
Mixed Cation Halide Perovskite under Environmental and Physical Stress

 ,
,  , , , , , and
, , , , , and 
Abstract
:1. Introduction
2. Results and Discussion
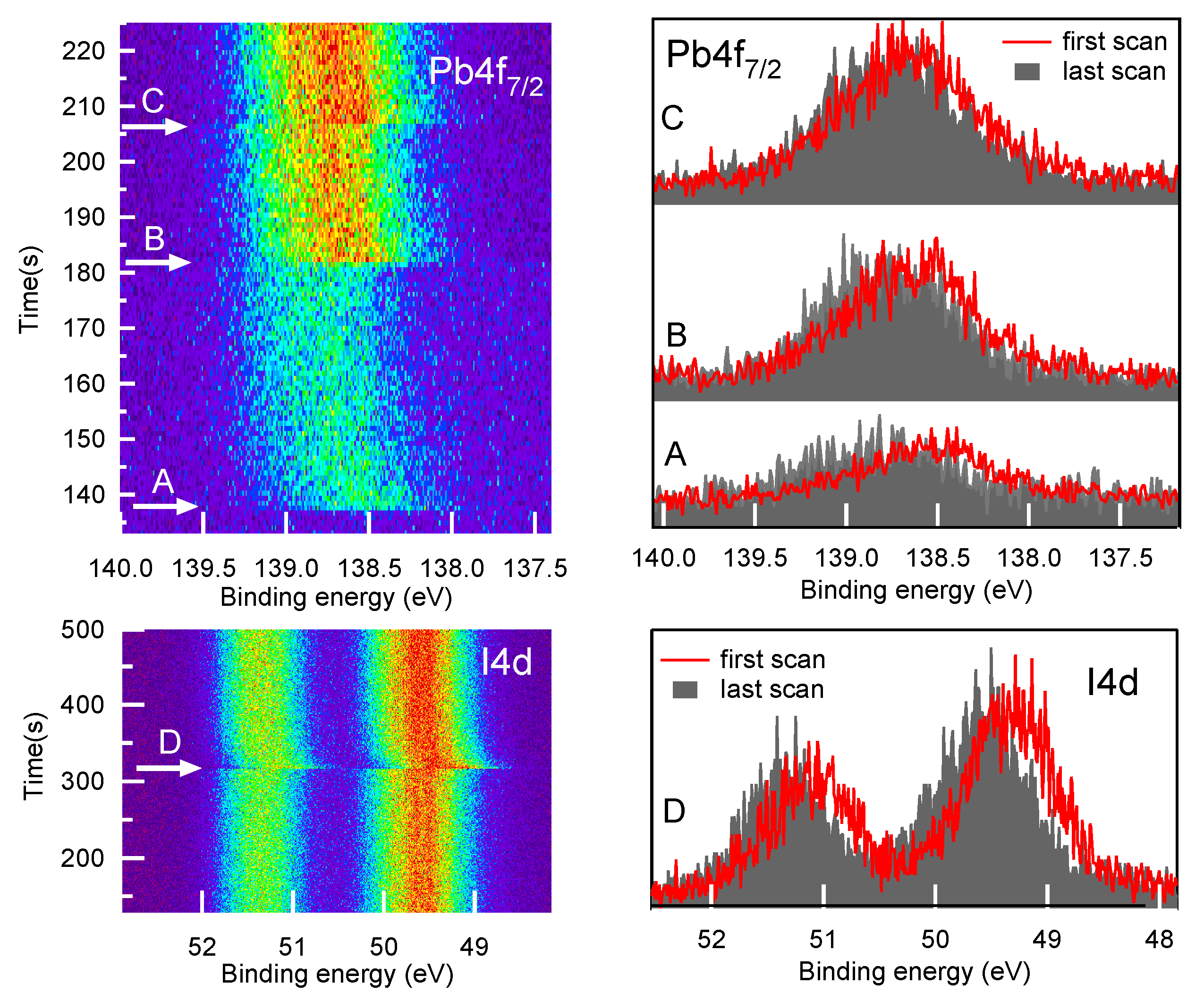
2.1. Fresh Samples
2.2. Aged Samples
2.2.1. Samples Kept in the Dark
2.2.2. Samples Kept in the Light
2.3. Thermal Stress
3. Materials and Methods
3.1. Perovskite Layer Deposition
3.2. XPS Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Best Research-Cell Efficiencies. Available online: https://www.nrel.gov/pv/cell-efficiency.html (accessed on 4 July 2021).
- Saliba, M.; Matsui, T.; Seo, J.Y.; Domanski, K.; Correa-Baena, J.P.; Nazeeruddin, M.K.; Zakeeruddin, S.M.; Tress, W.; Abate, A.; Hagfeldt, A.; et al. Cesium-containing triple cation perovskite solar cells: Improved stability, reproducibility and high efficiency. Energy Environ. Sci. 2016, 9, 1989–1997. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Mujahid, M.; Duan, Y.; Wang, Z.K.; Xue, J.; Yang, Y. A Review of Perovskites Solar Cell Stability. Adv. Funct. Mater. 2019, 29, 1808843. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhao, Y. Chemical stability and instability of inorganic halide perovskites. Energy Environ. Sci. 2019, 12, 1495–1511. [Google Scholar] [CrossRef]
- Misra, R.K.; Aharon, S.; Li, B.; Mogilyansky, D.; Visoly-Fisher, I.; Etgar, L.; Katz, E.A. Temperature- and Component-Dependent Degradation of Perovskite Photovoltaic Materials under Concentrated Sunlight. J. Phys. Chem. Lett. 2015, 6, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Ruess, R.; Benfer, F.; Böcher, F.; Stumpp, M.; Schlettwein, D. Stabilization of Organic–Inorganic Perovskite Layers by Partial Substitution of Iodide by Bromide in Methylammonium Lead Iodide. ChemPhysChem 2016, 17, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.H.; Im, S.H.; Heo, J.H.; Mandal, T.N.; Seok, S.I. Chemical Management for Colorful, Efficient, and Stable Inorganic–Organic Hybrid Nanostructured Solar Cells. Nano Lett. 2013, 13, 1764–1769. [Google Scholar] [CrossRef]
- Pont, S.; Bryant, D.; Lin, C.; Aristidou, N.; Wheeler, S.; Ma, X.; Godin, R.; Haque, S.A.; Durrant, J.R. Tuning CH3NH3Pb(I1-xBrx)3 perovskite oxygen stability in thin films and solar cells. J. Mater. Chem. A 2017, 5, 9553–9560. [Google Scholar] [CrossRef]
- Brennan, M.C.; Draguta, S.; Kamat, P.V.; Kuno, M. Light-Induced Anion Phase Segregation in Mixed Halide Perovskites. ACS Energy Lett. 2018, 3, 204–213. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Chen, C.; Li, C.; Chen, L.; Bista, S.S.; Liu, X.; Li, Y.; Awni, R.A.; Song, Z.; Yan, Y. Correlating Hysteresis and Stability with Organic Cation Composition in the Two-Step Solution-Processed Perovskite Solar Cells. ACS Appl. Mater. Interf. 2020, 12, 10588–10596. [Google Scholar] [CrossRef] [PubMed]
- Ruf, F.; Rietz, P.; Aygüler, M.F.; Kelz, I.; Docampo, P.; Kalt, H.; Hetterich, M. The Bandgap as a Moving Target: Reversible Bandgap Instabilities in Multiple-Cation Mixed-Halide Perovskite Solar Cells. ACS Energy Lett. 2018, 3, 2995–3001. [Google Scholar] [CrossRef]
- Andaji-Garmaroudi, Z.; Abdi-Jalebi, M.; Guo, D.; Macpherson, S.; Sadhanala, A.; Tennyson, E.M.; Ruggeri, E.; Anaya, M.; Galkowski, K.; Shivanna, R.; et al. A Highly Emissive Surface Layer in Mixed-Halide Multication Perovskites. Adv. Mater. 2019, 31, 1902374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Girolamo, D.; Phung, N.; Kosasih, F.U.; Di Giacomo, F.; Matteocci, F.; Smith, J.A.; Flatken, M.A.; Köbler, H.; Turren Cruz, S.H.; Mattoni, A.; et al. Ion Migration-Induced Amorphization and Phase Segregation as a Degradation Mechanism in Planar Perovskite Solar Cells. Adv. Energy Mater. 2020, 10, 2000310. [Google Scholar] [CrossRef]
- Yang, J.; Liu, X.; Zhang, Y.; Zheng, X.; He, X.; Wang, H.; Yue, F.; Braun, S.; Chen, J.; Xu, J.; et al. Comprehensive understanding of heat-induced degradation of triple-cation mixed halide perovskite for a robust solar cell. Nano Energy 2018, 54, 218–226. [Google Scholar] [CrossRef]
- Najafi, L.; Taheri, B.; Martín-García, B.; Bellani, S.; Di Girolamo, D.; Agresti, A.; Oropesa-Nuñez, R.; Pescetelli, S.; Vesce, L.; Calabrò, E.; et al. MoS2 Quantum Dot/Graphene Hybrids for Advanced Interface Engineering of a CH3NH3PbI3 Perovskite Solar Cell with an Efficiency of over 20%. ACS Nano 2018, 12, 10736–10754. [Google Scholar] [CrossRef] [Green Version]
- Agresti, A.; Pescetelli, S.; Palma, A.L.; Martín-García, B.; Najafi, L.; Bellani, S.; Moreels, I.; Prato, M.; Bonaccorso, F.; Di Carlo, A. Two-Dimensional Material Interface Engineering for Efficient Perovskite Large-Area Modules. ACS Energy Lett. 2019, 4, 1862–1871. [Google Scholar] [CrossRef]
- Agresti, A.; Pescetelli, S.; Taheri, B.; Del Rio Castillo, A.E.; Cinà, L.; Bonaccorso, F.; Di Carlo, A. Graphene-Perovskite Solar Cells Exceed 18% Eficiency: A Stability Study. ChemSusChem 2016, 9, 2609–2619. [Google Scholar] [CrossRef] [PubMed]
- Busby, Y.; Agresti, A.; Pescetelli, S.; Carlo, A.D.; Noel, C.; Pireaux, J.J.; Houssiau, L. Aging effects in interface-engineered perovskite solar cells with 2D nanomaterials: A depth profile analysis. Mater. Today Energy 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Kim, H.S.; Yang, B.; Stylianakis, M.M.; Kymakis, E.; Zakeeruddin, S.M.; Grätzel, M.; Hagfeldt, A. Reduced Graphene Oxide Improves Moisture and Thermal Stability of Perovskite Solar Cells. Cell Rep. Phys. Sci. 2020, 1, 100053. [Google Scholar] [CrossRef]
- Hadadian, M.; Smått, J.H.; Correa-Baena, J.P. The role of carbon-based materials in enhancing the stability of perovskite solar cells. Energy Environ. Sci. 2020, 13, 1377–1407. [Google Scholar] [CrossRef]
- Agresti, A.; Pescetelli, S.; Palma, A.L.; Del Rio Castillo, A.E.; Konios, D.; Kakavelakis, G.; Razza, S.; Cinà, L.; Kymakis, E.; Bonaccorso, F.; et al. Graphene Interface Engineering for Perovskite Solar Modules: 12.6% Power Conversion Efficiency over 50 cm2 Active Area. ACS Energy Lett. 2017, 2, 279–287. [Google Scholar] [CrossRef]
- Hantanasirisakul, K.; Gogotsi, Y. Electronic and Optical Properties of 2D Transition Metal Carbides and Nitrides (MXenes). Adv. Mater. 2018, 30, 1804779. [Google Scholar] [CrossRef]
- Agresti, A.; Pazniak, A.; Pescetelli, S.; Di Vito, A.; Rossi, D.; Pecchia, A.; Auf der Maur, M.; Liedl, A.; Larciprete, R.; Kuznetsov, V.; et al. Titanium-carbide MXenes for work function and interface engineering in perovskite solar cells. Nat. Mater. 2019, 18, 1228–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saranin, D.; Pescetelli, S.; Pazniak, A.; Rossi, D.; Liedl, A.; Yakusheva, A.; Luchnikov, L.; Podgorny, D.; Gostischev, P.; Didenko, S.; et al. Transition metal carbides (MXenes) for efficient NiO-based inverted perovskite solar cells. Nano Energy 2021, 82, 105771. [Google Scholar] [CrossRef]
- Béchu, S.; Ralaiarisoa, M.; Etcheberry, A.; Schulz, P. Photoemission Spectroscopy Characterization of Halide Perovskites. Adv. Energy Mat. 2020, 10, 1904007. [Google Scholar] [CrossRef]
- Das, C.; Wussler, M.; Hellmann, T.; Mayer, T.; Zimmermann, I.; Maheu, C.; Nazeeruddin, M.K.; Jaegermann, W. Surface, Interface, and Bulk Electronic and Chemical Properties of Complete Perovskite Solar Cells: Tapered Cross-Section Photoelectron Spectroscopy, a Novel Solution. ACS Appl. Mater. Interfaces 2020, 12, 40949–40957. [Google Scholar] [CrossRef] [PubMed]
- Ning, S.; Zhang, S.; Sun, J.; Li, C.; Zheng, J.; Khalifa, Y.M.; Zhou, S.; Cao, J.; Wu, Y. Ambient Pressure X-ray Photoelectron Spectroscopy Investigation of Thermally Stable Halide Perovskite Solar Cells via Post-Treatment. ACS Appl. Mater. Interfaces 2020, 12, 43705–43713. [Google Scholar] [CrossRef] [PubMed]
- Zhidkov, I.S.; Boukhvalov, D.W.; Akbulatov, A.F.; Frolova, L.A.; Finkelstein, L.D.; Kukharenko, A.I.; Cholakh, S.O.; Chueh, C.C.; Troshin, P.A.; Kurmaev, E.Z. XPS spectra as a tool for studying photochemical and thermal degradation in APbX3 hybrid halide perovskites. Nano Energy 2021, 79, 105421. [Google Scholar] [CrossRef]
- Tennyson, E.M.; Howard, J.M.; Roose, B.; Garrett, J.L.; Munday, J.N.; Abate, A.; Leite, M.S. The Effects of Incident Photon Energy on the Time-Dependent Voltage Response of Lead Halide Perovskites. Chem. Mater. 2019, 31, 8969–8976. [Google Scholar] [CrossRef]
- Hoke, E.T.; Slotcavage, D.J.; Dohner, E.R.; Bowring, A.R.; Karunadasa, H.I.; McGehee, M.D. Reversible photo-induced trap formation in mixed-halide hybrid perovskites for photovoltaics. Chem. Sci. 2015, 6, 613–617. [Google Scholar] [CrossRef] [Green Version]
- Boldyreva, A.G.; Akbulatov, A.F.; Tsarev, S.A.; Luchkin, S.Y.; Zhidkov, I.S.; Kurmaev, E.Z.; Stevenson, K.J.; Petrov, V.G.; Troshin, P.A. γ–Ray–Induced Degradation in the Triple—Cation Perovskite Solar Cells. J. Phys. Chem. Lett. 2019, 10, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Bischak, C.G.; Hetherington, C.L.; Wu, H.; Aloni, S.; Ogletree, D.F.; Limmer, D.T.; Ginsberg, N.S. Origin of Reversible Photoinduced Phase Separation in Hybrid Perovskites. Nano Lett. 2017, 17, 1028–1033. [Google Scholar] [CrossRef] [Green Version]
- Yeh, J. Atomic Calculation of Photoionization Cross-Sections and Asymmetry Parameters; Gordon and Breach Science Publishers: Langhorne, PE, USA, 1993. [Google Scholar]
- Steirer, K.X.; Schulz, P.; Teeter, G.; Stevanovic, V.; Yang, M.; Zhu, K.; Berry, J.J. Defect Tolerance in Methylammonium Lead Triiodide Perovskite. ACS Energy Lett. 2016, 1, 360–366. [Google Scholar] [CrossRef]
- Kerner, R.A.; Schloemer, T.H.; Schulz, P.; Berry, J.J.; Schwartz, J.; Sellinger, A.; Rand, B.P. Amine additive reactions induced by the soft Lewis acidity of Pb2+ in halide perovskites. Part I: Evidence for Pb–alkylamide formation. J. Mater. Chem. C 2019, 7, 5251–5259. [Google Scholar] [CrossRef]
- Philippe, B.; Saliba, M.; Correa-Baena, J.P.; Cappel, U.B.; Turren-Cruz, S.H.; Grätzel, M.; Hagfeldt, A.; Rensmo, H. Chemical Distribution of Multiple Cation (Rb+, Cs+, MA+, and FA+) Perovskite Materials by Photoelectron Spectroscopy. Chem. Mater. 2017, 29, 3589–3596. [Google Scholar] [CrossRef]
- Philippe, B.; Jacobsson, T.J.; Correa-Baena, J.P.; Jena, N.K.; Banerjee, A.; Chakraborty, S.; Cappel, U.B.; Ahuja, R.; Hagfeldt, A.; Odelius, M.; et al. Valence Level Character in a Mixed Perovskite Material and Determination of the Valence Band Maximum from Photoelectron Spectroscopy: Variation with Photon Energy. J. Phys. Chem C 2017, 121, 26655–26666. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, C.; Wang, S.; Liu, G.; Xia, H.; Tong, S.; He, J.; Niu, D.; Zhou, C.; Ding, K.; et al. Low-Temperature Processed, Efficient, and Highly Reproducible Cesium-Doped Triple Cation Perovskite Planar Heterojunction Solar Cells. Solar RRL 2018, 2, 1700209. [Google Scholar] [CrossRef]
- Endres, J.; Egger, D.A.; Kulbak, M.; Kerner, R.A.; Zhao, L.; Silver, S.H.; Hodes, G.; Rand, B.P.; Cahen, D.; Kronik, L.; et al. Valence and Conduction Band Densities of States of Metal Halide Perovskites: A Combined Experimental–Theoretical Study. J. Phys. Chem. Lett. 2016, 7, 2722–2729. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Kuklin, A.V.; Baev, A.; Ge, Y.; Ågren, H.; Zhang, H.; Prasad, P.N. Two-dimensional MXenes: From morphological to optical, electric, and magnetic properties and applications. Phys. Rep. 2020, 848, 1–58. [Google Scholar] [CrossRef]
- deQuilettes, D.W.; Zhang, W.; Burlakov, V.M.; Graham, D.J.; Leijtens, T.; Osherov, A.; Bulović, V.; Snaith, H.J.; Ginger, D.S.; Stranks, S.D. Photo-induced halide redistribution in organicâ-inorganic perovskite films. Nat. Commun. 2016, 7, 11683. [Google Scholar] [CrossRef]
- Ralaiarisoa, M.; Salzmann, I.; Zu, F.S.; Koch, N. Effect of Water, Oxygen, and Air Exposure on CH3NH3PbI(3-x)Clx Perovskite Surface Electronic Properties. Adv. Electr. Mat. 2018, 4, 1800307. [Google Scholar] [CrossRef]
- Smykalla, L.; Shukrynau, P.; Korb, M.; Lang, H.; Hietschold, M. Surface-confined 2D polymerization of a brominated copper-tetraphenylporphyrin on Au(111). Nanoscale 2015, 7, 4234–4241. [Google Scholar] [CrossRef] [Green Version]
- Cardenas, L.; Gutzler, R.; Lipton-Duffin, J.; Fu, C.; Brusso, J.L.; Dinca, L.E.; Vondráček, M.; Fagot-Revurat, Y.; Malterre, D.; Rosei, F.; et al. Synthesis and electronic structure of a two dimensional π–conjugated polythiophene. Chem. Sci. 2013, 4, 3263–3268. [Google Scholar] [CrossRef] [Green Version]
- Climent-Pascual, E.; Hames, B.C.; Moreno-Ramírez, J.S.; Álvarez, A.L.; Juarez-Perez, E.J.; Mas-Marza, E.; Mora-Seró, I.; de Andrés, A.; Coya, C. Influence of the substrate on the bulk properties of hybrid lead halide perovskite films. J. Mater. Chem. A 2016, 4, 18153–18163. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Manser, J.S.; Kamat, P.V.; Ptasinska, S. Evolution of Chemical Composition, Morphology, and Photovoltaic Efficiency of CH3NH3PbI3 Perovskite under Ambient Conditions. Chem. Mater. 2016, 28, 303–311. [Google Scholar] [CrossRef]
- Rocks, C.; Svrcek, V.; Maguire, P.; Mariotti, D. Understanding surface chemistry during MAPbI3 spray deposition and its effect on photovoltaic performance. J. Mater. Chem. C 2017, 5, 902–916. [Google Scholar] [CrossRef] [Green Version]
- Terpstra, H.J.; de Groot, R.A.; Haas, C. Electronic structure of the lead monoxides: Band-structure calculations and photoelectron spectra. Phys. Rev. B 1995, 52, 11690–11697. [Google Scholar] [CrossRef] [Green Version]
- Zu, F.S.; Amsalem, P.; Salzmann, I.; Wang, R.B.; Ralaiarisoa, M.; Kowarik, S.; Duhm, S.; Koch, N. Impact of White Light Illumination on the Electronic and Chemical Structures of Mixed Halide and Single Crystal Perovskites. Adv. Opt. Mater. 2017, 5, 1700139. [Google Scholar] [CrossRef]
- Yang, J.; Yuan, Z.; Liu, X.; Braun, S.; Li, Y.; Tang, J.; Gao, F.; Duan, C.; Fahlman, M.; Bao, Q. Oxygen- and Water-Induced Energetics Degradation in Organometal Halide Perovskites. ACS Appl. Mater. Interface 2018, 10, 16225–16230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.P.; Li, Y.Q.; Jin, T.Y.; Liu, Y.Q.; Bao, Q.Y.; O’Carroll, C.; Tang, J.X. In Situ Observation of Light Illumination-Induced Degradation in Organometal Mixed-Halide Perovskite Films. ACS Appl. Mater. Interface 2018, 10, 6737–6746. [Google Scholar] [CrossRef] [PubMed]
- Calloni, A.; Abate, A.; Bussetti, G.; Berti, G.; Yivlialin, R.; Ciccacci, F.; Duò, L. Stability of Organic Cations in Solution-Processed CH3NH3PbI3 Perovskites: Formation of Modified Surface Layers. J. Phys. Chem. C 2015, 119, 21329–21335. [Google Scholar] [CrossRef]
- Philippe, B.; Park, B.W.; Lindblad, R.; Oscarsson, J.; Ahmadi, S.; Johansson, E.M.J.; Rensmo, H. Chemical and Electronic Structure Characterization of Lead Halide Perovskites and Stability Behavior under Different Exposures—A Photoelectron Spectroscopy Investigation. Chem. Mater. 2015, 27, 1720–1731. [Google Scholar] [CrossRef]
- Choi, J.I.J.; Khan, M.E.; Hawash, Z.; Kim, K.J.; Lee, H.; Ono, L.K.; Qi, Y.; Kim, Y.H.; Park, J.Y. Atomic-scale view of stability and degradation of single-crystal MAPbBr3 surfaces. J. Mater. Chem. A 2019, 7, 20760–20766. [Google Scholar] [CrossRef]
- Conings, B.; Drijkoningen, J.; Gauquelin, N.; Babayigit, A.; D’Haen, J.; D’Olieslaeger, L.; Ethirajan, A.; Verbeeck, J.; Manca, J.; Mosconi, E.; et al. Intrinsic Thermal Instability of Methylammonium Lead Trihalide Perovskite. Adv. Energy Mater. 2015, 5, 1500477. [Google Scholar] [CrossRef]
- Misra, R.K.; Ciammaruchi, L.; Aharon, S.; Mogilyansky, D.; Etgar, L.; Visoly-Fisher, I.; Katz, E.A. Effect of Halide Composition on the Photochemical Stability of Perovskite Photovoltaic Materials. ChemSusChem 2016, 9, 2572–2577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, Y.; Li, Y.; Zhu, P.; Li, Q.; Gao, Y.; Tong, J.; Shi, L.; Zhou, Q.; Ling, C.; Chen, Q.; et al. Photo-oxidative degradation of methylammonium lead iodide perovskite: Mechanism and protection. J. Mater. Chem. A 2019, 7, 2275–2282. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Core Level | Assignment | Fresh | Dark-Aged | Light-Aged |
|---|---|---|---|---|
| (eV) | (eV) | (eV) | ||
| Pb4f | PV (Pb) | 138.76 | 138.76 | 139.41 *(Pb) |
| metallic Pb | 136.90 | 136.90 | – | |
| PbO | – | – | 138.04 * | |
| PbCO | – | – | 139.87 * | |
| I4d | PV (I) | 49.57 | 49.57 | 50.22 * (I) |
| Br3d | PV (Br) | 68.80 | 68.80 | 69.38 * (Br) |
| Br-C | – | – | 69.87 * | |
| N1s | FA | 400.92 | 400.92 | 401.03 |
| MA | 402.11 | 402.11 | 401.90 | |
| dissociated organic phase, N | 399.55 | 399.55 | 399.50 | |
| dissociated organic phase, N | – | – | 400.22 | |
| dissociated organic phase, N | – | – | 398.58 | |
| C1s | C-C, C-H (C) | 284.92 | 284.92 | 285.87 * (C) |
| MA | 286,60 | 286.60 | n.i. | |
| FA | 288,45 | 288.45 | n.i. | |
| C=O, O=C–O (C) | – | – | 289.49 * | |
| C–O, PbCO (C) | – | – | 287.61 * | |
| O1s | adsorbed water | – | 532.80 | – |
| C–O, PbO (O) | – | – | 530.59 * | |
| C=O, PbCO (O) | – | – | 532.46 * | |
| O=C–O (O) | – | – | 534.11 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larciprete, R.; Agresti, A.; Pescetelli, S.; Pazniak, H.; Liedl, A.; Lacovig, P.; Lizzit, D.; Tosi, E.; Lizzit, S.; Di Carlo, A. Mixed Cation Halide Perovskite under Environmental and Physical Stress. Materials 2021, 14, 3954. https://doi.org/10.3390/ma14143954
Larciprete R, Agresti A, Pescetelli S, Pazniak H, Liedl A, Lacovig P, Lizzit D, Tosi E, Lizzit S, Di Carlo A. Mixed Cation Halide Perovskite under Environmental and Physical Stress. Materials. 2021; 14(14):3954. https://doi.org/10.3390/ma14143954
Chicago/Turabian StyleLarciprete, Rosanna, Antonio Agresti, Sara Pescetelli, Hanna Pazniak, Andrea Liedl, Paolo Lacovig, Daniel Lizzit, Ezequiel Tosi, Silvano Lizzit, and Aldo Di Carlo. 2021. "Mixed Cation Halide Perovskite under Environmental and Physical Stress" Materials 14, no. 14: 3954. https://doi.org/10.3390/ma14143954
APA StyleLarciprete, R., Agresti, A., Pescetelli, S., Pazniak, H., Liedl, A., Lacovig, P., Lizzit, D., Tosi, E., Lizzit, S., & Di Carlo, A. (2021). Mixed Cation Halide Perovskite under Environmental and Physical Stress. Materials, 14(14), 3954. https://doi.org/10.3390/ma14143954