
A Perspective on Modelling Metallic Magnetic Nanoparticles in Biomedicine: From Monometals to Nanoalloys and Ligand-Protected Particles


Abstract
1. History of Use and Study of Metal Nanoparticles in Biomedicine
1.1. Biomedical Applications of mNPs
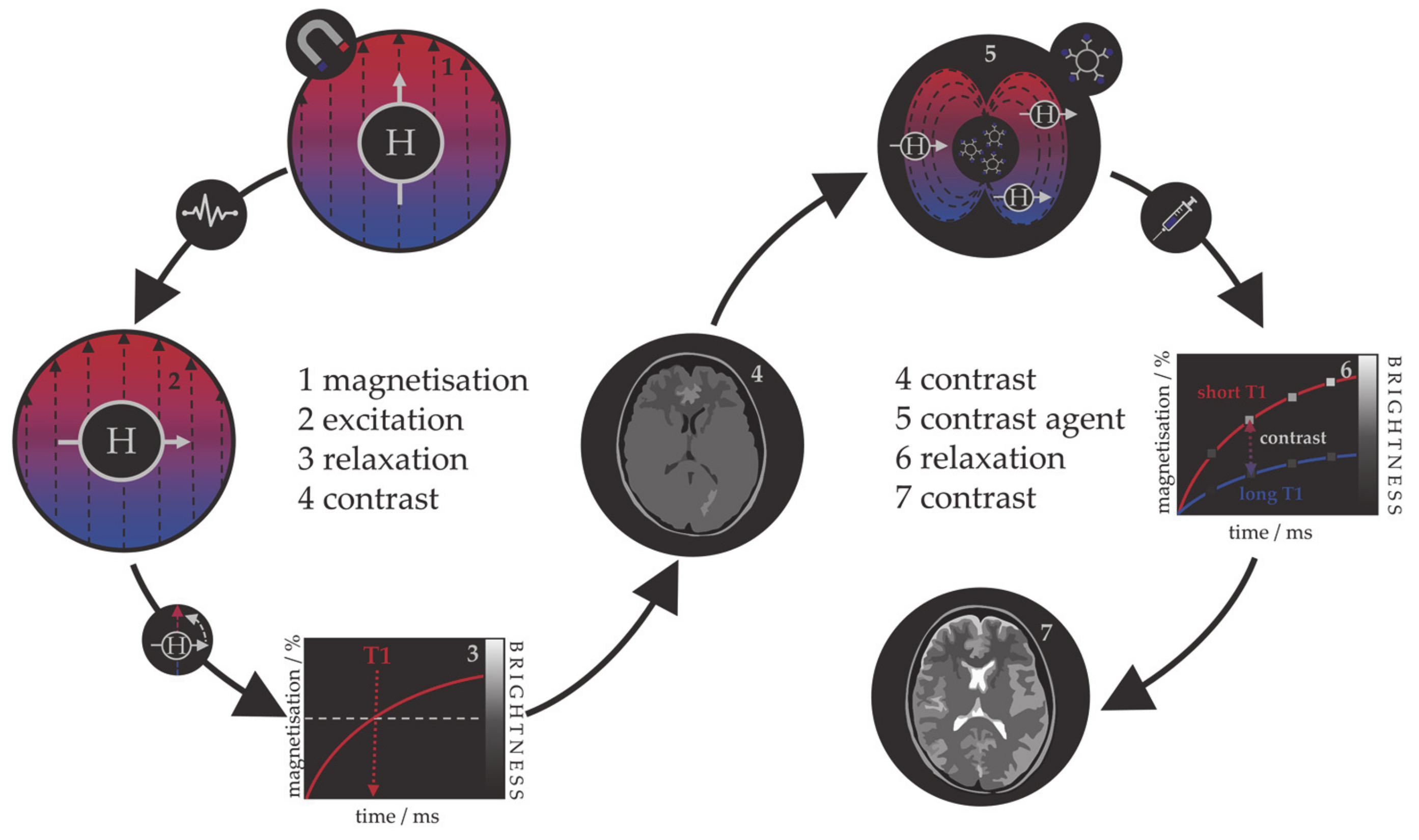
1.1.1. MRI

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Magnetic Core | Reported Coating Materials | Application | Ref |
|---|---|---|---|
| Metal | |||
| Fe | polymers | MRI, drug delivery | [45,46,47] |
| iron oxide | MRI, hyperthermia | [48] | |
| Au | drug delivery, photothermal therapy | [49] | |
| Co | organic acids | drug delivery, hyperthermia | [50] |
| polymers | MRI | [51] | |
| Au | MRI, gene transport, hyperthermia | [52,53,54,55] | |
| FeCo | graphite | MRI, optical imaging | [56,57] |
| CoFe2O4 | hyperthermia | [58] | |
| Au | MRI, medical labelling | [59] | |
| FePt | Au | MRI, photothermal therapy | [60,61] |
| organic acids/thiols | biosensors, MRI, CT | [62,63,64,65,66] | |
| SiW11O39 | hyperthermia | [67] | |
| polymer + SiO2 | drug delivery | [68] | |
| FeNi | polymers | hyperthermia | [69] |
| FeNiCo | propylene glycol | hyperthermia | [70] |
| Oxide | |||
| Fe3O4 | SiO2, TiO2 | MRI, photokilling agents | [71,72,73] |
| dextran, DMSA | MRI | [74] | |
| Au, Ag | MRI, immunosensor, photothermal therapy | [75,76,77,78] | |
| Fe2O3 | SiO2 | MRI, biolabelling | [79,80] |
| polymers | MRI, biolabelling, drug delivery, optical imaging | [81,82] | |
| MnFe2O4 | polymers and organic acids | MRI | [83,84] |
| CoFe2O4 | polymers and organic acids | MRI, drug delivery, hyperthermia | [84,85,86] |
| Au + PNA oligomers | biosensors, genomics | [87] | |
| NiFe2O4 | polymers and organic acids | drug delivery, hyperthermia | [84,88,89,90] |
| polysaccharides | MRI | [91] | |
| MnO | Au | MRI | [92] |
| polymers and organic acids | MRI | [81] | |
| SiO2 | biolabelling | [93] |
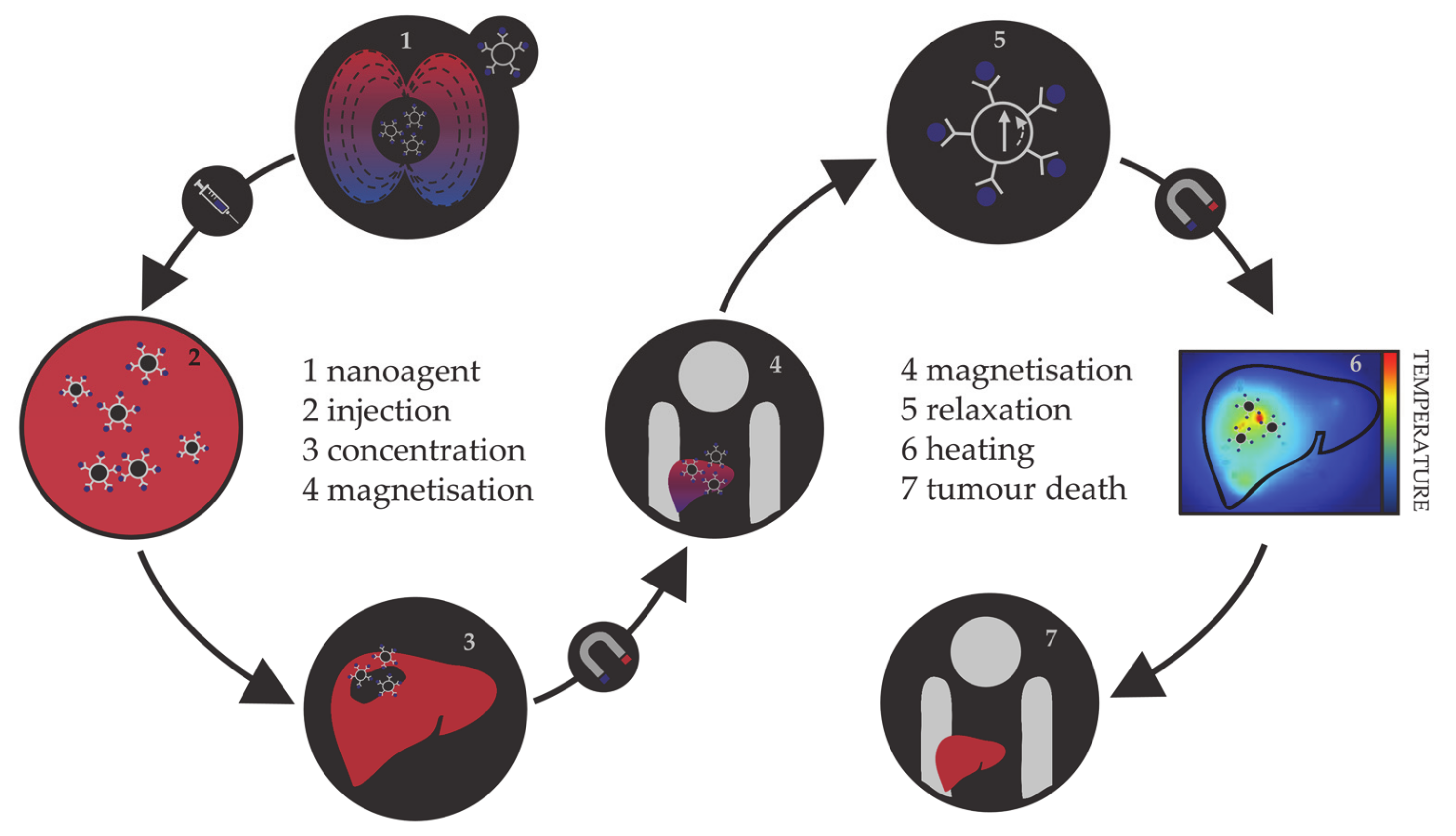
1.1.2. Magnetic Hyperthermia
1.1.3. Targeted Drug Delivery
2. Features of mNPs
2.1. Electronic and Magnetic Properties of mNPs
2.2. Biomedically Desired Properties of mNPs
2.2.1. Relevant Features in MRI
2.2.2. Relevant Features in Hyperthermia
2.2.3. Relevant Features in Drug Delivery
3. Computational Modelling of mNPs
3.1. Monometallic mNPs
3.1.1. Cluster Model
3.1.2. Surface Model
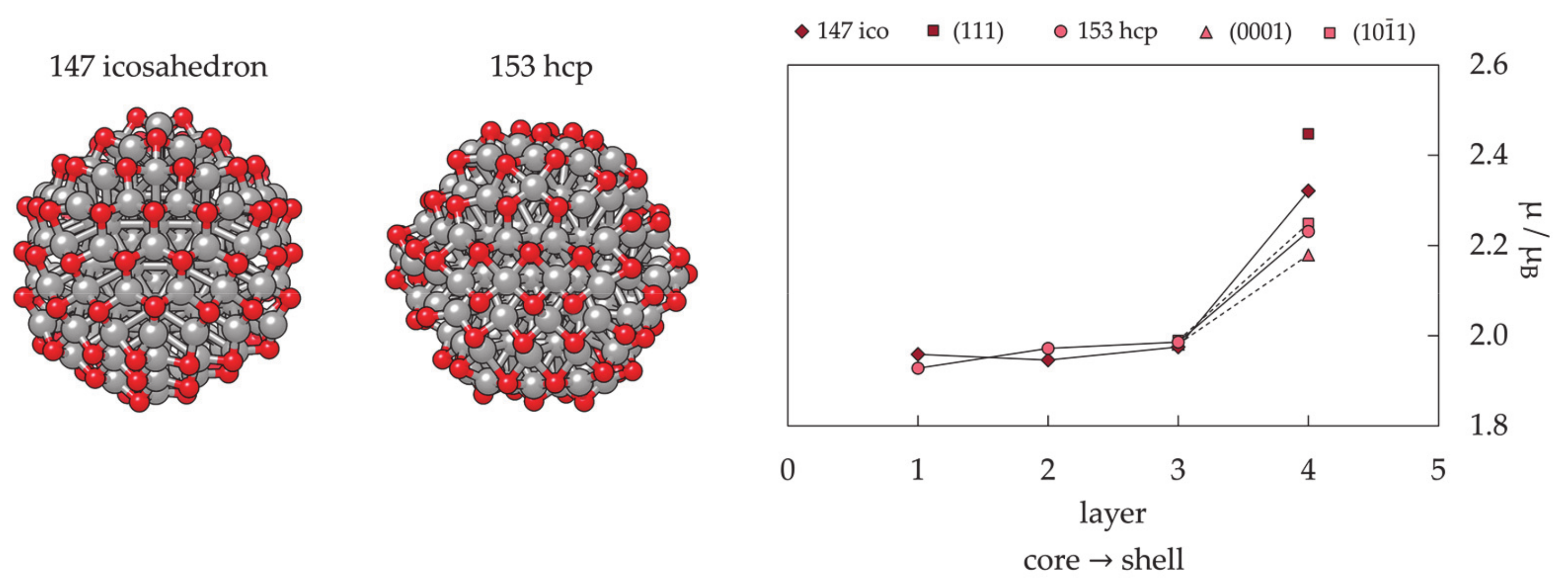
3.1.3. Nanoparticle Model

3.2. Protected mNPs: Ligand Effects on Magnetic Properties
3.3. Alloyed mNPs: Effects of Interfaces on Magnetic Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tabor, C.; Narayanan, R.; El-Sayed, M.A. Catalysis with transition metal nanoparticles in colloidal solution: Heterogeneous or homogeneous? Model Syst. Catal. Single Cryst. Support. Enzym. Mimics 2010, 395–414. [Google Scholar] [CrossRef]
- Garcia, M.A. Surface plasmons in metallic nanoparticles: Fundamentals and applications. J. Phys. D Appl. Phys. 2012, 45, 389501. [Google Scholar] [CrossRef]
- An, K.; Somorjai, G.A. Size and Shape Control of Metal Nanoparticles for Reaction Selectivity in Catalysis. ChemCatChem 2012, 4, 1512–1524. [Google Scholar] [CrossRef]
- Zhang, L.; Anderson, R.M.; Crooks, R.M.; Henkelman, G. Correlating structure and function of metal nanoparticles for catalysis. Surf. Sci. 2015, 640, 65–72. [Google Scholar] [CrossRef]
- Ko, S.H.; Park, I.; Pan, H.; Grigoropoulos, C.P.; Pisano, A.P.; Luscombe, C.K.; Fréchet, J.M.J. Direct nanoimprinting of metal nanoparticles for nanoscale electronics fabrication. Nano Lett. 2007, 7, 1869–1877. [Google Scholar] [CrossRef]
- Son, Y.; Yeo, J.; Moon, H.; Lim, T.W.; Hong, S.; Nam, K.H.; Yoo, S.; Grigoropoulos, C.P.; Yang, D.Y.; Ko, S.H. Nanoscale electronics: Digital fabrication by direct femtosecond laser processing of metal nanoparticles. Adv. Mater. 2011, 23, 3176–3181. [Google Scholar] [CrossRef]
- Liu, S.; Yuen, M.C.; White, E.L.; Boley, J.W.; Deng, B.; Cheng, G.J.; Kramer-Bottiglio, R. Laser Sintering of Liquid Metal Nanoparticles for Scalable Manufacturing of Soft and Flexible Electronics. ACS Appl. Mater. Interfaces 2018, 10, 28232–28241. [Google Scholar] [CrossRef]
- Sosa, I.O.; Noguez, C.; Barrera, R.G. Optical properties of metal nanoparticles with arbitrary shapes. J. Phys. Chem. B 2003, 107, 6269–6275. [Google Scholar] [CrossRef]
- Murphy, C.J.; Sau, T.K.; Gole, A.M.; Orendorff, C.J.; Gao, J.; Gou, L.; Hunyadi, S.E.; Li, T. Anisotropic metal nanoparticles: Synthesis, assembly, and optical applications. J. Phys. Chem. B 2005, 109, 13857–13870. [Google Scholar] [CrossRef]
- Zijlstra, P.; Orrit, M. Single metal nanoparticles: Optical detection, spectroscopy and applications. Rep. Prog. Phys. 2011, 74, 106401. [Google Scholar] [CrossRef]
- Kelly, K.L.; Coronado, E.; Zhao, L.L.; Schatz, G.C. The Optical Properties of Metal Nanoparticles: The Influence of Size, Shape, and Dielectric Environment. J. Phys. Chem. B 2003, 107, 668–677. [Google Scholar] [CrossRef]
- Wan, D.; Chen, H.L.; Tseng, S.C.; Wang, L.A.; Chen, Y.P. One-shot deep-UV pulsed-laser-induced photomodification of hollow metal nanoparticles for high-density data storage on flexible substrates. ACS Nano 2010, 4, 165–173. [Google Scholar] [CrossRef]
- Sun, X.; Huang, Y.; Nikles, D.E. FePt and CoPt magnetic nanoparticles film for future high density data storage media. Int. J. Nanotechnol. 2004, 1, 328–346. [Google Scholar] [CrossRef]
- Kang, M.; Baeg, K.J.; Khim, D.; Noh, Y.Y.; Kim, D.Y. Printed, flexible, organic nano-floating-gate memory: Effects of metal nanoparticles and blocking dielectrics on memory characteristics. Adv. Funct. Mater. 2013, 23, 3503–3512. [Google Scholar] [CrossRef]
- Liao, H.; Nehl, C.L.; Hafner, J.H. Biomedical applications of plasmon resonant metal nanoparticles. Nanomedicine 2006, 1, 201–208. [Google Scholar] [CrossRef]
- Zeisberger, M.; Dutz, S.; Müller, R.; Hergt, R.; Matoussevitch, N.; Bönnemann, H. Metallic cobalt nanoparticles for heating applications. J. Magn. Magn. Mater. 2007, 311, 224–227. [Google Scholar] [CrossRef]
- Cherukuri, P.; Glazer, E.S.; Curley, S.A. Targeted hyperthermia using metal nanoparticles. Adv. Drug Deliv. Rev. 2010, 62, 339–345. [Google Scholar] [CrossRef]
- Sharma, H.; Mishra, P.K.; Talegaonkar, S.; Vaidya, B. Metal nanoparticles: A theranostic nanotool against cancer. Drug Discov. Today 2015, 20, 1143–1151. [Google Scholar] [CrossRef]
- Rai, M.; Ingle, A.P.; Birla, S.; Yadav, A.; Santos, C.A. Dos Strategic role of selected noble metal nanoparticles in medicine. Crit. Rev. Microbiol. 2016, 42, 696–719. [Google Scholar] [CrossRef]
- Xia, Y.; Xiong, Y.; Lim, B.; Skrabalak, S.E. Shape-controlled synthesis of metal nanocrystals: Simple chemistry meets complex physics? Angew. Chem. Int. Ed. 2009, 48, 60–103. [Google Scholar] [CrossRef]
- Vajda, S.; Pellin, M.J.; Greeley, J.P.; Marshall, C.L.; Curtiss, L.A.; Ballentine, G.A.; Elam, J.W.; Catillon-Mucherie, S.; Redfern, P.C.; Mehmood, F.; et al. Subnanometre platinum clusters as highly active and selective catalysts for the oxidative dehydrogenation of propane. Nat. Mater. 2009, 8, 213–216. [Google Scholar] [CrossRef]
- Grassian, V.H. When size really matters: Size-dependent properties and surface chemistry of metal and metal oxide nanoparticles in gas and liquid phase environments. J. Phys. Chem. C 2008, 112, 18303–18313. [Google Scholar] [CrossRef]
- Carlson, C.; Hussein, S.M.; Schrand, A.M.; Braydich-Stolle, L.K.; Hess, K.L.; Jones, R.L.; Schlager, J.J. Unique cellular interaction of silver nanoparticles: Size-dependent generation of reactive oxygen species. J. Phys. Chem. B 2008, 112, 13608–13619. [Google Scholar] [CrossRef]
- Balamurugan, B.; Maruyama, T. Evidence of an enhanced interband absorption in Au nanoparticles: Size-dependent electronic structure and optical properties. Appl. Phys. Lett. 2005, 87, 143105. [Google Scholar] [CrossRef]
- Xiong, S.; Qi, W.; Cheng, Y.; Huang, B.; Wang, M.; Li, Y. Universal relation for size dependent thermodynamic properties of metallic nanoparticles. Phys. Chem. Chem. Phys. 2011, 13, 10652–10660. [Google Scholar] [CrossRef]
- Haldar, K.K.; Kundu, S.; Patra, A. Core-size-dependent catalytic properties of bimetallic Au/Ag core-shell nanoparticles. ACS Appl. Mater. Interfaces 2014, 6, 21946–21953. [Google Scholar] [CrossRef]
- Neubauer, N.; Palomaeki, J.; Karisola, P.; Alenius, H.; Kasper, G. Size-dependent ROS production by palladium and nickel nanoparticles in cellular and acellular environments - An indication for the catalytic nature of their interactions. Nanotoxicology 2015, 9, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Lian, C.; Hu, S.; Deng, Z.; Gong, J.; Li, M.; Liu, H.; Xing, M.; Zhang, J. Size-dependent activity and selectivity of carbon dioxide photocatalytic reduction over platinum nanoparticles. Nat. Commun. 2018, 9, 1252. [Google Scholar] [CrossRef] [PubMed]
- Mourdikoudis, S.; Pallares, R.M.; Thanh, N.T.K. Characterization techniques for nanoparticles: Comparison and complementarity upon studying nanoparticle properties. Nanoscale 2018, 10, 12871–12934. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Taylor, M.G.; Mascareno, A.; Mpourmpakis, G. Size-, Shape-, and Composition-Dependent Model for Metal Nanoparticle Stability Prediction. Nano Lett. 2018, 18, 2696–2704. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.G.; Austin, N.; Gounaris, C.E.; Mpourmpakis, G. Catalyst Design Based on Morphology- and Environment-Dependent Adsorption on Metal Nanoparticles. ACS Catal. 2015, 5, 6296–6301. [Google Scholar] [CrossRef]
- Kozlov, S.M.; Kovács, G.; Ferrando, R.; Neyman, K.M. How to determine accurate chemical ordering in several nanometer large bimetallic crystallites from electronic structure calculations. Chem. Sci. 2015, 6, 3868–3880. [Google Scholar] [CrossRef]
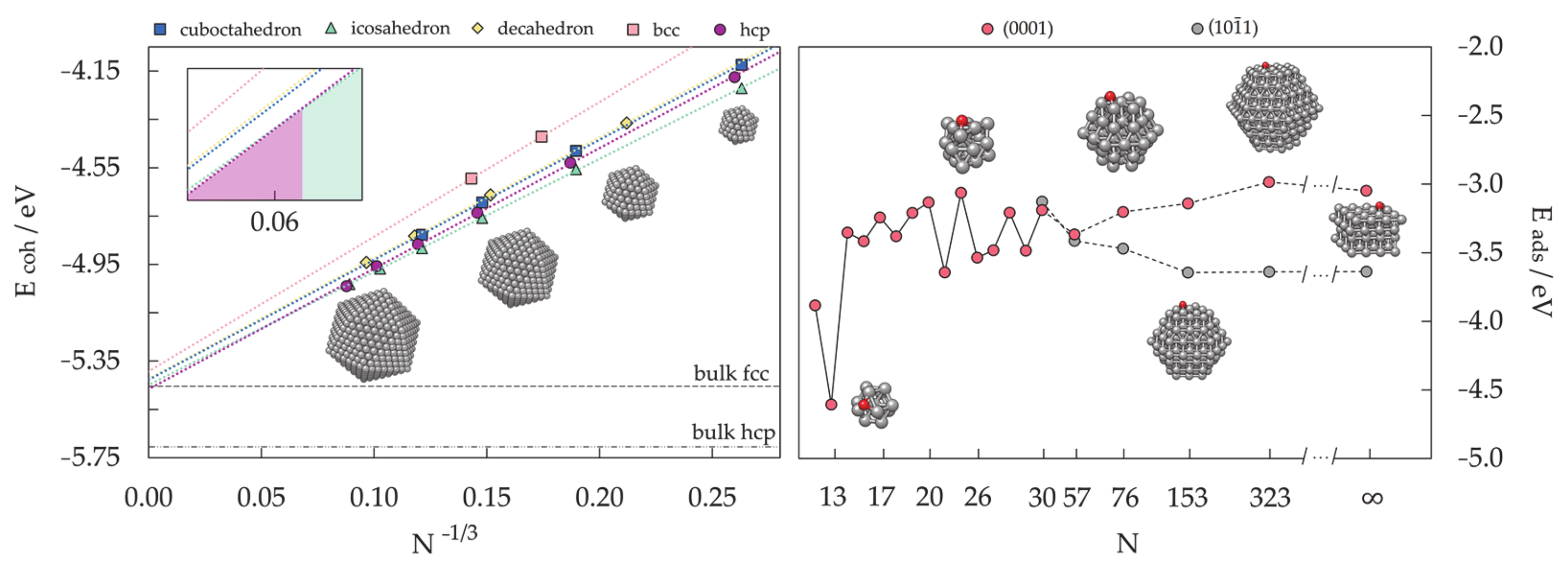
- Ferrando, R.; Fortunelli, A.; Rossi, G. Quantum effects on the structure of pure and binary metallic nanoclusters. Phys. Rev. B Condens. Matter Mater. Phys. 2005, 72, 085449. [Google Scholar] [CrossRef]
- Zhu, B.; Xu, Z.; Wang, C.; Gao, Y. Shape Evolution of Metal Nanoparticles in Water Vapor Environment. Nano Lett. 2016, 16, 2628–2632. [Google Scholar] [CrossRef]
- Sangaiya, P.; Jayaprakash, R. A Review on Iron Oxide Nanoparticles and Their Biomedical Applications. J. Supercond. Nov. Magn. 2018, 31, 3397–3413. [Google Scholar] [CrossRef]
- Lima-Tenório, M.K.; Gómez Pineda, E.A.; Ahmad, N.M.; Fessi, H.; Elaissari, A. Magnetic nanoparticles: In vivo cancer diagnosis and therapy. Int. J. Pharm. 2015, 493, 313–327. [Google Scholar] [CrossRef]
- Hervault, A.; Thanh, N.T.K. Magnetic nanoparticle-based therapeutic agents for thermo-chemotherapy treatment of cancer. Nanoscale 2014, 6, 11553–11573. [Google Scholar] [CrossRef]
- Laurent, S.; Dutz, S.; Häfeli, U.O.; Mahmoudi, M. Magnetic fluid hyperthermia: Focus on superparamagnetic iron oxide nanoparticles. Adv. Colloid Interface Sci. 2011, 166, 8–23. [Google Scholar] [CrossRef]
- Singh, A.; Sahoo, S.K. Magnetic nanoparticles: A novel platform for cancer theranostics. Drug Discov. Today 2014, 19, 474–481. [Google Scholar] [CrossRef]
- Périgo, E.A.; Hemery, G.; Sandre, O.; Ortega, D.; Garaio, E.; Plazaola, F.; Teran, F.J. Fundamentals and advances in magnetic hyperthermia. Appl. Phys. Rev. 2015, 2, 041302. [Google Scholar] [CrossRef]
- Hedayatnasab, Z.; Abnisa, F.; Daud, W.M.A.W. Review on magnetic nanoparticles for magnetic nanofluid hyperthermia application. Mater. Des. 2017, 123, 174–196. [Google Scholar] [CrossRef]
- Felder, R.C.; Parker, J.A. Principles of nuclear magnetic resonance imaging. Med. Instrum. 1985, 19, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Geva, T. Magnetic resonance imaging: Historical perspective. J. Cardiovasc. Magn. Reson. 2006, 8, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Grover, V.P.B.; Tognarelli, J.M.; Crossey, M.M.E.; Cox, I.J.; Taylor-Robinson, S.D.; McPhail, M.J.W. Magnetic Resonance Imaging: Principles and Techniques: Lessons for Clinicians. J. Clin. Exp. Hepatol. 2015, 5, 246–255. [Google Scholar] [CrossRef]
- Nattama, S.; Rahimi, M.; Wadajkar, A.S.; Koppolu, B.; Hua, J.; Nwariaku, F.; Nguyen, K.T. Characterization of polymer coated magnetic nanoparticles for targeted treatment of cancer. In Proceedings of the 2007 IEEE Dallas Engineering in Medicine and Biology Workshop, Dallas, TX, USA, 11–12 November 2007; 2007; pp. 35–38. [Google Scholar] [CrossRef]
- Rahimi, M.; Wadajkar, A.; Subramanian, K.; Yousef, M.; Cui, W.; Hsieh, J.T.; Nguyen, K.T. In vitro evaluation of novel polymer-coated magnetic nanoparticles for controlled drug delivery. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 672–680. [Google Scholar] [CrossRef]
- Mu, Q.; Yang, L.; Davis, J.C.; Vankayala, R.; Hwang, K.C.; Zhao, J.; Yan, B. Biocompatibility of polymer grafted core/shell iron/carbon nanoparticles. Biomaterials 2010, 31, 5083–5090. [Google Scholar] [CrossRef]
- Zhang, G.; Liao, Y.; Baker, I. Surface engineering of core/shell iron/iron oxide nanoparticles from microemulsions for hyperthermia. Mater. Sci. Eng. C 2010, 30, 92–97. [Google Scholar] [CrossRef]
- Jafari, T.; Simchi, A.; Khakpash, N. Synthesis and cytotoxicity assessment of superparamagnetic iron-gold core-shell nanoparticles coated with polyglycerol. J. Colloid Interface Sci. 2010, 345, 64–71. [Google Scholar] [CrossRef]
- Ansari, S.M.; Bhor, R.D.; Pai, K.R.; Sen, D.; Mazumder, S.; Ghosh, K.; Kolekar, Y.D.; Ramana, C.V. Cobalt nanoparticles for biomedical applications: Facile synthesis, physiochemical characterization, cytotoxicity behavior and biocompatibility. Appl. Surf. Sci. 2017, 414, 171–187. [Google Scholar] [CrossRef]
- Parkes, L.M.; Hodgson, R.; Lu, L.T.; Tung, L.D.; Robinson, I.; Fernig, D.G.; Thanh, N.T.K. Cobalt nanoparticles as a novel magnetic resonance contrast agent-relaxivities at 1.5 and 3 Tesla. Contrast Media Mol. Imaging 2008, 3, 150–156. [Google Scholar] [CrossRef]
- Lu, Y.; Zhao, Y.; Yu, L.; Dong, L.; Shi, C.; Hu, M.J.; Xu, Y.J.; Wen, L.P.; Yu, S.H. Hydrophilic Co@Au yolk/shell nanospheres: Synthesis, assembly, and application to gene delivery. Adv. Mater. 2010, 22, 1407–1411. [Google Scholar] [CrossRef]
- Hrubovčák, P.; Zeleňáková, A.; Zeleňák, V.; Kovác, J. Superparamagnetism in cobalt nanoparticles coated by protective gold layer. Acta Phys. Pol. A 2014, 126, 216–217. [Google Scholar] [CrossRef]
- Marbella, L.E.; Andolina, C.M.; Smith, A.M.; Hartmann, M.J.; Dewar, A.C.; Johnston, K.A.; Daly, O.H.; Millstone, J.E. Gold-cobalt nanoparticle alloys exhibiting tunable compositions, near-infrared emission, and high T2relaxivity. Adv. Funct. Mater. 2014, 24, 6532–6539. [Google Scholar] [CrossRef]
- Bouchard, L.; Anwar, M.S.; Liu, G.L.; Hann, B.; Xie, Z.H.; Gray, J.W.; Wang, X.; Pines, A.; Chen, F.F. Picomolar sensitivity MRI and photoacoustic imaging of cobalt nanoparticles. Proc. Natl. Acad. Sci. USA 2009, 106, 4085–4089. [Google Scholar] [CrossRef]
- Kosuge, H.; Sherlock, S.P.; Kitagawa, T.; Terashima, M.; Barral, J.K.; Nishimura, D.G.; Dai, H.; McConnell, M.V. FeCo/graphite nanocrystals for multi-modality imaging of experimental vascular inflammation. PLoS ONE 2011, 6, 14523. [Google Scholar] [CrossRef]
- Seo, W.S.; Lee, J.H.; Sun, X.; Suzuki, Y.; Mann, D.; Liu, Z.; Terashima, M.; Yang, P.C.; McConnell, M.V.; Nishimura, D.G.; et al. FeCo/graphitic-shell nanocrystals as advanced magnetic-resonance-imaging and near-infrared agents. Nat. Mater. 2006, 5, 971–976. [Google Scholar] [CrossRef]
- Habib, A.H.; Ondeck, C.L.; Chaudhary, P.; Bockstaller, M.R.; McHenry, M.E. Evaluation of iron-cobalt/ferrite core-shell nanoparticles for cancer thermotherapy. J. Appl. Phys. 2008, 103, 07A307. [Google Scholar] [CrossRef]
- Xu, Y.H.; Bai, J.; Wang, J.P. High-magnetic-moment multifunctional nanoparticles for nanomedicine applications. J. Magn. Magn. Mater. 2007, 311, 131–134. [Google Scholar] [CrossRef]
- Chen, C.L.; Kuo, L.R.; Lee, S.Y.; Hwu, Y.K.; Chou, S.W.; Chen, C.C.; Chang, F.H.; Lin, K.H.; Tsai, D.H.; Chen, Y.Y. Photothermal cancer therapy via femtosecond-laser-excited FePt nanoparticles. Biomaterials 2013, 34, 1128–1134. [Google Scholar] [CrossRef]
- Choi, J.S.; Jun, Y.W.; Yeon, S.I.; Kim, H.C.; Shin, J.S.; Cheon, J. Biocompatible heterostructured nanoparticles for multimodal biological detection. J. Am. Chem. Soc. 2006, 128, 15982–15983. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, J.; Tian, Q.; Hu, H.; Fang, Y.; Wu, H.; Yang, S. One-pot synthesis of amphiphilic superparamagnetic FePt nanoparticles and magnetic resonance imaging in vitro. J. Magn. Magn. Mater. 2010, 322, 973–977. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, P.C.; Guo, S.; Chou, P.T.; Deng, C.; Chou, S.W.; Yuan, Z.; Liu, T.M. Low-toxicity FePt nanoparticles for the targeted and enhanced diagnosis of breast tumors using few centimeters deep whole-body photoacoustic imaging. Photoacoustics 2020, 19, 100179. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.Y.; Zhou, Q.; Wang, M.; Zhu, Y.H.; Wu, Q.Z.; Yang, X.L. Water-soluble l-cysteine-coated FePt nanoparticles as dual MRI/CT imaging contrast agent for glioma. Int. J. Nanomed. 2015, 10, 2325–2333. [Google Scholar] [CrossRef][Green Version]
- Chou, S.W.; Shau, Y.H.; Wu, P.C.; Yang, Y.S.; Shieh, D.B.; Chen, C.C. In vitro and in vivo studies of fept nanoparticles for dual modal CT/MRI molecular imaging. J. Am. Chem. Soc. 2010, 132, 13270–13278. [Google Scholar] [CrossRef]
- Sun, H.; Chen, X.; Chen, D.; Dong, M.; Fu, X.; Li, Q.; Liu, X.; Wu, Q.; Qiu, T.; Wan, T.; et al. Influences of surface coatings and components of FePt nanoparticles on the suppression of glioma cell proliferation. Int. J. Nanomed. 2012, 7, 3295–3307. [Google Scholar] [CrossRef][Green Version]
- Seemann, K.M.; Luysberg, M.; Révay, Z.; Kudejova, P.; Sanz, B.; Cassinelli, N.; Loidl, A.; Ilicic, K.; Multhoff, G.; Schmid, T.E. Magnetic heating properties and neutron activation of tungsten-oxide coated biocompatible FePt core-shell nanoparticles. J. Control. Release 2015, 197, 131–137. [Google Scholar] [CrossRef]
- Fuchigami, T.; Kawamura, R.; Kitamoto, Y.; Nakagawa, M.; Namiki, Y. A magnetically guided anti-cancer drug delivery system using porous FePt capsules. Biomaterials 2012, 33, 1682–1687. [Google Scholar] [CrossRef]
- Salati, A.; Ramazani, A.; Almasi Kashi, M. Deciphering magnetic hyperthermia properties of compositionally and morphologically modulated FeNi nanoparticles using first-order reversal curve analysis. Nanotechnology 2019, 30, 025707. [Google Scholar] [CrossRef]
- Salati, A.; Ramazani, A.; Almasi Kashi, M. Tuning hyperthermia properties of FeNiCo ternary alloy nanoparticles by morphological and magnetic characteristics. J. Magn. Magn. Mater. 2020, 498, 166172. [Google Scholar] [CrossRef]
- Liu, H.M.; Wu, S.H.; Lu, C.W.; Yao, M.; Hsiao, J.K.; Hung, Y.; Lin, Y.S.; Mou, C.Y.; Yang, C.S.; Huang, D.M.; et al. Mesoporous silica nanoparticles improve magnetic labeling efficiency in human stem cells. Small 2008, 4, 619–626. [Google Scholar] [CrossRef]
- Tanaka, K.; Narita, A.; Kitamura, N.; Uchiyama, W.; Morita, M.; Inubushi, T.; Chujo, Y. Preparation for highly sensitive MRI contrast agents using core/shell type nanoparticles consisting of multiple SPIO cores with thin silica coating. Langmuir 2010, 26, 11759–11762. [Google Scholar] [CrossRef]
- Chen, W.-J.; Tsai, P.-J.; Chen, Y.-C. Functional Fe3O4/TiO2 Core/Shell Magnetic Nanoparticles as Photokilling Agents for Pathogenic Bacteria. Small 2008, 4, 485–491. [Google Scholar] [CrossRef]
- Estelrich, J.; Sánchez-Martín, M.J.; Busquets, M.A. Nanoparticles in magnetic resonance imaging: From simple to dual contrast agents. Int. J. Nanomed. 2015, 10, 1727–1741. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, Y.; Ge, S.; Song, X.; Huang, J.; Yan, M.; Yu, J. Core-shell Fe3O4-Au magnetic nanoparticles based nonenzymatic ultrasensitive electrochemiluminescence immunosensor using quantum dots functionalized graphene sheet as labels. Anal. Chim. Acta 2013, 770, 132–139. [Google Scholar] [CrossRef]
- Wang, X.; Wang, M.; Jiang, T.; Wang, F.; Qing, Y.; Bu, S.; Zhou, J. Dual-functional Fe3O4@SiO2@Ag triple core-shell microparticles as an effective SERS platform for adipokines detection. Colloids Surfaces A Physicochem. Eng. Asp. 2017, 535, 24–33. [Google Scholar] [CrossRef]
- Wang, L.; Bai, J.; Li, Y.; Huang, Y. Multifunctional nanoparticles displaying magnetization and near-IR absorption. Angew. Chem. Int. Ed. 2008, 47, 2439–2442. [Google Scholar] [CrossRef]
- Qiu, J.D.; Xiong, M.; Liang, R.P.; Peng, H.P.; Liu, F. Synthesis and characterization of ferrocene modified Fe3O4@Au magnetic nanoparticles and its application. Biosens. Bioelectron. 2009, 24, 2649–2653. [Google Scholar] [CrossRef]
- Caizer, C.; Hrianca, I. The temperature dependence of saturation magnetization of γ-Fe2O3/SiO2 magnetic nanocomposite. Ann. Phys. 2003, 12, 115–122. [Google Scholar] [CrossRef]
- Pinho, S.L.C.; Pereira, G.A.; Voisin, P.; Kassem, J.; Bouchaud, V.; Etienne, L.; Peters, J.A.; Carlos, L.; Mornet, S.; Geraldes, C.F.G.C.; et al. Fine tuning of the relaxometry of γ-Fe2O3@SiO2 nanoparticles by tweaking the silica coating thickness. ACS Nano 2010, 4, 5339–5349. [Google Scholar] [CrossRef]
- Park, J.Y.; Choi, E.S.; Baek, M.J.; Lee, G.H.; Woo, S.; Chang, Y. Water-soluble Ultra Small paramagnetic or superparamagnetic metal oxide nanoparticles for molecular MR imaging. Eur. J. Inorg. Chem. 2009, 2477–2481. [Google Scholar] [CrossRef]
- Schweiger, C.; Pietzonka, C.; Heverhagen, J.; Kissel, T. Novel magnetic iron oxide nanoparticles coated with poly(ethylene imine)-g-poly(ethylene glycol) for potential biomedical application: Synthesis, stability, cytotoxicity and MR imaging. Int. J. Pharm. 2011, 408, 130–137. [Google Scholar] [CrossRef]
- Zhao, Z.; Sun, C.; Bao, J.; Yang, L.; Wei, R.; Cheng, J.; Lin, H.; Gao, J. Surface manganese substitution in magnetite nanocrystals enhances: T1 contrast ability by increasing electron spin relaxation. J. Mater. Chem. B 2018, 6, 401–413. [Google Scholar] [CrossRef]
- Demirci Dönmez, C.E.; Manna, P.K.; Nickel, R.; Aktürk, S.; Van Lierop, J. Comparative Heating Efficiency of Cobalt-, Manganese-, and Nickel-Ferrite Nanoparticles for a Hyperthermia Agent in Biomedicines. ACS Appl. Mater. Interfaces 2019, 11, 6858–6866. [Google Scholar] [CrossRef]
- Amiri, S.; Shokrollahi, H. The role of cobalt ferrite magnetic nanoparticles in medical science. Mater. Sci. Eng. C 2013, 33, 1–8. [Google Scholar] [CrossRef]
- Dey, C.; Baishya, K.; Ghosh, A.; Goswami, M.M.; Ghosh, A.; Mandal, K. Improvement of drug delivery by hyperthermia treatment using magnetic cubic cobalt ferrite nanoparticles. J. Magn. Magn. Mater. 2017, 427, 168–174. [Google Scholar] [CrossRef]
- Pita, M.; Abad, J.M.; Vaz-Dominguez, C.; Briones, C.; Mateo-Martí, E.; Martín-Gago, J.A.; del Puerto Morales, M.; Fernández, V.M. Synthesis of cobalt ferrite core/metallic shell nanoparticles for the development of a specific PNA/DNA biosensor. J. Colloid Interface Sci. 2008, 321, 484–492. [Google Scholar] [CrossRef]
- Dumitrescu, A.M.; Slatineanu, T.; Poiata, A.; Iordan, A.R.; Mihailescu, C.; Palamaru, M.N. Advanced composite materials based on hydrogels and ferrites for potential biomedical applications. Colloids Surfaces A Physicochem. Eng. Asp. 2014, 455, 185–194. [Google Scholar] [CrossRef]
- Lasheras, X.; Insausti, M.; Gil De Muro, I.; Garaio, E.; Plazaola, F.; Moros, M.; De Matteis, L.; De La Fuente, J.; Lezama, L.M. Chemical Synthesis and Magnetic Properties of Monodisperse Nickel Ferrite Nanoparticles for Biomedical Applications. J. Phys. Chem. C 2016, 120, 3492–3500. [Google Scholar] [CrossRef]
- Menelaou, M.; Georgoula, K.; Simeonidis, K.; Dendrinou-Samara, C. Evaluation of nickel ferrite nanoparticles coated with oleylamine by NMR relaxation measurements and magnetic hyperthermia. Dalt. Trans. 2014, 43, 3626–3636. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Bae, H.; Iqbal, Y.; Rhee, I.; Hong, S.; Chang, Y.; Lee, J.; Sohn, D. Chitosan-coated nickel-ferrite nanoparticles as contrast agents in magnetic resonance imaging. J. Magn. Magn. Mater. 2015, 381, 151–157. [Google Scholar] [CrossRef]
- Liu, Y.; Lv, X.; Liu, H.; Zhou, Z.; Huang, J.; Lei, S.; Cai, S.; Chen, Z.; Guo, Y.; Chen, Z.; et al. Porous gold nanocluster-decorated manganese monoxide nanocomposites for microenvironment-activatable MR/photoacoustic/CT tumor imaging. Nanoscale 2018, 10, 3631–3638. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Momin, E.; Choi, J.; Yuan, K.; Zaidi, H.; Kim, J.; Park, M.; Lee, N.; McMahon, M.T.; Quinones-Hinojosa, A.; et al. Mesoporous silica-coated hollow manganese oxide nanoparticles as positive T1 contrast agents for labeling and MRI tracking of adipose-derived mesenchymal stem cells. J. Am. Chem. Soc. 2011, 133, 2955–2961. [Google Scholar] [CrossRef] [PubMed]
- Caravan, P.; Ellison, J.J.; McMurry, T.J.; Lauffer, R.B. Gadolinium(III) chelates as MRI contrast agents: Structure, dynamics, and applications. Chem. Rev. 1999, 99, 2293–2352. [Google Scholar] [CrossRef]
- Na, H.B.; Song, I.C.; Hyeon, T. Inorganic nanoparticles for MRI contrast agents. Adv. Mater. 2009, 21, 2133–2148. [Google Scholar] [CrossRef]
- Wahsner, J.; Gale, E.M.; Rodríguez-Rodríguez, A.; Caravan, P. Chemistry of MRI contrast agents: Current challenges and new frontiers. Chem. Rev. 2019, 119, 957–1057. [Google Scholar] [CrossRef]
- De Leõn-Rodríguez, L.M.; Martins, A.F.; Pinho, M.C.; Rofsky, N.M.; Sherry, A.D. Basic MR relaxation mechanisms and contrast agent design. J. Magnet. Reson. Imaging 2015, 42, 545–565. [Google Scholar] [CrossRef]
- Moreno-Romero, J.A.; Segura, S.; Mascaró, J.M.; Cowper, S.E.; Julià, M.; Poch, E.; Botey, A.; Herrero, C. Nephrogenic systemic fibrosis: A case series suggesting gadolinium as a possible aetiological factor. Br. J. Dermatol. 2007, 157, 783–787. [Google Scholar] [CrossRef]
- Hasebroock, K.M.; Serkova, N.J. Toxicity of MRI and CT contrast agents. Expert Opin. Drug Metab. Toxicol. 2009, 5, 403–416. [Google Scholar] [CrossRef]
- Khalkhali, M.; Rostamizadeh, K.; Sadighian, S.; Khoeini, F.; Naghibi, M.; Hamidi, M. The impact of polymer coatings on magnetite nanoparticles performance as MRI contrast agents: A comparative study. DARU J. Pharm. Sci. 2015, 23, 45. [Google Scholar] [CrossRef]
- Fernández-Barahona, I.; Muñoz-Hernando, M.; Ruiz-Cabello, J.; Herranz, F.; Pellico, J. Iron oxide nanoparticles: An alternative for positive contrast in magnetic resonance imaging. Inorganics 2020, 8, 28. [Google Scholar] [CrossRef]
- Tromsdorf, U.I.; Bruns, O.T.; Salmen, S.C.; Beisiegel, U.; Weller, H. A highly effective, nontoxic T1 MR contrast agent based on ultrasmall PEGylated iron oxide nanoparticles. Nano Lett. 2009, 9, 4434–4440. [Google Scholar] [CrossRef]
- Iqbal, M.Z.; Ma, X.; Chen, T.; Zhang, L.; Ren, W.; Xiang, L.; Wu, A. Silica-coated super-paramagnetic iron oxide nanoparticles (SPIONPs): A new type contrast agent of T1 magnetic resonance imaging (MRI). J. Mater. Chem. B 2015, 3, 5172–5181. [Google Scholar] [CrossRef]
- Alipour, A.; Soran-Erdem, Z.; Utkur, M.; Sharma, V.K.; Algin, O.; Saritas, E.U.; Demir, H.V. A new class of cubic SPIONs as a dual-mode T1 and T2 contrast agent for MRI. Magn. Reson. Imaging 2018, 49, 16–24. [Google Scholar] [CrossRef]
- Jeon, M.; Halbert, M.V.; Stephen, Z.R.; Zhang, M. Iron Oxide Nanoparticles as T1 Contrast Agents for Magnetic Resonance Imaging: Fundamentals, Challenges, Applications, and Prospectives. Adv. Mater. 2020, 33, 1906539. [Google Scholar] [CrossRef]
- Cao, Y.; Mao, Z.; He, Y.; Kuang, Y.; Liu, M.; Zhou, Y.; Zhang, Y.; Pei, R. Extremely Small Iron Oxide Nanoparticle-Encapsulated Nanogels as a Glutathione-Responsive T1Contrast Agent for Tumor-Targeted Magnetic Resonance Imaging. ACS Appl. Mater. Interfaces 2020, 12, 26973–26981. [Google Scholar] [CrossRef]
- Kiessling, F.; Mertens, M.E.; Grimm, J.; Lammers, T. Nanoparticles for imaging: Top or flop? Radiology 2014, 273, 10–28. [Google Scholar] [CrossRef]
- Wang, Y.-X.J. Superparamagnetic iron oxide based MRI contrast agents: Current status of clinical application. Quant. Imaging Med. Surg. 2011, 1, 35–40. [Google Scholar] [CrossRef]
- Wang, Y.-X.J. Current status of superparamagnetic iron oxide contrast agents for liver magnetic resonance imaging. World J. Gastroenterol. 2015, 21, 13400. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.M.; Huber, D.L.; Monson, T.C.; Esch, V.; Sillerud, L.O. Structural and magnetic characterization of superparamagnetic iron platinum nanoparticle contrast agents for magnetic resonance imaging. J. Vac. Sci. Technol. B Nanotechnol. Microelectron. Mater. Process. Meas. Phenom. 2012, 30, 02C101. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, E.E. Iron nanoparticles as potential magnetic carriers. J. Magn. Magn. Mater. 2001, 225, 17–20. [Google Scholar] [CrossRef]
- Hadjipanayis, C.G.; Bonder, M.J.; Balakrishnan, S.; Wang, X.; Mao, H.; Hadjipanayis, G.C. Metallic iron nanoparticles for MRI contrast enhancement and local hyperthermia. Small 2008, 4, 1925–1929. [Google Scholar] [CrossRef]
- Bergs, J.W.J.; Franken, N.A.P.; Haveman, J.; Geijsen, E.D.; Crezee, J.; van Bree, C. Hyperthermia, cisplatin and radiation trimodality treatment: A promising cancer treatment? A review from preclinical studies to clinical application. Int. J. Hyperth. 2007, 23, 329–341. [Google Scholar] [CrossRef]
- Chicheł, A.; Skowronek, J.; Kubaszewska, M.; Kanikowski, M. Hyperthermia - Description of a method and a review of clinical applications. Rep. Pract. Oncol. Radiother. 2007, 12, 267–275. [Google Scholar] [CrossRef]
- Saniei, N. Hyperthermia and cancer treatment. Heat Transf. Eng. 2009, 30, 915–917. [Google Scholar] [CrossRef]
- Thomas, L.A.; Dekker, L.; Kallumadil, M.; Southern, P.; Wilson, M.; Nair, S.P.; Pankhurst, Q.A.; Parkin, I.P. Carboxylic acid-stabilised iron oxide nanoparticles for use in magnetic hyperthermia. J. Mater. Chem. 2009, 19, 6529–6535. [Google Scholar] [CrossRef]
- Shah, R.R.; Davis, T.P.; Glover, A.L.; Nikles, D.E.; Brazel, C.S. Impact of magnetic field parameters and iron oxide nanoparticle properties on heat generation for use in magnetic hyperthermia. J. Magn. Magn. Mater. 2015, 387, 96–106. [Google Scholar] [CrossRef]
- Bauer, L.M.; Situ, S.F.; Griswold, M.A.; Samia, A.C.S. High-performance iron oxide nanoparticles for magnetic particle imaging-guided hyperthermia (hMPI). Nanoscale 2016, 8, 12162–12169. [Google Scholar] [CrossRef]
- Abenojar, E.C.; Wickramasinghe, S.; Bas-Concepcion, J.; Samia, A.C.S. Structural effects on the magnetic hyperthermia properties of iron oxide nanoparticles. Prog. Nat. Sci. Mater. Int. 2016, 26, 440–448. [Google Scholar] [CrossRef]
- Jeyadevan, B. Present status and prospects of magnetite nanoparticles-based hyperthermia. J. Ceram. Soc. Japan 2010, 118, 391–401. [Google Scholar] [CrossRef]
- Maier-Hauff, K.; Rothe, R.; Scholz, R.; Gneveckow, U.; Wust, P.; Thiesen, B.; Feussner, A.; Deimling, A.; Waldoefner, N.; Felix, R.; et al. Intracranial thermotherapy using magnetic nanoparticles combined with external beam radiotherapy: Results of a feasibility study on patients with glioblastoma multiforme. J. Neurooncol. 2007, 81, 53–60. [Google Scholar] [CrossRef]
- Johannsen, M.; Gneveckow, U.; Taymoorian, K.; Thiesen, B.; Waldöfner, N.; Scholz, R.; Jung, K.; Jordan, A.; Wust, P.; Loening, S.A. Morbidity and quality of life during thermotherapy using magnetic nanoparticles in locally recurrent prostate cancer: Results of a prospective phase I trial. Int. J. Hyperth. 2007, 23, 315–323. [Google Scholar] [CrossRef]
- Johannsen, M.; Gneveckow, U.; Thiesen, B.; Taymoorian, K.; Cho, C.H.; Waldöfner, N.; Scholz, R.; Jordan, A.; Loening, S.A.; Wust, P. Thermotherapy of Prostate Cancer Using Magnetic Nanoparticles: Feasibility, Imaging, and Three-Dimensional Temperature Distribution. Eur. Urol. 2007, 52, 1653–1662. [Google Scholar] [CrossRef]
- Dutz, S.; Hergt, R. Magnetic nanoparticle heating and heat transfer on a microscale: Basic principles, realities and physical limitations of hyperthermia for tumour therapy. Int. J. Hyperth. 2013, 29, 790–800. [Google Scholar] [CrossRef]
- Nieskoski, M.D.; Trembly, B.S. Comparison of a single optimized coil and a Helmholtz pair for magnetic nanoparticle hyperthermia. IEEE Trans. Biomed. Eng. 2014, 61, 1642–1650. [Google Scholar] [CrossRef]
- Ruuge, E.K.; Rusetski, A.N. Magnetic fluids as drug carriers: Targeted transport of drugs by a magnetic field. J. Magn. Magn. Mater. 1993, 122, 335–339. [Google Scholar] [CrossRef]
- Voltairas, P.A.; Fotiadis, D.I.; Michalis, L.K. Hydrodynamics of magnetic drug targeting. J. Biomech. 2002, 35, 813–821. [Google Scholar] [CrossRef]
- Grief, A.D.; Richardson, G. Mathematical modelling of magnetically targeted drug delivery. J. Magn. Magn. Mater. 2005, 293, 455–463. [Google Scholar] [CrossRef]
- Iacob, G.; Rotariu, O.; Strachan, N.J.C.; Häfeli, U.O. Magnetizable needles and wires - Modeling an efficient way to target magnetic microspheres in vivo. Biorheology 2004, 41, 599–612. [Google Scholar] [PubMed]
- Rosengart, A.J.; Kaminski, M.D.; Chen, H.; Caviness, P.L.; Ebner, A.D.; Ritter, J.A. Magnetizable implants and functionalized magnetic carriers: A novel approach for noninvasive yet targeted drug delivery. J. Magn. Magn. Mater. 2005, 293, 633–638. [Google Scholar] [CrossRef]
- Rotariu, O.; Strachan, N.J.C. Modelling magnetic carrier particle targeting in the tumor microvasculature for cancer treatment. J. Magn. Magn. Mater. 2005, 293, 639–646. [Google Scholar] [CrossRef]
- Yellen, B.B.; Forbes, Z.G.; Halverson, D.S.; Fridman, G.; Barbee, K.A.; Chorny, M.; Levy, R.; Friedman, G. Targeted drug delivery to magnetic implants for therapeutic applications. J. Magn. Magn. Mater. 2005, 293, 647–654. [Google Scholar] [CrossRef]
- Maier-Hauff, K.; Ulrich, F.; Nestler, D.; Niehoff, H.; Wust, P.; Thiesen, B.; Orawa, H.; Budach, V.; Jordan, A. Efficacy and safety of intratumoral thermotherapy using magnetic iron-oxide nanoparticles combined with external beam radiotherapy on patients with recurrent glioblastoma multiforme. J. Neurooncol. 2011, 103, 317–324. [Google Scholar] [CrossRef]
- Asín, L.; Ibarra, M.R.; Tres, A.; Goya, G.F. Controlled cell death by magnetic hyperthermia: Effects of exposure time, field amplitude, and nanoparticle concentration. Pharm. Res. 2012, 29, 1319–1327. [Google Scholar] [CrossRef]
- Skumiel, A.; Kaczmarek, K.; Flak, D.; Rajnak, M.; Antal, I.; Brząkała, H. The influence of magnetic nanoparticle concentration with dextran polymers in agar gel on heating efficiency in magnetic hyperthermia. J. Mol. Liq. 2020, 304, 112734. [Google Scholar] [CrossRef]
- Kumar, B.; Jalodia, K.; Kumar, P.; Gautam, H.K. Recent advances in nanoparticle-mediated drug delivery. J. Drug Deliv. Sci. Technol. 2017, 41, 260–268. [Google Scholar] [CrossRef]
- Hergt, R.; Dutz, S.; Müller, R.; Zeisberger, M. Magnetic particle hyperthermia: Nanoparticle magnetism and materials development for cancer therapy. J. Phys. Condens. Matter 2006, 18, S2919. [Google Scholar] [CrossRef]
- Vallejo-Fernandez, G.; Whear, O.; Roca, A.G.; Hussain, S.; Timmis, J.; Patel, V.; O’Grady, K. Mechanisms of hyperthermia in magnetic nanoparticles. J. Phys. D Appl. Phys. 2013, 46, 312001. [Google Scholar] [CrossRef]
- Deatsch, A.E.; Evans, B.A. Heating efficiency in magnetic nanoparticle hyperthermia. J. Magn. Magn. Mater. 2014, 354, 163–172. [Google Scholar] [CrossRef]
- Dennis, C.L.; Ivkov, R. Physics of heat generation using magnetic nanoparticles for hyperthermia. Int. J. Hyperth. 2013, 29, 715–729. [Google Scholar] [CrossRef]
- Datta, N.R.; Ordóñez, S.G.; Gaipl, U.S.; Paulides, M.M.; Crezee, H.; Gellermann, J.; Marder, D.; Puric, E.; Bodis, S. Local hyperthermia combined with radiotherapy and-/or chemotherapy: Recent advances and promises for the future. Cancer Treat. Rev. 2015, 41, 742–753. [Google Scholar] [CrossRef]
- Issels, R.D. Hyperthermia adds to chemotherapy. Eur. J. Cancer 2008, 44, 2546–2554. [Google Scholar] [CrossRef]
- Orel, V.; Shevchenko, A.; Romanov, A.; Tselepi, M.; Mitrelias, T.; Barnes, C.H.W.; Burlaka, A.; Lukin, S.; Shchepotin, I. Magnetic properties and antitumor effect of nanocomplexes of iron oxide and doxorubicin. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 47–55. [Google Scholar] [CrossRef]
- Hahn, G.M. Potential for therapy of drugs and hyperthermia. Cancer Res. 1979, 39, 2264–2268. [Google Scholar]
- Zhou, Z.; Sun, Y.; Shen, J.; Wei, J.; Yu, C.; Kong, B.; Liu, W.; Yang, H.; Yang, S.; Wang, W. Iron/iron oxide core/shell nanoparticles for magnetic targeting MRI and near-infrared photothermal therapy. Biomaterials 2014, 35, 7470–7478. [Google Scholar] [CrossRef]
- Estelrich, J.; Antònia Busquets, M. Iron oxide nanoparticles in photothermal therapy. Molecules 2018, 23, 1567. [Google Scholar] [CrossRef]
- Lee, C.W.; Wu, P.C.; Hsu, I.L.; Liu, T.M.; Chong, W.H.; Wu, C.H.; Hsieh, T.Y.; Guo, L.Z.; Tsao, Y.; Wu, P.T.; et al. New Templated Ostwald Ripening Process of Mesostructured FeOOH for Third-Harmonic Generation Bioimaging. Small 2019, 15, 1805086. [Google Scholar] [CrossRef]
- De Paula, L.B.; Primo, F.L.; Pinto, M.R.; Morais, P.C.; Tedesco, A.C. Combination of hyperthermia and photodynamic therapy on mesenchymal stem cell line treated with chloroaluminum phthalocyanine magnetic-nanoemulsion. J. Magn. Magn. Mater. 2015, 380, 372–376. [Google Scholar] [CrossRef]
- Di Corato, R.; Béalle, G.; Kolosnjaj-Tabi, J.; Espinosa, A.; Clément, O.; Silva, A.K.A.; Ménager, C.; Wilhelm, C. Combining magnetic hyperthermia and photodynamic therapy for tumor ablation with photoresponsive magnetic liposomes. ACS Nano 2015, 9, 2904–2916. [Google Scholar] [CrossRef]
- De Paula, L.B.; Primo, F.L.; Jardim, D.R.; Morais, P.C.; Tedesco, A.C. Development, characterization, and in vitro trials of chloroaluminum phthalocyanine-magnetic nanoemulsion to hyperthermia and photodynamic therapies on glioblastoma as a biological model. J. Appl. Phys. 2012, 111, 07B307. [Google Scholar] [CrossRef]
- Yang, K.; Wan, J.; Zhang, S.; Tian, B.; Zhang, Y.; Liu, Z. The influence of surface chemistry and size of nanoscale graphene oxide on photothermal therapy of cancer using ultra-low laser power. Biomaterials 2012, 33, 2206–2214. [Google Scholar] [CrossRef]
- Ma, X.; Cheng, Y.; Huang, Y.; Tian, Y.; Wang, S.; Chen, Y. PEGylated gold nanoprisms for photothermal therapy at low laser power density. RSC Adv. 2015, 5, 81682–81688. [Google Scholar] [CrossRef]
- Stern, J.M.; Kibanov Solomonov, V.V.; Sazykina, E.; Schwartz, J.A.; Gad, S.C.; Goodrich, G.P. Initial Evaluation of the Safety of Nanoshell-Directed Photothermal Therapy in the Treatment of Prostate Disease. Int. J. Toxicol. 2016, 35, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Winau, F.; Westphal, O.; Winau, R. Paul Ehrlich - In search of the magic bullet. Microbes Infect. 2004, 6, 786–789. [Google Scholar] [CrossRef] [PubMed]
- Widder, K.; Flouret, G.; Senyei, A. Magnetic microspheres: Synthesis of a novel parenteral drug carrier. J. Pharm. Sci. 1979, 68, 79–82. [Google Scholar] [CrossRef]
- Senyei, A.; Widder, K.; Czerlinski, G. Magnetic guidance of drug-carrying microspheres. J. Appl. Phys. 1978, 49, 3578–3583. [Google Scholar] [CrossRef]
- Mosbach, K.; Schröder, U. Preparation and application of magnetic polymers for targeting of drugs. FEBS Lett. 1979, 102, 112–116. [Google Scholar] [CrossRef]
- Kwon, G.S.; Okano, T. Polymeric micelles as new drug carriers. Adv. Drug Deliv. Rev. 1996, 21, 107–116. [Google Scholar] [CrossRef]
- López-Dávila, V.; Seifalian, A.M.; Loizidou, M. Organic nanocarriers for cancer drug delivery. Curr. Opin. Pharmacol. 2012, 12, 414–419. [Google Scholar] [CrossRef]
- Manatunga, D.C.; Godakanda, V.U.; de Silva, R.M.; de Silva, K.M.N. Recent developments in the use of organic–inorganic nanohybrids for drug delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, 12, e1605. [Google Scholar] [CrossRef]
- Zhang, R.; Olin, H. Carbon nanomaterials as drug carriers: Real time drug release investigation. Mater. Sci. Eng. C 2012, 32, 1247–1252. [Google Scholar] [CrossRef]
- Lim, D.J.; Sim, M.; Oh, L.; Lim, K.; Park, H. Carbon-based drug delivery carriers for cancer therapy. Arch. Pharm. Res. 2014, 37, 43–52. [Google Scholar] [CrossRef]
- Partha, R.; Conyers, J.L. Biomedical applications of functionalized fullerene-based nanomaterials. Int. J. Nanomed. 2009, 4, 261–275. [Google Scholar] [CrossRef]
- Rasheed, A.; Kumar C.K., A.; Sravanthi, V.V.N.S.S. Cyclodextrins as drug carrier molecule: A review. Sci. Pharm. 2008, 76, 567–598. [Google Scholar] [CrossRef]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release 2008, 132, 171–183. [Google Scholar] [CrossRef]
- MacDiarmid, J.A.; Brahmbhatt, H. Minicells: Versatile vectors for targeted drug or si/shRNA cancer therapy. Curr. Opin. Biotechnol. 2011, 22, 909–916. [Google Scholar] [CrossRef]
- Akash, M.S.H.; Rehman, K.; Parveen, A.; Ibrahim, M. Antibody-drug conjugates as drug carrier systems for bioactive agents. Int. J. Polym. Mater. Polym. Biomater. 2016, 65, 1–10. [Google Scholar] [CrossRef]
- Barbé, C.; Bartlett, J.; Kong, L.; Finnie, K.; Lin, H.Q.; Larkin, M.; Calleja, S.; Bush, A.; Calleja, G. Silica particles: A novel drug-delivery system. Adv. Mater. 2004, 16, 1959–1966. [Google Scholar] [CrossRef]
- Oussoren, C.; Storm, G. Liposomes to target the lymphatics by subcutaneous administration. Adv. Drug Deliv. Rev. 2001, 50, 143–156. [Google Scholar] [CrossRef]
- Städler, B.; Price, A.D.; Zelikin, A.N. A critical look at multilayered polymer capsules in biomedicine: Drug carriers, artificial organelles, and cell mimics. Adv. Funct. Mater. 2011, 21, 14–28. [Google Scholar] [CrossRef]
- Kataoka, K.; Harada, A.; Nagasaki, Y. Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv. Drug Deliv. Rev. 2012, 64, 37–48. [Google Scholar] [CrossRef]
- Moyano, D.F.; Goldsmith, M.; Solfiell, D.J.; Landesman-Milo, D.; Miranda, O.R.; Peer, D.; Rotello, V.M. Nanoparticle hydrophobicity dictates immune response. J. Am. Chem. Soc. 2012, 134, 3965–3967. [Google Scholar] [CrossRef]
- Corbo, C.; Molinaro, R.; Parodi, A.; Toledano Furman, N.E.; Salvatore, F.; Tasciotti, E. The impact of nanoparticle protein corona on cytotoxicity, immunotoxicity and target drug delivery. Nanomedicine 2016, 11, 81–100. [Google Scholar] [CrossRef]
- Jain, T.K.; Morales, M.A.; Sahoo, S.K.; Leslie-Pelecky, D.L.; Labhasetwar, V. Iron Oxide Nanoparticles for Sustained Delivery of Anticancer Agents. Mol. Pharm. 2005, 2, 194–205. [Google Scholar] [CrossRef]
- Estelrich, J.; Escribano, E.; Queralt, J.; Busquets, M.A. Iron oxide nanoparticles for magnetically-guided and magnetically-responsive drug delivery. Int. J. Mol. Sci. 2015, 16, 8070–8101. [Google Scholar] [CrossRef]
- Wahajuddin, S.A. Superparamagnetic iron oxide nanoparticles: Magnetic nanoplatforms as drug carriers. Int. J. Nanomed. 2012, 7, 3445–3471. [Google Scholar] [CrossRef]
- Khalid, K.; Tan, X.; Mohd Zaid, H.F.; Tao, Y.; Lye Chew, C.; Chu, D.T.; Lam, M.K.; Ho, Y.C.; Lim, J.W.; Chin Wei, L. Advanced in developmental organic and inorganic nanomaterial: A review. Bioengineered 2020, 11, 328–355. [Google Scholar] [CrossRef]
- Neuberger, T.; Schöpf, B.; Hofmann, H.; Hofmann, M.; Von Rechenberg, B. Superparamagnetic nanoparticles for biomedical applications: Possibilities and limitations of a new drug delivery system. J. Magn. Magn. Mater. 2005, 293, 483–496. [Google Scholar] [CrossRef]
- Lockman, P.R.; Mumper, R.J.; Khan, M.A.; Allen, D.D. Nanoparticle technology for drug delivery across the blood-brain barrier. Drug Dev. Ind. Pharm. 2002, 28, 1–13. [Google Scholar] [CrossRef]
- Saraiva, C.; Praça, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-mediated brain drug delivery: Overcoming blood-brain barrier to treat neurodegenerative diseases. J. Control. Release 2016, 235, 34–47. [Google Scholar] [CrossRef]
- Bao, Y.; Krishnan, K.M. Preparation of functionalized and gold-coated cobalt nanocrystals for biomedical applications. J. Magn. Magn. Mater. 2005, 293, 15–19. [Google Scholar] [CrossRef]
- Carpenter, E.E.; Sangregorio, C.; O’Connor, C.J. Effects of shell thickness on blocking temperature of nanocomposites of metal particles with gold shells. IEEE Trans. Magn. 1999, 35, 3496–3498. [Google Scholar] [CrossRef]
- Jung, J.S.; Chae, W.S.; McIntyre, R.A.; Seip, C.T.; Wiley, J.B.; O’Connor, C.J. Preparation and characterization of Ni nanoparticles in an MCM mesoporous material. Mater. Res. Bull. 1999, 34, 1353–1360. [Google Scholar] [CrossRef]
- Lin, X.M.; Sorensen, C.M.; Klabunde, K.J.; Hadjipanayis, G.C. Temperature Dependence of Morphology and Magnetic Properties of Cobalt Nanoparticles Prepared by an Inverse Micelle Technique. Langmuir 1998, 14, 7140–7146. [Google Scholar] [CrossRef]
- Yano, K.; Nandwana, V.; Chaubey, G.S.; Poudyal, N.; Kang, S.; Arami, H.; Griffis, J.; Liu, J.P. Synthesis and Characterization of Magnetic FePt/Au Core/Shell Nanoparticles. J. Phys. Chem. C 2009, 113, 13088–13091. [Google Scholar] [CrossRef]
- Koenig, S.H.; Kellar, K.E. Theory of 1/T1 and 1/T2 NMRD profiles of solutions of magnetic nanoparticles. Magn. Reson. Med. 1995, 34, 227–233. [Google Scholar] [CrossRef]
- Roch, A.; Muller, R.N.; Gillis, P. Theory of proton relaxation induced by superparamagnetic particles. J. Chem. Phys. 1999, 110, 5403–5411. [Google Scholar] [CrossRef]
- Zhou, Z.; Yang, L.; Gao, J.; Chen, X. Structure–Relaxivity Relationships of Magnetic Nanoparticles for Magnetic Resonance Imaging. Adv. Mater. 2019, 31, 1804567. [Google Scholar] [CrossRef]
- Devreux, M.; Henoumont, C.; Dioury, F.; Stanicki, D.; Boutry, S.; Larbanoix, L.; Ferroud, C.; Muller, R.N.; Laurent, S. Bimodal Probe for Magnetic Resonance Imaging and Photoacoustic Imaging Based on a PCTA-Derived Gadolinium(III) Complex and ZW800–1. Eur. J. Inorg. Chem. 2019, 45, 613–624. [Google Scholar] [CrossRef]
- De Haan, H.W. Mechanisms of proton spin dephasing in a system of magnetic particles. Magn. Reson. Med. 2011, 66, 1748–1758. [Google Scholar] [CrossRef]
- Shin, T.H.; Choi, Y.; Kim, S.; Cheon, J. Recent advances in magnetic nanoparticle-based multi-modal imaging. Chem. Soc. Rev. 2015, 44, 4501–4516. [Google Scholar] [CrossRef]
- Kostevšek, N. A review on the optimal design of magnetic nanoparticle-based T2 mri contrast agents. Magnetochemistry 2020, 6, 11. [Google Scholar] [CrossRef]
- Marashdeh, M.W.; Ababneh, B.; Lemine, O.M.; Alsadig, A.; Omri, K.; El Mir, L.; Sulieman, A.; Mattar, E. The significant effect of size and concentrations of iron oxide nanoparticles on magnetic resonance imaging contrast enhancement. Results Phys. 2019, 15, 102651. [Google Scholar] [CrossRef]
- Wabler, M.; Zhu, W.; Hedayati, M.; Attaluri, A.; Zhou, H.; Mihalic, J.; Geyh, A.; DeWeese, T.L.; Ivkov, R.; Artemov, D. Magnetic resonance imaging contrast of iron oxide nanoparticles developed for hyperthermia is dominated by iron content. Int. J. Hyperth. 2014, 30, 192–200. [Google Scholar] [CrossRef]
- Korchinski, D.J.; Taha, M.; Yang, R.; Nathoo, N.; Dunn, J.F. Iron Oxide as an Mri Contrast Agent for Cell Tracking: Supplementary Issue. Magn. Reson. Insights 2015, 8s1, MRI–S23557. [Google Scholar] [CrossRef]
- Labens, R.; Daniel, C.; Hall, S.; Xia, X.R.; Schwarz, T. Effect of intra-articular administration of superparamagnetic iron oxide nanoparticles (SPIONs) for MRI assessment of the cartilage barrier in a large animal model. PLoS ONE 2017, 12, e0190216. [Google Scholar] [CrossRef]
- Lu, J.; Yang, S.; Ng, K.M.; Su, C.H.; Yeh, C.S.; Wu, Y.N.; Shieh, D. Bin Solid-state synthesis of monocrystalline iron oxide nanoparticle based ferrofluid suitable for magnetic resonance imaging contrast application. Nanotechnology 2006, 17, 5812–5820. [Google Scholar] [CrossRef]
- Cho, S.J.; Jarrett, B.R.; Louie, A.Y.; Kauzlarich, S.M. Gold-coated iron nanoparticles: A novel magnetic resonance agent for T1 and T2 weighted imaging. Nanotechnology 2006, 17, 640–644. [Google Scholar] [CrossRef]
- Soukup, D.; Moise, S.; Céspedes, E.; Dobson, J.; Telling, N.D. In situ measurement of magnetization relaxation of internalized nanoparticles in live cells. ACS Nano 2015, 9, 231–240. [Google Scholar] [CrossRef]
- Pearce, J.; Giustini, A.; Stigliano, R.; Jack Hoopes, P. Magnetic Heating of Nanoparticles: The Importance of Particle Clustering to Achieve Therapeutic Temperatures. J. Nanotechnol. Eng. Med. 2013, 4, 011005. [Google Scholar] [CrossRef]
- Carrey, J.; Mehdaoui, B.; Respaud, M. Simple models for dynamic hysteresis loop calculations of magnetic single-domain nanoparticles: Application to magnetic hyperthermia optimization. J. Appl. Phys. 2011, 109, 083921. [Google Scholar] [CrossRef]
- Atkinson, W.J.; Brezovich, I.A.; Chakraborty, D.P. Usable Frequencies in Hyperthermia with Thermal Seeds. IEEE Trans. Biomed. Eng. 1984, BME 31, 70–75. [Google Scholar] [CrossRef]
- Lemine, O.M.; Omri, K.; Iglesias, M.; Velasco, V.; Crespo, P.; De La Presa, P.; El Mir, L.; Bouzid, H.; Yousif, A.; Al-Hajry, A. γ-Fe2O3 by sol-gel with large nanoparticles size for magnetic hyperthermia application. J. Alloys Compd. 2014, 607, 125–131. [Google Scholar] [CrossRef]
- Pilati, V.; Gomide, G.; Gomes, R.C.; Goya, G.F.; Depeyrot, J. Colloidal Stability and Concentration Effects on Nanoparticle Heat Delivery for Magnetic Fluid Hyperthermia. Langmuir 2021, 37, 1129–1140. [Google Scholar] [CrossRef]
- Gamarra, L.; Silva, A.C.; Oliveira, T.R.; Mamani, J.B.; Malheiros, S.M.F.; Malavolta, L.; Pavon, L.F.; Sibov, T.T.; Amaro, E., Jr.; Gamarra, L. Application of hyperthermia induced by superparamagnetic iron oxide nanoparticles in glioma treatment. Int. J. Nanomed. 2011, 6, 591–603. [Google Scholar] [CrossRef]
- Elsherbini, A.A.M.; El-Shahawy, A. Effect of SPIO nanoparticle concentrations on temperature changes for hyperthermia via MRI. J. Nanomater. 2013, 2013, 467878. [Google Scholar] [CrossRef]
- De La Presa, P.; Luengo, Y.; Multigner, M.; Costo, R.; Morales, M.P.; Rivero, G.; Hernando, A. Study of heating efficiency as a function of concentration, size, and applied field in γ-Fe2O3 nanoparticles. J. Phys. Chem. C 2012, 116, 25602–25610. [Google Scholar] [CrossRef]
- Kim, J.W.; Wang, J.; Kim, H.; Bae, S. Concentration-dependent oscillation of specific loss power in magnetic nanofluid hyperthermia. Sci. Rep. 2021, 11, 733. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Gu, H.C.; Wang, X.M. Magnetite ferrofluid with high specific absorption rate for application in hyperthermia. J. Magn. Magn. Mater. 2007, 311, 228–233. [Google Scholar] [CrossRef]
- Kekalo, K.; Baker, I.; Meyers, R.; Shyong, J. Magnetic Nanoparticles with High Specific Absorption Rate at Low Alternating Magnetic Field. Nano Life 2015, 05, 1550002. [Google Scholar] [CrossRef] [PubMed]
- Darwish, M.S.A. Effect of carriers on heating efficiency of oleic acid-stabilized magnetite nanoparticles. J. Mol. Liq. 2017, 231, 80–85. [Google Scholar] [CrossRef]
- Lin, X.M.; Sorensen, C.M.; Klabunde, K.J.; Hadjipanayis, G.C. Control of cobalt nanoparticle size by the germ-growth method in inverse micelle system: Size-dependent magnetic properties. J. Mater. Res. 1999, 14, 1542–1547. [Google Scholar] [CrossRef]
- Ibusuki, T.; Kojima, S.; Kitakami, O.; Shimada, Y. Magnetic anisotropy and behaviors of Fe nanoparticles. IEEE Trans. Magn. 2001, 37, 2223–2225. [Google Scholar] [CrossRef]
- Zhang, H.T.; Ding, J.; Chow, G.M. Morphological control of synthesis and anomalous magnetic properties of 3-D branched Pt nanoparticles. Langmuir 2008, 24, 375–378. [Google Scholar] [CrossRef]
- Sinclair, R.; Li, H.; Madsen, S.; Dai, H. HREM analysis of graphite-encapsulated metallic nanoparticles for possible medical applications. Ultramicroscopy 2013, 134, 167–174. [Google Scholar] [CrossRef]
- Skomski, R.; Balamurugan, B.; Manchanda, P.; Chipara, M.; Sellmyer, D.J. Size Dependence of Nanoparticle Magnetization. IEEE Trans. Magn. 2017, 53, 1–7. [Google Scholar] [CrossRef]
- Wang, C.; Han, X.; Zhang, X.; Hu, S.; Zhang, T.; Wang, J.; Du, Y.; Wang, X.; Xu, P. Controlled synthesis and morphology-dependent electromagnetic properties of hierarchical cobalt assemblies. J. Phys. Chem. C 2010, 114, 14826–14830. [Google Scholar] [CrossRef]
- Shao, H.; Huang, Y.; Lee, H.S.; Suh, Y.J.; Kim, C.O. Effect of PVP on the morphology of cobalt nanoparticles prepared by thermal decomposition of cobalt acetate. Curr. Appl. Phys. 2006, 6, 195–197. [Google Scholar] [CrossRef]
- Gruner, M.E.; Rollmann, G.; Entel, P.; Farle, M. Multiply twinned morphologies of FePt and CoPt nanoparticles. Phys. Rev. Lett. 2008, 100, 087203. [Google Scholar] [CrossRef] [PubMed]
- Simeonidis, K.; Martinez-Boubeta, C.; Iglesias, O.; Cabot, A.; Angelakeris, M.; Mourdikoudis, S.; Tsiaoussis, I.; Delimitis, A.; Dendrinou-Samara, C.; Kalogirou, O. Morphology influence on nanoscale magnetism of Co nanoparticles: Experimental and theoretical aspects of exchange bias. Phys. Rev. B Condens. Matter Mater. Phys. 2011, 84, 144430. [Google Scholar] [CrossRef]
- Moghimi, N.; Rahsepar, F.R.; Srivastava, S.; Heinig, N.; Leung, K.T. Shape-dependent magnetism of bimetallic FeNi nanosystems. J. Mater. Chem. C 2014, 2, 6370–6375. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Z.; Ma, L.; Li, A.; Xin, J.; Wei, R.; Lin, H.; Wang, R.; Chen, Z.; Gao, J. The Roles of Morphology on the Relaxation Rates of Magnetic Nanoparticles. ACS Nano 2018, 12, 4605–4614. [Google Scholar] [CrossRef]
- Essajai, R.; Benhouria, Y.; Rachadi, A.; Qjani, M.; Mzerd, A.; Hassanain, N. Shape-dependent structural and magnetic properties of Fe nanoparticles studied through simulation methods. RSC Adv. 2019, 9, 22057–22063. [Google Scholar] [CrossRef]
- Mujica-Martínez, C.A.; Arce, J.C. Mini-bandstructure tailoring in pi-conjugated periodic block copolymers using the envelope crystalline-orbital method. Int. J. Quantum Chem. J. 2010, 110, 2532–2540. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, G. Shape-dependent surface magnetism of Co-Pt and Fe-Pt nanoparticles from first principles. Phys. Rev. B 2017, 96, 224412. [Google Scholar] [CrossRef]
- Di Paola, C.; Baletto, F. Chemical order and magnetic properties in small Mx-2N2 nanoalloys. Eur. Phys. J. D 2013, 67, 49. [Google Scholar] [CrossRef]
- Liu, Z.; Lei, Y.; Wang, G. First-principles computation of surface segregation in L10 CoPt magnetic nanoparticles. J. Phys. Condens. Matter 2016, 28, 266002. [Google Scholar] [CrossRef]
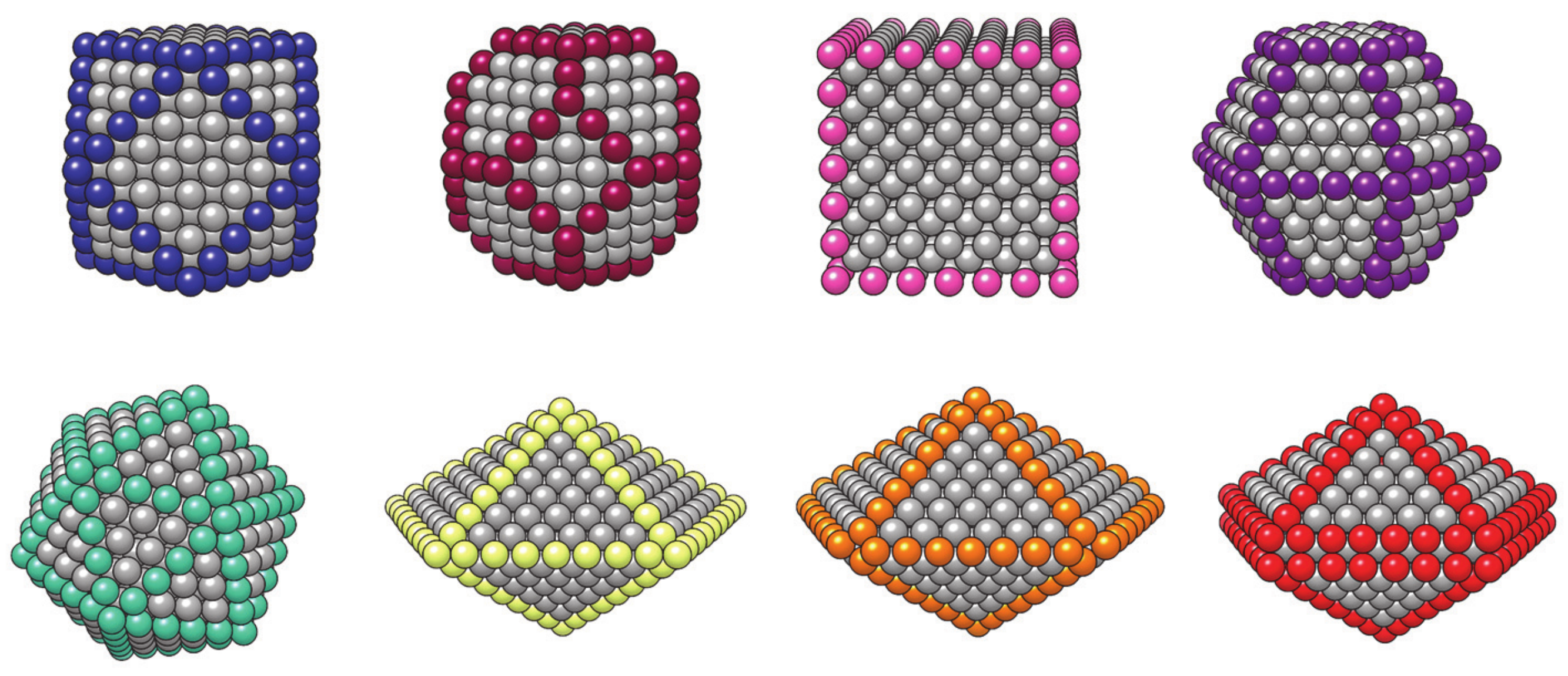
- Baletto, F.; Mottet, C.; Ferrando, R. Growth of Three-Shell Onionlike Bimetallic Nanoparticles. Phys. Rev. Lett. 2003, 90, 135504. [Google Scholar] [CrossRef]
- Lümmen, N.; Kraska, T. Investigation of the formation of iron nanoparticles from the gas phase by molecular dynamics simulation. Nanotechnology 2004, 15, 525–533. [Google Scholar] [CrossRef]
- Zeng, Q.; Jiang, X.; Yu, A.; Lu, G. Growth mechanisms of silver nanoparticles: A molecular dynamics study. Nanotechnology 2007, 18, 035708. [Google Scholar] [CrossRef]
- Grochola, G.; Russo, S.P.; Snook, I.K. On morphologies of gold nanoparticles grown from molecular dynamics simulation. J. Chem. Phys. 2007, 126, 164707. [Google Scholar] [CrossRef]
- Langlois, C.; Li, Z.L.; Yuan, J.; Alloyeau, D.; Nelayah, J.; Bochicchio, D.; Ferrando, R.; Ricolleau, C. Transition from core-shell to Janus chemical configuration for bimetallic nanoparticles. Nanoscale 2012, 4, 3381–3388. [Google Scholar] [CrossRef]
- Safaltın, Ş.; Gürmen, S. Molecular dynamics simulation of size, temperature, heating and cooling rates on structural formation of Ag-Cu-Ni ternary nanoparticles (Ag34-Cu33-Ni33). Comput. Mater. Sci. 2020, 183, 109842. [Google Scholar] [CrossRef]
- Han, Y.; Jiang, D.; Zhang, J.; Li, W.; Gan, Z.; Gu, J. Development, applications and challenges of ReaxFF reactive force field in molecular simulations. Front. Chem. Sci. Eng. 2016, 10, 16–38. [Google Scholar] [CrossRef]
- Deeth, R.J.; Anastasi, A.; Diedrich, C.; Randell, K. Molecular modelling for transition metal complexes: Dealing with d-electron effects. Coord. Chem. Rev. 2009, 253, 795–816. [Google Scholar] [CrossRef]
- Fracchia, F.; Del Frate, G.; Mancini, G.; Rocchia, W.; Barone, V. Force Field Parametrization of Metal Ions from Statistical Learning Techniques. J. Chem. Theory Comput. 2018, 14, 255–273. [Google Scholar] [CrossRef]
- Dasetty, S.; Meza-Morales, P.J.; Getman, R.B.; Sarupria, S. Simulations of interfacial processes: Recent advances in force field development. Curr. Opin. Chem. Eng. 2019, 23, 138–145. [Google Scholar] [CrossRef]
- Entel, P.; Gruner, M.E. Large-scale ab initio simulations of binary transition metal clusters for storage media materials. J. Phys. Condens. Matter 2009, 21, 064228. [Google Scholar] [CrossRef]
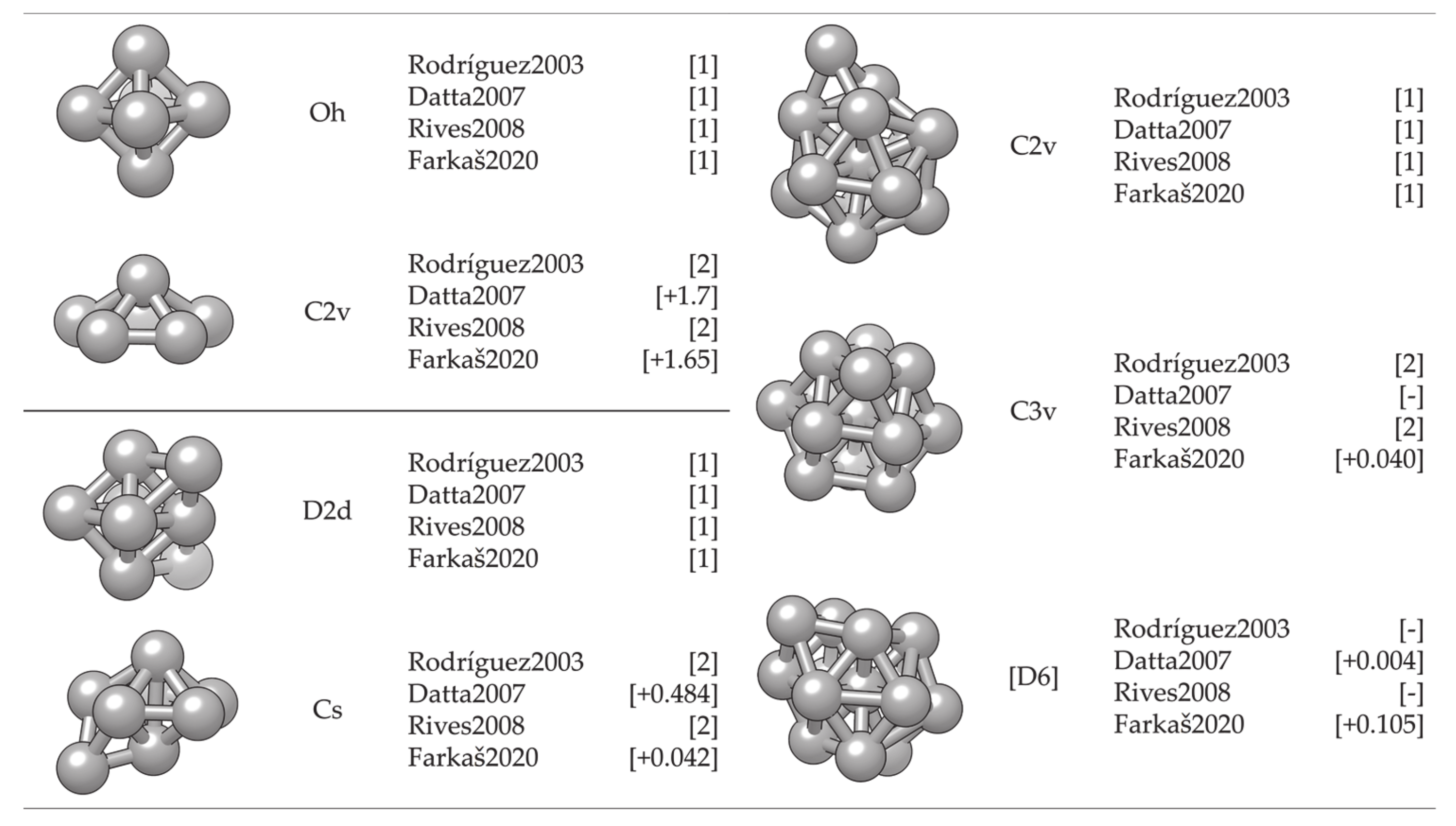
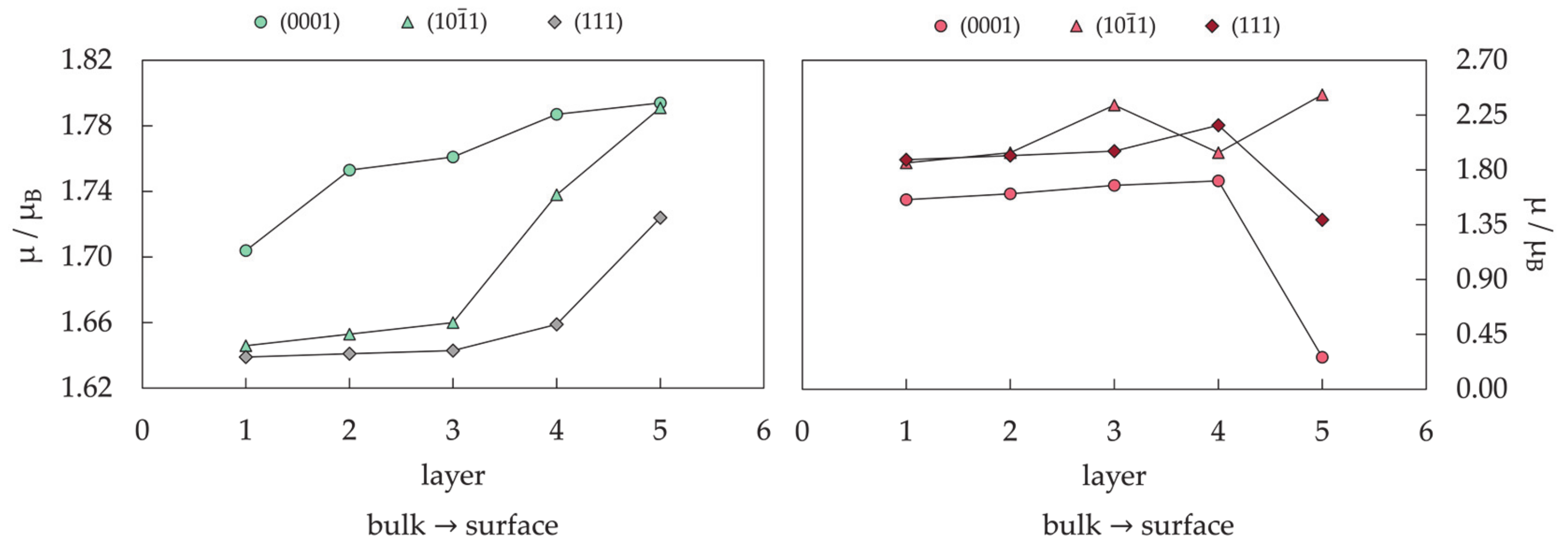
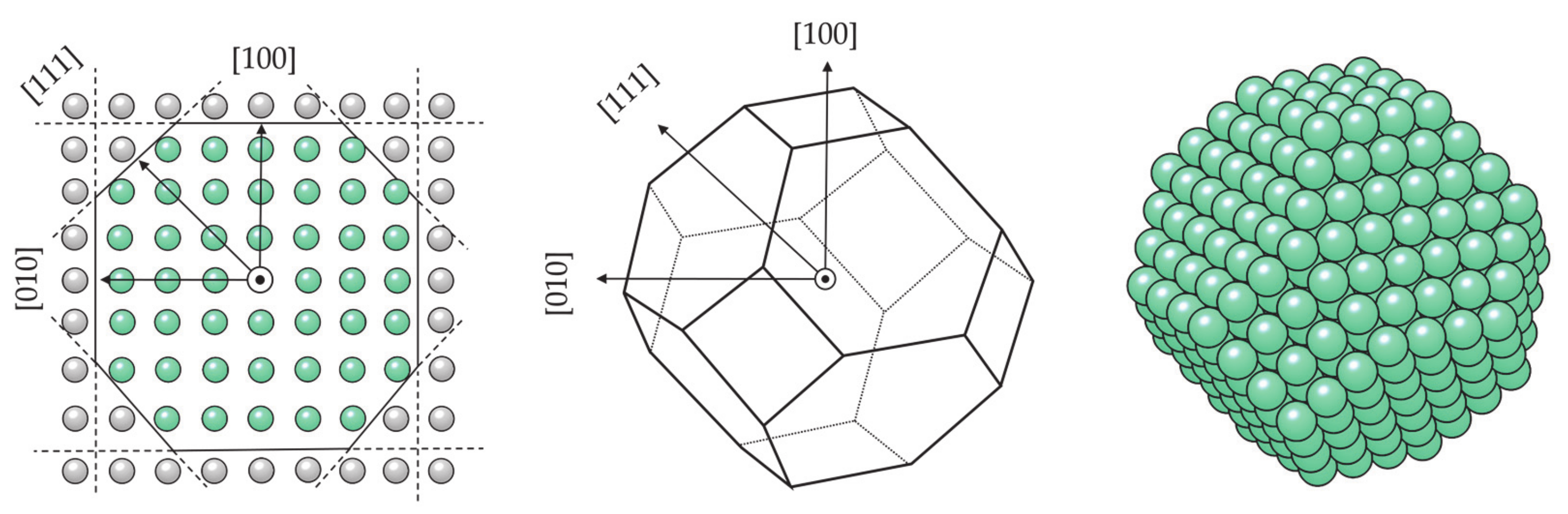
- Farkaš, B.; de Leeuw, N.H. Towards a morphology of cobalt nanoparticles: Size and strain effects. Nanotechnology 2020, 31, 195711. [Google Scholar] [CrossRef]
- Souto-Casares, J.; Sakurai, M.; Chelikowsky, J.R. Structural and magnetic properties of large cobalt clusters. Phys. Rev. B 2016, 93, 174418. [Google Scholar] [CrossRef]
- Fernando, A.; Weerawardene, K.L.D.M.; Karimova, N.V.; Aikens, C.M. Quantum Mechanical Studies of Large Metal, Metal Oxide, and Metal Chalcogenide Nanoparticles and Clusters. Chem. Rev. 2015, 115, 6112–6216. [Google Scholar] [CrossRef]
- Mark, L.O.; Zhu, C.; Medlin, J.W.; Heinz, H. Understanding the Surface Reactivity of Ligand-Protected Metal Nanoparticles for Biomass Upgrading. ACS Catal. 2020, 10, 5462–5474. [Google Scholar] [CrossRef]
- de Morais, R.F.; Kerber, T.; Calle-Vallejo, F.; Sautet, P.; Loffreda, D. Capturing Solvation Effects at a Liquid/Nanoparticle Interface by Ab Initio Molecular Dynamics: Pt201 Immersed in Water. Small 2016, 12, 5312–5319. [Google Scholar] [CrossRef]
- Kaddi, C.D.; Phan, J.H.; Wang, M.D. Computational nanomedicine: Modeling of nanoparticle-mediated hyperthermal cancer therapy. Nanomedicine 2013, 8, 1323–1333. [Google Scholar] [CrossRef]
- Nabil, M.; Decuzzi, P.; Zunino, P. Modelling mass and heat transfer in nano-based cancer hyperthermia. R. Soc. Open Sci. 2015, 2, 150447. [Google Scholar] [CrossRef]
- Ng, E.Y.K.; Kumar, S.D. Physical mechanism and modeling of heat generation and transfer in magnetic fluid hyperthermia through Néelian and Brownian relaxation: A review. Biomed. Eng. Online 2017, 16, 36. [Google Scholar] [CrossRef]
- Muscas, G.; Trohidou, K.N.; Peddis, D.; Vasilakaki, M.; Yaacoub, N.; Ntallis, N. Optimising the magnetic performance of Co ferrite nanoparticles via organic ligand capping. Nanoscale 2018, 10, 21244–21253. [Google Scholar] [CrossRef]
- Andreu, I.; Natividad, E. Accuracy of available methods for quantifying the heat power generation of nanoparticles for magnetic hyperthermia. Int. J. Hyperth. 2013, 29, 739–751. [Google Scholar] [CrossRef]
- Michalakis, J.; Georgatos, S.D.; De Bree, E.; Polioudaki, H.; Romanos, J.; Georgoulias, V.; Tsiftsis, D.D.; Theodoropoulos, P.A. Short-term exposure of cancer cells to micromolar doses of paclitaxel, with or without hyperthermia, induces long-term inhibition of cell proliferation and cell death in vitro. Ann. Surg. Oncol. 2007, 14, 1220–1228. [Google Scholar] [CrossRef]
- Yang, K.L.; Huang, C.C.; Chi, M.S.; Chiang, H.C.; Wang, Y.S.; Hsia, C.C.; Andocs, G.; Wang, H.E.; Chi, K.H. In vitro comparison of conventional hyperthermia and modulated electro-hyperthermia. Oncotarget 2016, 7, 84082–84092. [Google Scholar] [CrossRef]
- Mohamed, F.; Stuart, O.A.; Glehen, O.; Urano, M.; Sugarbaker, P.H. Docetaxel and hyperthermia: Factors that modify thermal enhancement. J. Surg. Oncol. 2004, 88, 14–20. [Google Scholar] [CrossRef]
- Rives, S.; Catherinot, A.; Dumas-Bouchiat, F.; Champeaux, C.; Videcoq, A.; Ferrando, R. Growth of Co isolated clusters in the gas phase: Experiment and molecular dynamics simulations. Phys. Rev. B Condens. Matter Mater. Phys. 2008, 77, 085407. [Google Scholar] [CrossRef]
- Rodríguez-López, J.L.; Aguilera-Granja, F.; Michaelian, K.; Vega, A. Structure and magnetism of cobalt clusters. Phys. Rev. B Condens. Matter Mater. Phys. 2003, 67, 174413. [Google Scholar] [CrossRef]
- Datta, S.; Kabir, M.; Ganguly, S.; Sanyal, B.; Saha-Dasgupta, T.; Mookerjee, A. Structure, bonding, and magnetism of cobalt clusters from first-principles calculations. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 76, 014429. [Google Scholar] [CrossRef]
- Harada, M.; Dexpert, H. Electronic structure of transition metal clusters from density functional theory. 1. Transition metal dimers. J. Phys. Chem. 1996, 100, 565–572. [Google Scholar] [CrossRef]
- Reddy, B.V.; Nayak, S.K.; Khanna, S.N.; Rao, B.K.; Jena, P. Physics of nickel clusters. 2. Electronic structure and magnetic properties. J. Phys. Chem. A 1998, 102, 1748–1759. [Google Scholar] [CrossRef]
- Derosa, P.A.; Seminario, J.M.; Balbuena, P.B. Properties of small bimetallic Ni-Cu clusters. J. Phys. Chem. A 2001, 105, 7917–7925. [Google Scholar] [CrossRef]
- Jain, P.K. A DFT-based study of the low-energy electronic structures and properties of small gold clusters. Struct. Chem. 2005, 16, 421–426. [Google Scholar] [CrossRef]
- Efremenko, I.; Sheintuch, M. DFT study of small bimetallic palladium-copper clusters. Chem. Phys. Lett. 2005, 401, 232–240. [Google Scholar] [CrossRef]
- Ma, Q.M.; Xie, Z.; Wang, J.; Liu, Y.; Li, Y.C. Structures, binding energies and magnetic moments of small iron clusters: A study based on all-electron DFT. Solid State Commun. 2007, 142, 114–119. [Google Scholar] [CrossRef]
- Zanti, G.; Peeters, D. DFT study of small palladium clusters Pdn and their interaction with a CO ligand (n = 1-9). Eur. J. Inorg. Chem. 2009, 3904–3911. [Google Scholar] [CrossRef]
- Katakuse, I.; Ichihara, T.; Fujita, Y.; Matsuo, T.; Sakurai, T.; Matsuda, H. Mass distributions of copper, silver and gold clusters and electronic shell structure. Int. J. Mass Spectrom. Ion Process. 1985, 67, 229–236. [Google Scholar] [CrossRef]
- de Heer, W.A.; Knight, W.D.; Chou, M.Y.; Cohen, M.L. Electronic Shell Structure and Metal Clusters. Solid State Phys. Adv. Res. Appl. 1987, 40, 93–181. [Google Scholar] [CrossRef]
- Martin, T.P.; Bergmann, T.; Göhlich, H.; Lange, T. Shell structure of clusters. J. Phys. Chem. 1991, 95, 6421–6429. [Google Scholar] [CrossRef]
- Jena, P.; Castleman, A.W. Clusters: A bridge across the disciplines of physics and chemistry. Proc. Natl. Acad. Sci. USA 2006, 103, 10560–10569. [Google Scholar] [CrossRef]
- Martins, M.; Wurth, W. Magnetic properties of supported metal atoms and clusters. J. Phys. Condens. Matter 2016, 28, 503002. [Google Scholar] [CrossRef]
- Zamudio-Bayer, V.; Hirsch, K.; Langenberg, A.; Ławicki, A.; Terasaki, A.; Von Issendorff, B.; Lau, J.T. Large orbital magnetic moments of small, free cobalt cluster ions Co+n with n ≤ 9. J. Phys. Condens. Matter 2018, 30, 464002. [Google Scholar] [CrossRef]
- Bucher, J.P.; Douglass, D.C.; Bloomfield, L.A. Magnetic properties of free cobalt clusters. Phys. Rev. Lett. 1991, 66, 3052–3055. [Google Scholar] [CrossRef]
- Jensen, P.J.; Bennemann, K.H. Magnetic properties of free ferromagnetic clusters in a Stern-Gerlach magnet. Comput. Mater. Sci. 1994, 2, 488–490. [Google Scholar] [CrossRef]
- Knickelbein, M.B. Magnetic moments of bare and benzene-capped cobalt clusters. J. Chem. Phys. 2006, 125, 044308. [Google Scholar] [CrossRef]
- Xu, X.; Yin, S.; Moro, R.; de Heer, W.A. Magnetic Moments and Adiabatic Magnetization of Free Cobalt Clusters. Phys. Rev. Lett. 2005, 95, 237209. [Google Scholar] [CrossRef]
- Nealon, G.L.; Donnio, B.; Greget, R.; Kappler, J.P.; Terazzi, E.; Gallani, J.L. Magnetism in gold nanoparticles. Nanoscale 2012, 4, 5244–5258. [Google Scholar] [CrossRef]
- Swart, I.; De Groot, F.M.F.; Weckhuysen, B.M.; Gruene, P.; Meijer, G.; Fielicke, A. H2 adsorption on 3d transition metal clusters: A combined infrared spectroscopy and density functional study. J. Phys. Chem. A 2008, 112, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Sauer, J. Molecular Models in ab Initio Studies of Solids and Surfaces: From Ionic Crystals and Semiconductors to Catalysts. Chem. Rev. 1989, 89, 199–255. [Google Scholar] [CrossRef]
- Wang, G.C.; Jiang, L.; Morikawa, Y.; Nakamura, J.; Cai, Z.S.; Pan, Y.M.; Zhao, X.Z. Cluster and periodic DFT calculations of adsorption and activation of CO2 on the Cu(hkl) surfaces. Surf. Sci. 2004, 570, 205–217. [Google Scholar] [CrossRef]
- Pessoa, A.M.; Fajín, J.L.C.; Gomes, J.R.B.; Cordeiro, M.N.D.S. Cluster and periodic DFT calculations of adsorption of hydroxyl on the Au(h k l) surfaces. J. Mol. Struct. THEOCHEM 2010, 946, 43–50. [Google Scholar] [CrossRef]
- Psofogiannakis, G.; St-Amant, A.; Ternan, M. Methane oxidation mechanism on Pt(111): A cluster model DFT study. J. Phys. Chem. B 2006, 110, 24593–24605. [Google Scholar] [CrossRef]
- Zhenming, H.; Boyd, R.J. Structure sensitivity and cluster size convergence for formate adsorption on copper surfaces: A DFT cluster model study. J. Chem. Phys. 2000, 112, 9562–9568. [Google Scholar] [CrossRef]
- Dehmani, Y.; Lgaz, H.; Alrashdi, A.A.; Lamhasni, T.; Abouarnadasse, S.; Chung, I.M. Phenol adsorption mechanism on the zinc oxide surface: Experimental, cluster DFT calculations, and molecular dynamics simulations. J. Mol. Liq. 2021, 324, 114993. [Google Scholar] [CrossRef]
- Tafreshi, S.S.; Roldan, A.; De Leeuw, N.H. Density Functional Theory Study of the Adsorption of Hydrazine on the Perfect and Defective Copper (100), (110), and (111) Surfaces. J. Phys. Chem. C 2014, 118, 26103–26114. [Google Scholar] [CrossRef]
- Liu, L.; Yu, M.; Hou, B.; Wang, Q.; Zhu, B.; Jia, L.; Li, D. Morphology evolution of fcc Ru nanoparticles under hydrogen atmosphere. Nanoscale 2019, 11, 8037–8046. [Google Scholar] [CrossRef]
- Zhao, P.; Cao, Z.; Liu, X.; Ren, P.; Cao, D.B.; Xiang, H.; Jiao, H.; Yang, Y.; Li, Y.W.; Wen, X.D. Morphology and Reactivity Evolution of HCP and FCC Ru Nanoparticles under CO Atmosphere. ACS Catal. 2019, 9, 2768–2776. [Google Scholar] [CrossRef]
- Shi, Q.; Sun, R. Adsorption manners of hydrogen on Pt(100), (110) and (111) surfaces at high coverage. Comput. Theor. Chem. 2017, 1106, 43–49. [Google Scholar] [CrossRef]
- Avanesian, T.; Dai, S.; Kale, M.J.; Graham, G.W.; Pan, X.; Christopher, P. Quantitative and Atomic-Scale View of CO-Induced Pt Nanoparticle Surface Reconstruction at Saturation Coverage via DFT Calculations Coupled with in Situ TEM and IR. J. Am. Chem. Soc. 2017, 139, 4551–4558. [Google Scholar] [CrossRef] [PubMed]
- Soon, A.; Wong, L.; Delley, B.; Stampfl, C. Morphology of copper nanoparticles in a nitrogen atmosphere: A first-principles investigation. Phys. Rev. B Condens. Matter Mater. Phys. 2008, 77, 125423. [Google Scholar] [CrossRef]
- Deshlahra, P.; Conway, J.; Wolf, E.E.; Schneider, W.F. Influence of Dipole–Dipole Interactions on Coverage-Dependent Adsorption: CO and NO on Pt(111). Langmuir 2012, 28, 8408–8417. [Google Scholar] [CrossRef] [PubMed]
- Bessarab, P.F.; Uzdin, V.M.; Jónsson, H. Effect of hydrogen adsorption on the magnetic properties of a surface nanocluster of iron. Phys. Rev. B Condens. Matter Mater. Phys. 2013, 88, 14–16. [Google Scholar] [CrossRef]
- Ram, S.; Lee, S.C.; Bhattacharjee, S. Adsorption energy scaling relation on bimetallic magnetic surfaces: Role of surface magnetic moments. Phys. Chem. Chem. Phys. 2020, 22, 17960–17968. [Google Scholar] [CrossRef]
- Pignocco, A.J.; Pellissier, G.E. Leed studies of oxygen adsorption and oxide formation on an (011) iron surface. Surf. Sci. 1967, 7, 261–278. [Google Scholar] [CrossRef]
- Brundle, C.R. Oxygen adsorption and thin oxide formation at iron surfaces: An XPS/UPS study. Surf. Sci. 1977, 66, 581–595. [Google Scholar] [CrossRef]
- Holloway, P.H. Chemisorption and Oxide Formation on Metals: Oxygen-Nickel Reaction. J. Vac. Sci. Technol. 1980, 18, 653–659. [Google Scholar] [CrossRef]
- Li, W.X.; Stampfl, C.; Scheffler, M. Subsurface oxygen and surface oxide formation at Ag(111): A density-functional theory investigation. Phys. Rev. B Condens. Matter Mater. Phys. 2003, 67, 045408. [Google Scholar] [CrossRef]
- Todorova, M.; Reuter, K.; Scheffler, M. Oxygen overlayers on Pd(111) studied by density functional theory. J. Phys. Chem. B 2004, 108, 14477–14483. [Google Scholar] [CrossRef]
- Shi, H.; Stampfl, C. First-principles investigations of the structure and stability of oxygen adsorption and surface oxide formation at Au(111). Phys. Rev. B 2007, 76, 075327. [Google Scholar] [CrossRef]
- Bridge, M.E.; Lambert, R.M. Oxygen chemisorption, surface oxidation, and the oxidation of carbon monoxide on cobalt (0001). Surf. Sci. 1979, 82, 413–424. [Google Scholar] [CrossRef]
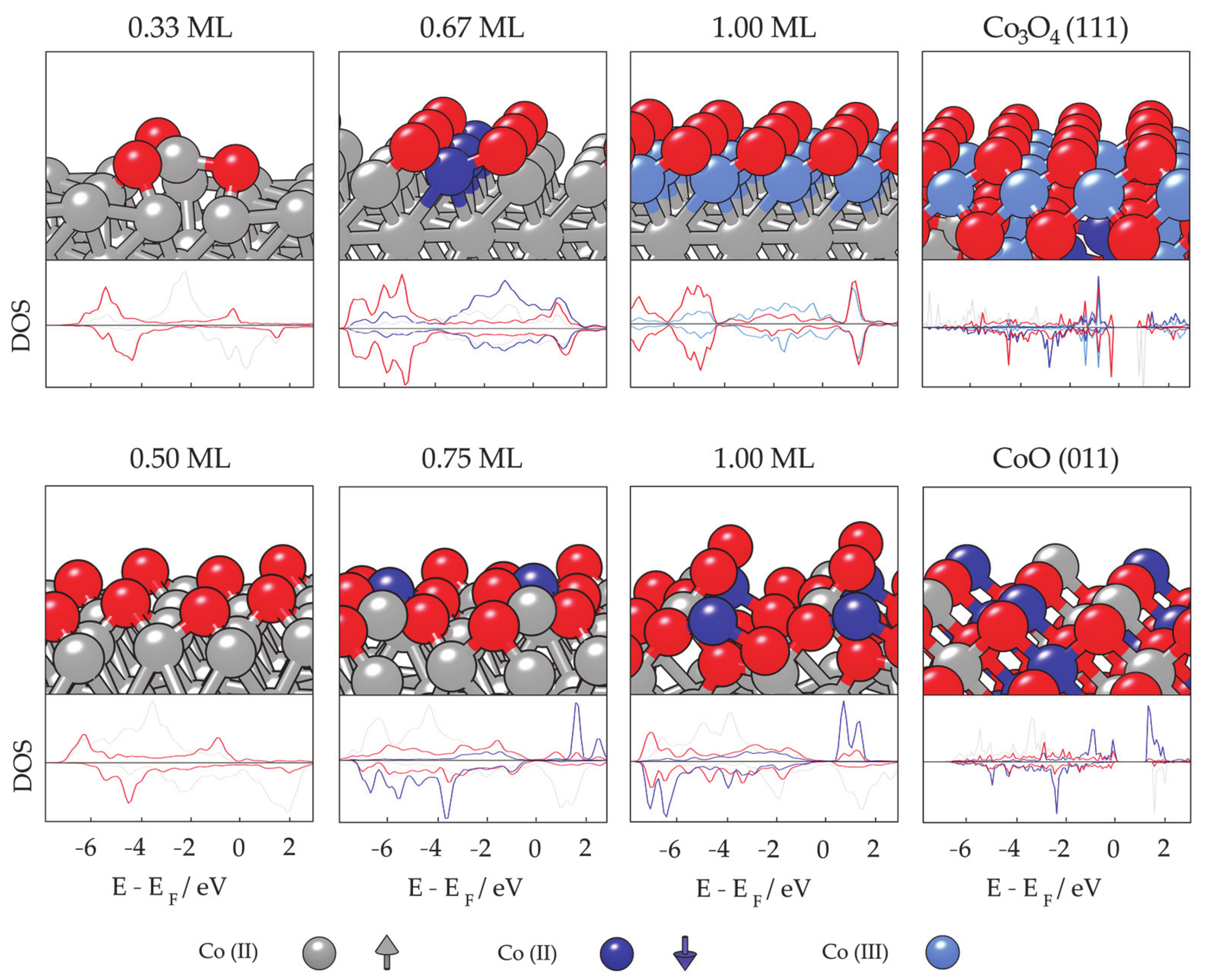
- Farkaš, B.; Santos-Carballal, D.; Cadi-Essadek, A.; de Leeuw, N.H. A DFT+U study of the oxidation of cobalt nanoparticles: Implications for biomedical applications. Materialia 2019, 7, 100381. [Google Scholar] [CrossRef]
- Wang, L.; Maxisch, T.; Ceder, G. Oxidation energies of transition metal oxides within the GGA+U framework. Phys. Rev. B Condens. Matter Mater. Phys. 2006, 73, 195107. [Google Scholar] [CrossRef]
- Toroker, M.C.; Kanan, D.K.; Alidoust, N.; Isseroff, L.Y.; Liao, P.; Carter, E.A. First principles scheme to evaluate band edge positions in potential transition metal oxide photocatalysts and photoelectrodes. Phys. Chem. Chem. Phys. 2011, 13, 16644–16654. [Google Scholar] [CrossRef]
- Getsoian, A.B.; Bell, A.T. The influence of functionals on density functional theory calculations of the properties of reducible transition metal oxide catalysts. J. Phys. Chem. C 2013, 117, 25562–25578. [Google Scholar] [CrossRef]
- Roth, W.L. Magnetic structures of MnO, FeO, CoO, and NiO. Phys. Rev. 1958, 110, 1333–1341. [Google Scholar] [CrossRef]
- Wdowik, U.D.; Parlinski, K. Lattice dynamics of CoO from first principles. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 75, 104306. [Google Scholar] [CrossRef]
- Roth, W.L. The magnetic structure of Co3O4. J. Phys. Chem. Solids 1964, 25, 1–10. [Google Scholar] [CrossRef]
- Scheerlinck, D.; Hautecler, S. Magnetic Interactions in Co3O4. Phys. Status Solidi 1976, 73, 223–228. [Google Scholar] [CrossRef]
- Lee, B.; Ignatiev, A.; Taylor, J.; Rabalais, J. Atomic structure sensitivity of XPS: The oxidation of cobalt. Solid State Commun. 1980, 33, 1205–1208. [Google Scholar] [CrossRef]
- Klingenberg, B.; Grellner, F.; Borgmann, D.; Wedler, G. Oxygen adsorption and oxide formation on Co(1120). Surf. Sci. 1993, 296, 374–382. [Google Scholar] [CrossRef]
- Van Vleck, J.H. On the anisotropy of cubic ferromagnetic crystals. Phys. Rev. 1937, 52, 1178–1198. [Google Scholar] [CrossRef]
- Brooks, H. Ferromagnetic Anisotropy and the Itinerant Electron Model. Phys. Rev. 1940, 58, 909–918. [Google Scholar] [CrossRef]
- Fletcher, G.C. Calculations of the first ferromagnetic anisotropy coefficient, gyromagnetic ratio and spectroscopic splitting factor for nickel. Proc. Phys. Soc. Sect. A 1954, 67, 505–519. [Google Scholar] [CrossRef]
- Daalderop, G.H.O.; Kelly, P.J.; Schuurmans, M.F.H. First-principles calculation of the magnetic anisotropy energy of (Co)n/(X)m multilayers. Phys. Rev. B 1990, 42, 7270–7273. [Google Scholar] [CrossRef]
- Daalderop, G.H.O.; Kelly, P.J.; Den Broeder, F.J.A. Prediction and confirmation of perpendicular magnetic anisotropy in Co/Ni multilayers. Phys. Rev. Lett. 1992, 68, 682–685. [Google Scholar] [CrossRef]
- Berger, A.; Hopster, H. Nonequilibrium Magnetization near the Reorientation Phase Transition of Fe/Ag(100) Films. Phys. Rev. Lett. 1996, 76, 519–522. [Google Scholar] [CrossRef]
- Pütter, S.; Millev, Y.T.; Ding, H.F.; Kirschner, J.; Oepen, H.P. Magnetic susceptibility: An easy approach to the spin-reorientation transition. Phys. Rev. B Condens. Matter Mater. Phys. 2001, 64, 092409. [Google Scholar] [CrossRef]
- O’Brien, W.; Droubay, T.; Tonner, B. Transitions in the direction of magnetism in Ni/Cu(001) ultrathin films and the effects of capping layers. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 9297–9303. [Google Scholar] [CrossRef]
- Młyńczak, E.; Aguilera, I.; Gospodarič, P.; Heider, T.; Jugovac, M.; Zamborlini, G.; Tusche, C.; Suga, S.; Feyer, V.; Blügel, S.; et al. Spin-polarized quantized electronic structure of Fe(001) with symmetry breaking due to the magnetization direction. Phys. Rev. B 2021, 103, 035134. [Google Scholar] [CrossRef]
- Bouhassoune, M.; Zimmermann, B.; Mavropoulos, P.; Wortmann, D.; Dederichs, P.H.; Blügel, S.; Lounis, S. Quantum well states and amplified spin-dependent Friedel oscillations in thin films. Nat. Commun. 2014, 5, 5558. [Google Scholar] [CrossRef]
- Mlynczak, E.; Eschbach, M.; Borek, S.; Minár, J.; Braun, J.; Aguilera, I.; Bihlmayer, G.; Döring, S.; Gehlmann, M.; Gospodaric, P.; et al. Fermi surface manipulation by external magnetic field demonstrated for a prototypical ferromagnet. Phys. Rev. X 2016, 6, 041048. [Google Scholar] [CrossRef]
- Pan, M.; He, K.; Zhang, L.; Jia, J.; Xue, Q.; Kim, W.; Qiu, Z.Q. Structure and magnetism of ultrathin Co film grown on Pt(100). J. Vac. Sci. Technol. A Vacuum Surf. Film 2005, 23, 790–795. [Google Scholar] [CrossRef]
- Yokoyama, T.; Matsumura, D.; Amemiya, K.; Kitagawa, S.; Suzuki, N.; Ohta, T. Spin reorientation transitions of ultrathin Co/Pd(111) films induced by chemisorption: X-ray magnetic circular dichroism study. J. Phys. Condens. Matter 2003, 15, S537. [Google Scholar] [CrossRef]
- Boukari, S.; Beaurepaire, E.; Bulou, H.; Carrière, B.; Deville, J.P.; Scheurer, F.; De Santis, M.; Baudoing-Savois, R. Influence of strain on the magnetocrystalline anisotropy in epitaxial Cr/Co/Pd(111) films. Phys. Rev. B Condens. Matter Mater. Phys. 2001, 64, 1444311–1444314. [Google Scholar] [CrossRef]
- Quirós, C.; Valvidares, S.M.; Robach, O.; Ferrer, S. Low-temperature growth favours hcp structure, flatness and perpendicular magnetic anisotropy of thin (1-5 nm) Co films on Pt(111). J. Phys. Condens. Matter 2005, 17, 5551–5561. [Google Scholar] [CrossRef]
- Zhang, H. Relativistic Density Functional Treatment of Magnetic Anisotropy. Ph.D. Thesis, Technische Universitat Dresden, Dresden, Germany, 2009. [Google Scholar]
- Paige, D.M.; Szpunar, B.; Tanner, B.K. The magnetocrystalline anisotropy of cobalt. J. Magn. Magn. Mater. 1984, 44, 239–248. [Google Scholar] [CrossRef]
- Dorantes-Dávila, J.; Dreyssé, H. Magnetic anisotropy of close-packed (111) ultrathin transition-metal films:Role of interlayer packing. Phys. Rev. B Condens. Matter Mater. Phys. 1997, 55, 15033–15042. [Google Scholar] [CrossRef]
- Chappert, C.; Bruno, P. Magnetic anisotropy in metallic ultrathin films and related experiments on cobalt films (invited). J. Appl. Phys. 1988, 64, 5736–5741. [Google Scholar] [CrossRef]
- Suzuki, T.; Weller, D.; Chang, C.A.; Savoy, R.; Huang, T.; Gurney, B.A.; Speriosu, V. Magnetic and magneto-optic properties of thick face-centered-cubic Co single-crystal films. Appl. Phys. Lett. 1994, 64, 2736–2738. [Google Scholar] [CrossRef]
- El Gabaly, F.; Gallego, S.; Muñoz, C.; Szunyogh, L.; Weinberger, P.; Klein, C.; Schmid, A.K.; McCarty, K.F.; De La Figuera, J. Imaging spin-reorientation transitions in consecutive atomic Co layers on Ru(0001). Phys. Rev. Lett. 2006, 96, 147202. [Google Scholar] [CrossRef]
- Przybylski, M.; Yan, L.; Zukrowski, J.; Nyvlt, M.; Shi, Y.; Winkelmann, A.; Barthel, J.; Waśniowska, M.; Kirschner, J. Topology-dependent interface contribution to magneto-optical response from ultrathin Co films grown on the (001), (110), and (111) surfaces of Pd. Phys. Rev. B Condens. Matter Mater. Phys. 2006, 73, 085413. [Google Scholar] [CrossRef]
- Szunyogh, L.; Újfalussy, B.; Blaas, C.; Pustogowa, U.; Sommers, C.; Weinberger, P. Oscillatory behavior of the magnetic anisotropy energy in multilayer systems. Phys. Rev. B Condens. Matter Mater. Phys. 1997, 56, 14036–14044. [Google Scholar] [CrossRef]
- Nicolas, G.; Dorantes-Dávila, J.; Pastor, G.M. Orbital polarization effects on the magnetic anisotropy and orbital magnetism of clusters, films, and surfaces: A comparative study within tight-binding theory. Phys. Rev. B Condens. Matter Mater. Phys. 2006, 74, 014415. [Google Scholar] [CrossRef]
- El Gabaly, F.; McCarty, K.F.; Schmid, A.K.; De La Figuera, J.; Muñoz, M.C.; Szunyogh, L.; Weinberger, P.; Gallego, S. Noble metal capping effects on the spin-reorientation transitions of Co/Ru(0001). New J. Phys. 2008, 10, 073024. [Google Scholar] [CrossRef][Green Version]
- Buruzs, Á.; Weinberger, P.; Szunyogh, L.; Udvardi, L.; Chleboun, P.I.; Fischer, A.M.; Staunton, J.B. Ab initio theory of temperature dependence of magnetic anisotropy in layered systems: Applications to thin Co films on Cu(100). Phys. Rev. B Condens. Matter Mater. Phys. 2007, 76, 064417. [Google Scholar] [CrossRef]
- Bruno, P. Magnetic surface anisotropy of cobalt and surface roughness effects within Neel’s model. J. Phys. F Met. Phys. 1988, 18, 1291–1298. [Google Scholar] [CrossRef]
- Campiglio, P.; Breitwieser, R.; Repain, V.; Guitteny, S.; Chacon, C.; Bellec, A.; Lagoute, J.; Girard, Y.; Rousset, S.; Sassella, A.; et al. Change of cobalt magnetic anisotropy and spin polarization with alkanethiolates self-assembled monolayers. New J. Phys. 2015, 17, 063022. [Google Scholar] [CrossRef][Green Version]
- Lehnert, A.; Dennler, S.; Błoński, P.; Rusponi, S.; Etzkorn, M.; Moulas, G.; Bencok, P.; Gambardella, P.; Brune, H.; Hafner, J. Magnetic anisotropy of Fe and Co ultrathin films deposited on Rh(111) and Pt(111) substrates: An experimental and first-principles investigation. Phys. Rev. B Condens. Matter Mater. Phys. 2010, 82, 094409. [Google Scholar] [CrossRef]
- Kebaili, A.S.; Blizak, S.; Bihlmayer, G.; Blügel, S. Magnetic properties of ultra-thin (Fe, Co) films coupled by Ir(001) spacers. Phys. B Condens. Matter 2020, 596, 412395. [Google Scholar] [CrossRef]
- Jia, H.; Zimmermann, B.; Hoffmann, M.; Sallermann, M.; Bihlmayer, G.; Blügel, S. Material systems for FM-/AFM-coupled skyrmions in Co/Pt-based multilayers. Phys. Rev. Mater. 2020, 4, 122405. [Google Scholar] [CrossRef]
- Di, N.; Kubal, J.; Zeng, Z.; Greeley, J.; Maroun, F.; Allongue, P. Influence of controlled surface oxidation on the magnetic anisotropy of Co ultrathin films. Appl. Phys. Lett. 2015, 106, 122405. [Google Scholar] [CrossRef]
- Dinghas, A. Über einen geometrischen Satz von Wulff für die Gleichgewichtsform von Kristallen. Z. Krist. Cryst. Mater. 1943, 105, 304–314. [Google Scholar] [CrossRef]
- Herring, C. Some Theorems on the Free Energies of Crystal Surfaces. Phys. Rev. 1951, 82, 87–93. [Google Scholar] [CrossRef]
- Fonseca, I. The Wulff Theorem Revisited. Proc. R. Soc. A Math. Phys. Eng. Sci. 1991, 432, 125–145. [Google Scholar] [CrossRef]
- Marks, L.D. Experimental studies of small particle structures. Rep. Prog. Phys. 1994, 57, 603–649. [Google Scholar] [CrossRef]
- Graoui, H.; Giorgio, S.; Enry, C.R. Effect of the interface structure on the high-temperature morphology of supported metal clusters. Philos. Mag. B Phys. Condens. Matter. Stat. Mech. Electron. Opt. Magn. Prop. 2001, 81, 1649–1658. [Google Scholar] [CrossRef]
- Mackay, A.L. A dense non-crystallographic packing of equal spheres. Acta Crystallogr. 1962, 15, 916–918. [Google Scholar] [CrossRef]
- Ino, S. Epitaxial Growth of Metals on Rocksalt Faces Cleaved in Vacuum. II. Orientation and Structure of Gold Particles Formed in Ultrahigh Vacuum. J. Phys. Soc. Jpn. 1966, 21, 346–362. [Google Scholar] [CrossRef]
- Ino, S. Stability of Multiply-Twinned Particles. J. Phys. Soc. Jpn. 1969, 27, 941–953. [Google Scholar] [CrossRef]
- Howie, A.; Marks, L.D. Multiply Twinned Particles. Oyobuturi 1984, 41, 388–397. [Google Scholar] [CrossRef]
- Teo, B.K.; Sloane, N.J.A. Magic Numbers in Polygonal and Polyhedral. Inorg. Chem. 1985, 4545–4558. [Google Scholar] [CrossRef]
- Kaatz, F.H.; Bultheel, A. Magic Mathematical Relationships for Nanoclusters. Nanoscale Res. Lett. 2019, 14, 150. [Google Scholar] [CrossRef]
- Cleveland, C.L.; Landman, U. The energetics and structure of nickel clusters: Size dependence. J. Chem. Phys. 1991, 94, 7376–7396. [Google Scholar] [CrossRef]
- Baletto, F.; Ferrando, R. Structural properties of nanoclusters: Energetic, thermodynamic, and kinetic effects. Rev. Mod. Phys. 2005, 77, 371–423. [Google Scholar] [CrossRef]
- Gafner, Y.Y.; Gafner, S.L.; Golonenko, Z.V.; Redel, L.V.; Khrustalev, V.I. Formation of structure in Au, Cu and Ni nanoclusters: MD simulations. IOP Conf. Ser. Mater. Sci. Eng. 2016, 110, 012015. [Google Scholar] [CrossRef]
- Rahm, J.M.; Erhart, P. Beyond Magic Numbers: Atomic Scale Equilibrium Nanoparticle Shapes for Any Size. Nano Lett. 2017, 17, 5775–5781. [Google Scholar] [CrossRef]
- Garden, A.L.; Pedersen, A.; Jónsson, H. Reassignment of “magic numbers” for Au clusters of decahedral and FCC structural motifs. Nanoscale 2018, 10, 5124–5132. [Google Scholar] [CrossRef]
- Liu, X.; Tian, D.; Meng, C. DFT study on the adsorption and dissociation of H2 on Pdn (n = 4, 6, 13, 19, 55) clusters. J. Mol. Struct. 2015, 1080, 105–110. [Google Scholar] [CrossRef]
- Yudanov, I.V.; Genest, A.; Schauermann, S.; Freund, H.J.; Rösch, N. Size Dependence of the adsorption energy of CO on metal nanoparticles: A DFT search for the minimum value. Nano Lett. 2012, 12, 2134–2139. [Google Scholar] [CrossRef]
- Laletina, S.S.; Mamatkulov, M.; Shor, E.A.; Kaichev, V.V.; Genest, A.; Yudanov, I.V.; Rösch, N. Size-Dependence of the Adsorption Energy of CO on Pt Nanoparticles: Tracing Two Intersecting Trends by DFT Calculations. J. Phys. Chem. C 2017, 121, 17371–17377. [Google Scholar] [CrossRef]
- Yang, Y.; Yu, H.; Cai, Y.; Ferrando, R.; Cheng, D. Origin of enhanced stability and oxygen adsorption capacity of medium-sized Pt-Ni nanoclusters. J. Phys. Condens. Matter 2018, 30, 285503. [Google Scholar] [CrossRef]
- Verga, L.G.; Aarons, J.; Sarwar, M.; Thompsett, D.; Russell, A.E.; Skylaris, C.K. DFT calculation of oxygen adsorption on platinum nanoparticles: Coverage and size effects. Faraday Discuss. 2018, 208, 497–522. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, J.-X.; Zijlstra, B.; Filot, I.A.W.; Zhou, Z.; Sun, S.; Hensen, E.J.M. Optimum Cu nanoparticle catalysts for CO2 hydrogenation towards methanol. Nano Energy 2018, 43, 200–209. [Google Scholar] [CrossRef]
- Zhang, R.; Xue, M.; Wang, B.; Ling, L. Acetylene selective hydrogenation over different size of Pd-modified Cu cluster catalysts: Effects of Pd ensemble and cluster size on the selectivity and activity. Appl. Surf. Sci. 2019, 481, 421–432. [Google Scholar] [CrossRef]
- Xia, Z.; Zhang, S.; Liu, F.; Ma, Y.; Qu, Y.; Wu, C. Size-Dependent Adsorption of Styrene on Pd Clusters: A Density Functional Theory Study. J. Phys. Chem. C 2019, 123, 2182–2188. [Google Scholar] [CrossRef]
- Farkaš, B.; Terranova, U.; de Leeuw, N.H. Binding modes of carboxylic acids on cobalt nanoparticles. Phys. Chem. Chem. Phys. 2020, 22, 985–996. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Martínez, J.I.; García-Lastra, J.M.; Sautet, P.; Loffreda, D. Fast Prediction of Adsorption Properties for Platinum Nanocatalysts with Generalized Coordination Numbers. Angew. Chemie Int. Ed. 2014, 53, 8316–8319. [Google Scholar] [CrossRef]
- Simmons, G.W.; Wang, Y.N.; Marcos, J.; Klier, K. Oxygen adsorption on palladium(100) surface: Phase transformations and surface reconstruction. J. Phys. Chem. 1991, 95, 4522–4528. [Google Scholar] [CrossRef]
- Koch, R.; Schwarz, E.; Schmidt, K.; Burg, B.; Christmann, K.; Rieder, K.H. Oxygen adsorption on Co(1010): Different reconstruction behavior of hcp (1010) and fcc(110). Phys. Rev. Lett. 1993, 71, 1047–1050. [Google Scholar] [CrossRef]
- Besenbacher, F.; Nørskov, J.K. Oxygen chemisorption on metal surfaces: General trends for Cu, Ni and Ag. Prog. Surf. Sci. 1993, 44, 5–66. [Google Scholar] [CrossRef]
- Jensen, F.; Besenbacher, F.; Laegsgaard, E.; Stensgaard, I. Surface reconstruction of Cu(110) induced by oxygen chemisorption. Phys. Rev. B 1990, 41, 10233–10236. [Google Scholar] [CrossRef]
- Merrick, I.; Inglesfield, J.E.; Ishida, H. Electronic structure and surface reconstruction of adsorbed oxygen on copper(001). Surf. Sci. 2004, 551, 158–170. [Google Scholar] [CrossRef]
- Deskins, N.A.; Lauterbach, J.; Thomson, K.T. Lifting the Pt{100} surface reconstruction through oxygen adsorption: A density functional theory analysis. J. Chem. Phys. 2005, 122, 184709. [Google Scholar] [CrossRef]
- Ciobîcǎ, I.M.; van Santen, R.A.; van Berge, P.J.; van de Loosdrecht, J. Adsorbate induced reconstruction of cobalt surfaces. Surf. Sci. 2008, 602, 17–27. [Google Scholar] [CrossRef]
- Oyarzún, S.; Tamion, A.; Tournus, F.; Dupuis, V.; Hillenkamp, M. Size effects in the magnetic anisotropy of embedded cobalt nanoparticles: From shape to surface. Sci. Rep. 2015, 5, 16–21. [Google Scholar] [CrossRef]
- Wernsdorfer, W.; Thirion, C.; Demoncy, N.; Pascard, H.; Mailly, D. Magnetisation reversal by uniform rotation (Stoner-Wohlfarth model) in FCC cobalt nanoparticles. J. Magn. Magn. Mater. 2002, 242–245, 132–138. [Google Scholar] [CrossRef]
- Balan, A.; Derlet, P.M.; Rodríguez, A.F.; Bansmann, J.; Yanes, R.; Nowak, U.; Kleibert, A.; Nolting, F. Direct observation of magnetic metastability in individual iron nanoparticles. Phys. Rev. Lett. 2014, 112, 107201. [Google Scholar] [CrossRef]
- Chen, J.P.; Sorensen, C.M.; Klabunde, K.J. Enhanced Magnetization of Nanoscale Colloidal Cobalt Particles. Phys. Rev. B 1995, 51, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Jamet, M.; Dupuis, V.; Mélinon, P.; Guiraud, G.; Pérez, A.; Wernsdorfer, W. Structure and magnetism of well defined cobalt nanoparticles embedded in a niobium matrix. Phys. Rev. B Condens. Matter Mater. Phys. 2000, 62, 493–499. [Google Scholar] [CrossRef]
- He, L.; Chen, C. Effect of temperature-dependent shape anisotropy on coercivity for aligned Stoner-Wohlfarth soft ferromagnets. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 75, 184424. [Google Scholar] [CrossRef]
- Wang, Z.W.; Palmer, R.E. Determination of the ground-state atomic structures of size-selected Au nanoclusters by electron-beam-induced transformation. Phys. Rev. Lett. 2012, 108, 245502. [Google Scholar] [CrossRef]
- Tournus, F.; Sato, K.; Epicier, T.; Konno, T.J.; Dupuis, V. Multi-L10 domain CoPt and FePt nanoparticles revealed by electron microscopy. Phys. Rev. Lett. 2013, 110, 055501. [Google Scholar] [CrossRef]
- Masini, F.; Hernández-Fernández, P.; Deiana, D.; Strebel, C.E.; McCarthy, D.N.; Bodin, A.; Malacrida, P.; Stephens, I.; Chorkendorff, I. Exploring the phase space of time of flight mass selected PtxY nanoparticles. Phys. Chem. Chem. Phys. 2014, 16, 26506–26513. [Google Scholar] [CrossRef]
- Barke, I.; Hartmann, H.; Rupp, D.; Flückiger, L.; Sauppe, M.; Adolph, M.; Schorb, S.; Bostedt, C.; Treusch, R.; Peltz, C.; et al. The 3D-architecture of individual free silver nanoparticles captured by X-ray scattering. Nat. Commun. 2015, 6, 6187. [Google Scholar] [CrossRef]
- Morel, R.; Brenac, A.; Portemont, C.; Deutsch, T.; Notin, L. Magnetic anisotropy in icosahedral cobalt clusters. J. Magn. Magn. Mater. 2007, 308, 296–304. [Google Scholar] [CrossRef]
- Sato, H.; Kitakami, O.; Sakurai, T.; Shimada, Y.; Otani, Y.; Fukamichi, K.; Introduction, I. Structure and magnetism of hcp-Co fine particles. J. Appl. Phys. 1997, 81, 1858. [Google Scholar] [CrossRef]
- Jamet, M.; Wernsdorfer, W.; Thirion, C.; Mailly, D.; Dupuis, V.; Mélinon, P.; Pérez, A. Magnetic Anisotropy of a Single Cobalt Nanocluster. Phys. Rev. Lett. 2001, 86, 4676–4679. [Google Scholar] [CrossRef]
- Farkaš, B.; De Leeuw, N.H. Effect of coverage on the magnetic properties of -COOH, -SH, and -NH2 ligand-protected cobalt nanoparticles. Nanoscale 2021. [Google Scholar] [CrossRef]
- Pachón, L.D.; Rothenberg, G. Transition-metal nanoparticles: Synthesis, stability and the leaching issue. Appl. Organomet. Chem. 2008, 22, 288–299. [Google Scholar] [CrossRef]
- Cao, A.; Lu, R.; Veser, G. Stabilizing metal nanoparticles for heterogeneous catalysis. Phys. Chem. Chem. Phys. 2010, 12, 13499–13510. [Google Scholar] [CrossRef]
- Albrecht, W.; Bladt, E.; Vanrompay, H.; Smith, J.D.; Skrabalak, S.E.; Bals, S. Thermal Stability of Gold/Palladium Octopods Studied in Situ in 3D: Understanding Design Rules for Thermally Stable Metal Nanoparticles. ACS Nano 2019, 13, 6522–6530. [Google Scholar] [CrossRef]
- Respaud, M.; Broto, J.M.; Rakoto, H.; Fert, A.R.; Thomas, L.; Barbara, B.; Verelst, M.; Snoeck, E.; Lecante, P.; Mosset, A.; et al. Surface effects on the magnetic properties of ultrafine cobalt particles. Phys. Rev. B 1998, 57, 2925–2935. [Google Scholar] [CrossRef]
- van Leeuwen, D.A.; van Ruitenbeek, J.M.; de Jongh, L.J.; Ceriotti, A.; Pacchioni, G.; Häberlen, O.D.; Rösch, N. Quenching of Magnetic Moments by Ligand-Metal Interactions in Nanosized Magnetic Metal Clusters. Phys. Rev. Lett. 1994, 73, 1432–1435. [Google Scholar] [CrossRef]
- Chen, W.; Zhou, W.; He, L.; Chen, C.; Guo, L. Surface magnetic states of Ni nanochains modified by using different organic surfactants. J. Phys. Condens. Matter 2010, 22, 126003. [Google Scholar] [CrossRef]
- Jo, C.; Il Lee, J. Spin polarization and charge transfer of Co nanoclusters coated with CO molecules. J. Magn. Magn. Mater. 2009, 321, 47–51. [Google Scholar] [CrossRef]
- Cotton, F.A.; Meyers, M.D. Magnetic and Spectral Properties of the Spin-Free 3d6 Systems Iron(II) and Cobalt(III) in Cobalt(III) Hexafluoride Ion: Probable Observation of Dynamic Jahn–Teller Effects. J. Am. Chem. Soc. 1960, 82, 5023–5026. [Google Scholar] [CrossRef]
- Cotton, F.A.; Goodgame, D.M.L.; Goodgame, M. The Electronic Structures of Tetrahedral Cobalt(II) Complexes. J. Am. Chem. Soc. 1961, 83, 4690–4699. [Google Scholar] [CrossRef]
- Aakesson, R.; Pettersson, L.G.M.; Sandstroem, M.; Wahlgren, U. Ligand Field Effects in the Hydrated Divalent and Trivalent Metal Ions of the First and Second Transition Periods. J. Am. Chem. Soc. 1994, 116, 8691–8704. [Google Scholar] [CrossRef]
- Hartmann, M.J.; Millstone, J.E.; Häkkinen, H. Surface Chemistry Controls Magnetism in Cobalt Nanoclusters. J. Phys. Chem. C 2016, 120, 20822–20827. [Google Scholar] [CrossRef]
- Hartmann, M.J.; Millstone, J.E.; Häkkinen, H. Ligand mediated evolution of size dependent magnetism in cobalt nanoclusters. Phys. Chem. Chem. Phys. 2018, 20, 4563–4570. [Google Scholar] [CrossRef]
- Kuznetsov, A.A.; Leontiev, V.G.; Brukvin, V.A.; Vorozhtsov, G.N.; Kogan, B.Y.; Shlyakhtin, O.A.; Yunin, A.M.; Tsybin, O.I.; Kuznetsov, O.A. Local radiofrequency-induced hyperthermia using CuNi nanoparticles with therapeutically suitable Curie temperature. J. Magn. Magn. Mater. 2007, 311, 197–203. [Google Scholar] [CrossRef]
- Wijaya, A.; Brown, K.A.; Alper, J.D.; Hamad-Schifferli, K. Magnetic field heating study of Fe-doped Au nanoparticles. J. Magn. Magn. Mater. 2007, 309, 15–19. [Google Scholar] [CrossRef]
- Liu, H.L.; Wu, J.H.; Min, J.H.; Kim, Y.K. Synthesis of monosized magnetic-optical AuFe alloy nanoparticles. J. Appl. Phys. 2008, 103, 07D529. [Google Scholar] [CrossRef]
- Srinoi, P.; Chen, Y.T.; Vittur, V.; Marquez, M.D.; Lee, T.R. Bimetallic nanoparticles: Enhanced magnetic and optical properties for emerging biological applications; 2018; Vol. 8; ISBN 1713743272. Appl. Sci. 2018, 8, 1106. [Google Scholar] [CrossRef]
- Farkaš, B.; Perry, C.B.; Jones, G.; De Leeuw, N.H. Adsorbate-Induced Segregation of Cobalt from PtCo Nanoparticles: Modeling Au Doping and Core AuCo Alloying for the Improvement of Fuel Cell Cathode Catalysts. J. Phys. Chem. C 2020, 124, 18321–18334. [Google Scholar] [CrossRef]
- Wu, X.; Sun, Y.; Wei, Z.; Chen, T. Influence of noble metal dopants (M = Ag, Au, Pd or Pt) on the stable structures of bimetallic Co-M clusters. J. Alloys Compd. 2017, 701, 447–455. [Google Scholar] [CrossRef]
- Hall, R.C. Magnetic anisotropy and magnetostriction of ordered and disordered Cobalt-iron alloys. J. Appl. Phys. 1960, 157, 10–12. [Google Scholar] [CrossRef]
- Weller, D.; Brändle, H.; Gorman, G.; Lin, C.J.; Notarys, H. Magnetic and magneto-optical properties of cobalt-platinum alloys with perpendicular magnetic anisotropy. Appl. Phys. Lett. 1992, 61, 2726–2728. [Google Scholar] [CrossRef]
- Weller, D.; Brändle, H.; Chappert, C. Relationship between Kerr effect and perpendicular magnetic anisotropy in Co1-xPtx and Co1-xPdx alloys. J. Magn. Magn. Mater. 1993, 121, 461–470. [Google Scholar] [CrossRef]
- Huang, J.C.A.; Hsu, A.C.; Lee, Y.H.; Wu, T.H.; Lee, C.H. Influence of crystal structure on the perpendicular magnetic anisotropy of an epitaxial CoPt alloy. J. Appl. Phys. 1999, 85, 5977–5979. [Google Scholar] [CrossRef]
- Lin, C.J.; Gorman, G.L. Evaporated CoPt alloy films with strong perpendicular magnetic anisotropy. Appl. Phys. Lett. 1992, 61, 1600–1602. [Google Scholar] [CrossRef]
- Hashimoto, S.; Ochiai, Y.; Aso, K. Perpendicular magnetic anisotropy in sputtered copd alloy films. Jpn. J. Appl. Phys. 1989, 28, 1596–1599. [Google Scholar] [CrossRef]
- Shick, A.B.; Mryasov, O.N. Coulomb correlations and magnetic anisotropy in ordered (formula presented) CoPt and FePt alloys. Phys. Rev. B Condens. Matter Mater. Phys. 2003, 67, 172407. [Google Scholar] [CrossRef]
- Staunton, J.B.; Ostanin, S.; Razee, S.S.A.; Gyorffy, B.; Szunyogh, L.; Ginatempo, B.; Bruno, E. Long-range chemical order effects upon the magnetic anisotropy of FePt alloys from an ab initio electronic structure theory. J. Phys. Condens. Matter 2004, 16, S5623. [Google Scholar] [CrossRef]
- Burkert, T.; Nordström, L.; Eriksson, O.; Heinonen, O. Giant magnetic anisotropy in tetragonal FeCo alloys. Phys. Rev. Lett. 2004, 93, 027203. [Google Scholar] [CrossRef]
- Turek, I.; Kudrnovský, J.; Carva, K. Magnetic anisotropy energy of disordered tetragonal Fe-Co systems from ab initio alloy theory. Phys. Rev. B Condens. Matter Mater. Phys. 2012, 86, 174430. [Google Scholar] [CrossRef]
- Eurin, P.; Pauleve, J. Influence of thermomagnetic treatments on the magnetic properties of Co-Pt 50-50 alloy. IEEE Trans. Magn. 1969, 5, 216–219. [Google Scholar] [CrossRef]
- Tournus, F.; Blanc, N.; Tamion, A.; Dupuis, V.; Epicier, T. Coalescence-free L10 ordering of embedded CoPt nanoparticles. J. Appl. Phys. 2011, 109, 2009–2012. [Google Scholar] [CrossRef]
- Sun, X.; Jia, Z.Y.; Huang, Y.H.; Harrell, J.W.; Nikles, D.E.; Sun, K.; Wang, L.M. Synthesis and magnetic properties of CoPt nanoparticles. J. Appl. Phys. 2004, 95, 6747–6749. [Google Scholar] [CrossRef]
- Shevchenko, E.V.; Talapin, D.V.; Schnablegger, H.; Kornowski, A.; Festin, Ö.; Svedlindh, P.; Haase, M.; Weller, H. Study of nucleation and growth in the organometallic synthesis of magnetic alloy nanocrystals: The role of nucleation rate in size control of CoPt3 nanocrystals. J. Am. Chem. Soc. 2003, 125, 9090–9101. [Google Scholar] [CrossRef]
- Nairan, A.; Khan, U.; Iqbal, M.; Khan, M.; Javed, K.; Riaz, S.; Naseem, S.; Han, X. Structural and magnetic response in bimetallic core/shell magnetic nanoparticles. Nanomaterials 2016, 6, 72. [Google Scholar] [CrossRef]
- Li, J.; Sharma, S.; Liu, X.; Pan, Y.T.; Spendelow, J.S.; Chi, M.; Jia, Y.; Zhang, P.; Cullen, D.A.; Xi, Z.; et al. Hard-Magnet L10 -CoPt Nanoparticles Advance Fuel Cell Catalysis. Joule 2019, 3, 124–135. [Google Scholar] [CrossRef]
- Li, G.; Wang, Q.; Li, D.; Lü, X.; He, J. Structure evolution during the cooling and coalesced cooling processes of Cu-Co bimetallic clusters. Phys. Lett. Sect. A Gen. At. Solid State Phys. 2008, 372, 6764–6769. [Google Scholar] [CrossRef]
- Mejía-Rosales, S.J.; Fernández-Navarro, C.; Pérez-Tijerina, E.; Blom, D.A.; Allard, L.F.; José-Yacamán, M. On the structure of Au/Pd bimetallic nanoparticles. J. Phys. Chem. C 2007, 111, 1256–1260. [Google Scholar] [CrossRef]
- Rodríguez-López, J.L.; Montejano-Carrizales, J.M.; José-Yacamán, M. Molecular dynamics study of bimetallic nanoparticles: The case of Au x Cu y alloy clusters. Appl. Surf. Sci. 2003, 219, 56–63. [Google Scholar] [CrossRef]
- Yeo, S.C.; Kim, D.H.; Shin, K.; Lee, H.M. Phase diagram and structural evolution of Ag-Au bimetallic nanoparticles: Molecular dynamics simulations. Phys. Chem. Chem. Phys. 2012, 14, 2791–2796. [Google Scholar] [CrossRef]
- Rodríguez-Proenza, C.A.; Palomares-Báez, J.P.; Chávez-Rojo, M.A.; García-Ruiz, A.F.; Azanza-Ricardo, C.L.; Santoveña-Uribe, A.; Luna-Bárcenas, G.; Rodríguez-López, J.L.; Esparza, R. Atomic surface segregation and structural characterization of PdPt bimetallic nanoparticles. Materials 2018, 11, 1882. [Google Scholar] [CrossRef]
- Rojas-Nunez, J.; Gonzalez, R.I.; Bringa, E.M.; Allende, S.; Sepúlveda, P.; Arancibia-Miranda, N.; Baltazar, S.E. Toward Controlled Morphology of FeCu Nanoparticles: Cu Concentration and Size Effects. J. Phys. Chem. C 2018, 122, 8528–8534. [Google Scholar] [CrossRef]
- Kumar, S. Structural Evolution of Iron-Copper (Fe-Cu) Bimetallic Janus Nanoparticles during Solidification: An Atomistic Investigation. J. Phys. Chem. C 2019, 124, 1053–1063. [Google Scholar] [CrossRef]
- Dannenberg, A.; Gruner, M.E.; Hucht, A.; Entel, P. Surface energies of stoichiometric FePt and CoPt alloys and their implications for nanoparticle morphologies. Phys. Rev. B Condens. Matter Mater. Phys. 2009, 80, 245438. [Google Scholar] [CrossRef]
- Okamoto, H.; Massalski, T.B.; Nishizawa, T.; Hasebe, M. The Au-Co (Gold-Cobalt) system. Bull. Alloy Phase Diagrams 1985, 6, 449–454. [Google Scholar] [CrossRef]
- Bochicchio, D.; Ferrando, R. Morphological instability of core-shell metallic nanoparticles. Phys. Rev. B Condens. Matter Mater. Phys. 2013, 87, 165435. [Google Scholar] [CrossRef]
- Nelli, D.; Ferrando, R. Core-shell: Vs. multi-shell formation in nanoalloy evolution from disordered configurations. Nanoscale 2019, 11, 13040–13050. [Google Scholar] [CrossRef]
- Bao, Y.; Calderon, H.; Krishnan, K.M. Synthesis and characterization of magnetic-optical Co-Au core-shell nanoparticles. J. Phys. Chem. C 2007, 111, 1941–1944. [Google Scholar] [CrossRef]
- Mandal, S.; Krishnan, K.M. Co core Au shell nanoparticles: Evolution of magnetic properties in the displacement reaction. J. Mater. Chem. 2007, 17, 372–376. [Google Scholar] [CrossRef]
- Mayoral, A.; Llamosa, D.; Huttel, Y. A novel Co@Au structure formed in bimetallic core@shell nanoparticles. Chem. Commun. 2015, 51, 8442–8445. [Google Scholar] [CrossRef]
- Zhang, Z.; Nenoff, T.M.; Huang, J.Y.; Berry, D.T.; Provencio, P.P. Room temperature synthesis of thermally tmmiscible Ag-Ni nanoalloys. J. Phys. Chem. C 2009, 113, 1155–1159. [Google Scholar] [CrossRef]
- Kuo, C.C.; Li, C.Y.; Lee, C.H.; Li, H.C.; Li, W.H. Huge inverse magnetization generated by faraday induction in nano-sized Au@Ni Core@Shell nanoparticles. Int. J. Mol. Sci. 2015, 16, 20139–20151. [Google Scholar] [CrossRef]
- Zhang, D.F.; Zhang, Q.; Huang, W.F.; Guo, L.; Chen, W.M.; Chu, W.S.; Chen, C.; Wu, Z.Y. Low-temperature fabrication of Au-Co cluster mixed nanohybrids with high magnetic moment of Co. ACS Appl. Mater. Interfaces 2012, 4, 5643–5649. [Google Scholar] [CrossRef]


















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farkaš, B.; de Leeuw, N.H. A Perspective on Modelling Metallic Magnetic Nanoparticles in Biomedicine: From Monometals to Nanoalloys and Ligand-Protected Particles. Materials 2021, 14, 3611. https://doi.org/10.3390/ma14133611
Farkaš B, de Leeuw NH. A Perspective on Modelling Metallic Magnetic Nanoparticles in Biomedicine: From Monometals to Nanoalloys and Ligand-Protected Particles. Materials. 2021; 14(13):3611. https://doi.org/10.3390/ma14133611
Chicago/Turabian StyleFarkaš, Barbara, and Nora H. de Leeuw. 2021. "A Perspective on Modelling Metallic Magnetic Nanoparticles in Biomedicine: From Monometals to Nanoalloys and Ligand-Protected Particles" Materials 14, no. 13: 3611. https://doi.org/10.3390/ma14133611
APA StyleFarkaš, B., & de Leeuw, N. H. (2021). A Perspective on Modelling Metallic Magnetic Nanoparticles in Biomedicine: From Monometals to Nanoalloys and Ligand-Protected Particles. Materials, 14(13), 3611. https://doi.org/10.3390/ma14133611