
Macromonomers as a Novel Way to Investigate and Tailor Silicon-Oxycarbide-Based Materials Obtained from Polymeric Preceramic Precursors


Abstract
:1. Introduction
2. Materials and Methods
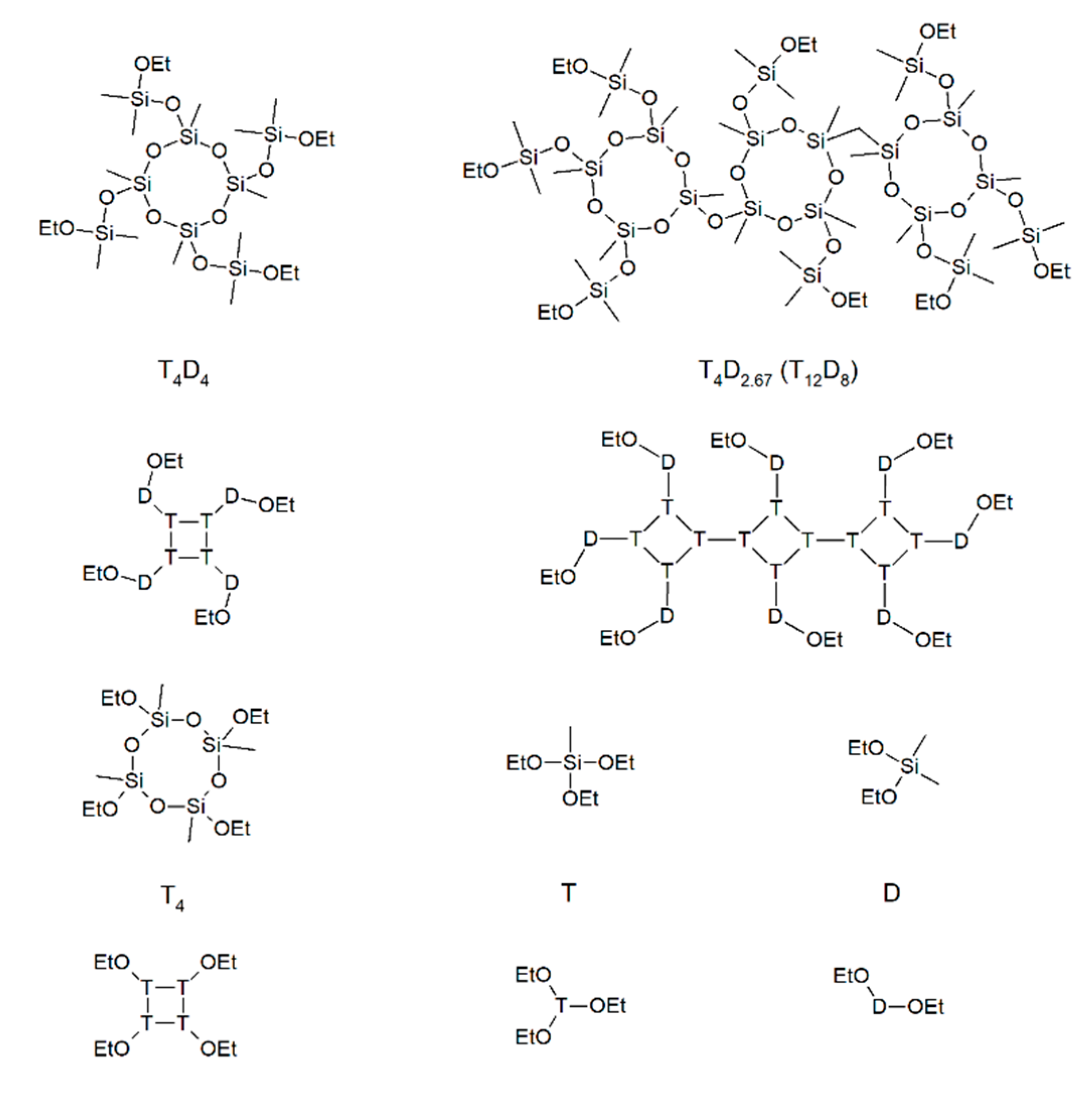
2.1. Synthesis of Macromonomer (T4D2.67)
2.1.1. Materials
2.1.2. Synthesis of 2,4,6,8-Tetrahydroxyl-2,4,6,8-Tetramethylcyclotetrasiloxane (T4OH)
2.1.3. Synthesis of Chlorodimethylsilane Modified 2,4,6,8-Tetrahydroxyl-2,4,6,8-Tetramethylcyclotetrasiloxane (T4M2.67H)
2.2. Synthesis of Preceramic Polymeric Precursors
2.3. Annealing of Silicon Oxycarbide Samples
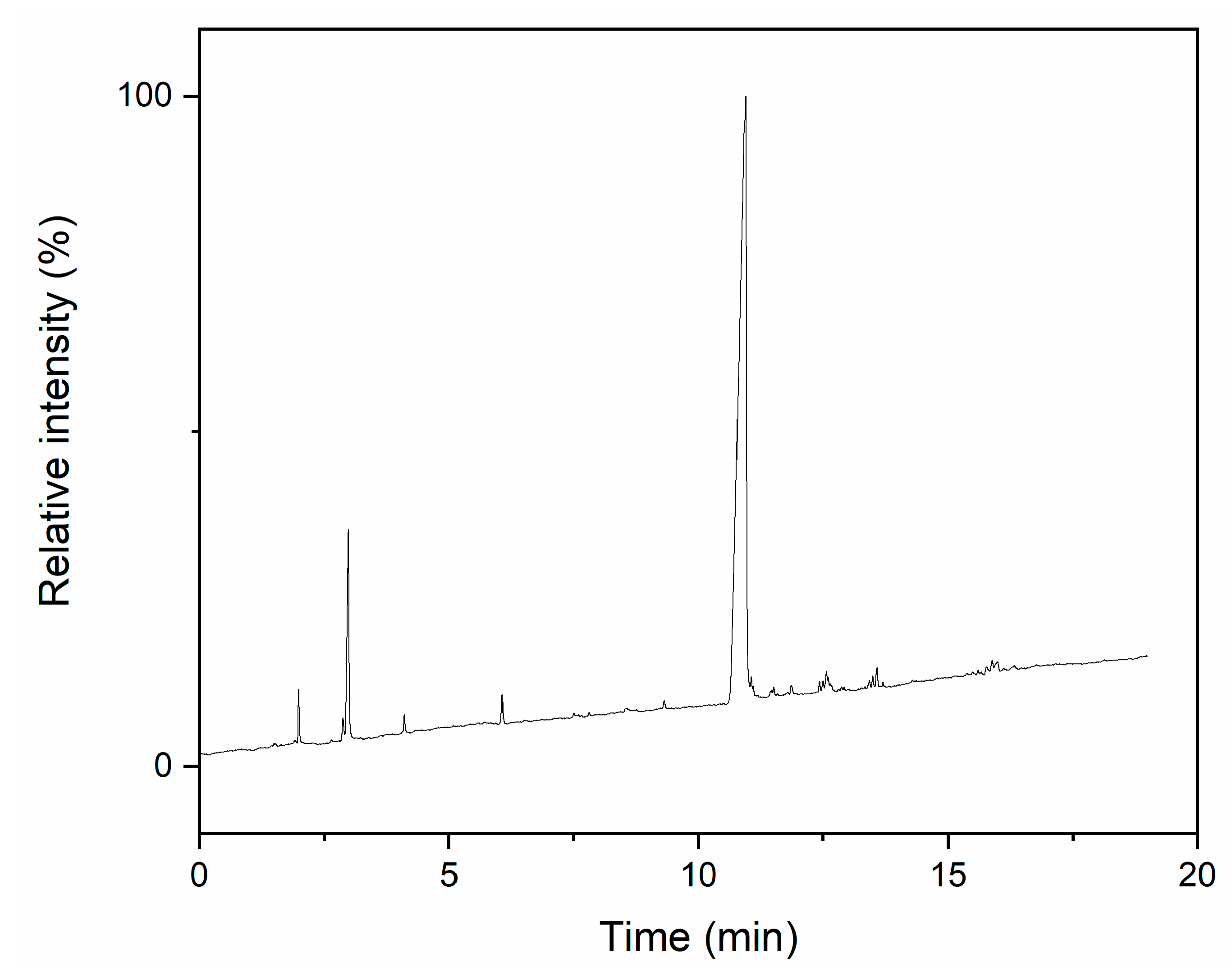
2.4. T4M2.67H Synthesis Progress and Product Structure Investigation
2.5. Precursors and Silicon Oxycarbide Structure Investigation
3. Results
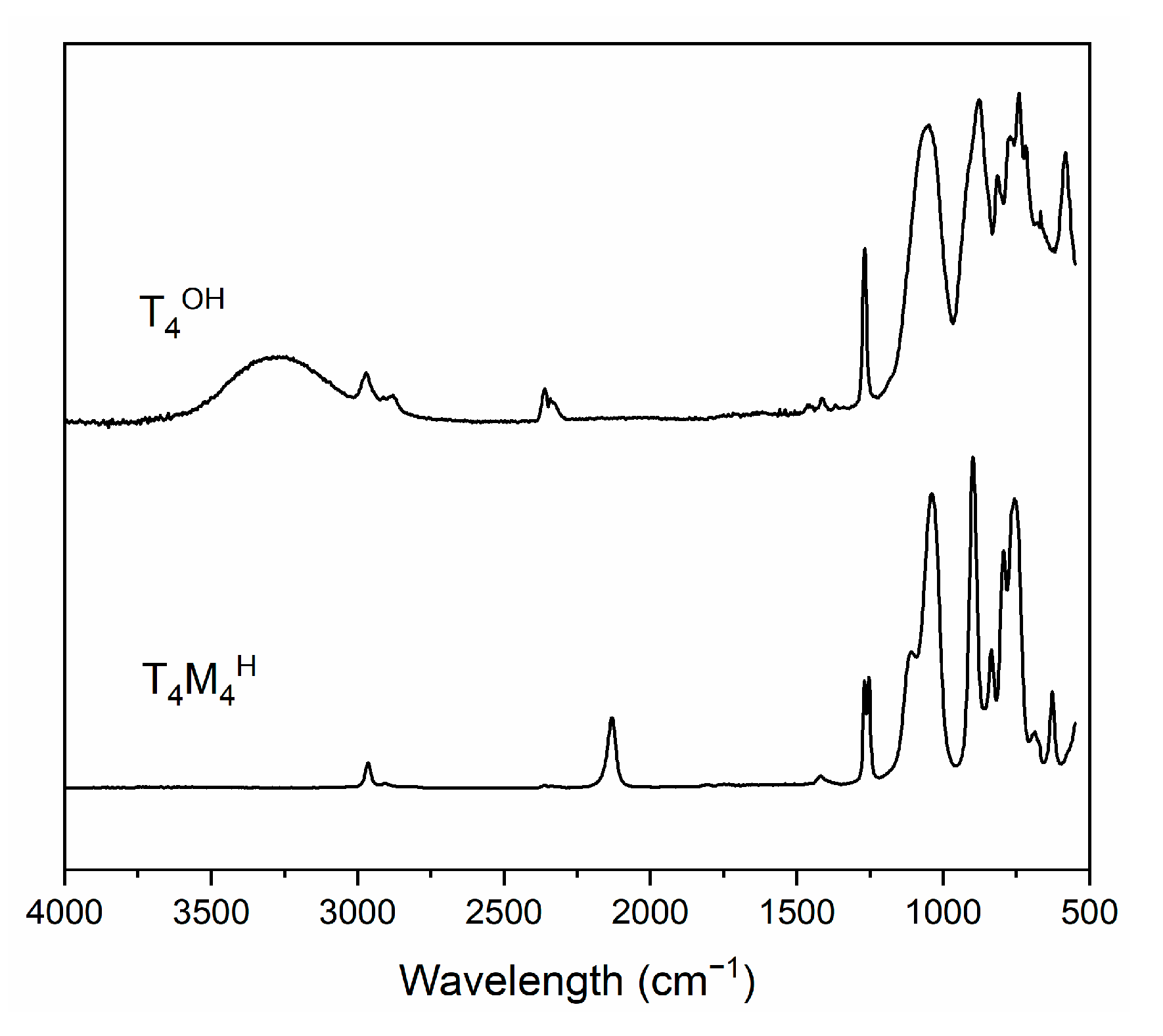
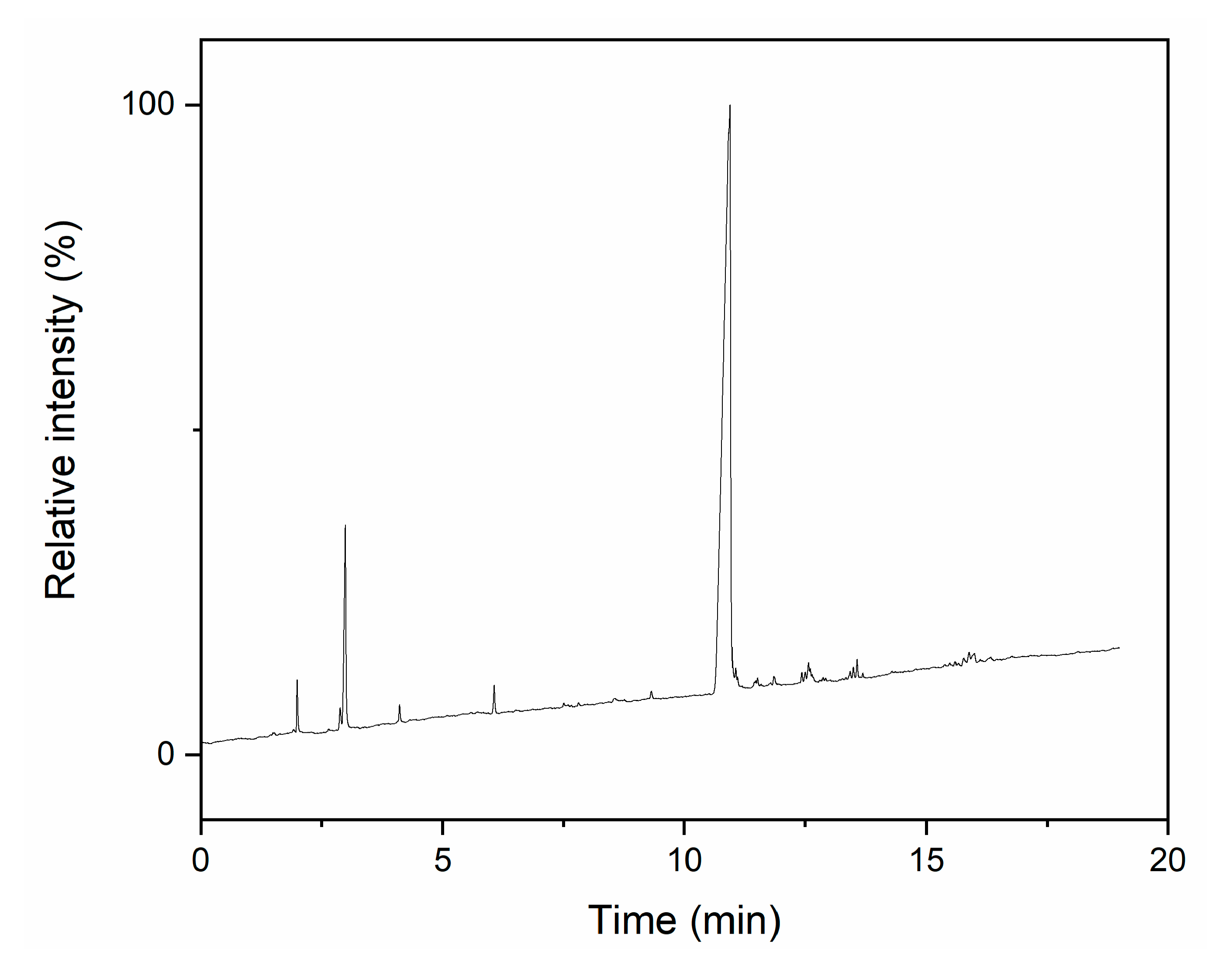
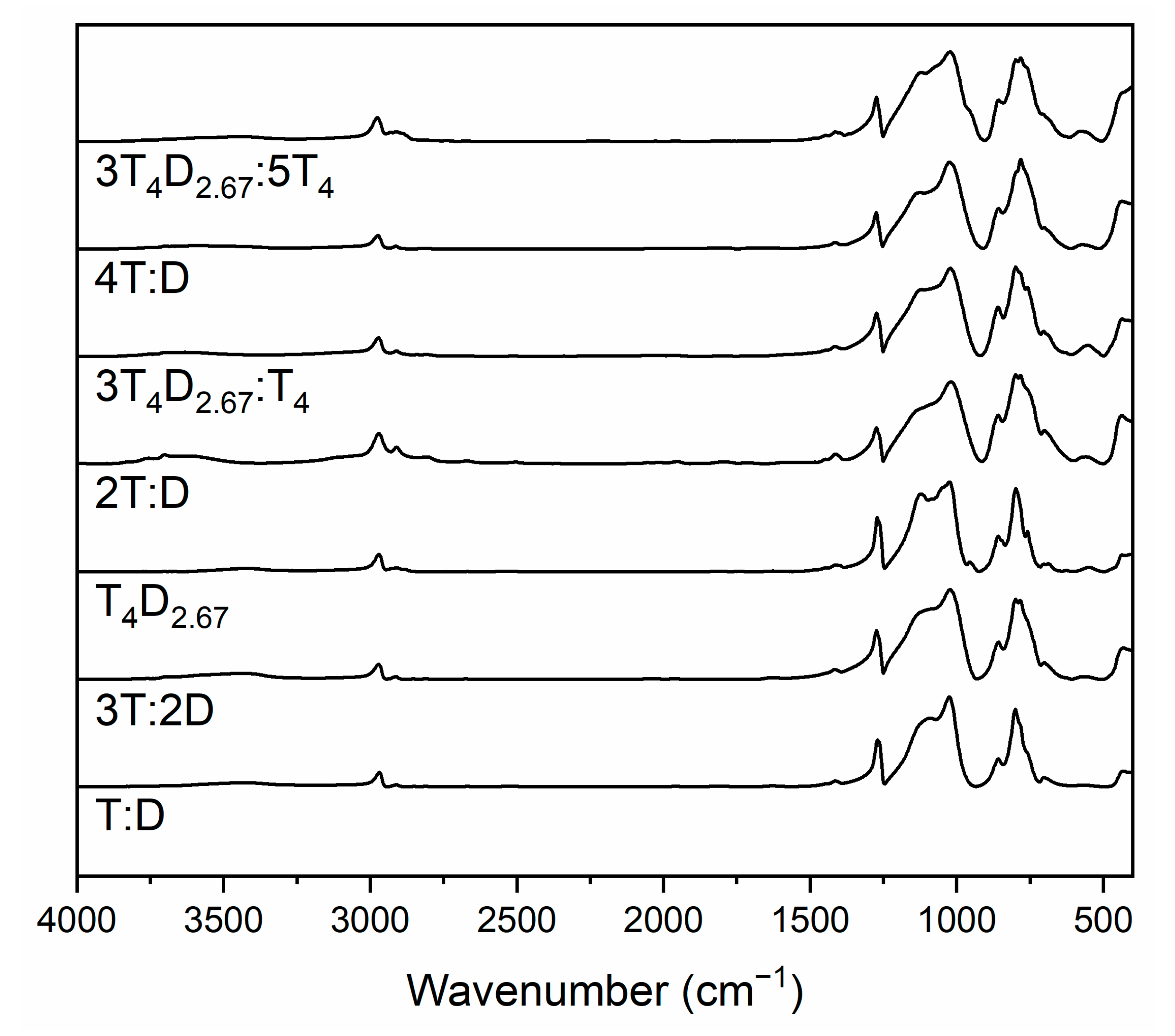
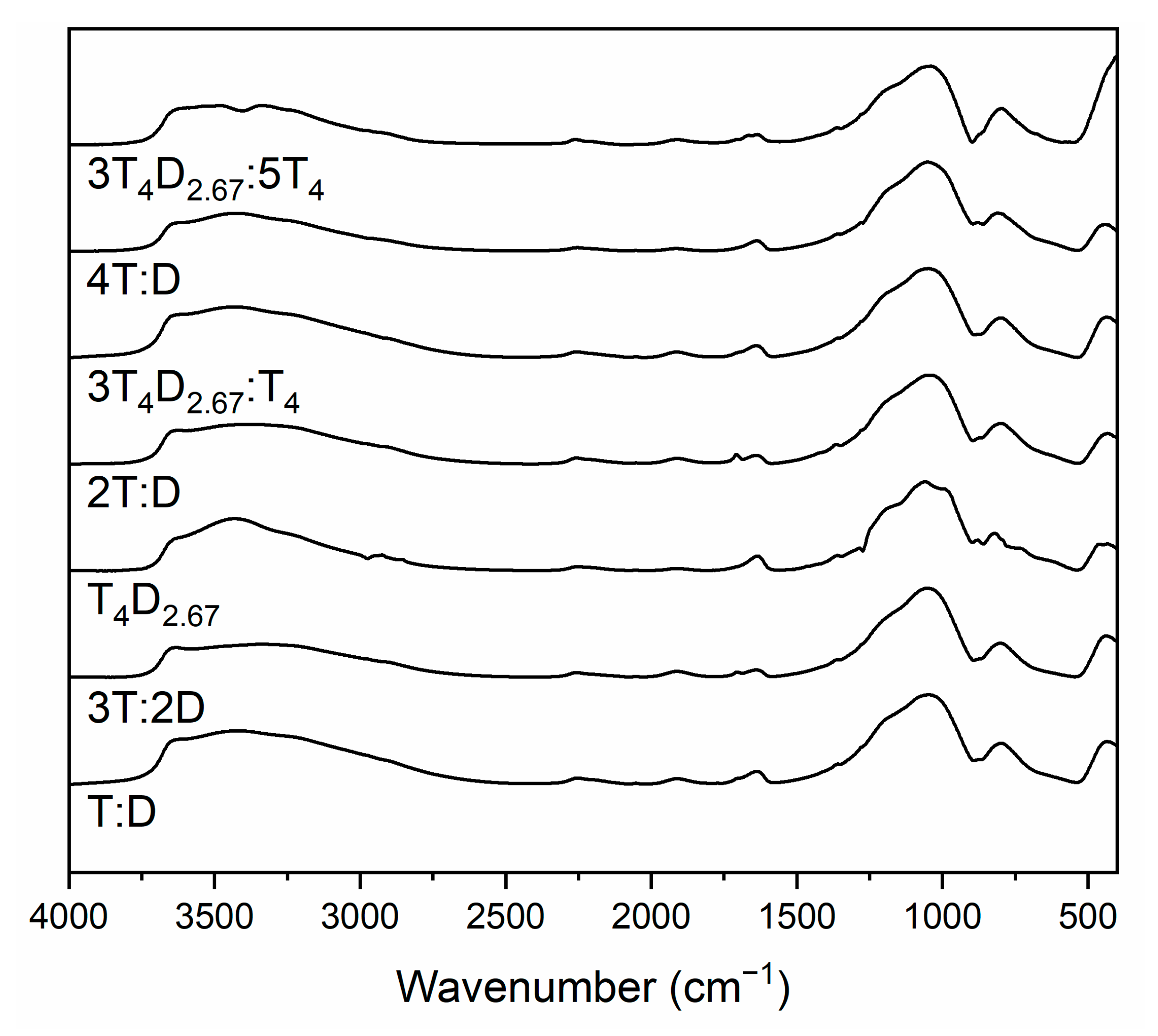
3.1. T4M2.67H Synthesis and Structure
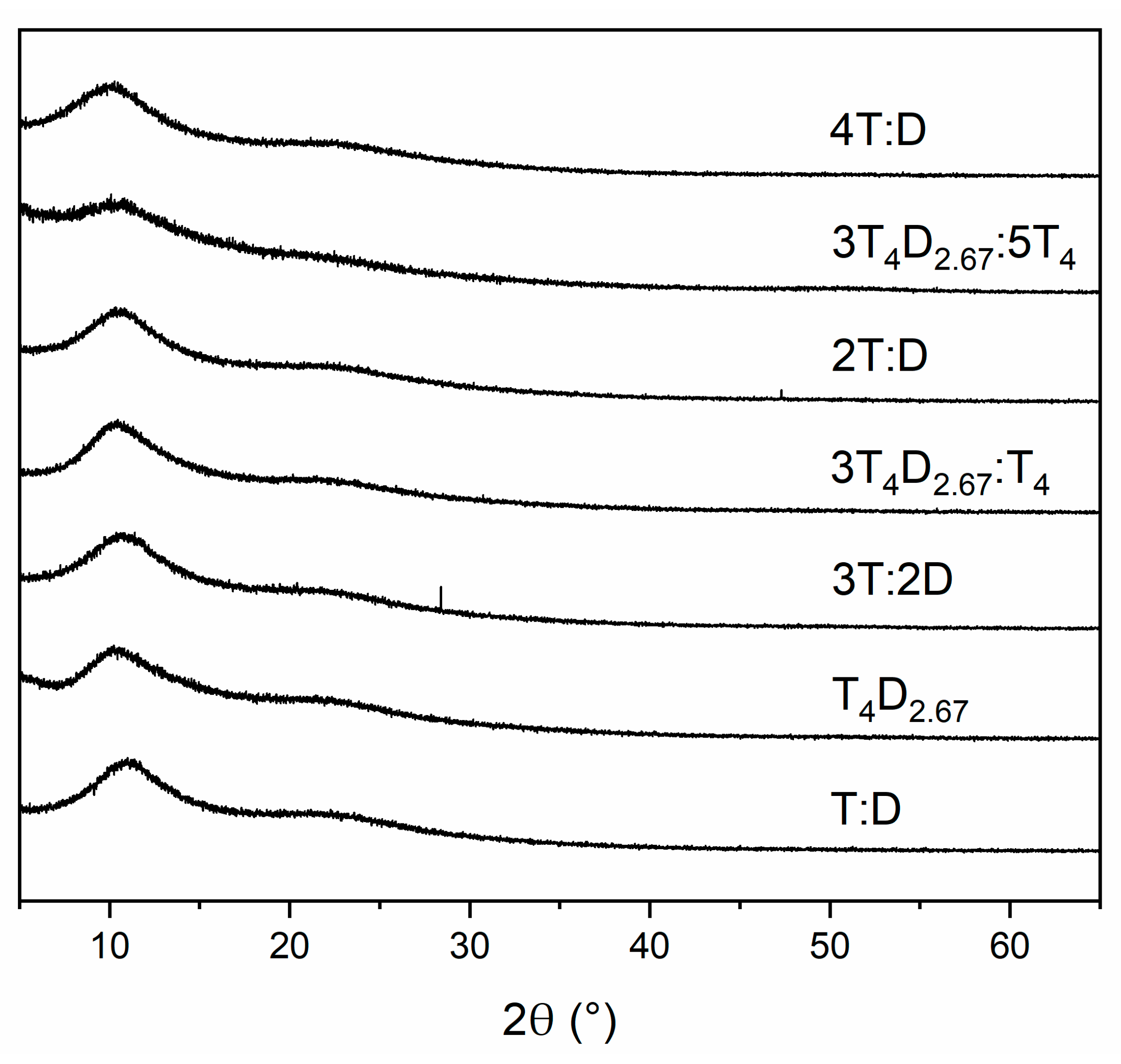
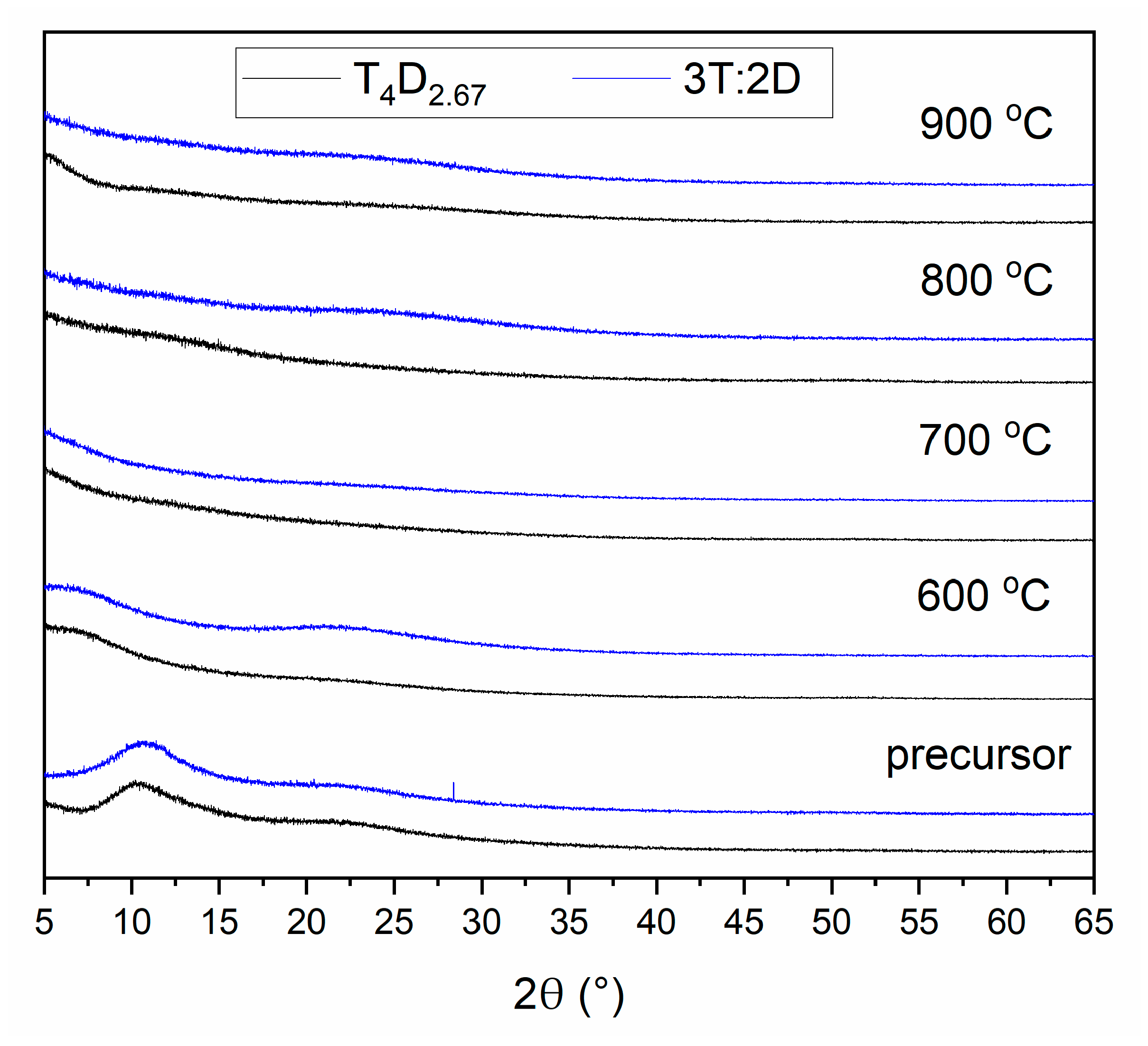
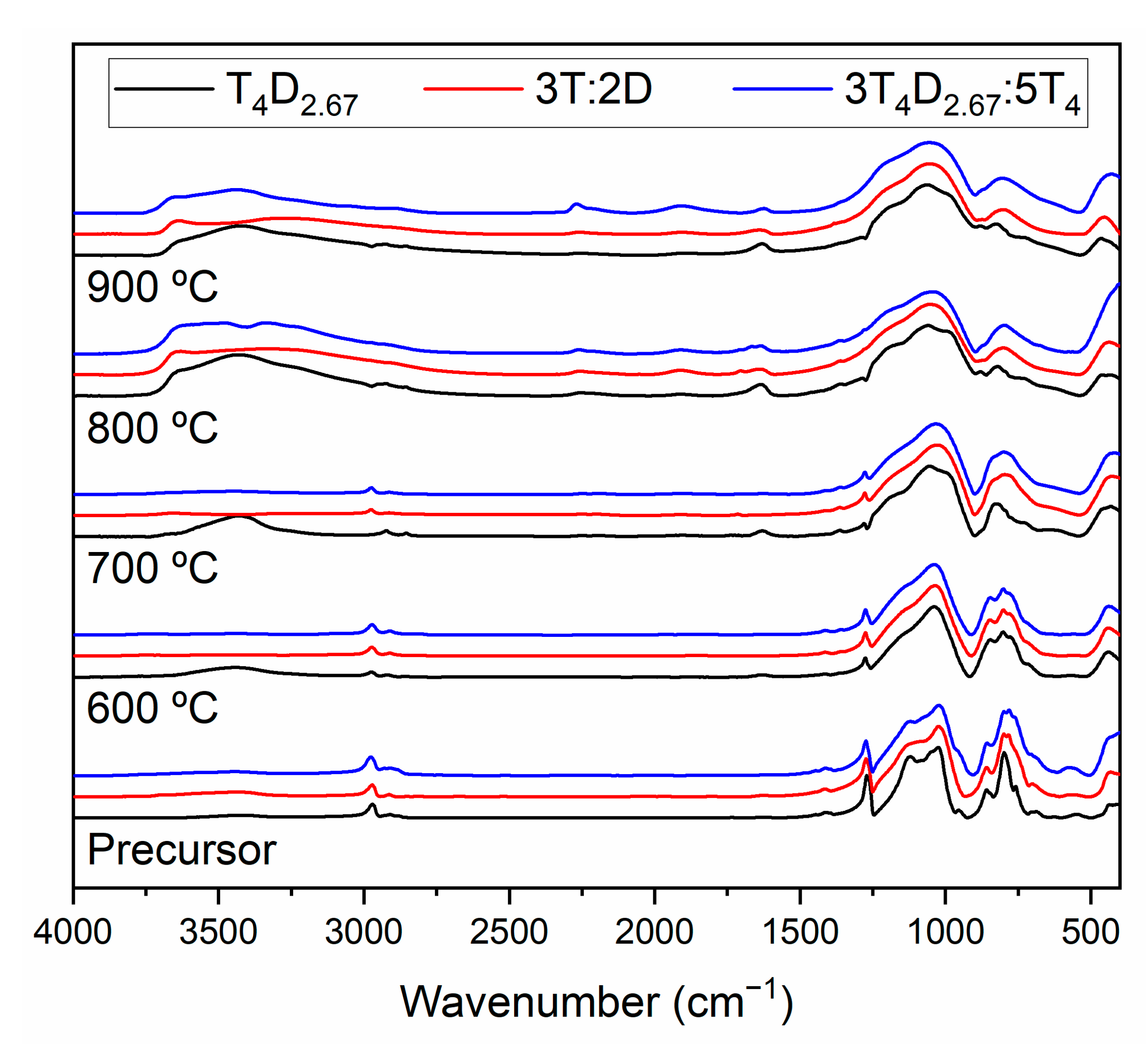
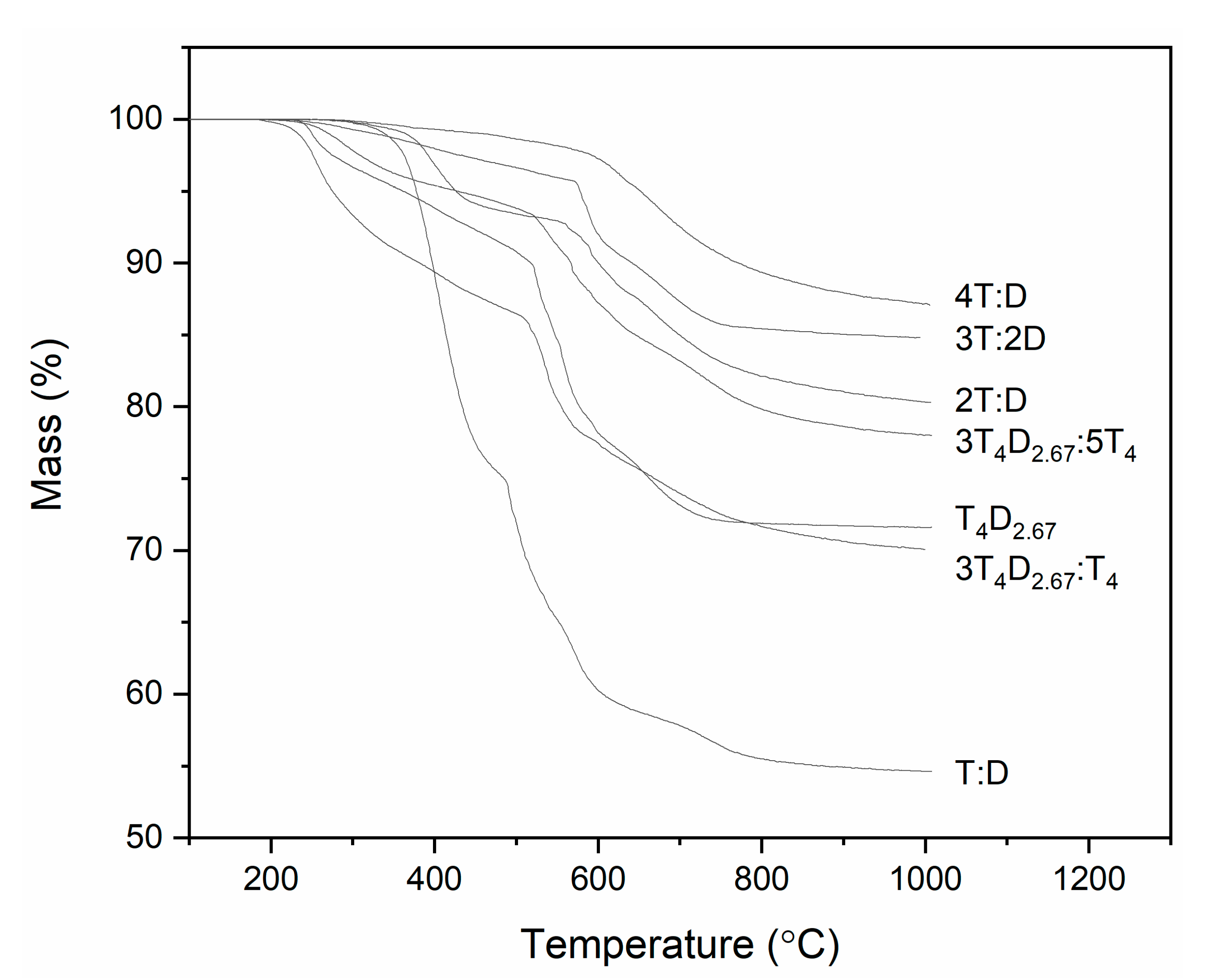
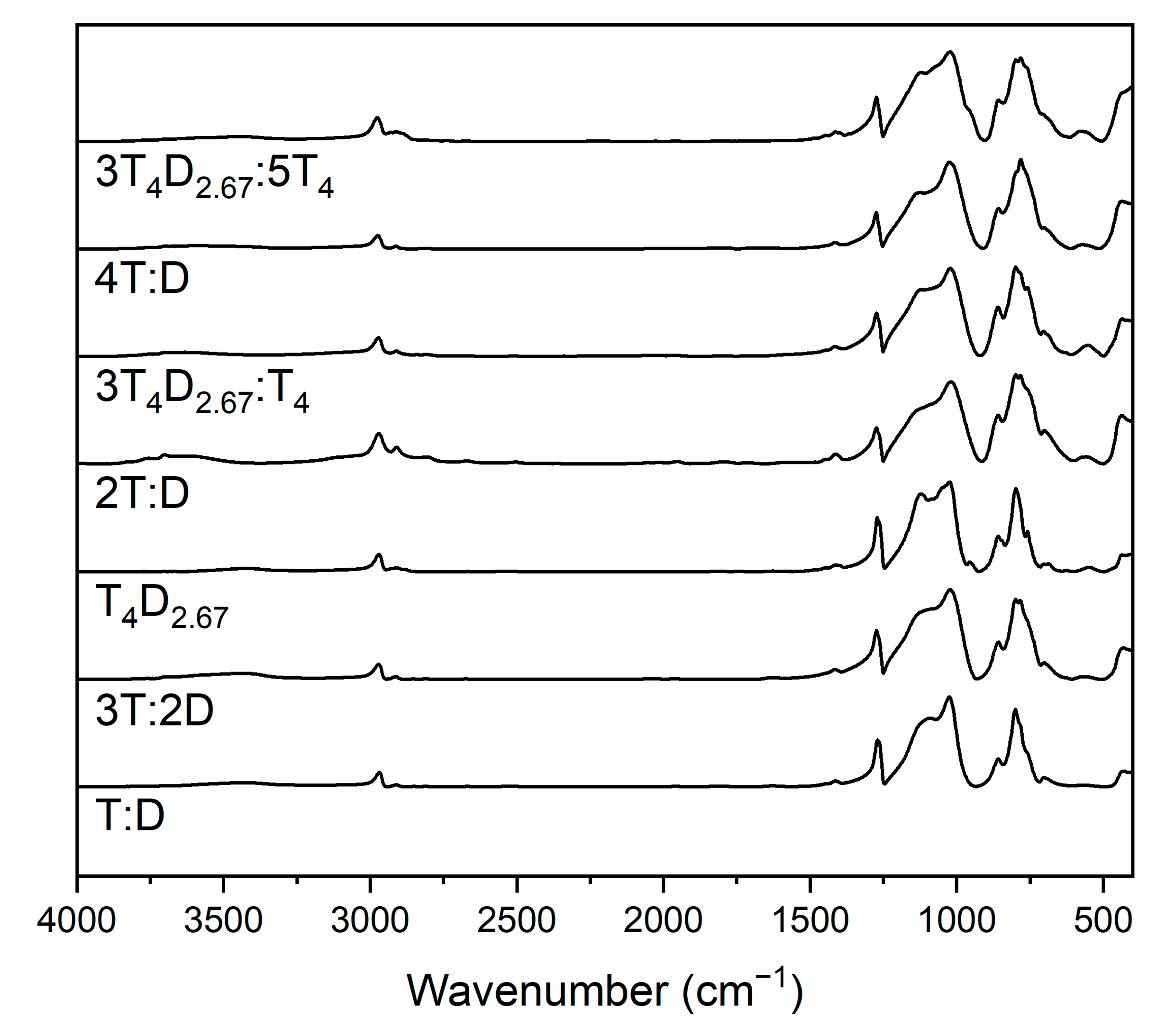
3.2. Structure of Silicon Oxycarbide Materials and Their Precursors
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saha, A.; Raj, R.; Williamson, D.L. A model for the nanodomains in polymer-derived SiCO. J. Am. Ceram. Soc. 2006, 89, 2188–2195. [Google Scholar] [CrossRef]
- Ming, K.; Gu, C.; Su, Q.; Wang, Y.; Zare, A.; Lucca, D.A.; Nastasi, M.; Wang, J. Strength and plasticity of amorphous silicon oxycarbide. J. Nucl. Mater. 2019, 516, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Szymanski, W.; Lipa, S.; Fortuniak, W.; Chojnowski, J.; Pospiech, P.; Mizerska, U.; Slomkowski, S.; Nyczyk-Malinowska, A.; Hasik, M. Silicon oxycarbide (SiOC) ceramic microspheres—Structure and mechanical properties by nanoindentation studies. Ceram. Int. 2019, 45, 11946–11954. [Google Scholar] [CrossRef]
- Nyczyk-Malinowska, A.; Niemiec, W.; Smoła, G.; Gaweł, R.; Szuwarzyński, M.; Grzesik, Z. Preparation and characterization of oxidation-resistant black glass (SiCO) coatings obtained by hydrosilylation of polysiloxanes. Surf. Coat. Tech. 2021, 407, 126760. [Google Scholar] [CrossRef]
- Guo, F.; Su, D.; Liu, Y.; Wang, J.; Yan, X.; Chen, J.; Chen, S. High acid resistant SiOC ceramic membranes for wastewater treatment. Ceram. Int. 2018, 44, 13444–13448. [Google Scholar] [CrossRef]
- Maheshwari, A.; Gopikrishnan, E.P.; Gangadhar, J.; Sujith, R. Highly conducting graphene dispersed silicon oxycarbide glasses. Mater. Chem. Phys. 2020, 239, 121963. [Google Scholar] [CrossRef]
- Leszczyński, J.; Nieroda, P.; Nieroda, J.; Zybała, R.; Król, M.; Łacz, A.; Kaszyca, K.; Mikuła, A.; Schmidt, M.; Sitarz, M.; et al. Si-O-C amorphous coatings for high temperature protection of In0.4Co4Sb12 skutterudite for thermoelectric applications. J. Appl. Phys. 2019, 125, 215113. [Google Scholar] [CrossRef]
- Martín-Palma, R.J.; Gago, R.; Torres-Costa, V.; Fernández-Hidalgo, P.; Kreissig, U.; Martínez Duart, J.M. Optical and compositional analysis of functional SiOxCy:H coatings on polymers. Thin Solid Films 2006, 515, 2493–2496. [Google Scholar] [CrossRef]
- Knozowski, D.; Graczyk-Zajac, M.; Trykowski, G.; Wilamowska-Zawłocka, M. Silicon oxycarbide-graphite electrodes for high-power energy storage devices. Materials 2020, 13, 4302. [Google Scholar] [CrossRef]
- Canuto de Almeida e Silva, T.; Bhowmick, G.D.; Ghangrekar, M.M.; Wilhelm, M.; Rezwan, K. SiOC-based polymer derived-ceramic porous anodes for microbial fuel cells. Biochem. Eng. J. 2019, 148, 29–36. [Google Scholar] [CrossRef]
- García, B.; Casado, E.; Tamayo, A. Synthesis and characterization of Ce/SiOC nanocomposites through the polymer derived ceramic method and evaluation of their catalytic activity. Ceram. Int. 2020, 46, 1362–1373. [Google Scholar] [CrossRef]
- Awin, E.W.; Lale, A.; Nair Hari Kumar, K.C.; Demirci, U.B.; Bernard, S.; Kumar, R. Novel Precursor-derived Meso-/Macroporous TiO2/SiOC nanocomposites with highly stable anatase nanophase providing visible light photocatalytic activity and superior adsorption of organic dyes. Materials 2018, 11, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arango-Ospina, M.; Xie, F.; Gonzalo-Juan, I.; Riedel, R.; Ionescu, E.; Boccaccini, A.R. Review: Silicon oxycarbide based materials for biomedical applications. Appl. Mater. Today 2020, 18, 100482. [Google Scholar] [CrossRef]
- Jang, J.; Warriam Sasikumar, P.V.; Navaee, F.; Hagelüken, L.; Blugan, G.; Brugger, J. Electrochemical performance of polymer-derived SiOC and SiTiOC ceramic electrodes for artificial cardiac pacemaker applications. Ceram. Int. 2021, 47, 7593–7601. [Google Scholar] [CrossRef]
- Nastasi, M.; Su, Q.; Price, L.; Colón Santana, J.A.; Chen, T.; Balerio, R.; Shao, L. Superior radiation tolerant materials: Amorphous silicon oxycarbide. J. Nucl. Mater. 2015, 461, 200–205. [Google Scholar] [CrossRef] [Green Version]
- Walkiewicz-Pietrzykowska, A.; Uznanski, P.; Wrobel, A.M. Silicon carbide, silicon carbonitride, and silicon oxycarbide thin films formed by remote hydrogen microwave plasma CVD. Curr. Org. Chem. 2017, 21, 2229–2239. [Google Scholar] [CrossRef]
- Yu, S.; Tu, R.; Goto, T. Preparation of SiOC nanocomposite films by laser chemical vapor deposition. J. Eur. Ceram. Soc. 2016, 36, 403–409. [Google Scholar] [CrossRef]
- Huang, K.; Elsayed, H.; Franchin, G.; Colombo, P. Complex SiOC ceramics from 2D structures by 3D printing and origami. Addit. Manuf. 2020, 33, 101144. [Google Scholar] [CrossRef]
- Venkatachalam, S.; Hourlier, D. Heat treatment of commercial Polydimethylsiloxane PDMS precursors: Part I. Towards conversion of patternable soft gels into hard ceramics. Ceram. Int. 2019, 45, 6255–6262. [Google Scholar] [CrossRef]
- Chauhan, P.K.; Sujith, R.; Parameshwaran, R.; Prasad, A.V.S.S. Role of polysiloxanes in the synthesis of aligned porous silicon oxycarbide ceramics. Ceram. Int. 2019, 45, 8150–8156. [Google Scholar] [CrossRef]
- Nyczyk-Malinowska, A.; Wójcik-Bania, M.; Gumuła, T.; Hasik, M.; Cypryk, M.; Olejniczak, Z. New precursors to SiCO ceramics derived from linear poly(vinylsiloxanes) of regular chain composition. J. Eur. Ceram. Soc. 2014, 34, 889–902. [Google Scholar] [CrossRef]
- Iwase, Y.; Fuchigami, T.; Horie, Y.; Daiko, Y.; Honda, S.; Iwamoto, Y. Formation and thermal behaviors of ternary silicon oxycarbides derived from silsesquioxane derivatives. Materials 2019, 12, 1721. [Google Scholar] [CrossRef] [Green Version]
- Xie, F.; Ionescu, E.; Arango-Ospina, M.; Riedel, R.; Boccaccini, A.R.; Gonzalo-Juan, I. Facile preparative access to bioactive silicon oxycarbides with tunable porosity. Materials 2019, 12, 3862. [Google Scholar] [CrossRef] [Green Version]
- Pantano, C.G.; Singh, A.K.; Zhang, H. Silicon oxycarbide glasses. J. Sol-Gel Sci. Technol. 1999, 14, 7–25. [Google Scholar] [CrossRef]
- Jeleń, P.; Szumera, M.; Gawęda, M.; Długoń, E.; Sitarz, M. Thermal evolution of ladder-like silsesquioxanes during formation of black glasses. J. Therm. Anal. Calorim. 2017, 130, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Lu, K.; Ma, R. Effects of different polymer precursors on the characteristics of SiOC bulk ceramics. Appl. Phys. A Mater. 2019, 125, 395. [Google Scholar] [CrossRef]
- Havelcová, M.; Strachota, A.; Černý, M.; Sucharda, Z.; Šlouf, M. Effect of the dimethylsilyloxy co-monomer “d” on the chemistry of polysiloxane pyrolysis to SiOC. J. Anal. Appl. Pyrol. 2016, 117, 30–45. [Google Scholar] [CrossRef]
- Niemiec, W.; Szczygieł, P.; Jeleń, P.; Handke, M. IR investigation on silicon oxycarbide structure obtained from precursors with 1:1 silicon to carbon atoms ratio and various carbon atoms distribution. J. Mol. Struct. 2018, 1164, 217–226. [Google Scholar] [CrossRef]
- Lee, H.S.; Choi, S.-S.; Baek, K.-Y.; Hong, S.M.; Lee, E.C.; Lee, J.-C.; Hwang, S.S. Synthesis and structure characterization of ladder-like polymethylsilsesquioxane (PMSQ) by isolation of stereoisomer. Eur. Polym. J. 2012, 48, 1073–1081. [Google Scholar] [CrossRef]
- Pakhomov, A.A.; Kononevich, Y.N.; Stukalova, M.V.; Svidchenko, E.A.; Surin, N.M.; Cherkaev, G.V.; Shchegolikhina, O.I.; Martynov, V.I.; Muzafarov, A.M. Synthesis and photophysical properties of a new BODIPY-based siloxane dye. Tetrahedron Lett. 2016, 57, 979–982. [Google Scholar] [CrossRef]
- Jeleń, P.; Bik, M.; Nocuń, M.; Gawęda, M.; Długoń, E.; Sitarz, M. Free carbon phase in SiOC glasses derived from ladder-like silsesquioxanes. J. Mol. Struct. 2016, 1126, 172–176. [Google Scholar] [CrossRef]
- Ermakova, E.N.; Sysoev, S.V.; Nikolaev, R.E.; Długoń, E.; Handke, B. Thermal properties of some organosilicon precursors for chemical vapor deposition. J. Therm. Anal. Calorim. 2016, 126, 609–616. [Google Scholar] [CrossRef]
- Sarada, K.; Muraleedharan, K. Thermal degradation and optical properties of SiC-infused polystyrene nanocomposites. J. Therm. Anal. Calorim. 2016, 126, 1809–1819. [Google Scholar] [CrossRef]
- Yun, S.; Luo, H.; Gao, Y. Superhydrophobic silica aerogel microspheres from methyltrimethoxysilane: Rapid synthesis via ambient pressure drying and excellent absorption properties. RSC Adv. 2014, 4, 4535–4542. [Google Scholar] [CrossRef]
- Enríquez, E.; Fernández, J.F.; De La Rubia, M.A. Highly conductive coatings of carbon black/silica composites obtained by a sol–gel process. Carbon 2012, 50, 4409–4417. [Google Scholar] [CrossRef] [Green Version]
- Sorarù, G.D.; Karakuscu, A.; Boissiere, C.; Babonneau, F. On the shrinkage during pyrolysis of thin films and bulk components: The case of a hybrid silica gel precursor for SiOC glasses. J. Eur. Ceram. Soc. 2012, 32, 627–632. [Google Scholar] [CrossRef]
- Oh, T.; Kim, C.H.; Jung, C.S. Relationship between the binding energy and boundary condition in SiOC film for ILD application. GDC 2011, 261, 213–218. [Google Scholar] [CrossRef]
- Handke, M.; Handke, B.; Kowalewska, A.; Jastrzebski, W. New polysilsesquioxane materials of ladder-like structure. J. Mol. Struct. 2009, 924–926, 254–263. [Google Scholar] [CrossRef]
- Handke, M.; Jastrzebski, W.; Kowalewska, A.; Mozgawa, W. Spectroscopic study of preceramic polymers (xerogels) obtained by hydrolytic condensation of ethoxycyclosiloxanes. J. Mol. Struct. 2009, 924–926, 248–253. [Google Scholar] [CrossRef]
- Navamathavan, R.; Kim, C.-Y.; Jung, A.-S.; Choi, C.-K.; Lee, H.-J. Deposition and characterization of porous low-dielectric-constant SiOC(-H) thin films deposited from TES/O2 precursors by using plasma-enhanced chemical vapor deposition. J. Korean Phys. Soc. 2008, 53, 351–356. [Google Scholar] [CrossRef]
- Ward, L.J.; Schofield, W.C.E.; Badyal, J.P.S.; Goodwin, A.J.; Merlin, P.J. Atmospheric pressure glow discharge deposition of polysiloxane and SiOx films. Langmuir 2003, 19, 2110–2114. [Google Scholar] [CrossRef]
- Kwang-Man, L.; Chang-Young, K.; Chi-Kyu, C.; Navamathavan, R. Characteristics of SiOC(-H) thin films prepared by using plasma-enhanced atomic layer deposition. J. Korean Phys. Soc. 2011, 59, 3074–3079. [Google Scholar]
- Segatelli, M.G.; Pires, A.T.N.; Yoshida, I.V.P. Synthesis and structural characterization of carbon-rich SiCxOy derived from a Ni-containing hybrid polymer. J. Eur. Ceram. Soc. 2008, 28, 2247–2257. [Google Scholar] [CrossRef]
- Dřínek, V.; Bastl, Z.; Šubrt, J.; Yabe, A.; Pola, J. IR laser-induced reactive ablation of silicon monoxide in hydrogen and water atmosphere. J. Mater. Chem. 2002, 12, 1800–1805. [Google Scholar] [CrossRef]
- Sitarz, M.; Jastrzȩbski, W.; Jeleń, P.; Długoń, E.; Gawȩda, M. Preparation and structural studies of black glasses based on ladder-like silsesquioxanes. Spectrochim. Acta A 2014, 132, 884–888. [Google Scholar] [CrossRef]
- Lubas, M.; Sitarz, M.; Fojud, Z.; Jurga, S. Structure of multicomponent SiO2–Al2O3–Fe2O3–MgO glasses for preparation of fibrous insulating materials. J. Mol. Struct. 2005, 744–747, 615–619. [Google Scholar] [CrossRef]
- Bréquel, H.; Parmentier, J.; Walter, S.; Badheka, R.; Trimmel, G.; Masse, S.; Latournerie, J.; Dempsey, P.; Turquat, C.; Desmartin-Chomel, A.; et al. Systematic structural characterization of the high-temperature behavior of nearly stoichiometric silicon oxycarbide glasses. Chem. Mater. 2004, 16, 2585–2598. [Google Scholar] [CrossRef]
- Takahashi, T.; Münstedt, H.; Colombo, P.; Modesti, M. Thermal evolution of a silicone resin/polyurethane blend from preceramic to ceramic foam. J. Mater. Sci. 2001, 36, 1627–1639. [Google Scholar] [CrossRef]
- Anisimov, A.A.; Kononevich, Y.N.; Buzin, M.I.; Peregudov, A.S.; Shchegolikhina, O.I.; Muzafarov, A.M. Convenient synthesis of new Si-H and Si-vinyl functionalized stereospecific 8-, 12- and 24-membered cyclosiloxanes. Macroheterocycles 2016, 9, 442–452. [Google Scholar] [CrossRef] [Green Version]
- Handke, M.; Sitarz, M.; Długoń, E. Amorphous SiCxOy coatings from ladder-like polysilsesquioxanes. J. Mol. Struct. 2011, 993, 193–197. [Google Scholar] [CrossRef]
- Smith, A.L. Infrared spectra-structure correlations for organosilicon compounds. Spectrochim. Acta 1960, 16, 87–105. [Google Scholar] [CrossRef]
- Lewis, R.N.; McElhaney, R.N.; Pohle, W.; Mantsch, H.H. Components of the Carbonyl Stretching Band in the Infrared Spectra of Hydrated 1,2-Diacylglycerolipid Bilayers: A Reevaluation. Biophys. J. 1994, 67, 2367–2375. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Annealing Step | |||
|---|---|---|---|
| Sample | I | II | III |
| T:D | 25.06% | 15.74% | 4.38% |
| T4D2.67 | 9.90% | 13.89% | 4.49% |
| 3T:2D | 4.06% | 6.90% | 4.09% |
| 3T4D2.67:T4 | 14.05% | 8.82% | 5.49% |
| 2T:D | 6.41% | 6.75% | 6.21% |
| 3T4D2.67:5T4 | 6.07% | 10.85% | 4.89% |
| 4T:D | 1.92% | 7.03% | 3.69% |
| Sample | Carbon Atoms per Silicon Atom in the Material | ||
|---|---|---|---|
| Theoretical | Experimental | ||
| 700 °C | 800 °C | ||
| T:D | 1.50 | - | 0.64 ± 0.04 |
| T4D2.67 | 1.40 | 0.82 ± 0.05 | 0.74 ± 0.21 |
| 3T:2D | 1.40 | 0.85 ± 0.05 | 0.68 ± 0.02 |
| 3T4D2.67:T4 | 1.33 | 0.70 ± 0.22 | 0.64 ± 0.03 |
| 2T:D | 1.33 | 0.86 ± 0.18 | 0.69 ± 0.06 |
| 3T4D2.67:5T4 | 1.20 | - | 0.69 ± 0.03 |
| 4T:D | 1.20 | - | 0.94 ± 0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Owińska, M.; Matoga, P.; Jeleń, P.; Długoń, E.; Handke, B.; Niemiec, W. Macromonomers as a Novel Way to Investigate and Tailor Silicon-Oxycarbide-Based Materials Obtained from Polymeric Preceramic Precursors. Materials 2021, 14, 3703. https://doi.org/10.3390/ma14133703
Owińska M, Matoga P, Jeleń P, Długoń E, Handke B, Niemiec W. Macromonomers as a Novel Way to Investigate and Tailor Silicon-Oxycarbide-Based Materials Obtained from Polymeric Preceramic Precursors. Materials. 2021; 14(13):3703. https://doi.org/10.3390/ma14133703
Chicago/Turabian StyleOwińska, Maria, Paulina Matoga, Piotr Jeleń, Elżbieta Długoń, Bartosz Handke, and Wiktor Niemiec. 2021. "Macromonomers as a Novel Way to Investigate and Tailor Silicon-Oxycarbide-Based Materials Obtained from Polymeric Preceramic Precursors" Materials 14, no. 13: 3703. https://doi.org/10.3390/ma14133703
APA StyleOwińska, M., Matoga, P., Jeleń, P., Długoń, E., Handke, B., & Niemiec, W. (2021). Macromonomers as a Novel Way to Investigate and Tailor Silicon-Oxycarbide-Based Materials Obtained from Polymeric Preceramic Precursors. Materials, 14(13), 3703. https://doi.org/10.3390/ma14133703