
Potassium Complexes of Quercetin-5′-Sulfonic Acid and Neutral O-Donor Ligands: Synthesis, Crystal Structure, Thermal Analysis, Spectroscopic Characterization and Physicochemical Properties

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of KQSA Complexes
2.3. Methods
3. Results and Discussion
3.1. Morphology of Crystals
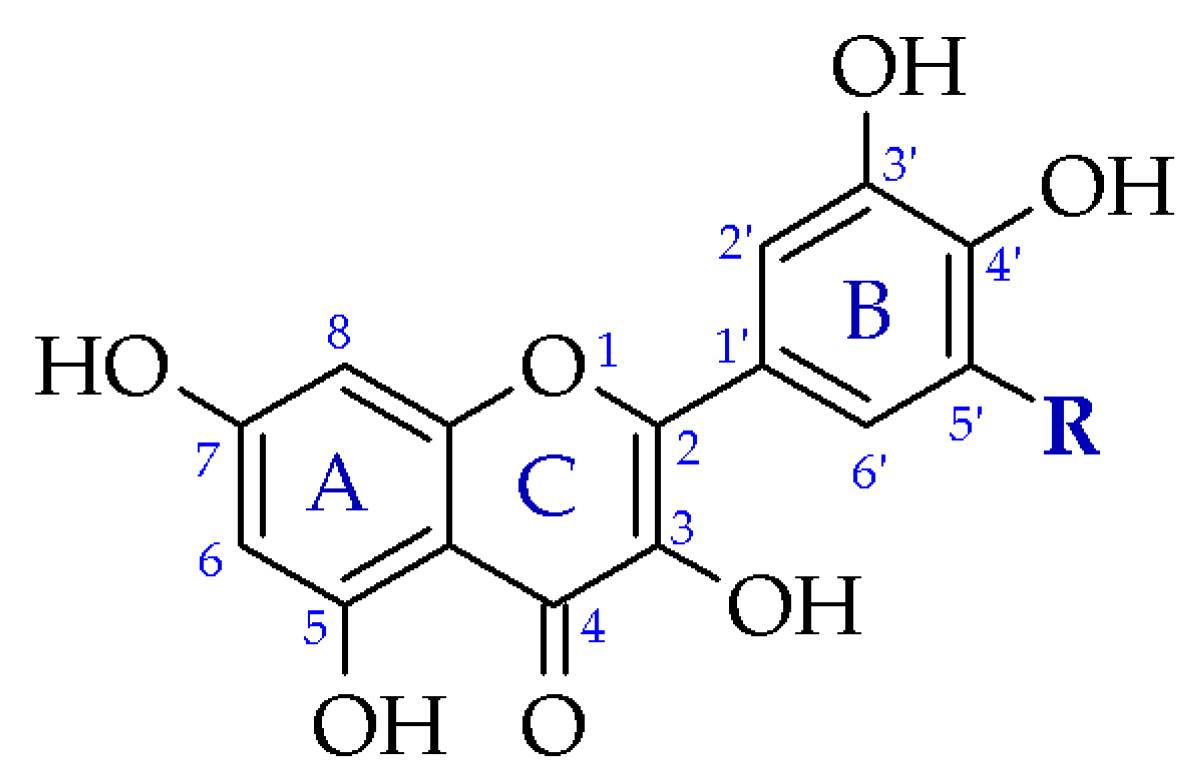
3.2. Crystal Structure of Compound 1
3.3. Thermal Analysis of Complex 1
3.3.1. Thermal Analysis in Helium Atmosphere
3.3.2. Thermal Analysis in Air Atmosphere
3.4. Dissolution Tests of Complex 1
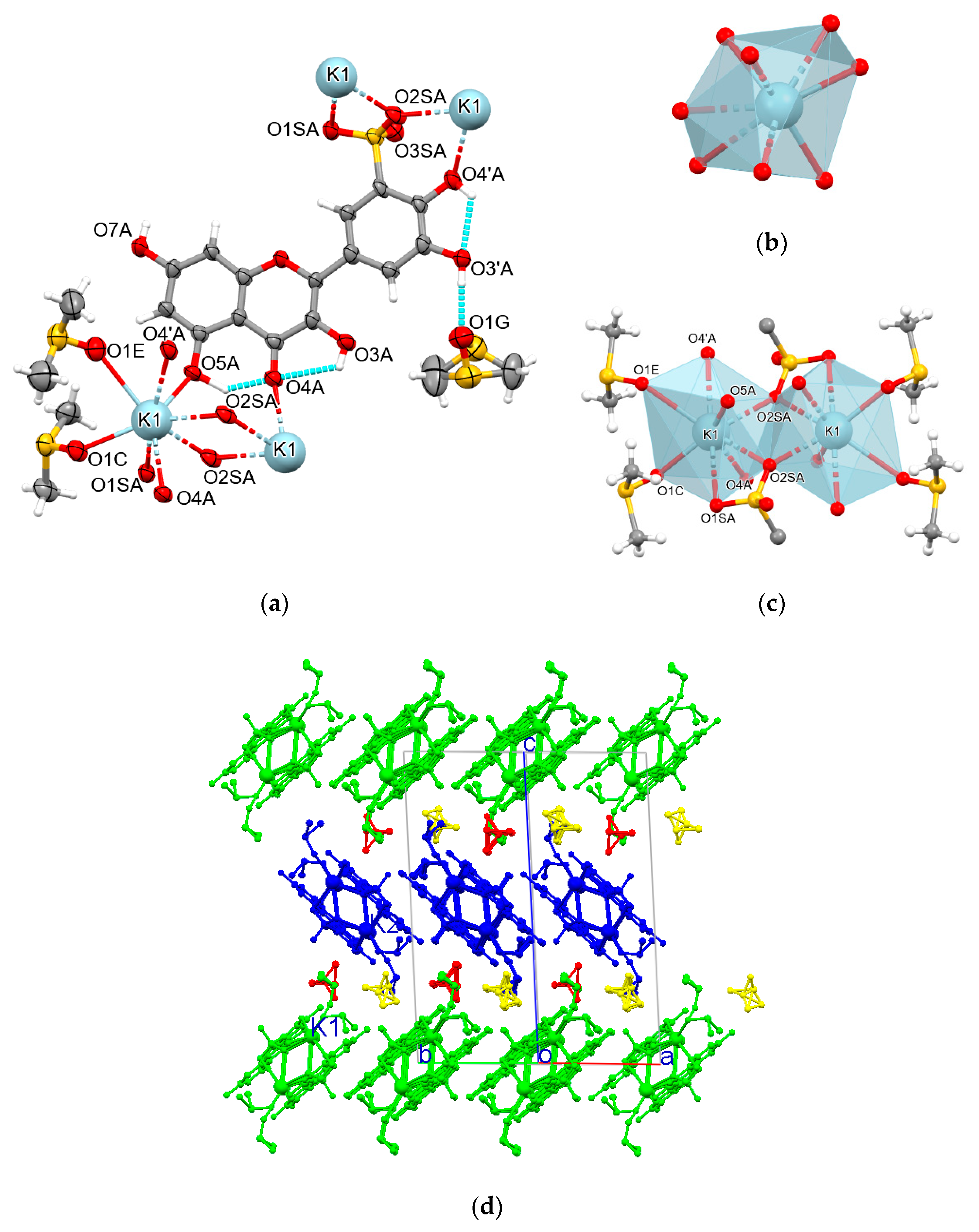
3.5. Crystal Structure of Compound 3
3.6. FT-IR and FT-Raman Spectroscopy
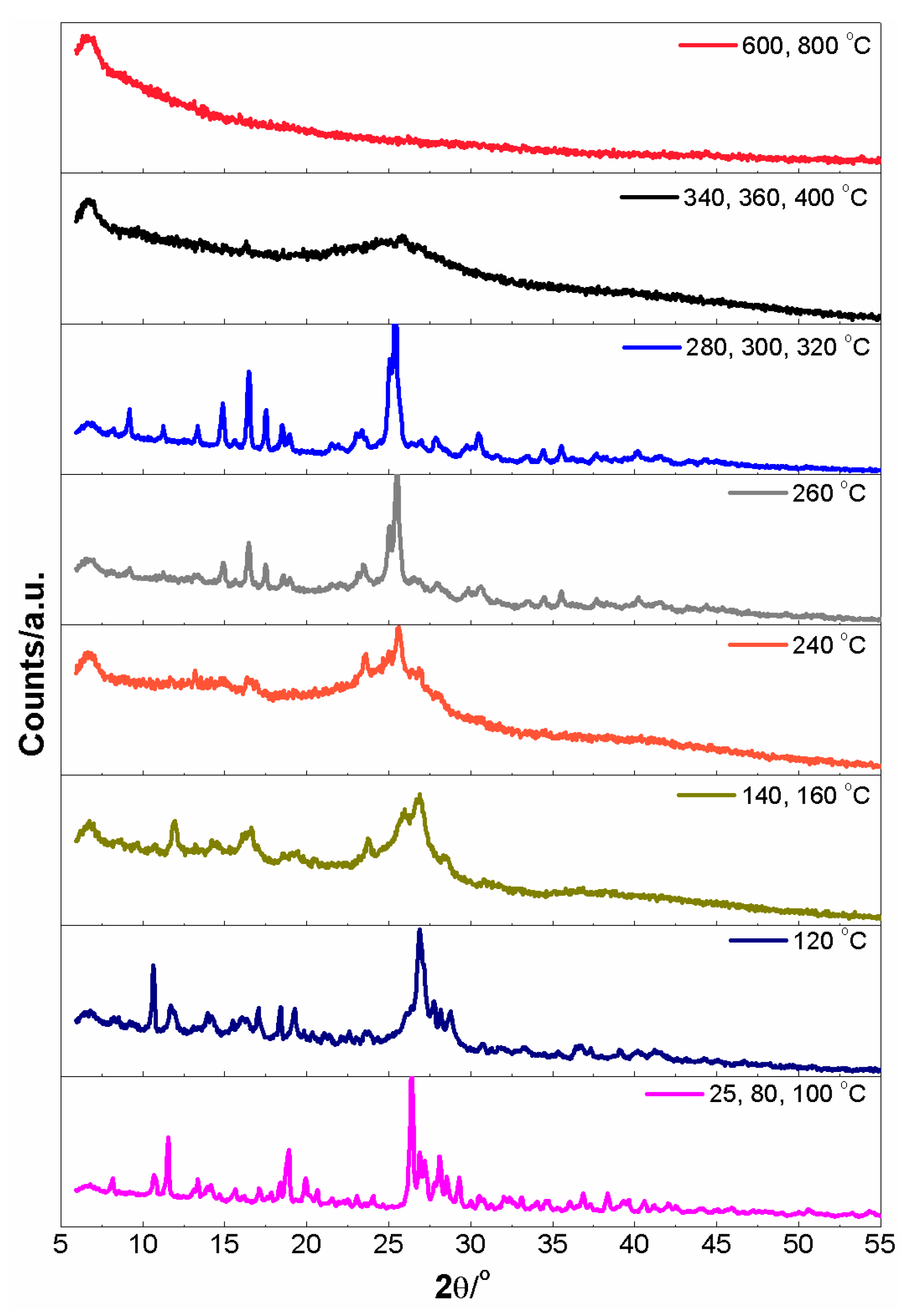
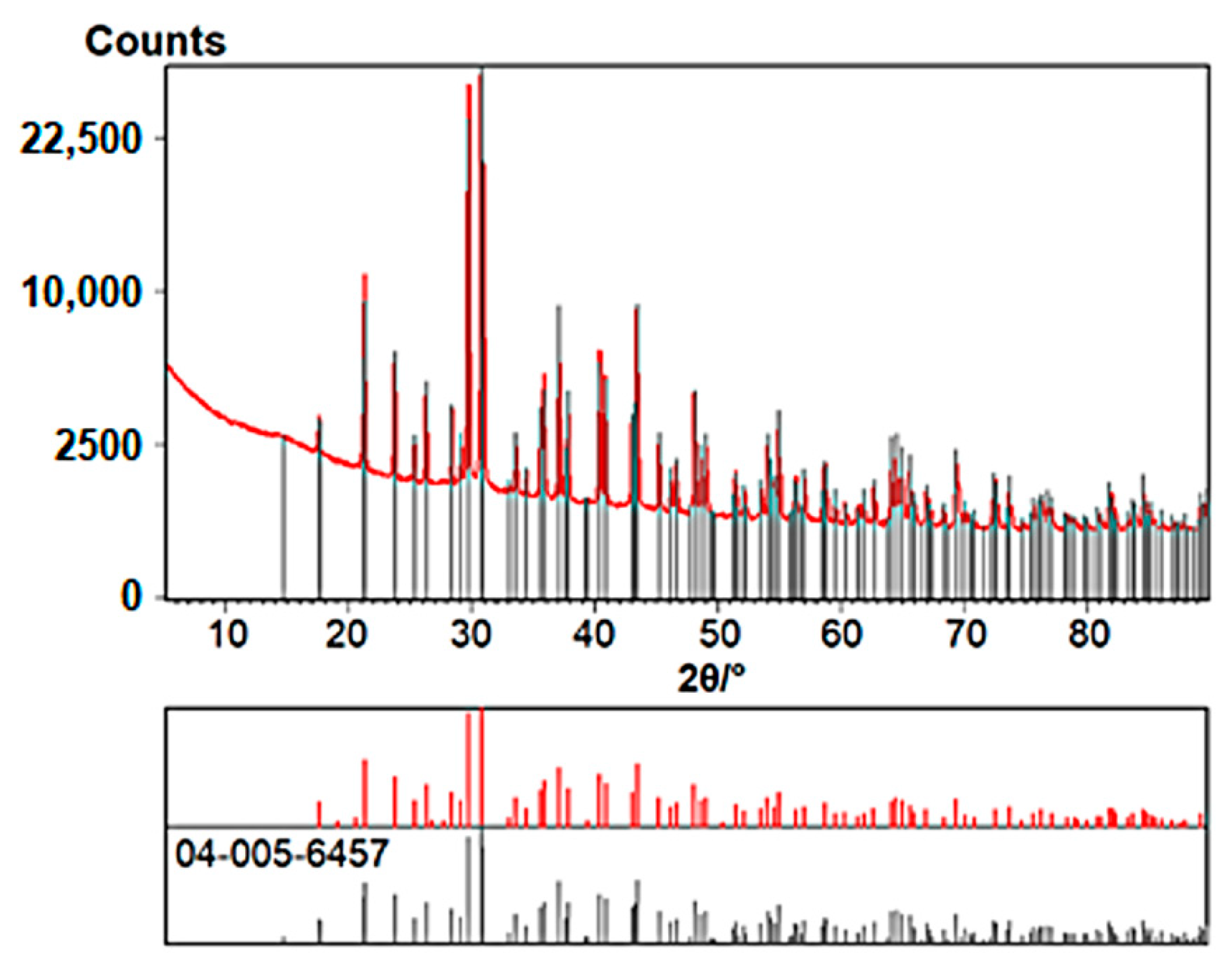
3.7. Desolvation of Crystals 1 and 3 in a Vacuum
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khater, M.; Ravishankar, D.; Greco, F.; Osborn, H.M. Metal complexes of flavonoids: Their synthesis, characterization and enhanced antioxidant and anticancer activities. Future Med. Chem. 2019, 11, 2845–2867. [Google Scholar] [CrossRef]
- Kasprzak, M.M.; Erxleben, A.; Ochocki, J. Properties and applications of flavonoid metal complexes. RSC Adv. 2015, 5, 45853–45877. [Google Scholar] [CrossRef]
- Selvaraj, S.; Krishnaswamy, S.; Devashya, V.; Sethuraman, S.; Krishnan, U.M. Flavonoid-metal ion complexes: A novel class of therapeutic agents. Med. Res. Rev. 2014, 34, 677–702. [Google Scholar] [CrossRef]
- Uivarosi, V.; Munteanu, A.C. Flavonoid Complexes as Promising Anticancer Metallodrugs. In Flavonoids—From Biosynthesis to Human Health; IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef]
- Caddeo, C.; Gabriele, M.; Fernàndez-Busquets, X.; Valenti, D.; Fadda, A.M.; Pucci, L.; Manconi, M. Antioxidant activity of quercetin in Eudragit-coated liposomes for intestinal delivery. Int. J. Pharm. 2019, 565, 64–69. [Google Scholar] [CrossRef]
- Piovezana Bossolani, G.D.; Silva, B.T.; Colombo Martins Perles, J.V.; Lima, M.M.; Vieira Frez, F.C.; Garcia de Souza, S.R.; Sehaber-Sierakowski, C.C.; Bersani-Amado, C.A.; Zanoni, J.N. Rheumatoid arthritis induces enteric neurodegeneration and jejunal inflammation, and quercetin promotes neuroprotective and anti-inflammatory actions. Life Sci. 2019, 238, 116956. [Google Scholar] [CrossRef]
- Güran, M.; Şanlıtürk, G.; Kerküklü, N.R.; Altundağ, E.M.; Süha Yalçın, A. Combined effects of quercetin and curcumin on anti-inflammatory and antimicrobial parameters in vitro. Eur. J. Pharmacol. 2019, 859, 172486. [Google Scholar] [CrossRef]
- Kundur, S.; Prayag, A.; Selvakumar, P.; Nguyen, H.; McKee, L.; Cruz, C.; Srinivasan, A.; Shoyele, S.; Lakshmikuttyamma, A. Synergistic anticancer action of quercetin and curcumin against triple-negative breast cancer cell lines. J. Cell Physiol. 2019, 234, 11103–11118. [Google Scholar] [CrossRef]
- Ding, Y.; Li, C.; Zhang, Y.; Ma, P.; Zhao, T.; Che, D.; Cao, J.; Wang, J.; Liu, R.; Zhang, T.; et al. Quercetin as a Lyn kinase inhibitor inhibits IgE-mediated allergic conjunctivitis. Food Chem. Toxicol. 2020, 135, 110924. [Google Scholar] [CrossRef]
- Wang, Y.; Tao, B.; Wan, Y.; Sun, Y.; Wang, L.; Sun, J.; Li, C. Drug delivery based pharmacological enhancement and current insights of quercetin with therapeutic potential against oral diseases. Biomed. Pharmacother. 2020, 128, 110372. [Google Scholar] [CrossRef]
- Woźnicka, E.; Kuźniar, A.; Nowak, D.; Nykiel, E.; Kopacz, M.; Gruszecka, J.; Golec, K. Comparative study on the antibacterial activity of some flavonoids and their sulfonic derivatives. Acta Pol. Pharm. Drug Res. 2013, 70, 567–571. [Google Scholar]
- Król, W.; Dworniczak, S.; Pietsz, G.; Czuba, Z.P.; Kunicka, M.; Kopacz, M.; Nowak, D. Synthesis and tumoricidal activity evaluation of new morin and quercetin sulfonic derivatives. Acta Pol. Pharm. 2002, 59, 77–79. [Google Scholar]
- Chlebda, E.; Magdalan, J.; Merwid-Lad, A.; Trocha, M.; Kopacz, M.; Kuźniar, A.; Nowak, D.; Szelag, A. Influence of water-soluble flavonoids, quercetin-5′-sulfonic acid sodium salt and morin-5′-sulfonic acid sodium salt, on antioxidant parameters in the subacute cadmium intoxication mouse model. Exp. Toxicol. Pathol. 2010, 62, 105–108. [Google Scholar] [CrossRef]
- Trocha, M.; Merwid-Lad, A.; Szuba, A.; Sozański, T.; Magdalan, J.; Szelag, A.; Kopacz, M.; Kuźniar, A.; Nowak, D. Effect of quercetin-5′-sulfonic acid sodium salt on SOD activity and ADMA/DDAH pathway in extracorporeal liver perfusion in rats. Adv. Clin. Exp. Med. 2012, 21, 423–431. [Google Scholar]
- Magdalan, J.; Szelag, A.; Chlebda, E.; Merwid-Lad, A.; Trocha, M.; Kopacz, M.; Kuźniar, A.; Nowak, D. Quercetin-5′-sulfonic acid solution salt and morin-5′-sulfonic acid sodium salt as antidotes in the subacute cadmium intoxication in mice. Pharmacol. Rep. 2007, 59, 210–216. [Google Scholar]
- Mazur, L.; Tokarska, J.; Koziol, A.E.; Kopacz, M. Ammonium quercetin-5′-sulfonate formamide solvate. Acta Crystallogr. 2004, E60, o779–o781. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Chen, P.; Han, T.; Zhao, W. Sodium quercetin-8-sulfonate trihydrate. Acta Crystallogr. 2010, E66, m1036–m1037. [Google Scholar] [CrossRef]
- Liu, B.; Yang, B.L. Syntheses and crystal structures of [Na(H2O)1/2]X and NH2(CH2CH3)2X and antioxidant activity of the former. Chin. J. Struct. Chem. 2009, 28, 1112–1120. [Google Scholar]
- Liu, B.; Yang, B.L. Poly[(μ-5,7-dihydr-oxy-4-oxo-2-phenyl-4H-chromene-8-sulfonato)potassium(I)]. Acta Crystallogr. 2009, E65, m66–m77. [Google Scholar] [CrossRef]
- Liu, B.; Yang, B.L. Hexaaqua (5,7-dihydroxy-4-oxo-2-phenyl-4H-chromene-8-sulfonato)calcium(II) 5,7-dihydroxy-4-oxo-2-phenyl-4H-chromene-8-sulfonate trihydrate. Acta Crystallogr. 2008, E64, m1569–m1570. [Google Scholar] [CrossRef]
- Zhang, Z.T.; Shi, J.; He, Y.; Guo, Y.N. Self-assembly and crystal structure of a barium sulfonate chrysin coordination polimer. Inorg. Chem. Commun. 2006, 9, 579–581. [Google Scholar] [CrossRef]
- Li, W.W.; Zhang, Z.T. Synthesis, crystal structure and antitumor activity of tectochrysin-6-sulfonate. Chin. J. Struct. Chem. 2018, 1, 97. [Google Scholar]
- Li, W.W.; Zhang, Z.T. Hexaqua-cobalt(II) bis (5-hydroxy-7-methoxy-4-oxo-2-phenyl -4H-chromene-6-sulfonate) tetrahydrate. Acta Crystallogr. 2008, C64, m176–m178. [Google Scholar] [CrossRef]
- Yun, H. Tetraaqua(7-hydroxy-5-oxidoflavone-6-sulfonato-kappa-2O4,O5)nickel(II) dimethylformamide solvate monohydrate. Acta Crystallogr. 2006, E62, m469–m471. [Google Scholar] [CrossRef]
- Zhang, Z.T.; Shi, J. Self-assembly and crystal structure of a three-dimensional copper(II) complex. J. Coord. Chem. 2007, 60, 1485–1495. [Google Scholar] [CrossRef]
- Li, W.W.; Zheng, M.Y.; Gao, Y.H.; Zhang, Z.T. Argentum 5-Hydroxy-7-methoxy-2-phenyl-4H-chromen-4-one-6-sulfonate:Synthesis, Crystal Structure and Antitumor Activity. Chin. J. Struct. Chem. 2020, 39, 1898–1905. [Google Scholar]
- Pieniazek, E.; Kalembkiewicz, J.; Dranka, M.; Woznicka, E. Syntheses, crystal structures and antioxidant study of Zn(II) complexes with morin-5′-sulfonic acid (MSA). J. Inorg. Biochem. 2014, 141, 180–187. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, Z. Crystal Structure and Photoluminescence of a Tetranuclear Cadmium Complex. Chin. J. Chem. 2012, 30, 1057–1062. [Google Scholar] [CrossRef]
- Jiang, R.W.; He, Z.D.; But, P.P.H.; Chan, Y.M.; Mak, T.C. A novel 1: 1 Complex of potassium mikanin-3-O-sulfate with methanol. Chem. Pharm. Bull. 2001, 49, 1166–1169. [Google Scholar] [CrossRef][Green Version]
- Liu, Q.G.; Zhang, Z.T.; Xue, D. Synthesis, crystal structure and activity of sulfated daidzein. Chem. J. Chin. U 2003, 24, 820–825. [Google Scholar]
- Zhang, X.L.; Zhang, Z.T.; Liang, Y. Synthesis and single crystal X-ray structure of [Na(H2O)2(C18H15O6SO3)]2. J. Chem. Crystallogr. 2008, 38, 861–865. [Google Scholar] [CrossRef]
- Zhang, Z.T.; Guo, Y.N.; Liu, Q.G. Syntheses and crystal structures of [Na(H2O)] (C17H13O6SO3)·2H2O and [Ni(H2O)6](C17H13O6SO3)2·4H2O. Chin. J. Chem. 2004, 22, 971–977. [Google Scholar] [CrossRef]
- Wang, Q.Y.; Zhang, Z.T. Synthesis and crystal structure of [Na(H2O)1.5][Na(H2O)3.5] X2·2H2O (X = 4′-methoxy-7-hydroxyisoflavone-3′-sulfonate). J. Chem. Crystallogr. 2011, 41, 1467–1471. [Google Scholar] [CrossRef]
- Zhang, Z.T.; Zhang, Z.T.; Wang, Q.Y.; He, Y.; Wang, X.B.; Xue, D.; Zheng, J.B. Syntheses, crystal structures and biological activity of bimethylation daidzein sulfonates. Chem. J. Chin. U 2005, 26, 2247–2253. [Google Scholar]
- Woźnicka, E.; Pieniążek, E.; Zapała, L.; Byczyński, Ł.; Trojnar, I.; Kopacz, M. New sulfonic derivatives of quercetin as complexing reagents: Synthesis, spectral, and thermal characterization. J. Therm. Anal. Calorim. 2015, 120, 351–361. [Google Scholar] [CrossRef]
- Czerwonka, A.; Maciołek, U.; Kałafut, J.; Mendyk, E.; Kuźniar, A.; Rzeski, W. Anticancer effects of sodium and potassium quercetin-5′-sulfonates through inhibition of proliferation, induction of apoptosis, and cell cycle arrest in the HT-29 human adenocarcinoma cell line. Bioorg. Chem. 2020, 94, 103426. [Google Scholar] [CrossRef]
- Masoud, M.S.; El-Enein, S.A.A.; Ramadan, A.M.; Goher, A.S. Thermal properties of some biologically active 5-(p-substituted phenylazo)-6-aminouracil complexes. J. Anal. Appl. Pyrol. 2008, 81, 45–51. [Google Scholar] [CrossRef]
- Ekawa, B.; Nunes, W.D.G.; Teixeira, J.A.; Cebim, M.A.; Ionashiro, E.Y.; Caires, F.J. New complexes of light lanthanides with the valsartan in the solid state: Thermal and spectroscopic studies. J. Anal. Appl. Pyrol. 2018, 135, 299–309. [Google Scholar] [CrossRef]
- Jendrzejewska, I.; Goryczka, T.; Pietrasik, E.; Klimontko, J.; Jampilek, J. X-ray and Thermal Analysis of Selected Drugs Containing Acetaminophen. Molecules 2020, 25, 5909. [Google Scholar] [CrossRef]
- Qi, S. Advances in Delivery Science and Technology. In Analytical Techniques in the Pharmaceutical Sciences; Müllertz, A., Perrie, Y., Rades, T., Eds.; Springer: New York, NY, USA, 2016; pp. 363–387. [Google Scholar]
- Maciołek, U.; Mendyk, E.; Kosińska, M.; Sternik, D.; Drewniak, M.; Kozioł, A.E. Thermal study, identification of intermediate solid products and evolved gas analysis (EGA) during pyrolysis and oxidative decomposition of sodium complex of quercetin-5′-sulfonic acid (Na-5′-QSA). J. Anal. Appl. Pyrolysis 2020, 150, 104881. [Google Scholar] [CrossRef]
- Rigaku. CRYSALIS Software System; Rigaku: Oxford, UK, 2016. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Sherje, A.; Desai, K. Spectrophotometric Determination of Poorly Water Soluble Drug Rosiglitazone Using Hydrotropic Solubilization technique. Indian J. Pharm. Sci. 2011, 73, 579–582. [Google Scholar] [CrossRef]
- Czakis-Sulikowska, D.; Radwańska-Doczekalska, J.; Czyłkowska, A.; Gołuchowska, J. TG-MS, TG, DTG and DTA methods in study of thermal decomposition of some d-metal complexes with 4,4′-bpy and propionates. J. Therm. Anal. Calorim. 2004, 78, 501–511. [Google Scholar] [CrossRef]
- Risoluti, R.; Piazzese, D.; Napoli, A.; Materazzi, S. Study of [2-(2′-pyridyl)imidazole] complexes to confirm two main characteristic thermoanalytical behaviors of transition metal complexes based on imidazole derivatives. J. Anal. Appl. Pyrolysis. 2016, 117, 82–87. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Webster, F.X.; Kiemle, D.J. Spectrometric Identification of Organic Compound, 7th ed.; John Wiley and Sons: Hoboken, NJ, USA, 2005. [Google Scholar]
- Anoop, M.R.; Jisha, K.R.; Suma, S.; Sudarsanakumar, M.R. Synthesis, spectral characterization, thermal and biological studies of lanthanide(III) complexes of oxyphenbutazone. J. Rare Earths 2014, 32, 43–51. [Google Scholar] [CrossRef]
- Guerra, R.B.; Gálico, D.A.; Holanda, B.B.C.; Bannach, G. Solid-state thermal and spectroscopic studies of the antiinflammatory drug sulindac using UV–Vis, MIR, NIR, DSC, simultaneous TG–DSC, and the coupled techniques TG-EGA-MIR and DSC–optical microscopy. J. Therm. Anal. Calorim. 2016, 123, 2523–2530. [Google Scholar] [CrossRef]
- Otto, K.; Bombicz, P.; Madarász, J.; Acik, I.O.; Krunks, M.; Pokol, G. Structure and evolved gas analyses (TG/DTA-MS and TG-FTIR) of mer-trichlorotris(thiourea)-indium(III), a precursor for indium sulfide thin films. J. Therm. Anal. Calorim. 2011, 105, 83–91. [Google Scholar] [CrossRef]
- Franzé, J.A.; Carvalho, T.F.; Gaglieri, C.; Caires, F.J.; Bannach, G.; Castro, R.C.; Treu-Filho, O.; Ionashiro, M.; Mendes, R.A. Synthesis, characterization, thermal and spectroscopic studies and bioactivity of complexes of meloxicam with some bivalent transition metals. J. Therm. Anal. Calorim. 2017, 127, 1393–1405. [Google Scholar] [CrossRef]
- Madarász, J. Evolved gas analyses on a mixed valence copper(I,II) complex salt with thiosulfate and ammonia by in situ TG-EGA-FTIR and TG/DTA-EGA-MS. J. Therm. Anal. Calorim. 2009, 97, 111–116. [Google Scholar] [CrossRef]
- Hanuza, J. Vibrational Dynamics of the Bent Symmetric and Asymmetric Oxygen Bridges and Terminal Metal=Oxygen Bond. Bull. Pol. Acad. Sci. Chem. 1994, 42, 255–267. [Google Scholar]
- Xu, G.R.; In, M.Y.; Yuan, Y.; Lee, J.J.; Kim, S. In situ Spectroelectrochemical Study of Quercetin Oxidation and Complexation with Metal Ions in Acidic Solutions. Bull. Korean Chem. Soc. 2007, 28, 889–892. [Google Scholar] [CrossRef]
- Symonowicz, M.; Kolanek, M. Flavonoids and Their Properties to form Chelate Complexes. Biotechnol. Food Sci. 2012, 76, 35–41. [Google Scholar]
- Teslova, T.; Corredor, C.; Livingstone, R.; Spataru, R.; Birke, R.L.; Lombardi, J.R.; Cañamares, M.V.; Leona, M. Raman and surface-enhanced Raman spectra of flavone and several hydroxy derivatives. J. Raman Spectrosc. 2007, 38, 802–818. [Google Scholar] [CrossRef]
- Rossi, M.; Rickles, L.F.; Halpin, W.A. The crystal and molecular structure of quercetin: A biologically active and naturally occurring flavonoid. Bioorg. Chem. 1986, 14, 55–69. [Google Scholar] [CrossRef]
- Numata, Y.; Tanaka, H. Quantitative analysis of quercetin using Raman spectroscopy. Food Chem. 2011, 126, 751–755. [Google Scholar] [CrossRef]
- Hanuza, J.; Godlewska, P.; Kucharska, E.; Ptak, M.; Kopacz, M.; Mączka, M.; Hermanowicz, K.; Macalik, L. Molecular Structure and Vibrational Spectra of Quercetin and Quercetin-5′-Sulfonic Acid A. Vib. Spectrosc. 2017, 88, 94–105. [Google Scholar] [CrossRef]
- Dimitrić Marković, J.M.; Marković, Z.S.; Krstić, J.B.; Milenković, D.; Lučić, B.; Amić, D. Interpretation of the IR and Raman spectra of morin by density functional theory and comparative analysis. Vib. Spectrosc. 2013, 64, 1–9. [Google Scholar] [CrossRef]
- Pompeu, D.R.; Larondelle, Y.; Rogez, H.; Abbas, O.; Pierna, J.A.F.; Baeten, V. Characterization and discrimination of phenolic compounds using Fourier transform Raman spectroscopy and chemometric tools. Biotechnol. Agron. Soc. Environ. 2018, 22, 13–28. [Google Scholar] [CrossRef]
- Heneczkowski, M.; Kopacz, M.; Nowak, D.; Kuźniar, A. Infrared spectrum analysis of some flavonoids. Acta Pol. Pharm. 2001, 58, 415–420. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cation | CSD Refcode/SO3− Substitution | Coordination Sphere | Coordinating SO3− Group | Type of Complex | Reference |
|---|---|---|---|---|---|
| Na | DURTEQ/C8 | CN = 6, NaO6 | yes | 1D polymer | [17] |
| SULFOV/C8 | CN = 5, NaO5 CN = 5, NaO5 | yes | 3D polymer | [18] | |
| K | QOLLAF/C8 | CN = 5, KO5 | yes | 3D polymer | [19] |
| Ca * | POMDUR/C8 | CN = 7, CaO7 | yes | mononuclear | [20] |
| Ba | PEHROK/C6 | CN = 10, BaO10 | yes | 1D polymer | [21] |
| Mn * | YELTAN/C6 | CN = 6, MnO6 | no | mononuclear | [22] |
| Co * | WIZGIW/C6 | CN = 6, CoO6 | no | mononuclear | [23] |
| Ni * | WEPPAJ/C6 | CN = 6, NiO6 | no | mononuclear | [24] |
| YELSUG/C6 | CN = 6, NiO6 | no | mononuclear | [22] | |
| Cu * | YIGZOE/C6 | CN = 5, CuO5 | no | mononuclear | [25] |
| Ag | PAGVUS/C6 | CN = 4, AgO4 CN = 4, AgO4 | yes | tetranuclear | [26] |
| Zn | ZUJTIJ/C5′ | CN = 6, ZnO6 | yes | 1D polymer | [27] |
| Cd | XESPES/C6 | CN = 7, CdO7 CN = 7, CdO7 CN = 7, CdO7 CN = 7, CdO7 | yes | tetranuclear | [28] |
| Na, Zn | ZUJGUI/C5′ | CN = 6, ZnO6 CN = 6, NaO6 | no yes | 1D polymer 1D polymer | [27] |
| K-O | Symmetry Transformations | d(K-O) [Å] |
|---|---|---|
| * K(1)-O(4’)#2 | #1 −x, −y + 1, −z + 1 #2 −x, −y, −z + 2 #3 x − 1, y − 1, z + 1 | 3.016(2) |
| *$ K(1)-O(4’) | 3.101(2) | |
| $ K(1)-O(2S) | 2.754(2) | |
| K(1)-O(1S)#2 | 2.754(2) | |
| K(1)-O(4)#1 | 2.713(2) | |
| K(1)-O(5)#3 | 2.759(2) | |
| K(1)-O(1W) | 3.034(2) | |
| K(1)-O(2W) | 3.029(2) |
| D-H...A | d(D-H) [Å] | d(H...A) [Å] | d(H...A) [Å] | <(DHA) [°] |
|---|---|---|---|---|
| O(1W)-H(2W1)…O(2W) | 0.89(5 | 2.05(5) | 2.849(3) | 150(3) |
| O(1W)-H(1W1)…O(2S) | 0.83(4) | 2.03(4) | 2.766(2) | 147(3) |
| O(4’)-H(4’)...O(3’) | 0.77(3) | 2.21(3) | 2.653(2) | 118(2) |
| O(3)-H(3)...O(4) | 0.84(3) | 2.29(3) | 2.718(2) | 112(2) |
| O(5)-H(5)...O(4) | 0.88(3) | 1.78(3) | 2.610(2) | 156(3) |
| O(2W)-H(1W2)...O(3S)#a | 0.83(4) | 2.02(4) | 2.829(2) | 164(3) |
| O(2W)-H(2W2)...O(3S)#b | 0.92(4) | 1.90(4) | 2.814(2) | 173(3) |
| O(3’)-H(3’)...O(1W)#c | 0.86(3) | 1.78(3) | 2.628(2) | 169(3) |
| O(4’)-H(4’)...O(7)#d | 0.77(3) | 2.09(3) | 2.764(2) | 146(3) |
| O(7)-H(7)...O(1S)#e | 0.83(3) | 1.98(3) | 2.783(2) | 161(3) |
| O(3)-H(3)...O(2W)#f | 0.84(3) | 2.03(3) | 2.823(2) | 158(2) |
| Medium | Stage | tpeak [°C] | Thermogravimetry | ||
|---|---|---|---|---|---|
| Temp.Range; TDTG(mx) [°C] | Δm [%] Found (calc.) | Residue [%] Found (calc.) | |||
| air | I | 154 (−) | 25−300; 144 | 8.24 (7.89) | 19.31 (19.09) 0.5 K2SO4 |
| 280 (+) | |||||
| II | 367 (+) | 300−400; 351 | 20.00 | ||
| III | 453 (+) | 400−600; 449 | 52.45 | ||
| IV | 1062 (−) | 800−1400; 1215 | 14.13 | 5.18 | |
| helium | I | 130 (−) | 25−300; 127 | 8.13 (7.89) | 42.79 (42.72) 0.5 K2C2 + 12C |
| 280 (+) | |||||
| II | 350 (−) | 300−700; 350 | 49.08 | ||
| III | 700−1420; - | 29.30 | 13.49 | ||
| Solvent | QUE·2H2O | 1 | ||
|---|---|---|---|---|
| [g/L] | [mol/L] | [g/L] | [mol/L] | |
| Water | 0.002 | 5.91·10−6 | 0.09 | 1.97 × 10−4 |
| Acetone | 3.69 | 1.10·10−2 | 0.25 | 5.48 × 10−4 |
| Ethanol | 8.11 | 2.40·10−2 | 0.06 | 1.31 × 10−4 |
| DMSO | 685 | 2.03 | 167 | 0.37 |
| K(1)-O | Symmetry Transformations | d(K-O) [Å] | K(2)-O | Symmetry Transformations | d(K-O) [Å] |
|---|---|---|---|---|---|
| K(1)-O(1C) | #1 1 − x, 1 − y, −z #2 2 − x, 2 − y, −z #3 x + 1, y + 1, z | 2.672(5) | K(2)-O(5B) | #4 1 − x, 2 – y, 1 – z #5 −x, 1 − y, 1 – z #6 x − 1, y − 1, z | 2.685(5) |
| K(1)-O(1E) | 3.037(7) | K(2)-O(3SB)#4 | 2.657(5) | ||
| K(1)-O(5A) | 2.730(5) | K(2)-O(4’B)#4 | 2.934(5) | ||
| * K(1)-O(2SA)#3 | 2.807(6) | K(2)-O(4B)#5 | 2.725(5) | ||
| *$$ K(1)-O(2SA)#1 | 2.661(5) | K(2)-O(3SB)#6 | 2.796(6) | ||
| $ K(1)-O(4’A)#1 | 2.835(5) | K(2)-O(1SB)#6 | 3.019(5) | ||
| $ K(1)-O(1SA)#3 | 3.167(6) | K(2)-O(1F) | 2.681(6) | ||
| K(1)-O(4A)#2 | 2.675(5) | K(2)-O(1D) | 2.962(7) |
| D-H...A | d(D-H) [Å] | d(H...A) [Å] | d(H...A) [Å] | <(DHA) [°] |
|---|---|---|---|---|
| O(3A)-H(3A)...O(4A) | 0.84 | 2.23 | 2.688(6) | 115 |
| O(5A)-H(5A)...O(4A) | 1.19 | 1.65 | 2.671(7) | 139 |
| O(4′A)-H(4′A)...O(3′A) | 0.90 | 2.14 | 2.695(7) | 112 |
| O(7A)-H(7A)...O(3SA)#a | 0.79 | 1.90 | 2.673(7) | 163 |
| O(3A)-H(3A)...O(1C)#b | 0.84 | 2.04 | 2.770(7) | 144 |
| O(4′A)-H(4′A)...O(1E)#c | 0.90 | 1.98 | 2.653(7) | 130 |
| O(3′A)-H(3′A)...O(1G) | 0.84 | 1.82 | 2.661(8) | 177 |
| O(3B)-H(3B)...O(4B) | 0.96 | 2.06 | 2.687(7) | 121 |
| O(5B)-H(5B)...O(4B) | 0.93 | 1.86 | 2.665(7) | 144 |
| O(7B)-H(7B)...O(2SB)#d | 0.88 | 1.84 | 2.697(7) | 165 |
| O(4′B)-H(4′B)...O(1D)#e | 0.88 | 1.90 | 2.649(8) | 142 |
| O(3B)-H(3B)...O(1F)#f | 0.96 | 2.05 | 2.796(7) | 133 |
| O(3’B)-H(3’B)...O(1H)#e | 0.84 | 1.85 | 2.684(9) | 172 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maciołek, U.; Mendyk, E.; Kosińska-Pezda, M.; Kamiński, D.M.; Kozioł, A.E. Potassium Complexes of Quercetin-5′-Sulfonic Acid and Neutral O-Donor Ligands: Synthesis, Crystal Structure, Thermal Analysis, Spectroscopic Characterization and Physicochemical Properties. Materials 2021, 14, 6798. https://doi.org/10.3390/ma14226798
Maciołek U, Mendyk E, Kosińska-Pezda M, Kamiński DM, Kozioł AE. Potassium Complexes of Quercetin-5′-Sulfonic Acid and Neutral O-Donor Ligands: Synthesis, Crystal Structure, Thermal Analysis, Spectroscopic Characterization and Physicochemical Properties. Materials. 2021; 14(22):6798. https://doi.org/10.3390/ma14226798
Chicago/Turabian StyleMaciołek, Urszula, Ewaryst Mendyk, Małgorzata Kosińska-Pezda, Daniel M. Kamiński, and Anna E. Kozioł. 2021. "Potassium Complexes of Quercetin-5′-Sulfonic Acid and Neutral O-Donor Ligands: Synthesis, Crystal Structure, Thermal Analysis, Spectroscopic Characterization and Physicochemical Properties" Materials 14, no. 22: 6798. https://doi.org/10.3390/ma14226798
APA StyleMaciołek, U., Mendyk, E., Kosińska-Pezda, M., Kamiński, D. M., & Kozioł, A. E. (2021). Potassium Complexes of Quercetin-5′-Sulfonic Acid and Neutral O-Donor Ligands: Synthesis, Crystal Structure, Thermal Analysis, Spectroscopic Characterization and Physicochemical Properties. Materials, 14(22), 6798. https://doi.org/10.3390/ma14226798