
Rational Design and Characterization of Symmetry-Breaking Organic Semiconductors in Polymer Solar Cells: A Theory Insight of the Asymmetric Advantage

 and
and
Abstract
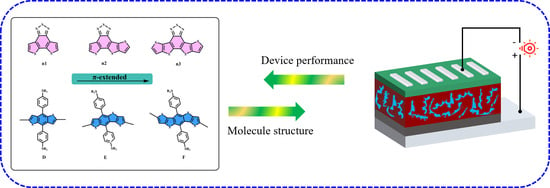
:
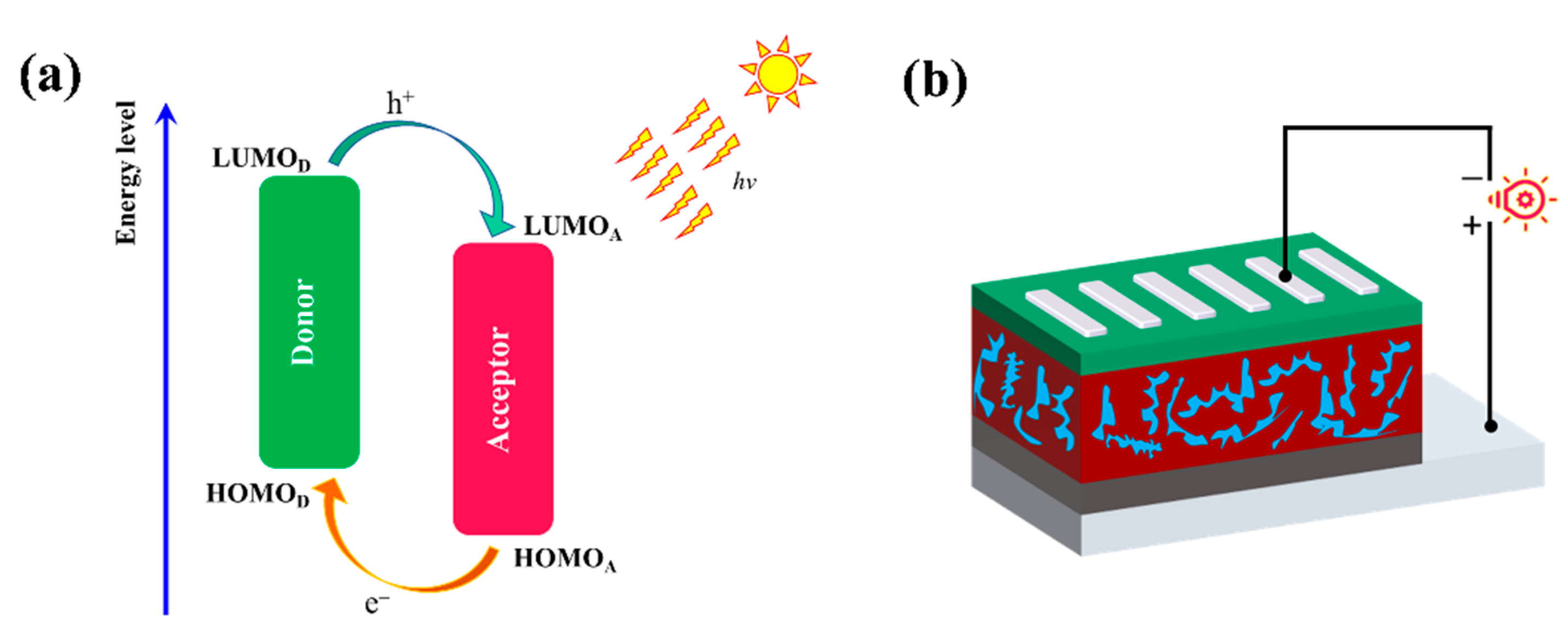
1. Introduction
2. Computational Methods
3. Results and Discussion
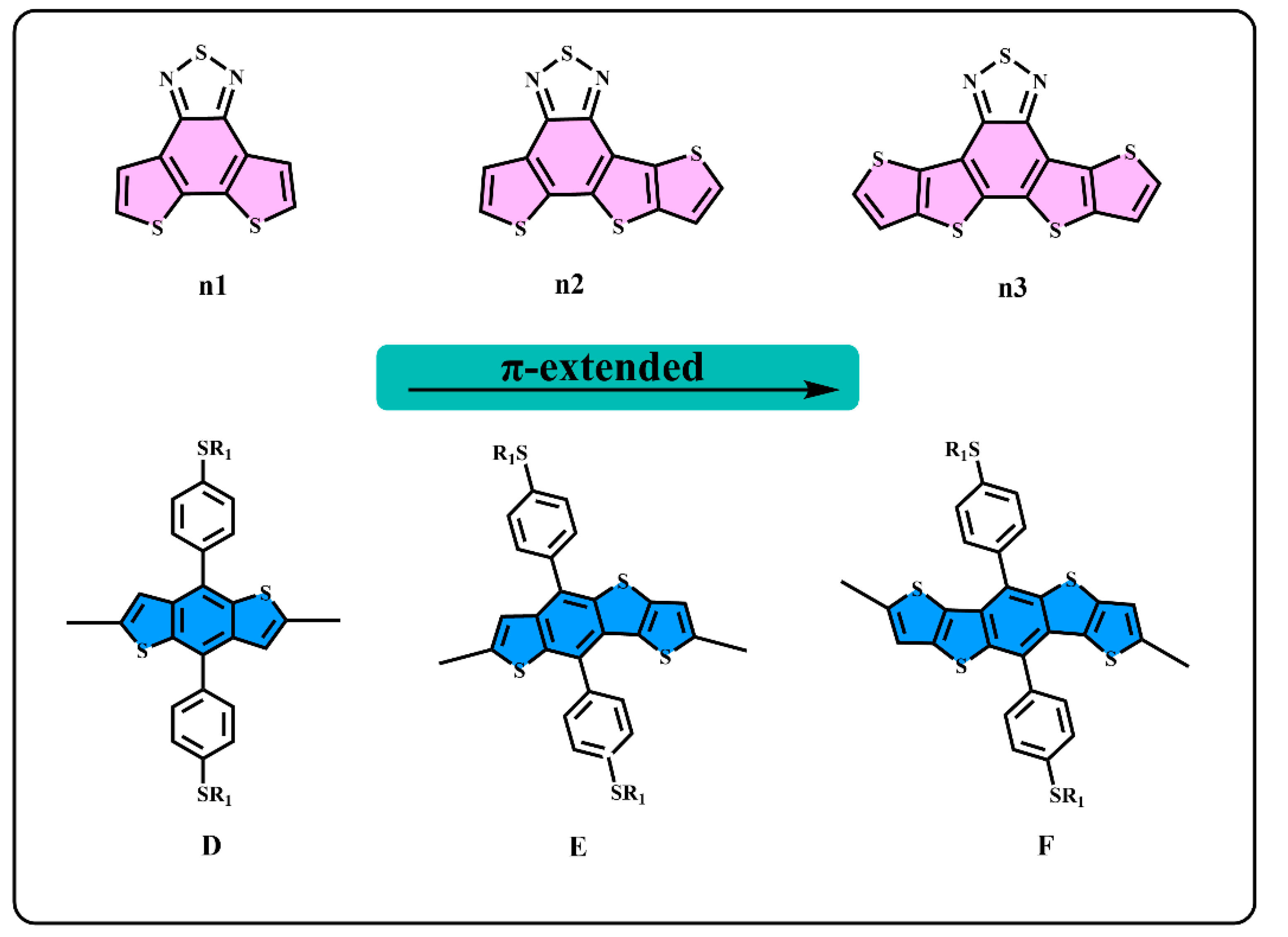
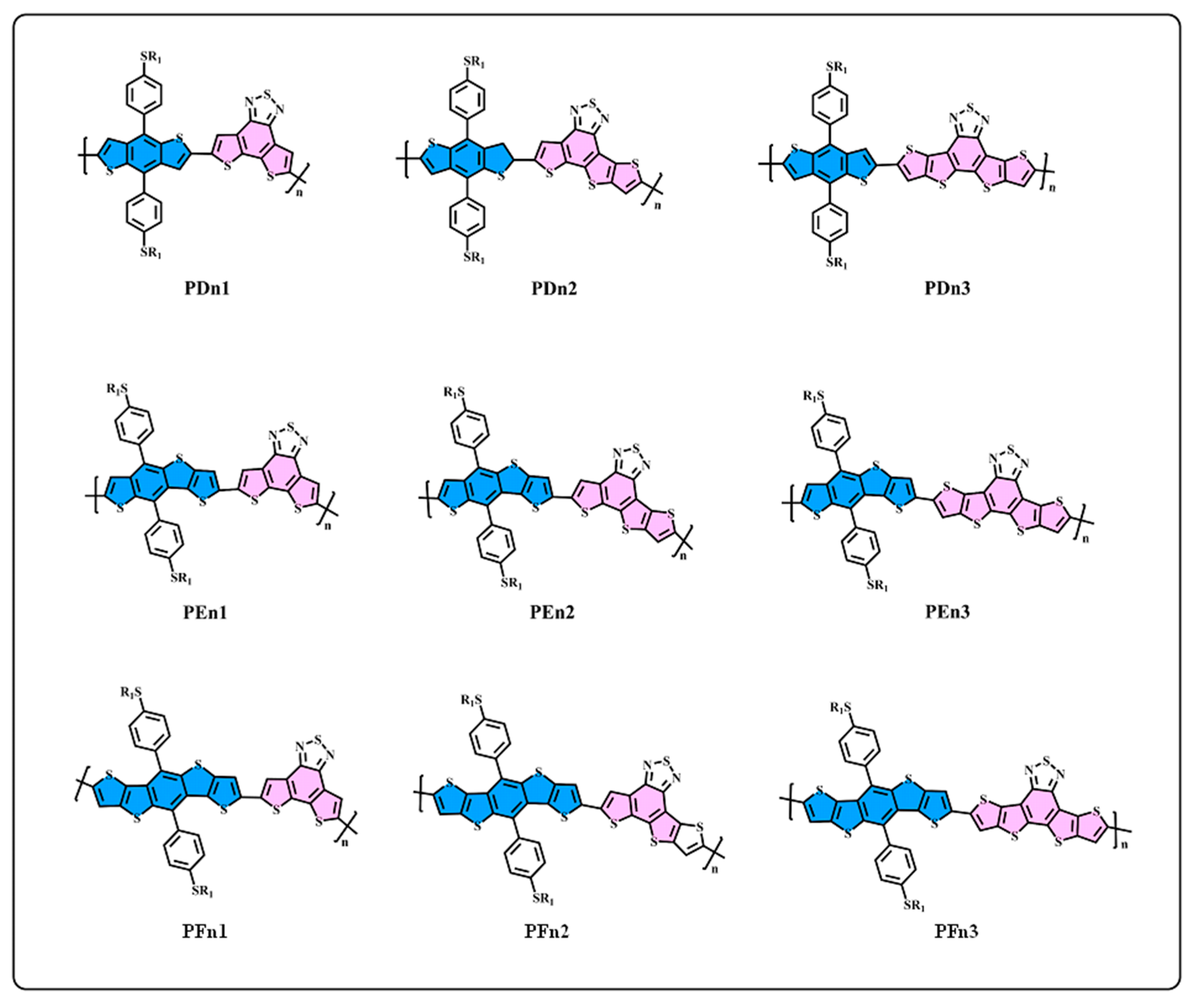
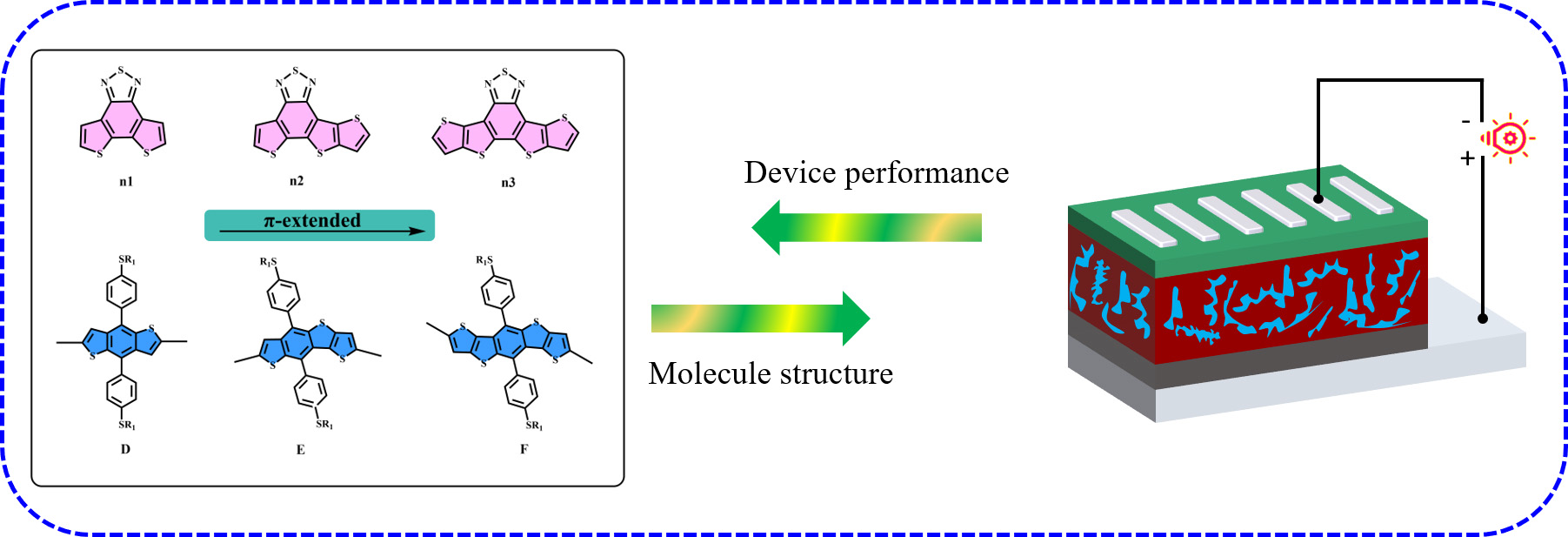
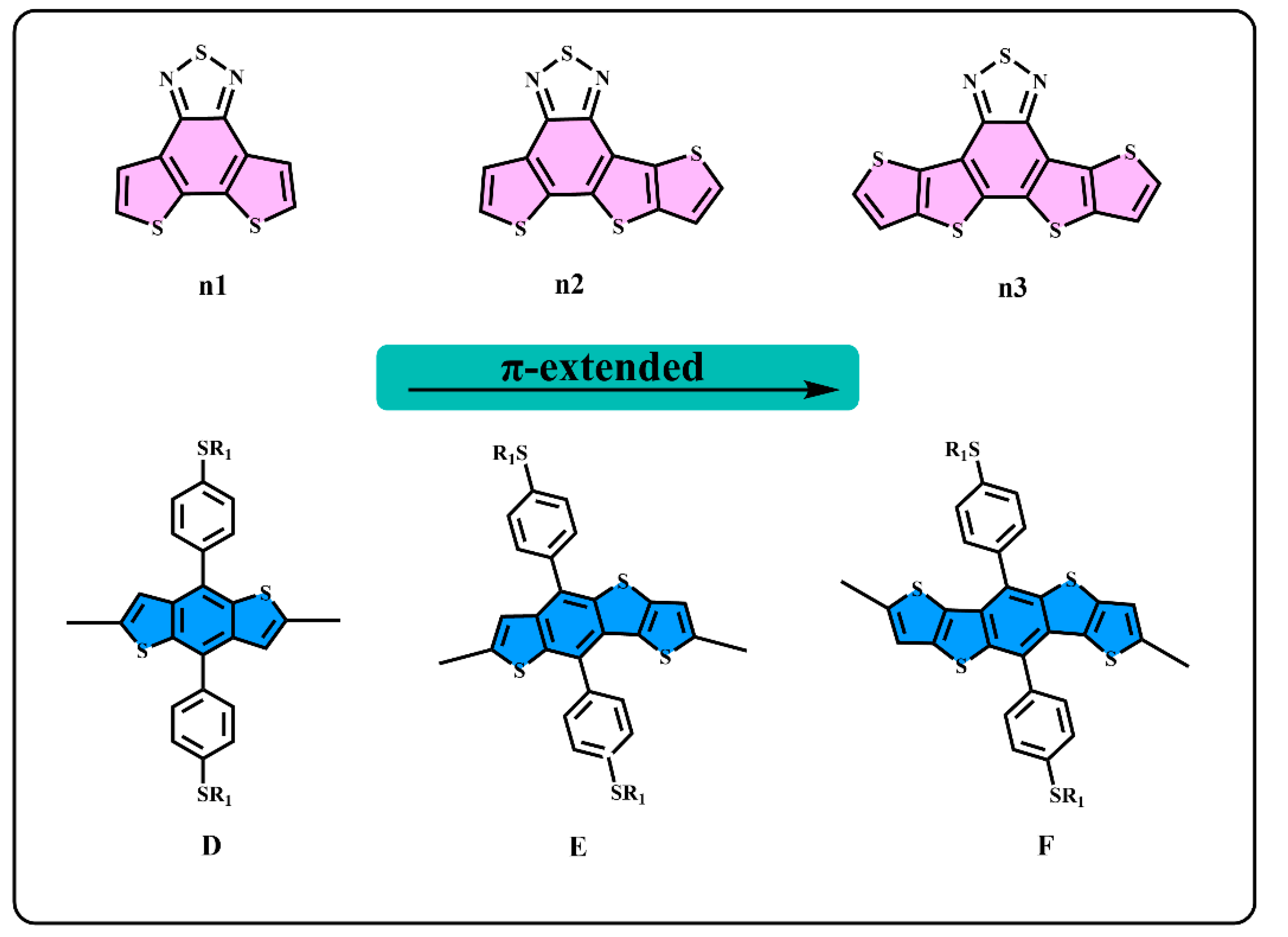
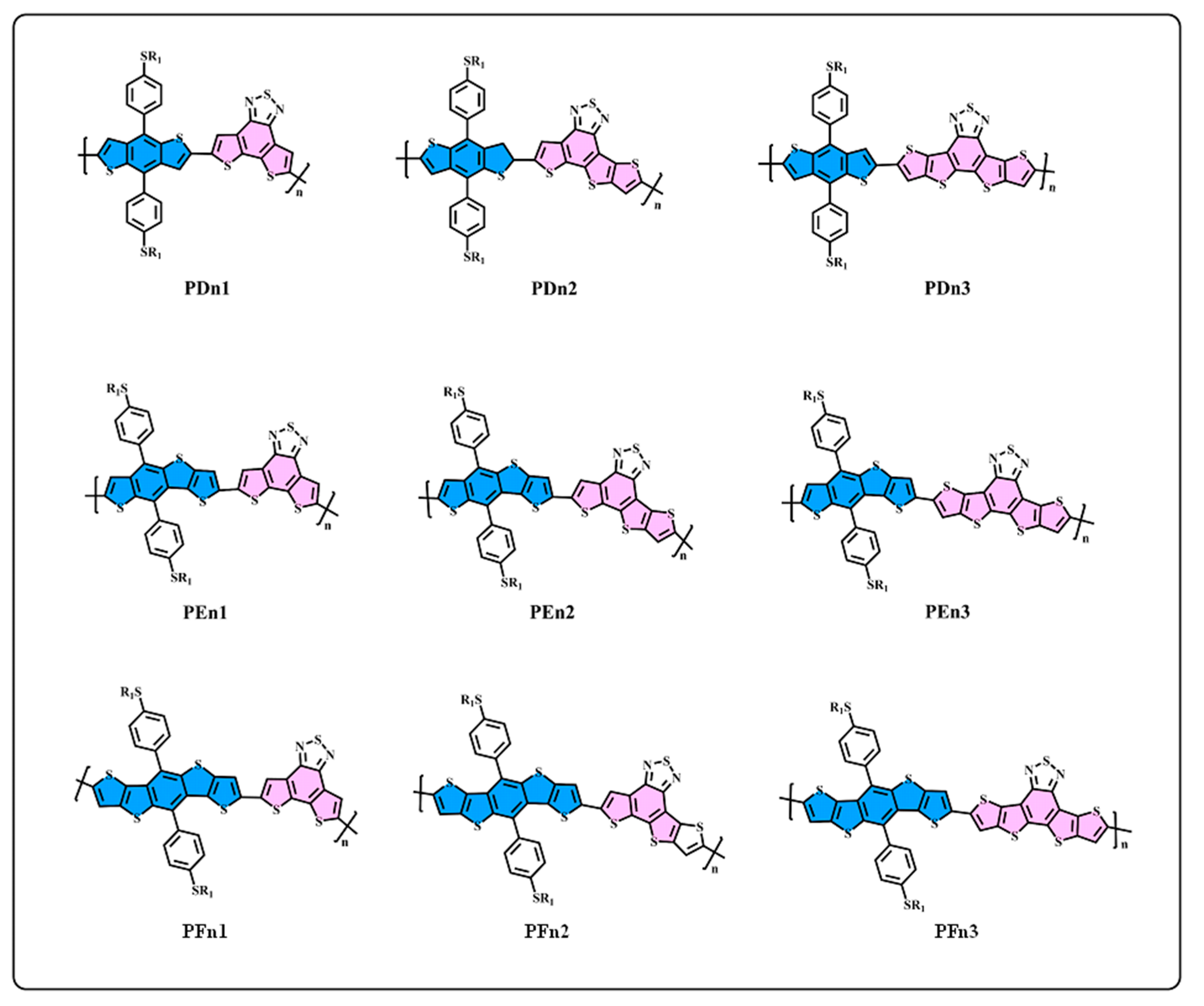
3.1. Molecule Design
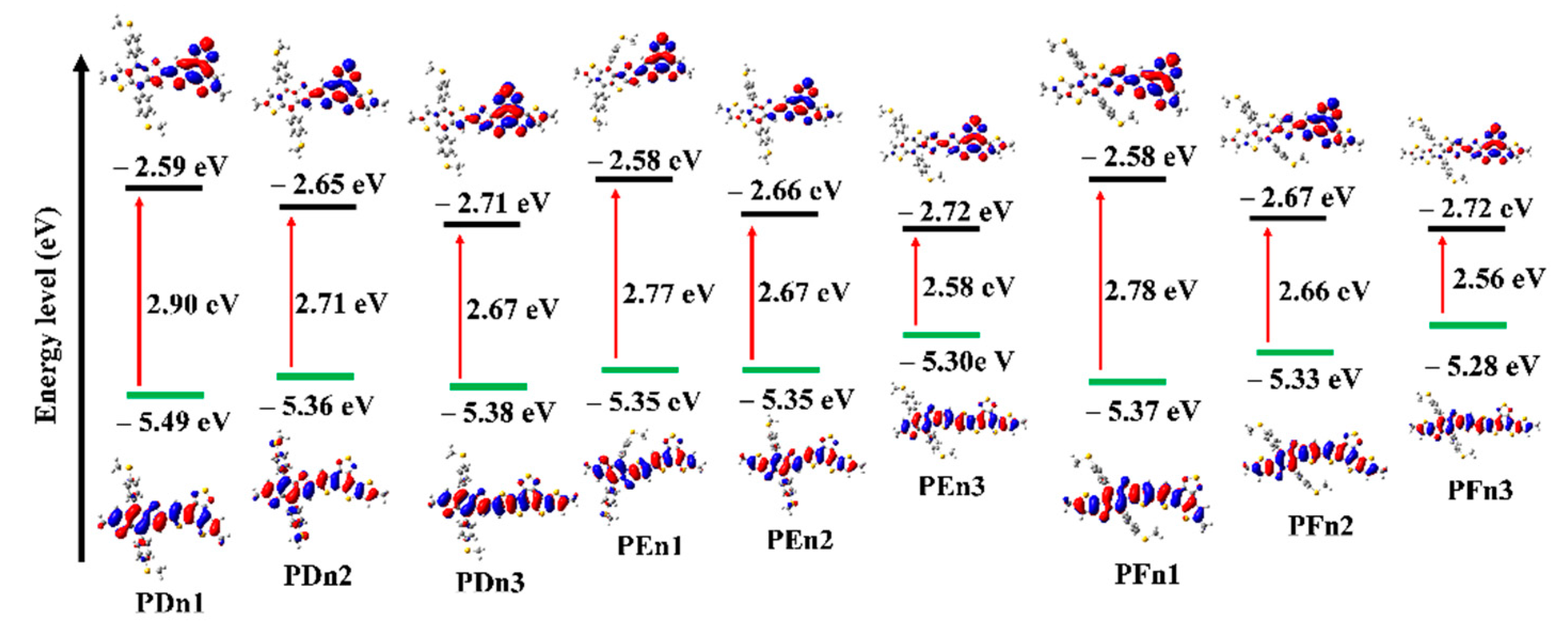
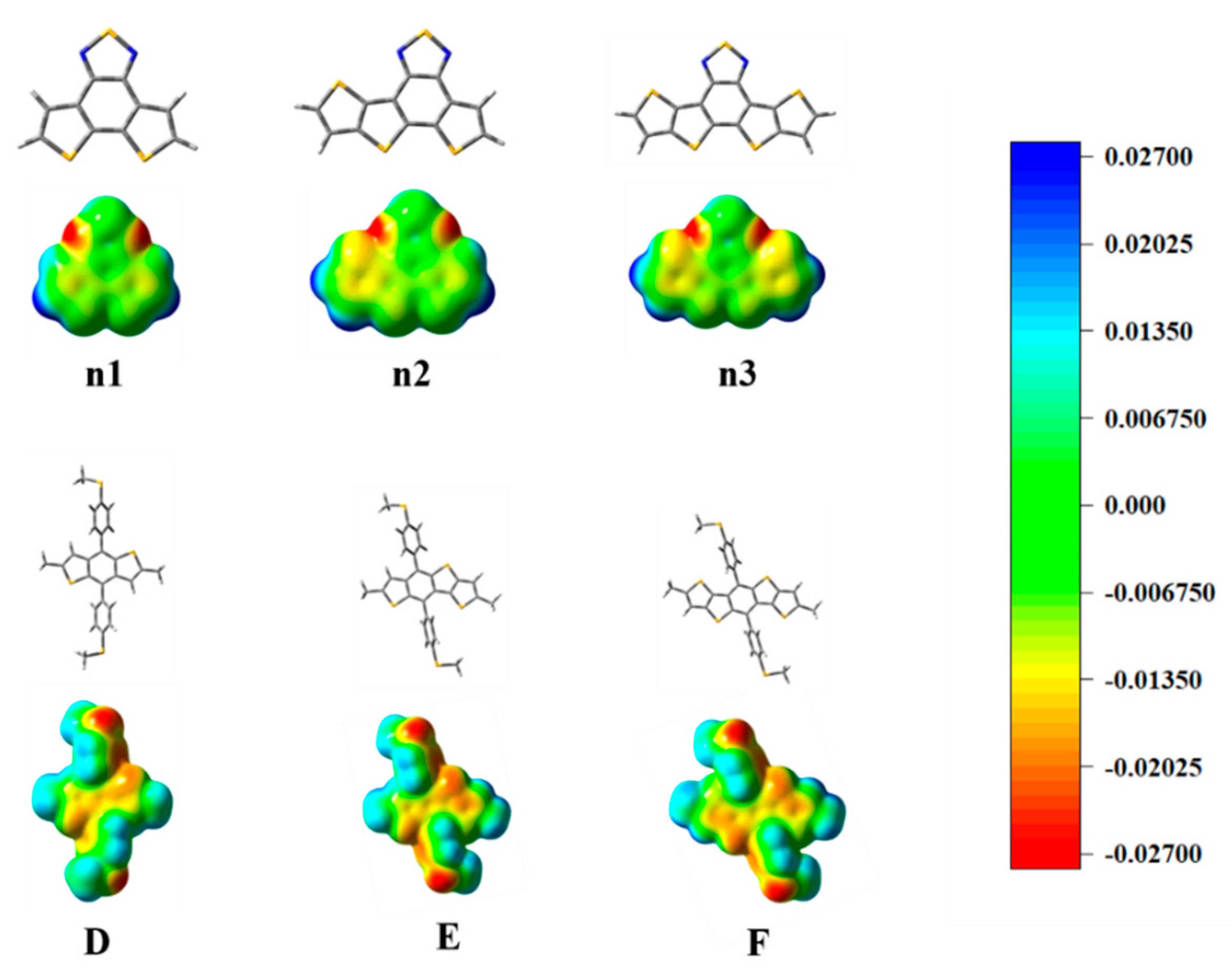
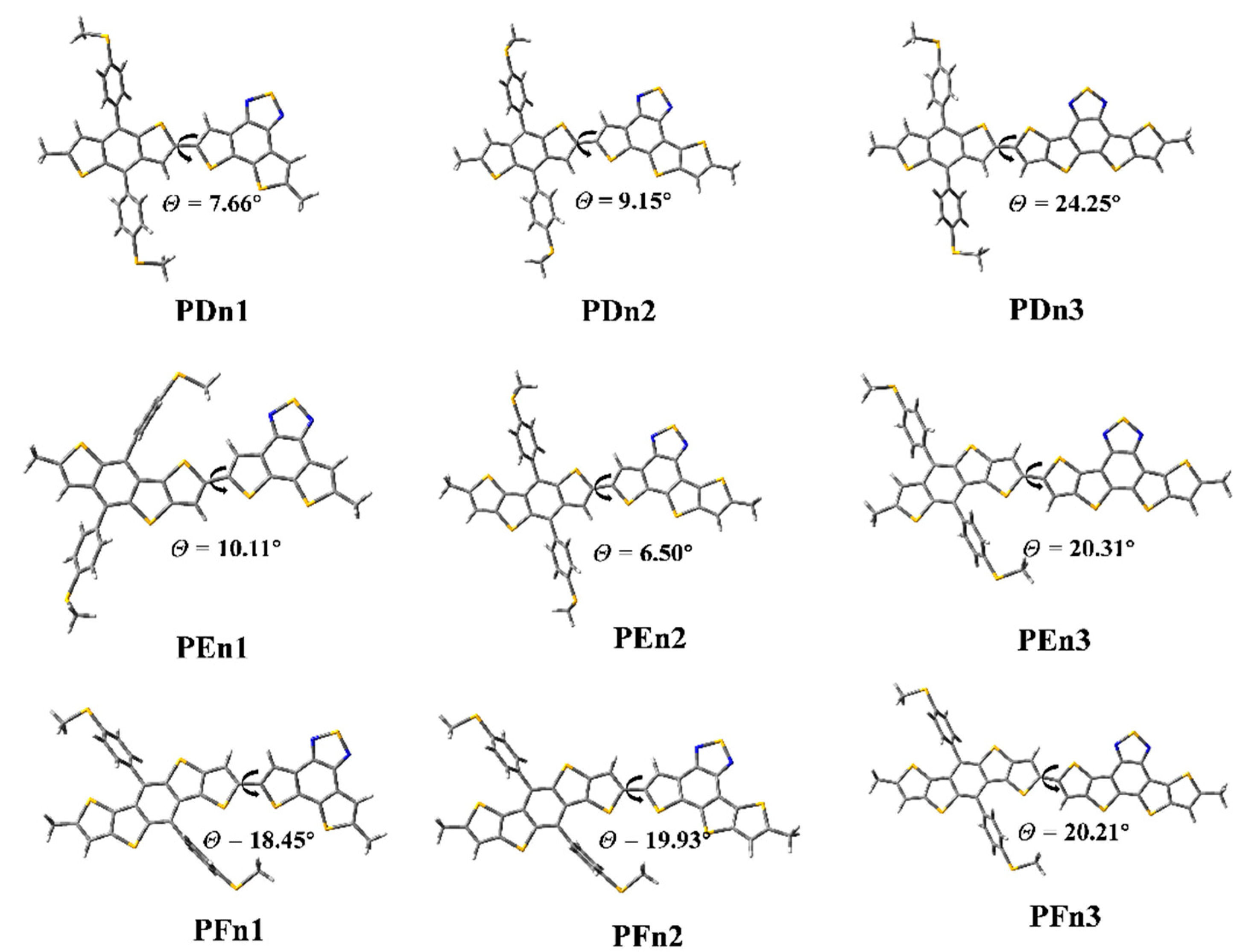
3.2. Geometric Optimization and Electronic Structure
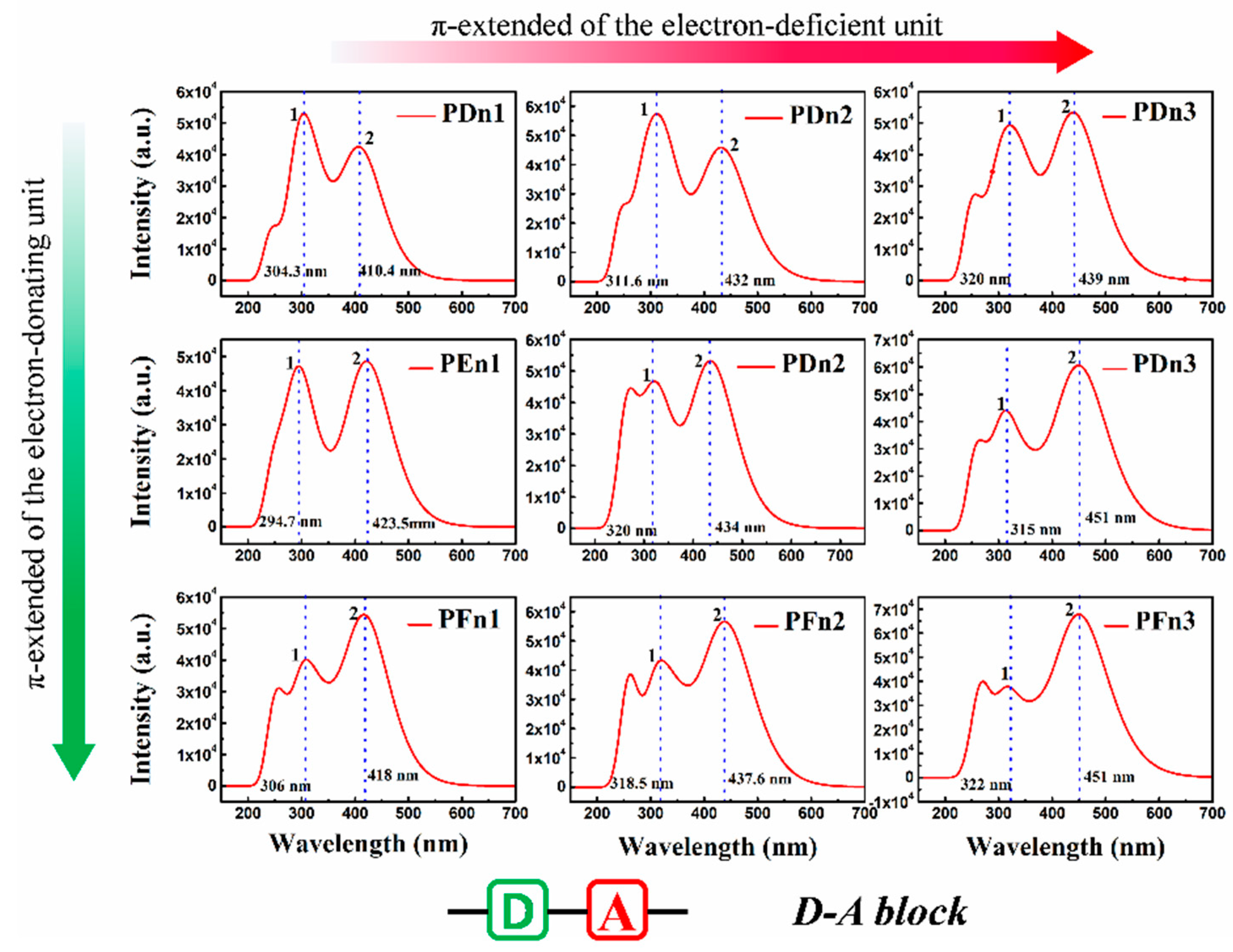
3.3. Spectral Properties
3.4. Ionization Potential (IP) and Electron Affinity (EA)
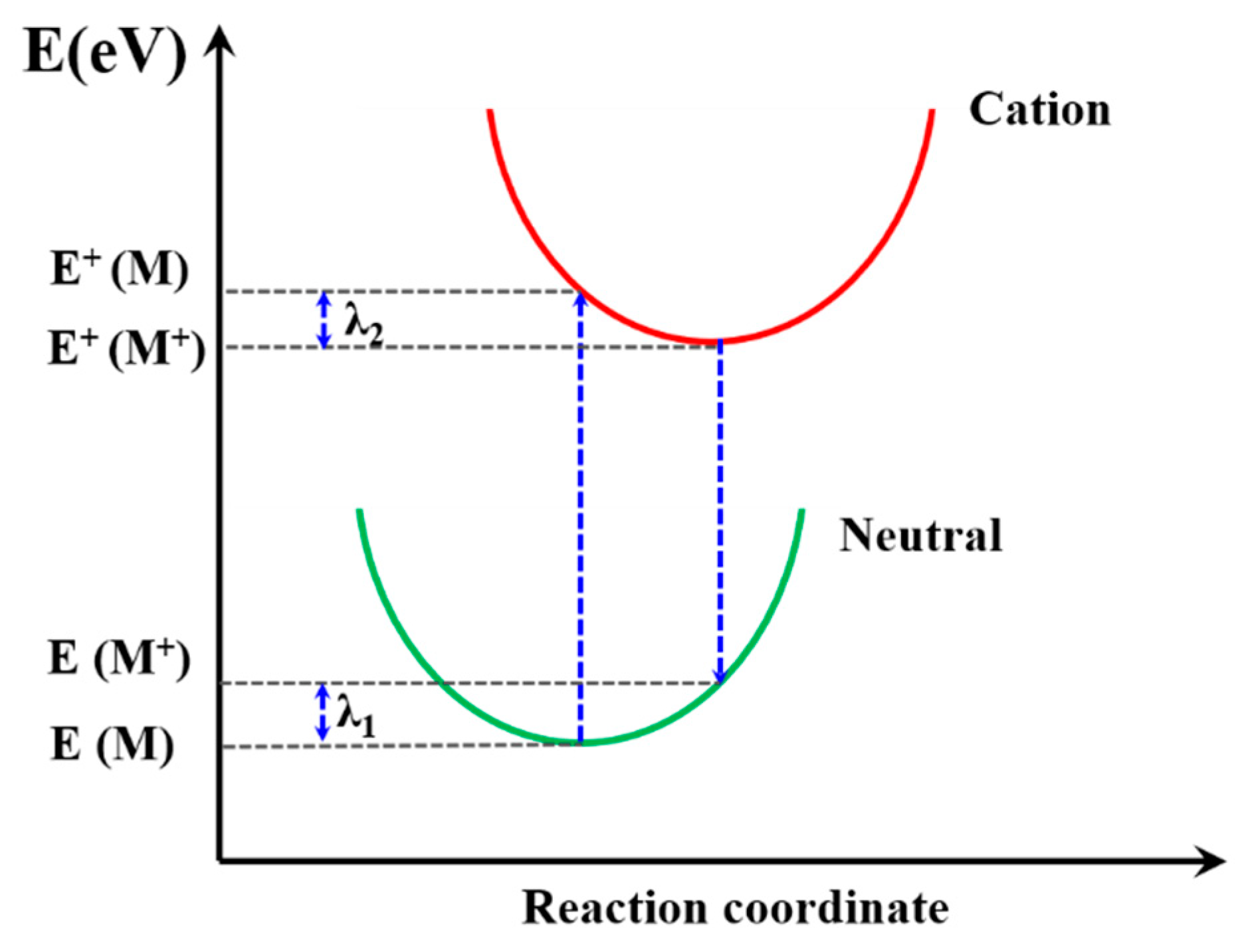
3.5. Reorganization Energy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, G.; Shrotriya, V.; Huang, J.; Yao, Y.; Moriarty, T.; Emery, K.; Yang, Y. High-efficiency solution processable polymer pho-tovoltaic cells by self-organization of polymer blends. Nat. Mater. 2005, 4, 864–868. [Google Scholar] [CrossRef]
- Zhang, Y.; Hau, S.K.; Yip, H.-L.; Sun, Y.; Acton, O.; Jen, A. Efficient Polymer Solar Cells Based on the Copolymers of Benzodithiophene and Thienopyrroledione. Chem. Mater. 2010, 22, 2696–2698. [Google Scholar] [CrossRef]
- Peet, J.; Kim, J.Y.; Coates, N.E.; Ma, W.L.; Moses, D.; Heeger, A.J.; Bazan, G.C. Efficiency enhancement in low-bandgap polymer solar cells by processing with alkane dithiols. Mater. Sustain. Energy 2010, 6, 47–50. [Google Scholar] [CrossRef]
- Zhang, F.; Mammo, W.; Andersson, L.M.; Admassie, S.; Andersson, M.R.; Inganäs, O. Low-Bandgap Alternating Fluorene Copolymer/Methanofullerene Heterojunctions in Efficient Near-Infrared Polymer Solar Cells. Adv. Mater. 2006, 18, 2169–2173. [Google Scholar] [CrossRef]
- Tamayo, A.B.; Walker, B.; Nguyen, T.-Q. A Low Band Gap, Solution Processable Oligothiophene with a Diketopyrrolopyrrole Core for Use in Organic Solar Cells. J. Phys. Chem. C 2008, 112, 11545–11551. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.; Wang, L.; Lan, L.; Luo, C.; Zhuang, W.; Peng, J.; Cao, Y. High-performance polymer heterojunction solar cells of a polysilafluorene derivative. Appl. Phys. Lett. 2008, 92, 33307. [Google Scholar] [CrossRef]
- Liu, X.; Ma, R.; Wang, Y.; Du, S.; Tong, J.; Shi, X.; Li, J.; Bao, X.; Xia, Y.; Liu, T.; et al. Significantly Boosting Efficiency of Polymer Solar Cells by Employing a Nontoxic Halogen-Free Additive. ACS Appl. Mater. Interfaces 2021, 13, 11117–11124. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Gao, J.; Hummelen, J.C.; Wudl, F.; Heeger, A.J. Polymer Photovoltaic Cells: Enhanced Efficiencies via a Network of Internal Donor-Acceptor Heterojunctions. Science 1995, 270, 1789–1791. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Cao, Y.; Gao, J.; Wang, D.; Yu, G.; Heeger, A.J. Electrochemical properties of luminescent polymers and polymer light-emitting electrochemical cells. Synth. Met. 1999, 99, 243–248. [Google Scholar] [CrossRef]
- Liang, Z.; Tong, J.; Li, H.; Wang, Y.; Wang, N.; Li, J.; Yang, C.; Xia, Y. The comprehensive utilization of the synergistic effect of fullerene and non-fullerene acceptors to achieve highly efficient polymer solar cells. J. Mater. Chem. A 2019, 7, 15841–15850. [Google Scholar] [CrossRef]
- Li, J.; Liang, Z.; Li, X.; Li, H.; Wang, Y.; Qin, J.; Tong, J.; Yan, L.; Bao, X.; Xia, Y. Insights into Excitonic Dynamics of Terpol-ymer-Based High-Efficiency Nonfullerene Polymer Solar Cells: Enhancing the Yield of Charge Separation States. ACS Appl. Mater. Interfaces 2020, 12, 8475–8484. [Google Scholar] [CrossRef]
- Li, X.M.; Liang, Z.Z.; Wang, H.; Qiao, S.L.; Liu, Z.L.; Jiang, H.X.; Chen, W.C.; Yang, R.Q. Fluorinated D1((0.5))-A-D2((0.5))-A model terpolymer: Ultrafast charge separation kinetics and electron transfer at the fluorinated D/A interface for power con-version. J. Mater. Chem. A 2020, 8, 1360–1367. [Google Scholar] [CrossRef]
- Liu, X.; Liang, Z.; Du, S.; Tong, J.; Li, J.; Zhang, R.; Shi, X.; Yan, L.; Bao, X.; Xia, Y. Non-Halogenated Polymer Donor-Based Organic Solar Cells with a Nearly 15% Efficiency Enabled by a Classic Ternary Strategy. ACS Appl. Energy Mater. 2021, 4, 1774–1783. [Google Scholar] [CrossRef]
- Liu, X.; Du, S.; Fu, Z.; Chen, C.; Tong, J.; Li, J.; Zheng, N.; Zhang, R.; Xia, Y. Ternary solar cells via ternary polymer donors and third component PC71BM to optimize morphology with 13.15% efficiency. Sol. Energy 2021, 222, 18–26. [Google Scholar] [CrossRef]
- Fu, Z.; Liu, X.; Wang, Y.; Du, S.; Tong, J.; Li, J.; Zhang, R.; Yang, C.; Xia, Y. Enhance the efficiency of polymer solar cells through regulating phase segregation and improving charge transport via non-toxic halogen-free additive. Sol. Energy 2021, 218, 375–382. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, C.; Peng, Y.; Wang, N.; Liu, X.; Du, S.; Tong, J.; Li, J.; Xia, Y. An alcohol-soluble small molecule as efficient cathode interfacial layer materials for polymer solar cells. Opt. Mater. 2021, 113, 110909. [Google Scholar] [CrossRef]
- Wang, E.; Ma, Z.; Zhang, Z.; Vandewal, K.; Henriksson, P.; Inganäs, O.; Zhang, F.; Andersson, M.R. An Easily Accessible Isoindigo-Based Polymer for High-Performance Polymer Solar Cells. J. Am. Chem. Soc. 2011, 133, 14244–14247. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, S.; He, C.; Zhang, J.; Yao, H.; Yang, Y.; Zhang, Y.; Zhao, W.; Hou, J. New Wide Band Gap Donor for Efficient Fullerene-Free All-Small-Molecule Organic Solar Cells. J. Am. Chem. Soc. 2017, 139, 1958–1966. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Huo, L.; Chandrabose, S.; Chen, K.; Han, G.; Qi, F.; Meng, X.; Xie, D.; Ma, W.; Yi, Y.; et al. Optimized Fibril Network Morphology by Precise Side-Chain Engineering to Achieve High-Performance Bulk-Heterojunction Organic Solar Cells. Adv. Mater. 2018, 30, e1707353. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Cui, Y.; Qian, D.; Ponseca, C.S.; Honarfar, A.; Xu, Y.; Xin, J.; Chen, Z.; Hong, L.; Gao, B.; et al. 14.7% Efficiency Organic Photovoltaic Cells Enabled by Active Materials with a Large Electrostatic Potential Difference. J. Am. Chem. Soc. 2019, 141, 7743–7750. [Google Scholar] [CrossRef]
- Zhou, C.; Chen, Z.; Zhang, G.; McDowell, C.; Luo, P.; Jia, X.; Ford, M.J.; Wang, M.; Bazan, G.C.; Huang, F.; et al. Toward High Efficiency Polymer Solar Cells: Rearranging the Backbone Units into a Readily Accessible Random Tetrapolymer. Adv. Energy Mater. 2018, 8, 1701668–1701676. [Google Scholar] [CrossRef]
- Fan, B.; Zhang, D.; Li, M.; Zhong, W.; Zeng, Z.; Ying, L.; Huang, F.; Cao, Y. Achieving over 16% efficiency for single-junction organic solar cells. Sci. China Ser. B Chem. 2019, 62, 746–752. [Google Scholar] [CrossRef]
- Zhang, M.; Guo, X.; Ma, W.; Ade, H.; Hou, J. A Large-Bandgap Conjugated Polymer for Versatile Photovoltaic Applications with High Performance. Adv. Mater. 2015, 27, 4655–4660. [Google Scholar] [CrossRef]
- Liu, Q.S.; Jiang, Y.F.; Jin, K.; Qin, J.Q.; Xu, J.G.; Li, W.T.; Xiong, J.; Liu, J.F.; Xiao, Z.; Sun, K.; et al. 18% Efficiency organic solar cells. Sci. Bull. 2020, 65, 272–275. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Champion, R.D.; Jenekhe, S.A. Conjugated donor-acceptor copolymer semiconductors with large intramolecular charge transfer: Synthesis, optical properties, electrochemistry, and field effect carrier mobility of thienopyrazine-based co-polymers. Macromolecules 2006, 39, 8712–8719. [Google Scholar] [CrossRef]
- Darling, S.B. Block copolymers for photovoltaics. Energy Environ. Sci. 2009, 2, 1266–1273. [Google Scholar] [CrossRef]
- Zhang, Y.; Zou, J.; Yip, H.-L.; Chen, K.-S.; Zeigler, D.F.; Sun, Y.; Jen, A.K.-Y. Indacenodithiophene and Quinoxaline-Based Conjugated Polymers for Highly Efficient Polymer Solar Cells. Chem. Mater. 2011, 23, 2289–2291. [Google Scholar] [CrossRef]
- Li, Y.W.; Xue, L.L.; Li, H.; Li, Z.F.; Xu, B.; Wen, S.P.; Tian, W.J. Energy Level and Molecular Structure Engineering of Conju-gated Donor-Acceptor Copolymers for Photovoltaic Applications. Macromolecules 2009, 42, 4491–4499. [Google Scholar] [CrossRef]
- Kang, T.E.; Kim, K.-H.; Kim, B.J. Design of terpolymers as electron donors for highly efficient polymer solar cells. J. Mater. Chem. A 2014, 2, 15252–15267. [Google Scholar] [CrossRef]
- Lee, J.; Lee, S.M.; Chen, S.; Kumari, T.; Kang, S.; Cho, Y.; Yang, C. Organic Photovoltaics with Multiple Donor–Acceptor Pairs. Adv. Mater. 2019, 31, e1804762. [Google Scholar] [CrossRef]
- Wang, X.; Han, J.; Huang, D.; Wang, J.; Xie, Y.; Liu, Z.; Li, Y.; Yang, C.; Zhang, Y.; He, Z.; et al. Optimized Mo-lecular Packing and Nonradiative Energy Loss Based on Terpolymer Methodology Combining Two Asymmetric Segments for High-Performance Polymer Solar Cells. ACS Appl. Mater. Interfaces 2020, 12, 20393–20403. [Google Scholar] [CrossRef]
- Luo, Z.; Ma, R.; Liu, T.; Yu, J.; Xiao, Y.; Sun, R.; Xie, G.; Yuan, J.; Chen, Y.; Chen, K.; et al. Fine-Tuning Energy Levels via Asymmetric End Groups Enables Polymer Solar Cells with Efficiencies over 17%. Joule 2020, 4, 1236–1247. [Google Scholar] [CrossRef]
- Li, S.X.; Zhan, L.L.; Jin, Y.Z.; Zhou, G.Q.; Lau, T.K.; Qin, R.; Shi, M.M.; Li, C.Z.; Zhu, H.M.; Lu, X.H.; et al. Asymmetric Electron Acceptors for High-Efficiency and Low-Energy-Loss Organic Photovoltaics. Adv. Mater. 2020, 32, 2001160–2001169. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Du, Z.; Dou, K.; Jiang, H.; Gao, C.; Han, L.; Yang, R. A Maverick Asymmetrical Backbone with Distinct Flanked Twist Angles Modulating the Molecular Aggregation and Crystallinity for High Performance Nonfullerene Solar Cells. Adv. Energy Mater. 2018, 9, 1802530–1802540. [Google Scholar] [CrossRef]
- Liu, D.Y.; Wang, J.Y.; Gu, C.Y.; Li, Y.H.; Bao, X.C.; Yang, R.Q. Stirring Up Acceptor Phase and Controlling Morphology via Choosing Appropriate Rigid Aryl Rings as Lever Arms in Symmetry-Breaking Benzodithiophene for High-Performance Full-erene and Fullerene-Free Polymer Solar Cells. Adv. Mater. 2018, 30, 1705870–1705878. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, M.; Guo, Y.; Lu, H.; Song, J.; Bo, Z.; Wang, H. Dibenzopyran-Based Wide Band Gap Conjugated Copolymers: Structural Design and Application for Polymer Solar Cells. ACS Appl. Mater. Interfaces 2016, 8, 31348–31358. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.Y.; Zhu, Q.Q.; Gu, C.Y.; Wang, J.Y.; Qiu, M.; Chen, W.C.; Bao, X.C.; Sun, M.L.; Yang, R.Q. High-Performance Photo-voltaic Polymers Employing Symmetry-Breaking Building Blocks. Adv. Mater. 2016, 28, 8490–8498. [Google Scholar] [CrossRef]
- Wang, M.; Cai, D.; Yin, Z.; Chen, S.-C.; Du, C.-F.; Zheng, Q. Asymmetric-Indenothiophene-Based Copolymers for Bulk Heterojunction Solar Cells with 9.14% Efficiency. Adv. Mater. 2016, 28, 3359–3365. [Google Scholar] [CrossRef]
- Mo, D.Z.; Wang, H.; Chen, H.; Qu, S.W.; Chao, P.J.; Yang, Z.; Tian, L.L.; Su, Y.A.; Gao, Y.; Yang, B.; et al. Chlorination of Low-Band-Gap Polymers: Toward High-Performance Polymer Solar Cells. Chem. Mater. 2017, 29, 2819–2830. [Google Scholar] [CrossRef]
- Feng, S.; Zhang, C.; Liu, Y.; Bi, Z.; Zhang, Z.; Xu, X.; Ma, W.; Bo, Z. Fused-Ring Acceptors with Asymmetric Side Chains for High-Performance Thick-Film Organic Solar Cells. Adv. Mater. 2017, 29, 1703527–1703533. [Google Scholar] [CrossRef]
- Guo, Y.-Q.; Wang, Y.; Song, L.-C.; Liu, F.; Wan, X.; Zhang, H.; Chen, Y. Small Molecules with Asymmetric 4-Alkyl-8-alkoxybenzo[1,2-b:4,5-b′]dithiophene as the Central Unit for High-Performance Solar Cells with High Fill Factors. Chem. Mater. 2017, 29, 3694–3703. [Google Scholar] [CrossRef]
- Song, J.; Li, C.; Ye, L.; Koh, C.; Cai, Y.; Wei, D.; Woo, H.Y.; Sun, Y. Extension of indacenodithiophene backbone conjugation enables efficient asymmetric A–D–A type non-fullerene acceptors. J. Mater. Chem. A 2018, 6, 18847–18852. [Google Scholar] [CrossRef]
- Chochos, C.L.; Avgeropoulos, A.; Lidorikis, E. Theoretical study of phenyl-substituted indacenodithiophene copolymers for high performance organic photovoltaics. J. Chem. Phys. 2013, 138, 064901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, H.B.; Sun, S.L.; Geng, Y.; Wu, Y.; Su, Z.M. Density functional theory characterization and design of high-performance diarylamine-fluorene dyes with different pi spacers for dye-sensitized solar cells. J. Mater. Chem. 2012, 22, 568–576. [Google Scholar] [CrossRef]
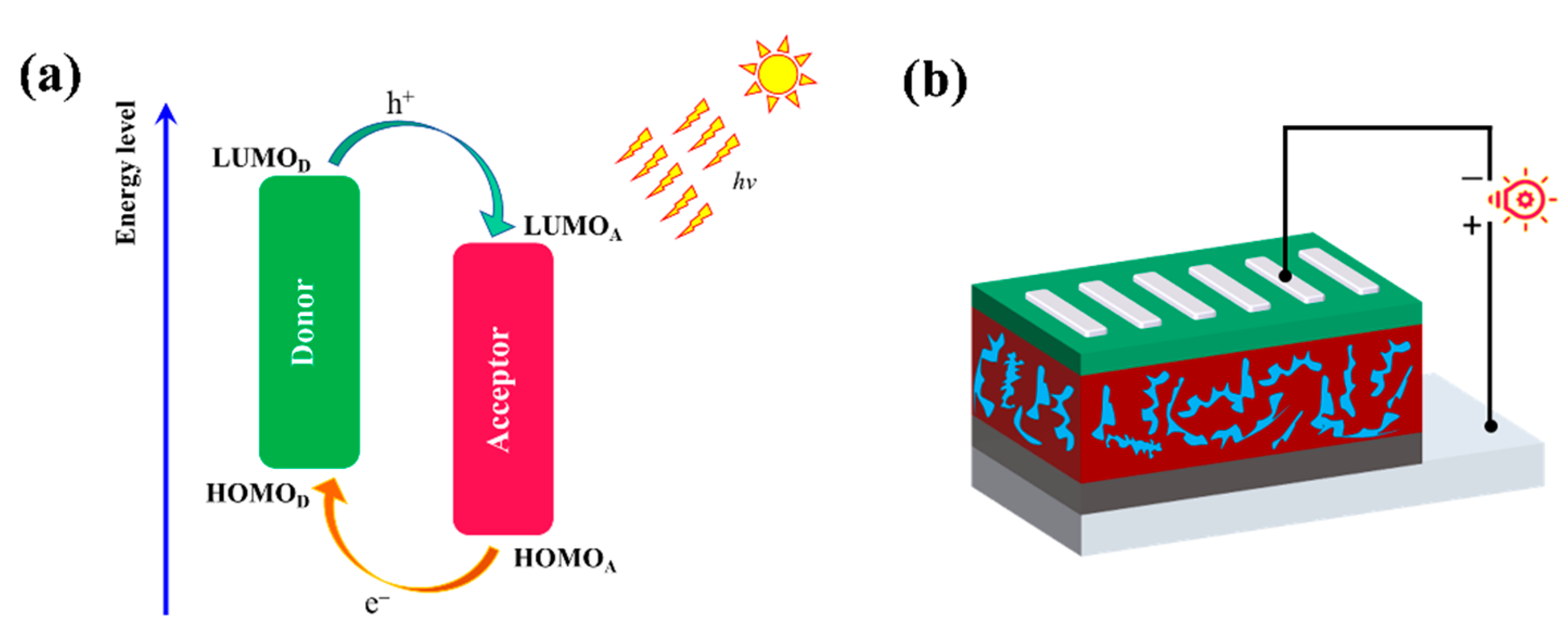
- Bredas, J.L.; Norton, J.E.; Cornil, J.; Coropceanu, V. Molecular understanding of organic solar cells: The challenges. ACC Chem. Res. 2009, 42, 1691–1699. [Google Scholar] [CrossRef]
- Ahmed, S.; Dutta, R.; Kalita, D.J. Strategical designing of diketopyrrolopyrrole-thiophene based donor-acceptor type organic oligomers and study their transport properties: A DFT/TD-DFT perspective. Chem. Phys. Lett. 2019, 730, 14–25. [Google Scholar] [CrossRef]
- Liang, Y.J.; Zhao, Z.W.; Geng, Y.; Pan, Q.Q.; Gu, H.Y.; Zhao, L.; Zhang, M.; Wu, S.X.; Su, Z.M. Can we utilize the higher Frenkel exciton state in biazulene diimides-based non-fullerene acceptors to promote charge separation at the donor/acceptor interface? New J. Chem. 2020, 44, 9767–9774. [Google Scholar] [CrossRef]
- Mabrouk, A.; Alimi, K.; Molinie, P.; Nguyen, T.P. A combined experimental and theoretical study on the effect of doping and interface form;ation on Ppv-ether copolymer. J. Phys. Chem. B 2006, 110, 1141–1150. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Khalid, M.; Khera, R.A.; Jabeen, S.; Langer, P.; Iqbal, J. Designing 2D fused ring materials for small molecules organic solar cells. Comput. Theor. Chem. 2020, 1183, 112848. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01.; Gaussian, Inc: Wallingford, CT, USA, 2013. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, T.; Chen, Q. An sp-hybridized all-carboatomic ring, cyclo[18]carbon: Electronic structure, electronic spectrum, and optical nonlinearity. Carbon 2020, 165, 461–467. [Google Scholar] [CrossRef]
- Wang, Y.; Michinobu, T. Benzothiadiazole and its pi-extended, heteroannulated derivatives: Useful acceptor building blocks for high-performance donor-acceptor polymers in organic electronics. J. Mater. Chem. C 2016, 4, 6200–6214. [Google Scholar] [CrossRef] [Green Version]
- Mei, C.-Y.; Liang, L.; Zhao, F.-G.; Wang, J.-T.; Yu, L.-F.; Li, Y.-X.; Li, W.-S. A Family of Donor–Acceptor Photovoltaic Polymers with Fused 4,7-Dithienyl-2,1,3-benzothiadiazole Units: Effect of Structural Fusion and Side Chains. Macromolecules 2013, 46, 7920–7931. [Google Scholar] [CrossRef]
- Li, X.; Huang, G.; Zheng, N.; Li, Y.; Kang, X.; Qiao, S.; Jiang, H.; Chen, W.; Yang, R. High-Efficiency Polymer Solar Cells Over 13.9% With a High V OC Beyond 1.0 V by Synergistic Effect of Fluorine and Sulfur. Sol. RRL 2019, 3, 1900005–1900011. [Google Scholar] [CrossRef]
- Zhang, Z.; Yu, J.; Yin, X.; Hu, Z.; Jiang, Y.; Sun, J.; Zhou, J.; Zhang, F.; Russell, T.P.; Liu, F.; et al. Conformation Locking on Fused-Ring Electron Acceptor for High-Performance Nonfullerene Organic Solar Cells. Adv. Funct. Mater. 2018, 28, 1705095–1705102. [Google Scholar] [CrossRef]
- Wu, L.-N.; Yin, H.; Li, M.-Y.; Sun, G.-Y.; Jin, G.-D. Density functional theory analysis for the limitations of fluoranthene-fused imide based small molecule acceptor materials in photovoltaic performance. Comput. Theor. Chem. 2019, 1156, 37–42. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, J.; Zhang, Z.-G.; Bai, H.; Li, Y.; Zhu, D.; Zhan, X. An Electron Acceptor Challenging Fullerenes for Efficient Polymer Solar Cells. Adv. Mater. 2015, 27, 1170–1174. [Google Scholar] [CrossRef]
- Wang, Z.; Ivanov, M.; Gao, Y.; Bussotti, L.; Foggi, P.; Zhang, H.; Russo, N.; Dick, B.; Zhao, J.; DI Donato, M.; et al. Spin–Orbit Charge-Transfer Intersystem Crossing (ISC) in Compact Electron Donor–Acceptor Dyads: ISC Mechanism and Application as Novel and Potent Photodynamic Therapy Reagents. Chem. A Eur. J. 2019, 26, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Vandewal, K.; Tvingstedt, K.; Gadisa, A.; Inganas, O.; Manca, J.V. On the origin of the open-circuit voltage of polymer-fullerene solar cells. Nat. Mater. 2009, 8, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Köse, M.E. Evaluation of Acceptor Strength in Thiophene Coupled Donor–Acceptor Chromophores for Optimal Design of Organic Photovoltaic Materials. J. Phys. Chem. A 2012, 116, 12503–12509. [Google Scholar] [CrossRef]
- Zaier, R.; Hajaji, S.; Kozaki, M.; Ayachi, S. DFT and TD-DFT studies on the electronic and optical properties of linear pi-conjugated cyclopentadithiophene (CPDT) dimer for efficient blue OLED. Optic. Mater. 2019, 91, 108–114. [Google Scholar] [CrossRef]
- Tripathi, A.; Prabhakar, C. Impact of replacement of the central benzene ring in anthracene by a heterocyclic ring on electronic excitations and reorganization energies in anthratetrathiophene molecules. J. Chin. Chem. Soc. 2018, 65, 918–924. [Google Scholar] [CrossRef]
- Luo, D.M.; Jin, R.F. Theoretical characterisation and design of D-pi-A star-shaped molecules with triphenylamine as core and diketopyrrolopyrroles as arms for organic solar cells. Mol. Phys. 2019, 117, 1825–1832. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
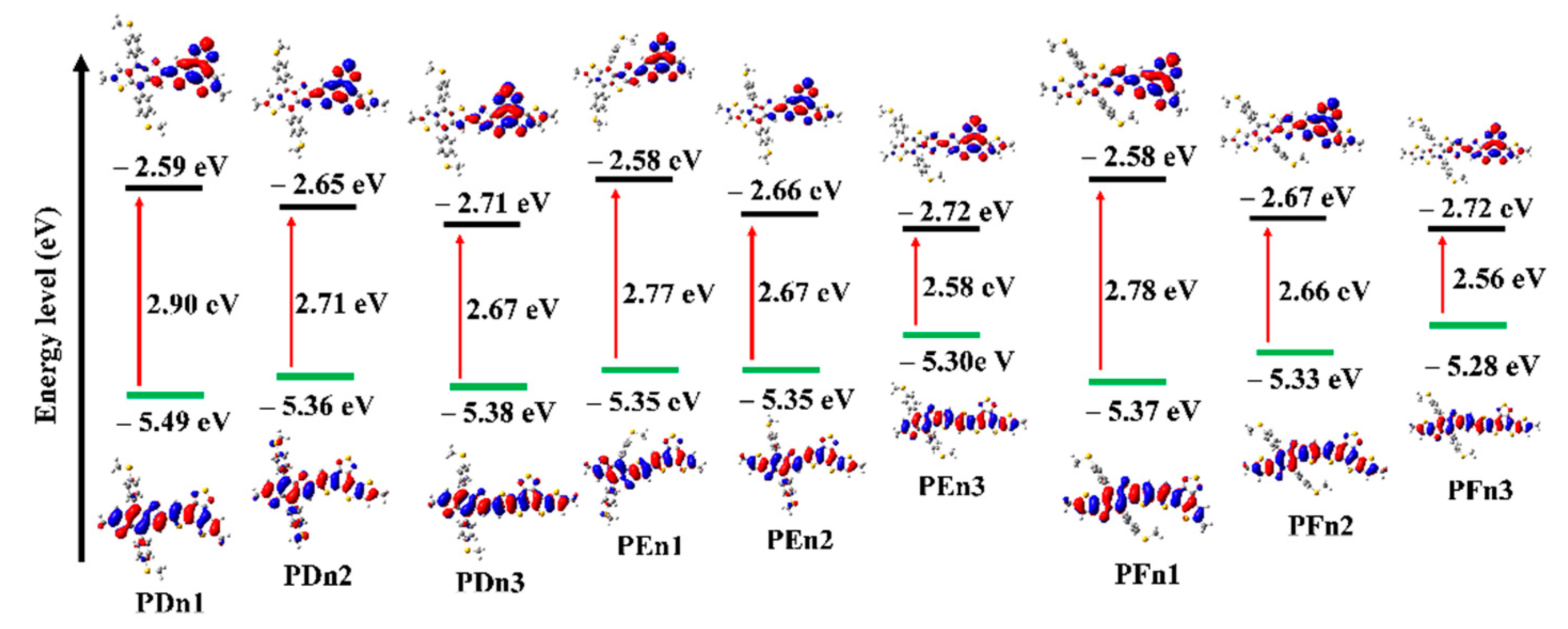
| Molecule | θ (°) | HOMO (eV) | LUMO (eV) | Eg (eV) | VOC (V) |
|---|---|---|---|---|---|
| PDn1 | 7.66 | −5.49 | −2.59 | 2.90 | 1.35 |
| PDn2 | 9.15 | −5.36 | −2.65 | 2.71 | 1.22 |
| PDn3 | 24.25 | −5.38 | −2.71 | 2.67 | 1.24 |
| PEn1 | 10.11 | −5.35 | −2.58 | 2.77 | 1.21 |
| PEn2 | 6.50 | −5.33 | −2.66 | 2.67 | 1.19 |
| PEn3 | 20.31 | −5.30 | −2.72 | 2.58 | 1.16 |
| PFn1 | 18.45 | −5.37 | −2.58 | 2.78 | 1.23 |
| PFn2 | 19.93 | −5.33 | −2.67 | 2.66 | 1.19 |
| PFn3 | 20.21 | −5.28 | −2.72 | 2.56 | 1.14 |
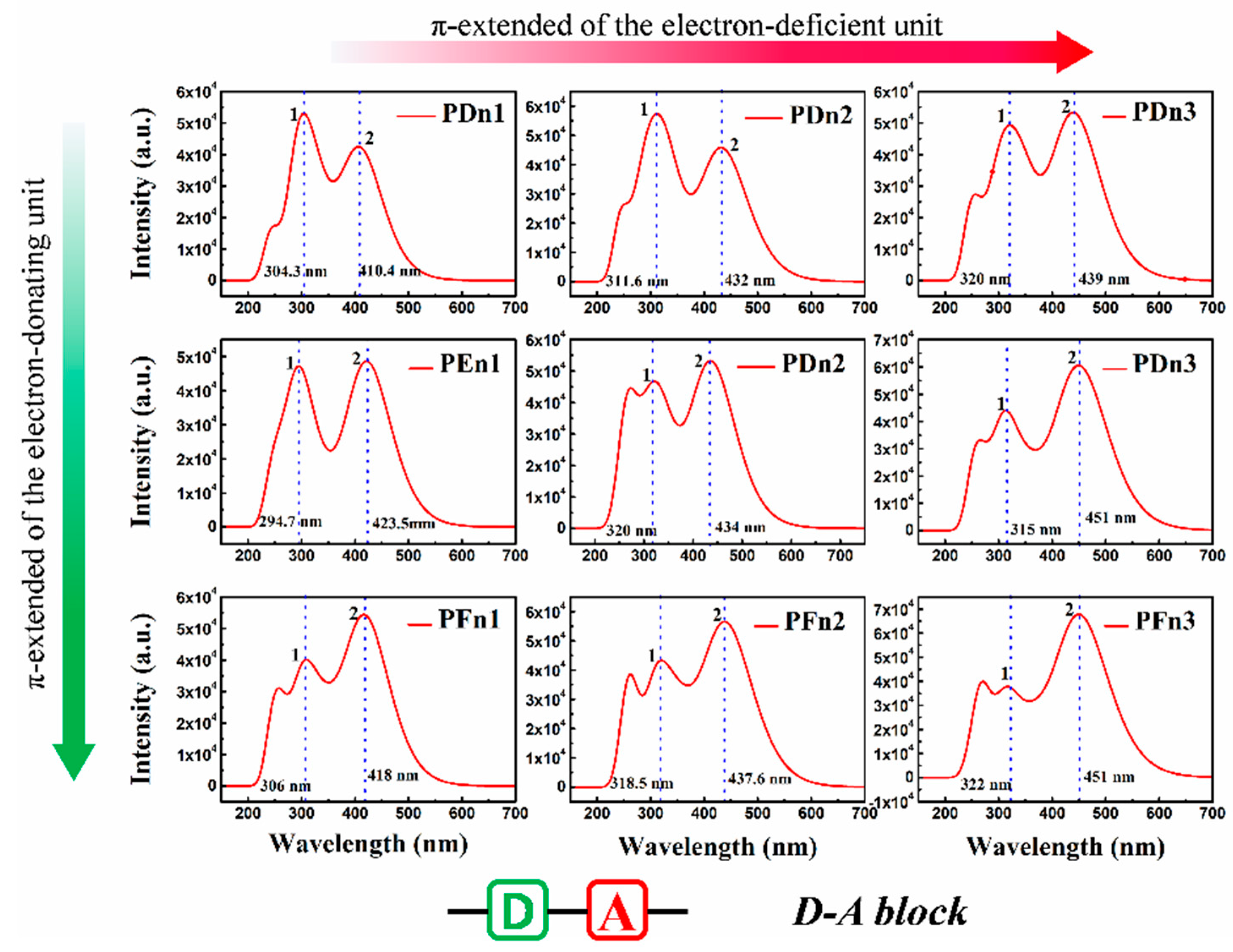
| Molecule | State | Composition a | E (eV) | λabs (nm) | Eb (eV) | f | τ (ns) | LHE |
|---|---|---|---|---|---|---|---|---|
| PDn1 | S0→S1 | H→L (78.3%) | 3.02 | 410.40 | 0.12 | 1.0063 | 2.51 | 0.901 |
| S0→S2 | H→L + 1 (55.6%) | 3.66 | 339.06 | |||||
| PDn2 | S0→S1 | H→L (75.0%) | 2.85 | 434.39 | 0.14 | 1.1058 | 2.56 | 0.922 |
| S0→S2 | H→L + 1 (47.0%) | 3.55 | 349.30 | |||||
| PDn3 | S0→S1 | H→L (79.7%) | 2.80 | 442.02 | 0.13 | 1.2878 | 2.27 | 0.948 |
| S0→S2 | H→L + 1(61.7%) | 3.52 | 352.15 | |||||
| PEn1 | S0→S1 | H→L (77.0%) | 2.93 | 423.32 | 0.16 | 1.1767 | 2.28 | 0.933 |
| S0→S2 | H→L + 1 (71.3%) | 3.55 | 348.97 | |||||
| PEn2 | S0→S1 | H→L (76.7%) | 2.83 | 438.25 | 0.16 | 1.2817 | 2.24 | 0.947 |
| S0→S2 | H→L + 1 (55.2%) | 3.49 | 354.74 | |||||
| PEn3 | S0→S1 | H→L (79.6%) | 2.74 | 452.55 | 0.16 | 1.4560 | 2.10 | 0.965 |
| S0→S2 | H→L + 1 (66.8%) | 3.38 | 366.57 | |||||
| PFn1 | S0→S1 | H→L (76.4%) | 2.95 | 420.78 | 0.17 | 1.2804 | 2.07 | 0.947 |
| S0→S2 | H→L + 1 (67.9%) | 3.53 | 350.71 | |||||
| PFn2 | S0→S1 | H→L (79.4%) | 2.81 | 441.75 | 0.15 | 1.3501 | 2.16 | 0.955 |
| S0→S2 | H→L + 1 (66.3%) | 3.45 | 358.92 | |||||
| PFn3 | S0→S1 | H→L (76.7%) | 2.73 | 454.02 | 0.17 | 1.6127 | 1.92 | 0.975 |
| S0→S2 | H→L + 1 (65.8%) | 3.33 | 372.74 |
| Molecule | IP (eV) | EA (eV) | λ1 (eV) | λ2 (eV) | λ (eV) |
| PDn1 | 5.56 | 2.34 | 0.086 | 0.393 | 0.479 |
| PDn2 | 5.40 | 2.40 | 0.068 | 0.112 | 0.180 |
| PDn3 | 5.44 | 2.46 | 0.064 | 0.378 | 0.442 |
| PEn1 | 5.42 | 2.33 | 0.058 | 0.291 | 0.349 |
| PEn2 | 5.38 | 2.42 | 0.070 | 0.111 | 0.181 |
| PEn3 | 5.37 | 2.48 | 0.084 | 0.350 | 0.434 |
| PFn1 | 5.44 | 2.35 | 0.125 | 0.406 | 0.531 |
| PFn2 | 5.39 | 2.43 | 0.126 | 0.392 | 0.518 |
| PFn3 | 5.34 | 2.49 | 0.094 | 0.350 | 0.443 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, Z.; Yan, L.; Si, J.; Gong, P.; Li, X.; Liu, D.; Li, J.; Hou, X. Rational Design and Characterization of Symmetry-Breaking Organic Semiconductors in Polymer Solar Cells: A Theory Insight of the Asymmetric Advantage. Materials 2021, 14, 6723. https://doi.org/10.3390/ma14216723
Liang Z, Yan L, Si J, Gong P, Li X, Liu D, Li J, Hou X. Rational Design and Characterization of Symmetry-Breaking Organic Semiconductors in Polymer Solar Cells: A Theory Insight of the Asymmetric Advantage. Materials. 2021; 14(21):6723. https://doi.org/10.3390/ma14216723
Chicago/Turabian StyleLiang, Zezhou, Lihe Yan, Jinhai Si, Pingping Gong, Xiaoming Li, Deyu Liu, Jianfeng Li, and Xun Hou. 2021. "Rational Design and Characterization of Symmetry-Breaking Organic Semiconductors in Polymer Solar Cells: A Theory Insight of the Asymmetric Advantage" Materials 14, no. 21: 6723. https://doi.org/10.3390/ma14216723
APA StyleLiang, Z., Yan, L., Si, J., Gong, P., Li, X., Liu, D., Li, J., & Hou, X. (2021). Rational Design and Characterization of Symmetry-Breaking Organic Semiconductors in Polymer Solar Cells: A Theory Insight of the Asymmetric Advantage. Materials, 14(21), 6723. https://doi.org/10.3390/ma14216723