Adv. Sci. 2020, 7, 1903243. [Google Scholar] [CrossRef]
- Visconti, R.; Della Monica, R.; Grieco, D. Cell cycle checkpoint in cancer: A therapeutically targetable double-edged sword. J. Exp. Clin. Cancer Res. 2016, 35, 1–8. [Google Scholar] [CrossRef]
- Yuan, J.; Gao, H.; Ching, C.-B. Comparative protein profile of human hepatoma HepG2 cells treated with graphene and single-walled carbon nanotubes: An iTRAQ-coupled 2D LC–MS/MS proteome analysis. Toxicol. Lett. 2011, 207, 213–221. [Google Scholar] [CrossRef]
- Prieto-Bermejo, R.; Hernández-Hernández, Á. The Importance of NADPH Oxidases and Redox Signaling in Angiogenesis. Antioxidants 2017, 6, 32. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, B.; Zhou, L.; Shi, Y.; Li, Z.; Xia, Y.; Tian, J. Imaging Dendrimer-Grafted Graphene Oxide Mediated Anti-miR-21 Delivery with an Activatable Luciferase Reporter. ACS Appl. Mater. Interfaces 2016, 8, 9014–9021. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Gurunathan, S.; Han, J.W.; Eppakayala, V.; Kim, J.-H. Green synthesis of graphene and its cytotoxic effects in human breast cancer cells. Int. J. Nanomed. 2013, 8, 1015–1027. [Google Scholar] [CrossRef]
- Krizkova, S.; Heger, Z.; Zalewska, M.; Moulick, A.; Adam, V.; Kizek, R. Nanotechnologies in protein microarrays. Nanomedicine 2015, 10, 2743–2755. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Dodig-Crnković, T.; Schwenk, J.M.; Tao, S.-C. Current applications of antibody microarrays. Clin. Proteom. 2018, 15, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. et Biophys. Acta (BBA) Bioenerg. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Karch, J.; Kwong, J.Q.; Burr, A.R.; Sargent, M.A.; Elrod, J.W.; Peixoto, P.M.; Martínez-Caballero, S.; Osinska, H.; Cheng, E.H.-Y.; Robbins, J.; et al. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. eLife 2013, 2, e00772. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W. Molecular Mechanisms That Differentiate Apoptosis from Programmed Necrosis. Toxicol. Pathol. 2012, 41, 227–234. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef]
- Nicosia, A.; Cavallaro, G.; Costa, S.; Utzeri, M.A.; Cuttitta, A.; Giammona, G.; Mauro, N. Carbon Nanodots for On Demand Chemophotothermal Therapy Combination to Elicit Necroptosis: Overcoming Apoptosis Resistance in Breast Cancer Cell Lines. Cancers 2020, 12, 3114. [Google Scholar] [CrossRef]
- Sepand, M.R.; Ranjbar, S.; Kempson, I.M.; Akbariani, M.; Muganda, W.C.A.; Müller, M.; Ghahremani, M.H.; Raoufi, M. Targeting non-apoptotic cell death in cancer treatment by nanomaterials: Recent advances and future outlook. Nanomed.: Nanotechnol. Biol. Med. 2020, 29, 102243. [Google Scholar] [CrossRef]
- Sharifi, M.; Hosseinali, S.H.; Saboury, A.A.; Szegezdi, E.; Falahati, M. Involvement of planned cell death of necroptosis in cancer treatment by nanomaterials: Recent advances and future perspectives. J. Control. Release 2019, 299, 121–137. [Google Scholar] [CrossRef]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef]
- Velez, A.M.A.; Howard, M.S. Tumor-suppressor genes, cell cycle regulatory checkpoints, and the skin. North Am. J. Med. Sci. 2015, 7, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Luk, S.C.-W.; Siu, S.W.-F.; Lai, C.-K.; Wu, Y.-J.; Pang, S.-F. Cell Cycle Arrest by a Natural Product via G2/M Checkpoint. Int. J. Med. Sci. 2005, 2, 64–69. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Master Mix Volume/15 µL Reaction |
|---|---|
| 100 mM dNTPs (with dTTP) | 0.15 |
| MultiScribe™ Reverse Transcriptase, 50 U/μL | 1.00 |
| 10× Reverse Transcription Buffer | 1.50 |
| RNase Inhibitor, 20 U/μL | 0.19 |
| Nuclease-free water | 4.16 |
| Total | 7.00 |
| Component | Volume (µL)/20 µL Reaction |
|---|---|
| TaqMan gene expression Assay (20×) | 1.00 |
| Product from RT reaction (Minimum 1:15 Dilution) | 1.33 |
| TaqMan 2× Universal PCR Master Mix, No AmpErase UNGa | 10.00 |
| Nuclease-free water | 7.67 |
| Gene | Primer Sequences |
|---|---|
| Bad | F-5′-CAGGGGCCTCGTTATCGG-3′ R-5′-GGACTCTGGATCAGACCTCA-3′ |
| Bax | F-5′-ATGTTTTCTGACGGCAACTTC-3′ R-5′-AGTCCAATGTCCAGCCCAT-3′ |
| Caspase-3 | F-5′-TGTTTGTGTGCTTCTGAGCC-3′ R-5′-CACGCCATGTCATCATCAAC-3′ |
| Cytochrome c | F-5′-GCTACTCCTACCTATCTCCC-3′ R-5′-TGTGGTCGTTACCTAGAAGG-3′ |
| p21 | F-5′-TGGAGACTCTCAGGGTCGAAA-3′ R-5′-GGCGTTTGGAGTGGTAGAAATC-3′ |
| p53 | F-5′-ATGTTTTGCCAACTGGCCAAG-3′ R-5′-TGAGCAGCGCTCATGGTG-3’ |
| Bcl-2 | F-5′-ATGTGTGTGGAGACCGTCAA-3′ R-5′-GCCGTACAGTTCCACAAAGG-3′ |
| Bcl-xL | F-5′-CAGAGCTTTGAACAGGTAG-3′ R-5′-GCTCTCGGGTGCTGTATTG-3′ |
| HSP70 | F-5′-AGGCCGACAAGAAGAAGGTGCT-3′ R-5′-TGGTACAGTCCGCTGATGATGG-3′ |
| GAPDH | F-5′-GGCAAATTCAACGGCACAGT-3′ R-5′-AGATGGTGATGGGCTTCCC-3′ |
| Nanocomposites | ζ-Potential (mV) |
|---|---|
| GO | −29.6 ± 2.304 |
| GOP | −9.92 ± 2.112 |
| GOP–PCA | −15.5 ± 1.872 |
| GOP–PCA–FA | −17.3 ± 2.007 |
| Nanocomposites | Loading Content (%) | Encapsulation Efficiency (%) |
|---|---|---|
| GOP–PCA | 35.10% | 97.03% |
| GOP–PCA–FA | 41.06% | 97.17% |
| Apoptotic Protein | Signal Intensity (Arbitrary Unit) | Fold Change | |
|---|---|---|---|
| (Treated/Untreated) | |||
| Untreated | GOP–PCA–FA-Treated | ||
| Pro-apoptotic proteins | |||
| BAD | 7823.15 ± 113.13 | 19,711.53 ± 154.16 * | 2.53 |
| BAX | 7047.33 ± 102.91 | 19,790.15 ± 206.50 ** | 2.81 |
| Pro-Caspase-3 | 8224.25 ± 193.75 | 15,682.80 ± 100.84 * | 1.91 |
| Cytochrome c | 7177.67 ± 387.15 | 19,050.79 ± 394.31 ** | 2.62 |
| p21 | 2682.75 ± 232.20 | 10,412.01 ± 136.61 *** | 3.88 |
| Phospho-p53 (S15) | 2723.11 ± 269.43 | 10,851.03 ± 198.33 *** | 3.99 |
| Anti-apoptotic proteins | |||
| Bcl-2 | 10,791.18 ± 241.87 | 2847.35 ± 183.76 *** | 0.26 |
| Bcl-xL | 9645.18 ± 103.31 | 2833.87 ± 129.91 *** | 0.29 |
| HSP70 | 22,100.67 ± 175.72 | 9006.71 ± 414.04 ** | 0.41 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buskaran, K.; Bullo, S.; Hussein, M.Z.; Masarudin, M.J.; Mohd Moklas, M.A.; Fakurazi, S. Anticancer Molecular Mechanism of Protocatechuic Acid Loaded on Folate Coated Functionalized Graphene Oxide Nanocomposite Delivery System in Human Hepatocellular Carcinoma. Materials 2021, 14, 817. https://doi.org/10.3390/ma14040817
Buskaran K, Bullo S, Hussein MZ, Masarudin MJ, Mohd Moklas MA, Fakurazi S. Anticancer Molecular Mechanism of Protocatechuic Acid Loaded on Folate Coated Functionalized Graphene Oxide Nanocomposite Delivery System in Human Hepatocellular Carcinoma. Materials. 2021; 14(4):817. https://doi.org/10.3390/ma14040817
Chicago/Turabian StyleBuskaran, Kalaivani, Saifullah Bullo, Mohd Zobir Hussein, Mas Jaffri Masarudin, Mohamad Aris Mohd Moklas, and Sharida Fakurazi. 2021. "Anticancer Molecular Mechanism of Protocatechuic Acid Loaded on Folate Coated Functionalized Graphene Oxide Nanocomposite Delivery System in Human Hepatocellular Carcinoma" Materials 14, no. 4: 817. https://doi.org/10.3390/ma14040817
APA StyleBuskaran, K., Bullo, S., Hussein, M. Z., Masarudin, M. J., Mohd Moklas, M. A., & Fakurazi, S. (2021). Anticancer Molecular Mechanism of Protocatechuic Acid Loaded on Folate Coated Functionalized Graphene Oxide Nanocomposite Delivery System in Human Hepatocellular Carcinoma. Materials, 14(4), 817. https://doi.org/10.3390/ma14040817