
Phase Composition of Silica Fume—Portland Cement Systems Formed under Hydrothermal Curing Evaluated by FTIR, XRD, and TGA


 , and
, and
Abstract
1. Introduction
2. Material and Methods
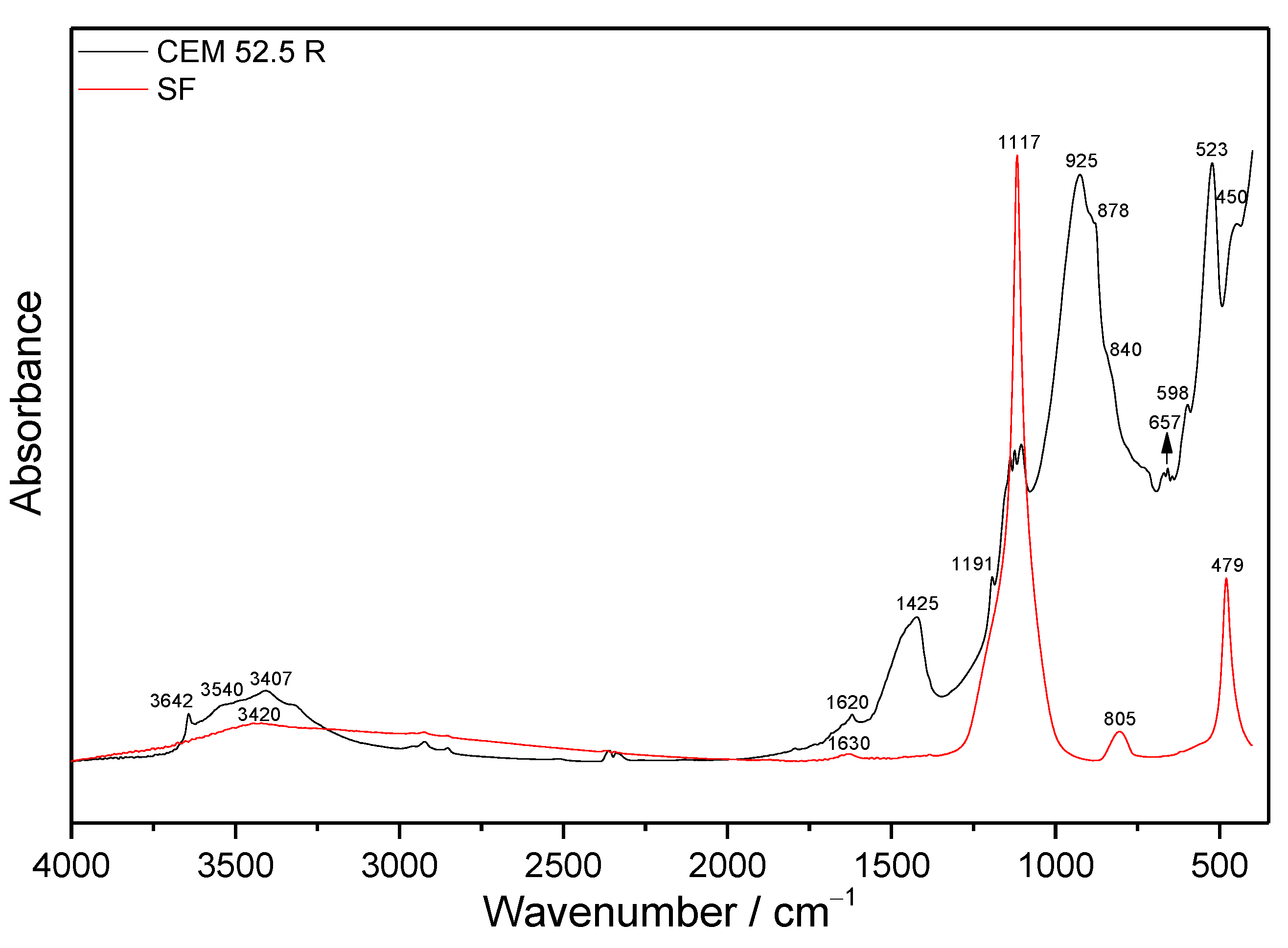
Structural Characterization of Raw and Modified Cement by FTIR Spectroscopy
3. Results and Discussion
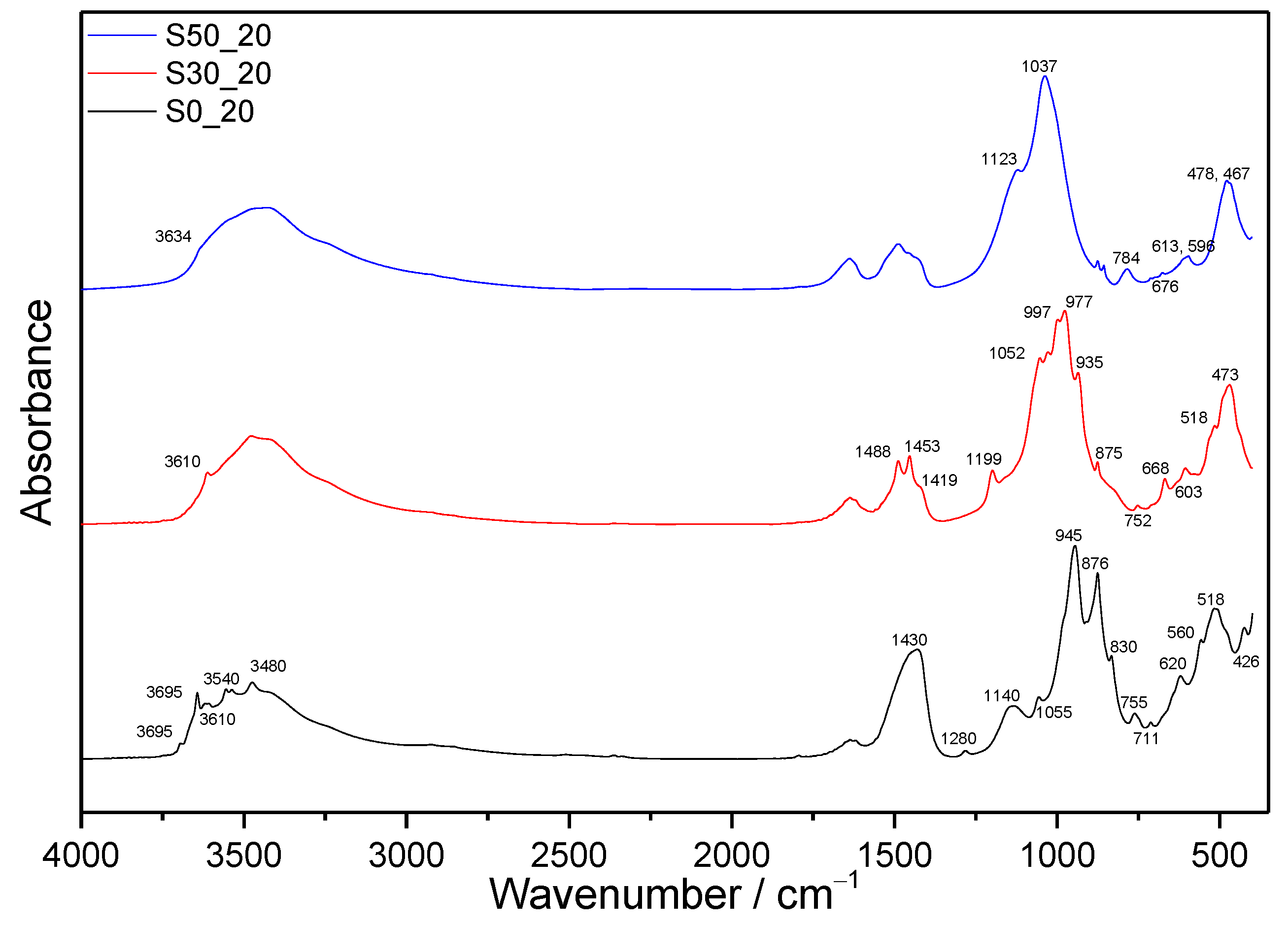
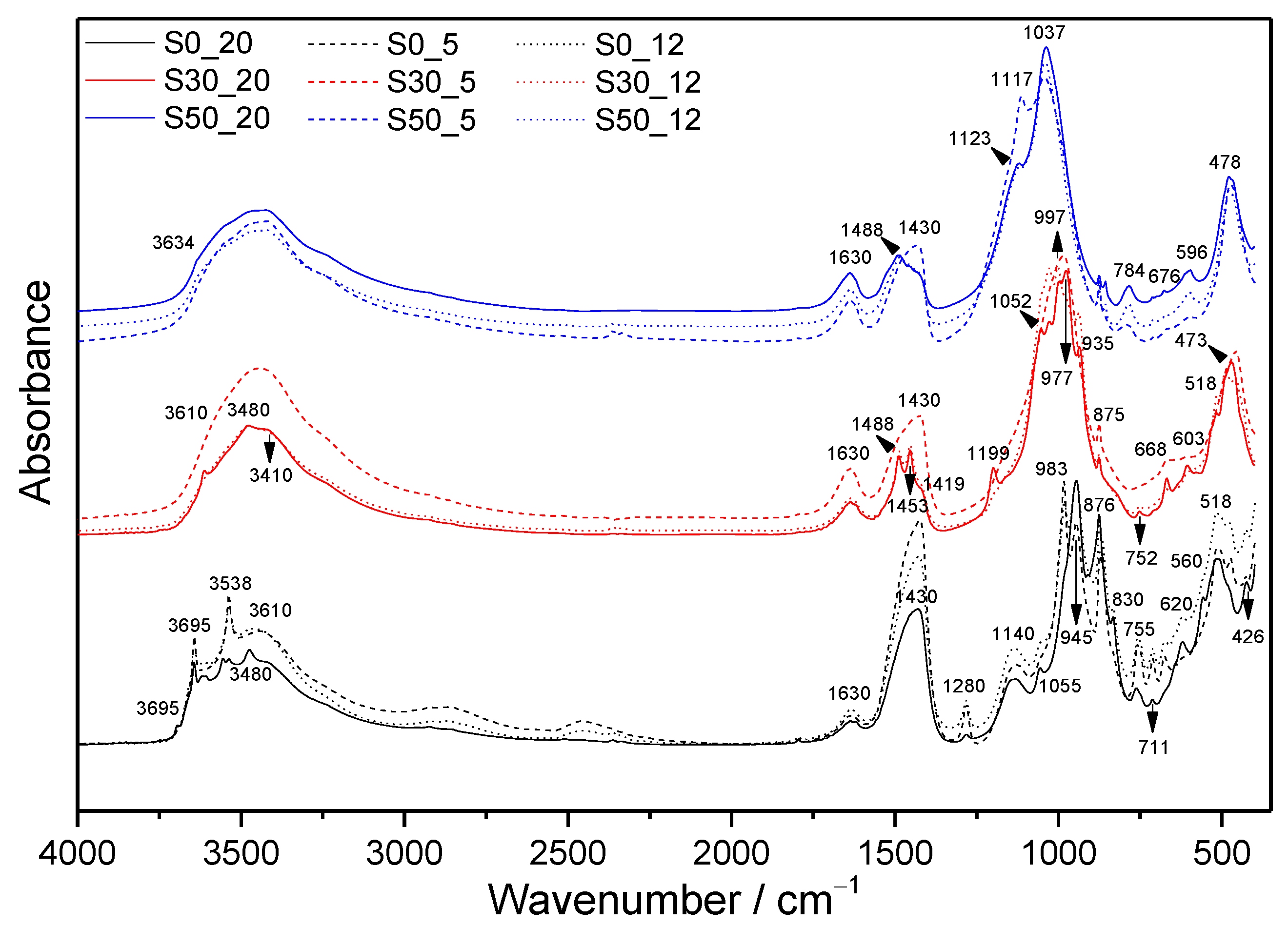
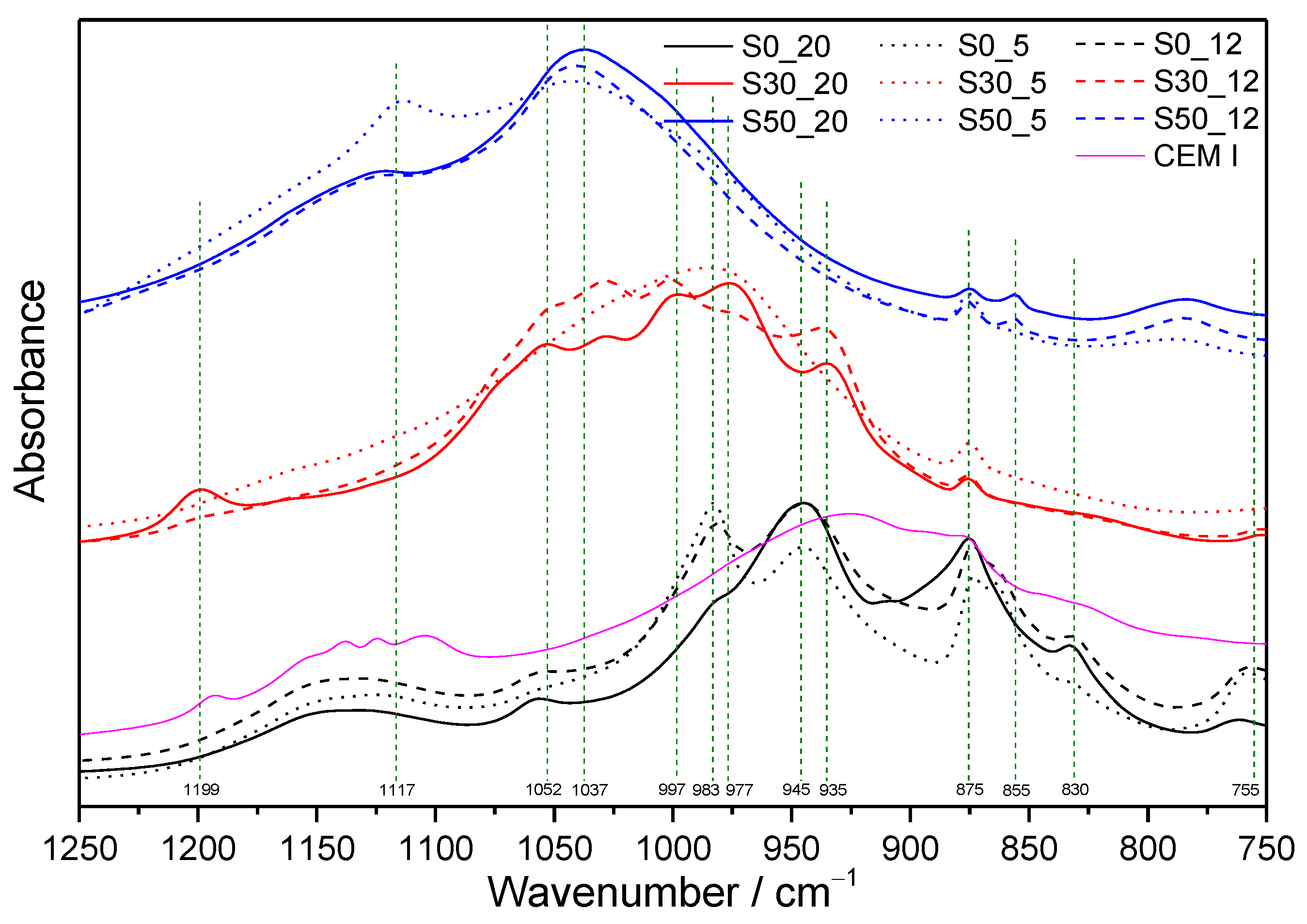
3.1. FTIR Spectroscopy
3.1.1. Samples without SF Addition
3.1.2. Samples with SF Addition
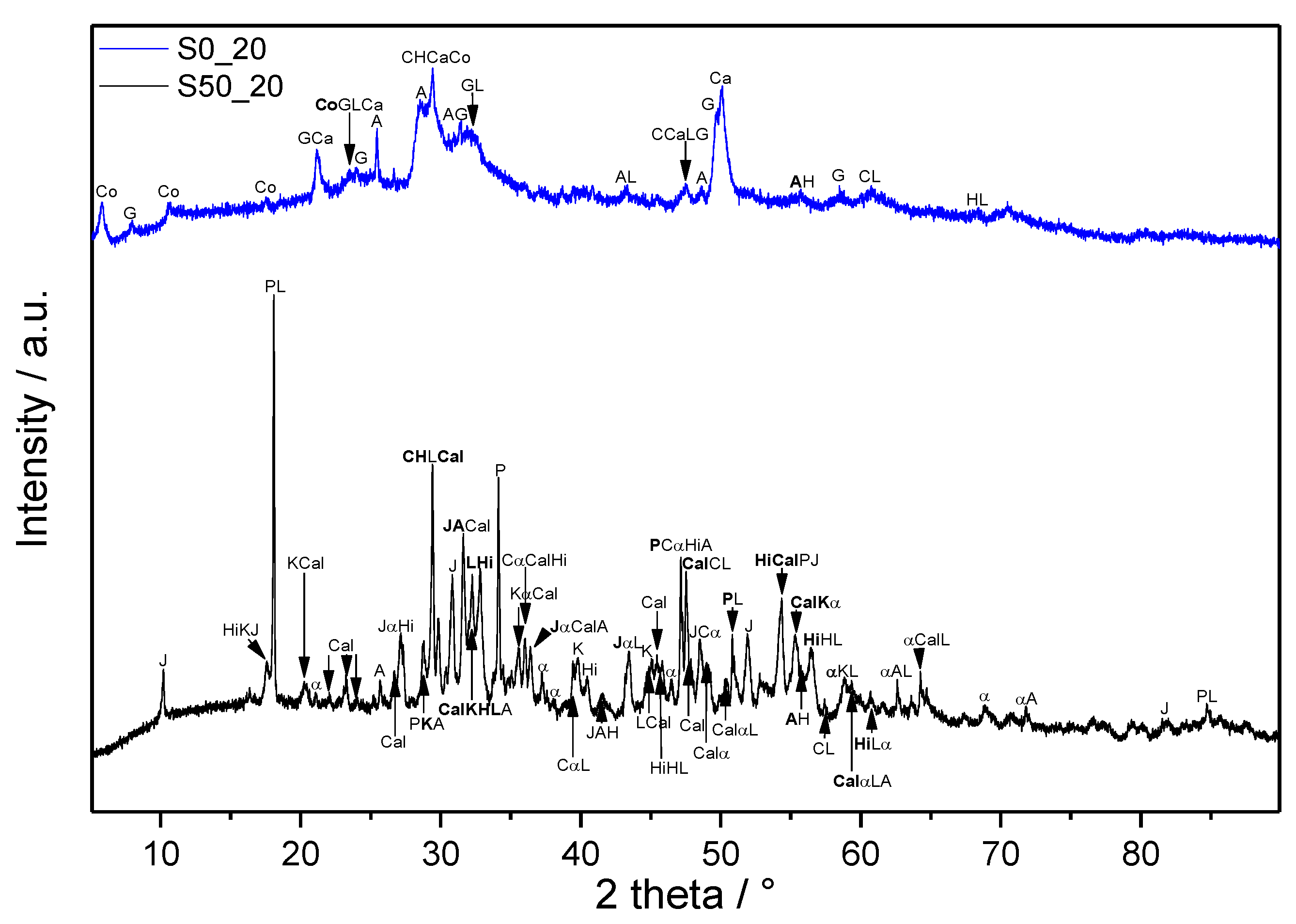
3.2. XRD
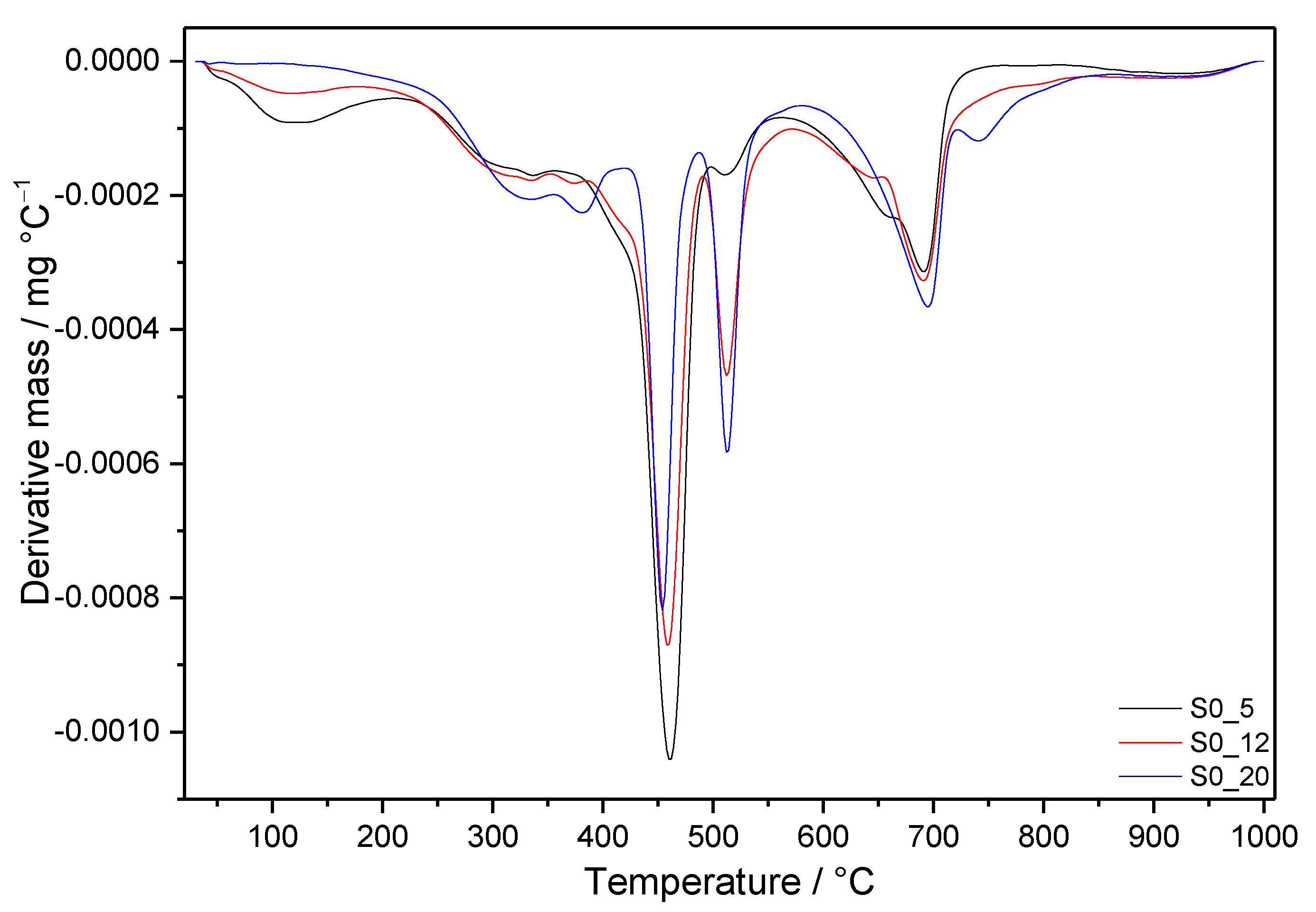
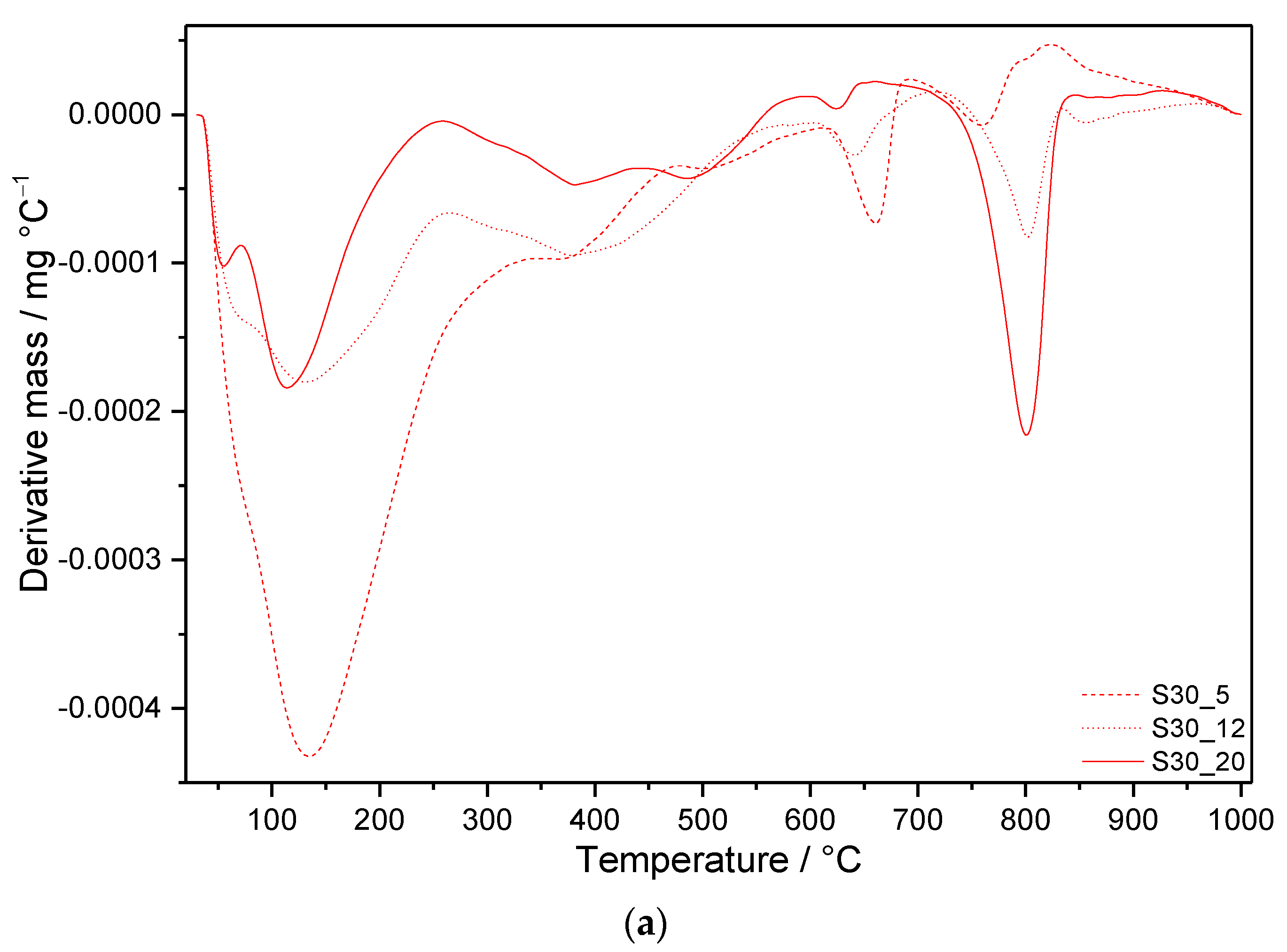
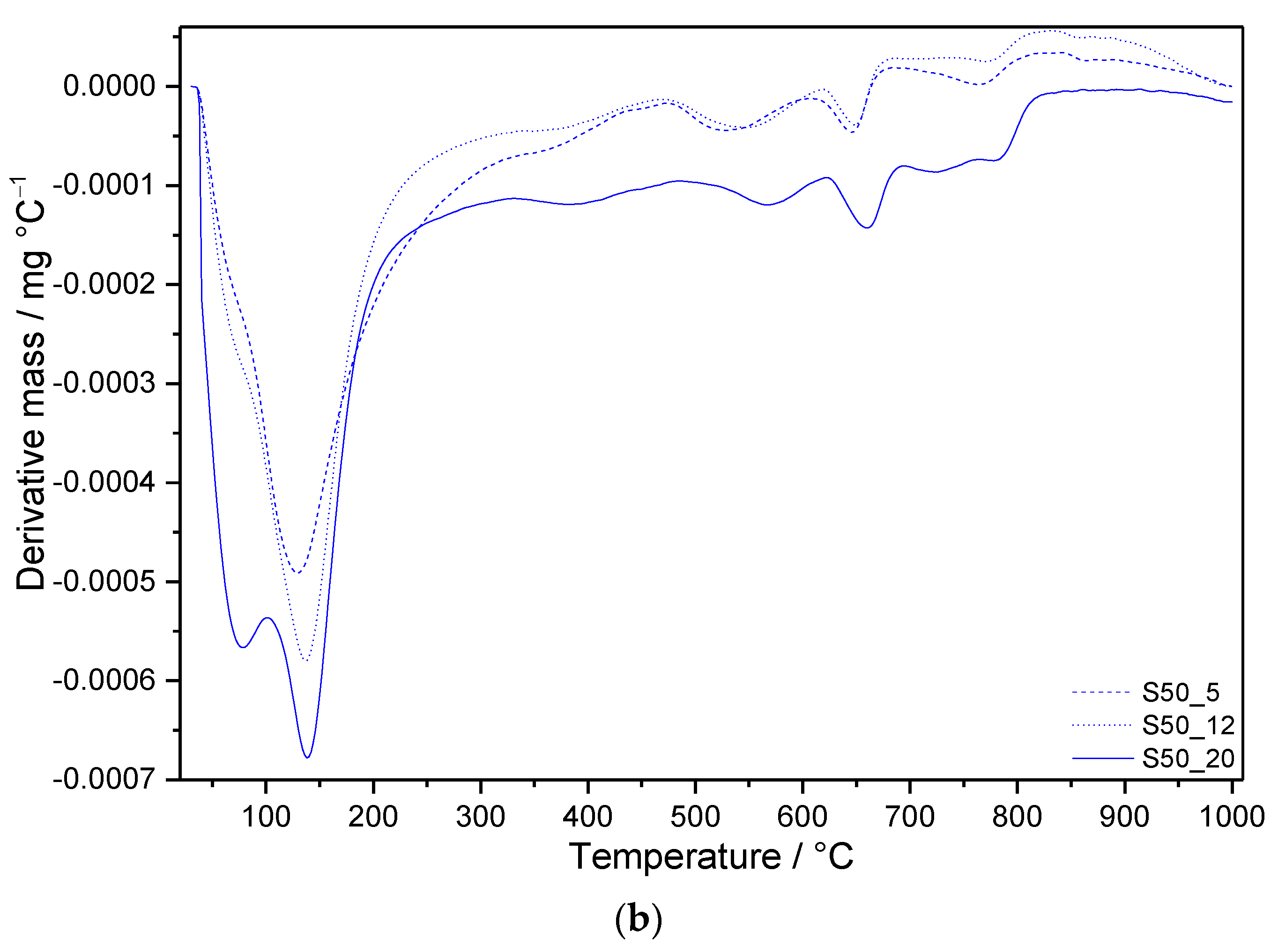
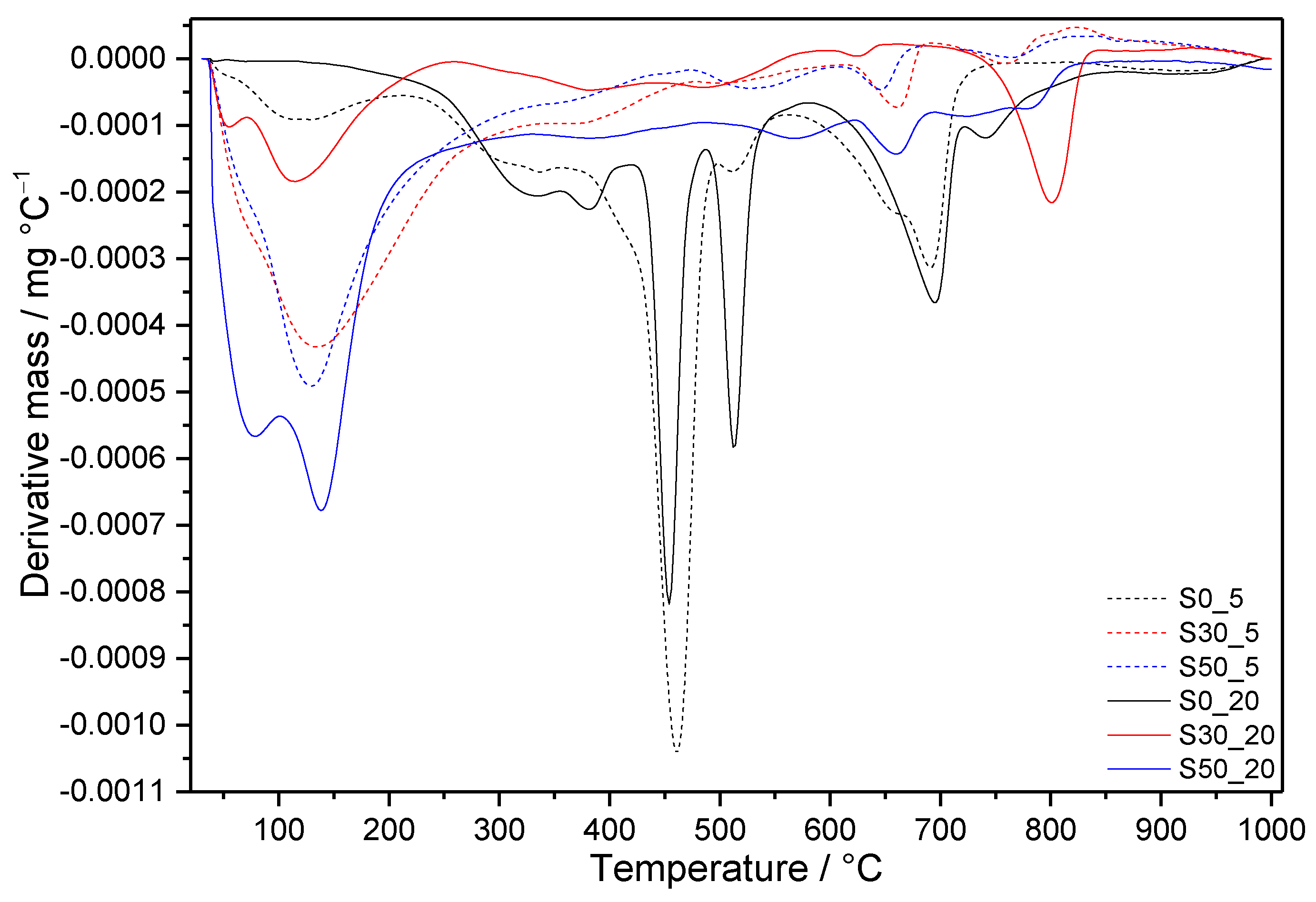
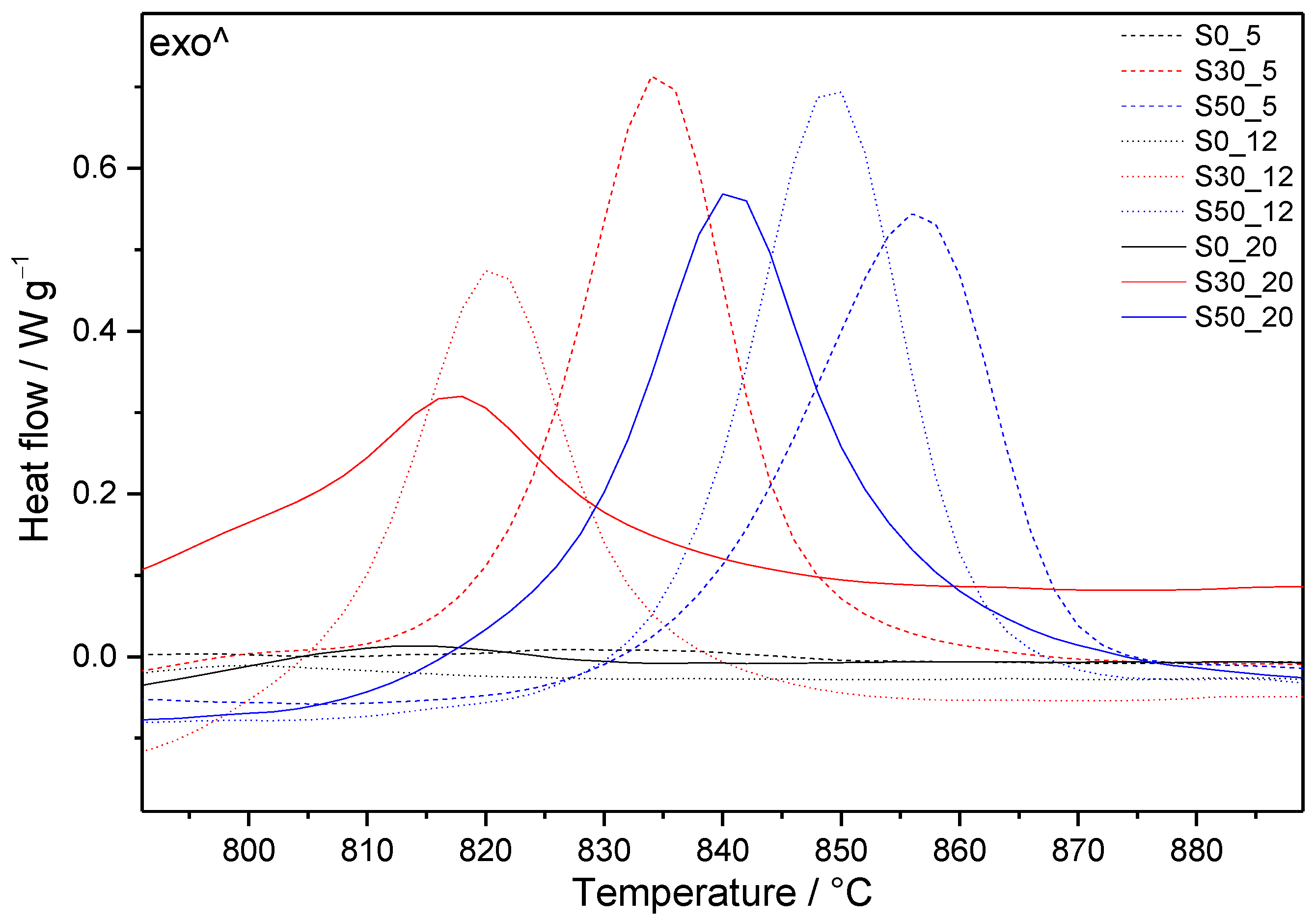
3.3. Thermal Analysis
3.3.1. Samples without SF Addition
3.3.2. Samples with SF Addition
3.4. A Consideration of the Changes Taking Place in the Systems
4. Conclusions
- -
- Calciochondrite, katoite, hibschite, α-C2SH, jaffeite, portlandite, anhydrite, and calcite were identified in the hydrothermally cured pure cement pastes.
- -
- In case of two modest hydrothermal regimes, poorly crystalline C-S-H was still present in the samples without SF addition.
- -
- Whilst the amount of α-C2SH decreased, the amount of jaffeite increased with increasing temperature and pressure.
- -
- The transformation of primary hydration products to α-C2SH and jaffeite was pre-vented by SF additions. In case of its lower substitution level (30 mass%), only the traces of jaffeite were revealed by the performed analyses. Higher substitution led to the total suppression of the formation of these phases.
- -
- Portlandite was already depleted by the lower replacement of cement, which was related to the lower carbonation degree in particular samples. In addition to calcite, other modifications of calcium carbonate were identified at higher temperatures and pressures.
- -
- 30 mass% SF resulted in the formation of stable tobermorite, whilst calcium hydrogen silicate, gyrolite, and smaller amounts of cowlesite were identified as the main crystalline phases in the samples with 50 mass% SF.
- -
- The main advantage of the implementation of the FTIR technique, in comparison to the other ones, lies in the possibility to follow the changes in the silicate hydrates structure. Especially in the region of silicate stretching vibrations, their shifts can clearly denote polymerization of the chains or decomposition and higher condensation degree of the formed phases accompanying transformation and crystallization of primary hydration products at very high temperatures.
- -
- In the region of higher wavenumbers, the replacement of [SiO4]4− tetrahedra by 4OH−, for example, the formation of different hydrogrossular phases, can be observed.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A | Al2O3 |
| C | CaO |
| C-A-H | Calcium Aluminate Hydrate |
| C-A-S-H | Calcium Alumina Silicate Hydrate |
| C-S-H | Calcium Silicate Hydrate |
| CO2 | |
| F | Fe2O3 |
| FTIR | Fourier Transform Infrared Spectroscopy |
| H | H2O |
| M | MgO |
| S | SiO2 |
| SF | Silica Fume |
| SO3 | |
| TGA | Thermogravimetric Analysis |
| XRD | X-Ray Diffraction |
References
- Cong, X.D.; Kirkpatrick, R.J. Effects of the temperature and relative humidity on the structure of C-S-H gel. Cem. Concr. Res. 1995, 25, 1237–1245. [Google Scholar] [CrossRef]
- Richardson, I.G. The calcium silicate hydrates. Cem. Concr. Res. 2008, 38, 137–158. [Google Scholar] [CrossRef]
- Števula, L.; Harman, M.; Horváth, I.; Putyera, K. The mineral gyrolite and its stability under hydrothermal conditions. Ceram. Silikaty 1990, 34, 315–330. [Google Scholar]
- Bahafid, S.; Ghabezloo, S.; Duc, M.; Faure, P.; Sulem, J. Effect of the hydration temperature on the microstructure of Class G cement: C-S-H composition and density. Cem. Concr. Res. 2017, 95, 270–281. [Google Scholar] [CrossRef]
- Jeong, Y.J.; Youm, K.S.; Yun, S. Effect of nano-silica and curing conditions on the reaction rate of class G well cement exposed to geological CO2-sequestration conditions. Cem. Concr. Res. 2018, 109, 208–216. [Google Scholar] [CrossRef]
- Chen, J.J.; Thomas, J.J.; Taylor, H.F.W.; Jennings, H.M. Solubility and structure of calcium silicate hydrate. Cem. Concr. Res. 2004, 34, 1499–1519. [Google Scholar] [CrossRef]
- Shirinyan, A.S.; Gusak, A.M.; Wautelet, M. Phase diagram versus diagram of solubility: What is the difference for nanosystems? Acta Mater. 2005, 53, 5025–5032. [Google Scholar] [CrossRef]
- Spycher, N.; Pruess, K. CO2-H2O Mixtures in the Geological Sequestration of CO2. II. Partitioning in Chloride Brines at 12–100 °C and up to 600 bar. Geochim. Cosmochim. Acta 2005, 69, 3309–3320. [Google Scholar] [CrossRef]
- Wilberforce, T.; Baroutaji, A.; El Hassan, Z.; Thompson, J.; Soudan, B.; Olabi, A.G. Prospects and challenges of concentrated solar photovoltaics and enhanced geothermal energy technologies. Sci. Total Environ. 2019, 659, 851–861. [Google Scholar] [CrossRef]
- Richardson, I.G. The nature of C-S-H in hardened cements. Cem. Concr. Res. 1999, 29, 1131–1147. [Google Scholar] [CrossRef]
- Hope, B.B. Autoclaved concrete containing flyash. Cem. Concr. Res. 1981, 11, 227–233. [Google Scholar] [CrossRef]
- Qomi, M.J.A.; Krakowiak, K.J.; Bauchy, M.; Stewart, K.L.; Shahsavari, R.; Jagannathan, D.; Brommer, D.B.; Baronnet, A.; Buehler, M.J.; Yip, S.; et al. Combinatorial molecular optimization of cement hydrates. Nat. Commun. 2014, 5, 4960. [Google Scholar] [CrossRef]
- Strunz, H. Mineralogische Tabellen, 8th ed.; Akad. Verl.-Ges. Geest u. Portig: Leipzig, Germany, 1982; pp. 1–621. [Google Scholar]
- Garbev, K.; Beuchle, G.; Schweike, U.; Merz, D.; Dregert, O.; Stemmermann, P. Preparation of a novel cementitious material from hydrothermally synthesized C-S-H phases. J. Am. Ceram. Soc. 2014, 97, 2298–2307. [Google Scholar] [CrossRef]
- Baltakys, K.; Siauciunas, R. The influence of γ-Al2O3 and Na2O on the formation of calcium silicate hydrates in the CaO-quartz-H2O system. Mater. Sci. 2007, 25, 185–198. [Google Scholar]
- Black, L.; Garbev, K.; Stemmermann, P.; Hallam, K.R.; Allen, G.C. X-ray photoelectron study of oxygen bonding in crystalline C-S-H phases. Phys. Chem. Miner. 2004, 31, 337–346. [Google Scholar] [CrossRef]
- Dharmawardhana, C.C.; Misra, A.; Ching, W.Y. Quantum Mechanical Metric for Internal Cohesion in Cement Crystals. Sci. Rep. 2014, 4, 7332. [Google Scholar] [CrossRef]
- Jennings, H.M. A model for the microstructure of calcium silicate hydrate in cement paste. Cem. Concr. Res. 2000, 30, 101–116. [Google Scholar] [CrossRef]
- Cong, X.D.; Kirkpatrick, R.J. 17O and 29Si MAS NMR study of β-C2S hydration and the structure of calcium-silicate hydrates. Cem. Concr. Res. 1993, 23, 1065–1077. [Google Scholar] [CrossRef]
- Dharmawardhana, C.C.; Misra, A.; Aryal, S.; Rulis, P.; Ching, W.Y. Role of interatomic bonding in the mechanical anisotropy and interlayer cohesion of CSH crystals. Cem. Concr. Res. 2013, 52, 123–130. [Google Scholar] [CrossRef]
- Jacobsen, J.; Rodrigues, M.S.; Telling, M.T.; Beraldo, A.L.; Santos, S.F.; Aldridge, L.P.; Bordallo, H.N. Nano-scale hydrogen-bond network improves the durability of greener cements. Sci. Rep. 2013, 3, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Berenguer, R.A.; Lima, N.B.; Lima, V.M.E.; Estolano, A.M.L.; Póvoas, Y.V.; Lima, N.B.D. The role of hydrogen bonds on the mechanical properties of cement-based mortars applied to concrete surfaces. Cem. Concr. Compos. 2021, 115, 103848. [Google Scholar] [CrossRef]
- Pushcharovsk, D.Y.; Yamnova, N.A.; Egorov-Tismenko, Y.K. The crystal structure of jaffeite and related compounds. Acta Cryst. 1993, A49, c275. [Google Scholar] [CrossRef]
- Black, L.; Garbev, K.; Stumm, A. Structure, bonding and morphology of hydrothermally synthesised xonotlite. Adv. Appl. Ceram. 2009, 108, 137–144. [Google Scholar] [CrossRef]
- Garbev, K.; Gasharova, B.; Beuchle, G.; Kreisz, S.; Stemmermann, P. First observation of α-Ca2[SiO3(OH)](OH)–Ca6[Si2O7][SiO4](OH)2 phase transformation upon thermal treatment in air. J. Am. Ceram. Soc. 2008, 91, 263–271. [Google Scholar] [CrossRef]
- Meducin, F.; Bresson, B.; Lequeux, N.; de Noirfontaine, M.N.; Zanni, H. Calcium silicate hydrates investigated by solid-state high resolution 1H and 29Si nuclear magnetic resonance. Cem. Concr. Res. 2007, 37, 631–638. [Google Scholar] [CrossRef]
- Zhu, X.P.; Qian, C.; He, B.; Chen, Q.; Jiang, Z.W. Experimental study on the stability of C-S-H nanostructures with varying bulk CaO/SiO2 ratios under cryogenic attack. Cem. Concr. Res. 2020, 135, 106114. [Google Scholar] [CrossRef]
- Mercury, J.M.R.; Pena, P.; De Aza, A.H.; Turrillas, X.; Sobrados, I.; Sanz, J. Solid-state 27Al and 29Si NMR investigations on Si-substituted hydrogarnets. Acta Mater. 2007, 55, 1183–1191. [Google Scholar] [CrossRef]
- Blanc, P.; Bourbon, X.; Lassin, A.; Gaucher, E.C. Chemical model for cement-based materials: Temperature dependence of thermodynamic functions for nanocrystalline and crystalline C-S-H phases. Cem. Concr. Res. 2010, 40, 851–866. [Google Scholar] [CrossRef]
- Palou, M.T.; Kuzielová, E.; Žemlicka, M.; Boháč, M.; Novotný, R. The effect of curing temperature on the hydration of binary Portland cement. J. Therm. Anal. Calorim. 2016, 125, 1301–1310. [Google Scholar] [CrossRef]
- Kuzielová, E.; Žemlicka, M.; Másilko, J.; Palou, M.T. Pore structure development of blended G-oil well cement submitted to hydrothermal curing conditions. Geothermics 2017, 68, 86–93. [Google Scholar] [CrossRef]
- Kuzielová, E.; Žemlicka, M.; Másilko, J.; Palou, M.T. Effect of additives on the performance of Dyckerhoff cement, Class G, submitted to simulated hydrothermal curing. J. Therm. Anal. Calorim. 2018, 133, 63–76. [Google Scholar] [CrossRef]
- Kuzielová, E.; Žemlicka, M.; Másilko, J.; Palou, M.T. Development of G-oil well cement phase composition during long therm hydrothermal curing. Geothermics 2019, 80, 129–137. [Google Scholar] [CrossRef]
- Madej, J.; Madejová, J.; Jakubeková, D. IR spectroscopic study of hydration of condensed silica fume modified cement pastes. Ceram. Silikáty 1990, 34, 131–141. [Google Scholar]
- Fernández-Carrasco, L.; Torrens-Martín, D.; Morales, L.M.; Martínez-Ramírez, S. Infrared Spectroscopy in the Analysis of Building and Construction Materials. In Infrared Spectroscopy–Materials Science, Engineering and Technology; Theophanides, T., Ed.; InTech: Rijeka, Croatia, 2012; pp. 369–382. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, D.; Ma, X.; Zhang, G.; Jia, Y.; Hatada, K. Spectroscopic and DFT study on the species and local structure of arsenate incorporated in gypsum lattice. Chem. Geol. 2017, 460, 46–53. [Google Scholar] [CrossRef]
- Liu, Y. Raman, Mid-IR, and NIR spectroscopic study of calcium sulfates and mapping gypsum abundances in Columbus Crater, Mars. Planet. Space Sci. 2018, 163, 35–41. [Google Scholar] [CrossRef]
- Mendes, A.; Gates, W.P.; Sanjayan, J.G.; Collins, F. NMR, XRD, IR and synchrotron NEXAFS spectroscopic studies of OPC and OPC/slag cement paste hydrates. Mater. Struct. 2011, 44, 1773–1791. [Google Scholar] [CrossRef]
- Bensted, J.; Varma, S.P. Some applications of infrared and Raman spectroscopy in cement chemistry. Part 3-hydration of Portland cement and its constituents. Cem. Technol. 1974, 5, 440–445. [Google Scholar]
- Passaglia, E.; Rinaldi, R. Katoite, a new member of the Ca3Al2(SiO4)3-Ca3Al2(OH)12 series and a new nomenclature for the hydrogrossular group of minerals. Bull. Mineral. 1984, 107, 605–618. [Google Scholar] [CrossRef]
- McMillan, P.F. Water Solubility and Speciation Models. In Volatiles in Magmas; Carroll, M.R., Holloway, J.R., Eds.; De Gruyter: Berlin, Boston, 1994; Volume 30, pp. 131–156. [Google Scholar] [CrossRef]
- Kljajević, L.M.; Nenadović, S.S.; Nenadović, M.T.; Bundaleski, N.K.; Todorović, B.Ž.; Pavlović, V.B.; Rakočević, Z.L. Structural and chemical properties of thermally treated geopolymer samples. Ceram. Int. 2017, 43, 6700–6708. [Google Scholar] [CrossRef]
- Speakman, K.; Taylor, H.F.W.; Bennett, J.M.; Gard, J.A. Hydrothermal reactions of γ-dicalcium silicate. J. Chem. Soc. A 1967, 1052–1060. [Google Scholar] [CrossRef]
- Allahverdi, A.; Pilehvar, S.; Mahinroosta, M. Influence of curing conditions on the mechanical and physical properties of chemically-activated phosphorous slag cement. Powder Technol. 2016, 288, 132–139. [Google Scholar] [CrossRef]
- Sugama, T.; Pyatina, T. Alkali-Activated Cement Composites for High Temperature Geothermal Wells; Scientific Research Publishing Inc.: Irvine, CA, USA, 2017; pp. 21–212. [Google Scholar]
- del Bosque, I.S.; Martínez-Ramírez, S.; Blanco-Varela, M.T. FTIR study of the effect of temperature and nanosilica on the nano structure of C–S–H gel formed by hydrating tricalcium silicate. Constr. Build. Mater. 2014, 52, 314–323. [Google Scholar] [CrossRef]
- del Bosque, I.S.; Martín-Pastor, M.; Martínez-Ramírez, S.; Blanco-Varela, M.T. Effect of temperature on C3S and C3S plus nanosilica hydration and C–S–H structure. J. Am. Ceram. Soc. 2013, 96, 957–965. [Google Scholar] [CrossRef]
- Kuzielová, E.; Žemlička, M.; Novotný, R.; Palou, M.T. Simultaneous effect of silica fume, metakaolin and ground granulated blast-furnace slag on the hydration of multicomponent cementitious binders. J. Therm. Anal. Calorim. 2019, 136, 1527–1537. [Google Scholar] [CrossRef]
- Rossen, J.E.; Lothenbach, B.; Scrivener, K.L. Composition of C–S–H in pastes with increasing levels of silica fume addition. Cem. Concr. Res. 2015, 75, 14–22. [Google Scholar] [CrossRef]
- Alonso, C.; Fernandez, L. Dehydration and rehydration processes of cement paste exposed to high temperature environments. J. Mater. Sci. 2004, 39, 3015–3024. [Google Scholar] [CrossRef]
- Yu, P.; Kirkpatrick, R.J.; Poe, B.; McMillan, P.F.; Cong, X. Structure of calcium silicate hydrate (C-S-H): Near-, Mid-, and Far-infrared spectroscopy. J. Am. Cera. Soc. 1999, 82, 742–748. [Google Scholar] [CrossRef]
- Mostafa, N.Y.; Shaltout, A.A.; Omar, H.; Abo-El-Enein, S.A. Hydrothermal synthesis and characterization of aluminium and sulfate substituted 1.1 nm tobermorites. J. Alloy. Comp. 2009, 467, 332–337. [Google Scholar] [CrossRef]
- Baltakys, K.; Siauciunas, R. Influence of gypsum additive on the gyrolite formation process. Cem. Concr. Res. 2010, 40, 376–383. [Google Scholar] [CrossRef]
- Kasperaviciute, V.; Baltakys, K.; Siauciunas, R. The sorption properties of gyrolite for copper ions. Ceram. Silikáty 2008, 52, 95–101. [Google Scholar]
- Garbev, K. Struktur, Eigenschaften und Quantitative Rietveldanalyse von Hydrothermal Kristallisierten Calciumsilikathydraten (CSH-Phasen). Ph.D. Thesis, Universität Heidelberg, Heidelberg, Germany, 2003. [Google Scholar] [CrossRef]
- Chukanov, N.V. Infrared Spectra of Mineral Species: Extended Library; Springer: Dordrecht, The Netherlands; Springer: Heidelberg, Germany, 2014; pp. 21–1701. [Google Scholar] [CrossRef]
- Yanagisawa, K.; Hu, X.; Onda, A.; Kajiyoshi, K. Hydration of β-dicalcium silicate at high temperatures under hydrothermal conditions. Cem. Concr. Res. 2006, 36, 810–816. [Google Scholar] [CrossRef]
- Scrivener, K.; Snellings, R.; Lothenbach, B. A Practical Guide to Microstructural Analysis of Cementitious Materials, 1st ed.; CRC Press: Boca Raton, FL, USA, 2016; pp. 1–560. [Google Scholar] [CrossRef]
- Thiery, M.; Villain, G.; Dangla, P.; Platret, G. Investigation of the carbonation front shape on cementitious materials: Effects of the chemical kinetics. Cem. Concr. Res. 2007, 37, 1047–1058. [Google Scholar] [CrossRef]
- Stumm, A.; Garbev, K.; Beuchle, G.; Black, L.; Stemmermann, P.; Nüesch, R. Incorporation of zinc into calcium silicate hydrates, Part I: Formation of CSH (I) with C/S= 2/3 and its isochemical counterpart gyrolite. Cem. Concr. Res. 2005, 35, 1665–1675. [Google Scholar] [CrossRef]
- Shaw, S.; Henderson, C.M.B.; Komanschek, B.U. Dehydration/recrystallization mechanisms, energetics, and kinetics of hydrated calcium silicate minerals: An in situ TGA/DSC and synchrotron radiation SAXS/WAXS study. Chem. Geol. 2000, 167, 141–159. [Google Scholar] [CrossRef]
- Palou, M.T.; Šoukal, F.; Boháč, M.; Šiler, P.; Ifka, T.; Živica, V. Performance of G-Oil Well cement exposed to elevated hydrothermal curing conditions. J. Therm. Anal. Calorim. 2014, 118, 865–874. [Google Scholar] [CrossRef]
- Palou, M.T.; Kuzielová, E.; Novotný, R.; Šoukal, F.; Žemlička, M. Blended cements consisting of Portland cement–slag–silica fume–metakaolin system. J. Therm. Anal. Calorim. 2016, 125, 1025–1034. [Google Scholar] [CrossRef]
- Lothenbach, B. Thermodynamic equilibrium calculations in cementitious systems. Mater. Struct. 2010, 43, 1413–1433. [Google Scholar] [CrossRef]
- Dauzères, A.; De Windt, L.; Bartier, D.; Sammaljärvi, J.; Barnichon, J.D.; Techer, I.; Detilleux, V. Impact of a 70 °C temperature on an ordinary Portland cement paste/claystone interface: An in situ experiment. Cem. Concr. Res. 2016, 83, 164–178. [Google Scholar] [CrossRef]
- Houston, J.R.; Maxwell, R.S.; Carroll, S.A. Transformation of meta-stable calcium silicate hydrates to tobermorite: Reaction kinetics and molecular structure from XRD and NMR spectroscopy. Geochem. Trans. 2009, 10, 1–14. [Google Scholar] [CrossRef]
- Galvánková, L.; Másilko, J.; Solný, T.; Štěpánková, E. Tobermorite synthesis under hydrothermal conditions. Procedia Eng. 2016, 151, 100–107. [Google Scholar] [CrossRef]
- Wang, H.Y.; Shie, J.J. Effect of Autoclave Curing on the Compressive Strength and Elastic Modulus of Lightweight Aggregate Concrete. J. ASTM Int. 2009, 6, 1–11. [Google Scholar] [CrossRef]
- Siauciunas, R.; Baltakys, K. Formation of gyrolite during hydrothermal synthesis in the mixtures of CaO and amorphous SiO2 or quartz. Cem. Concr. Res. 2004, 34, 2029–2036. [Google Scholar] [CrossRef]
- Taylor, H.F.W. The Calcium Silicate Hydrates. In The Chemistry of Cements; Taylor, H.F.W., Ed.; Academic Press: New York City, NY, USA, 1964; Volume 1, pp. 168–232. [Google Scholar]
- Meller, N.; Kyritsis, K.; Hall, C. The mineralogy of the CaO–Al2O3–SiO2–H2O (CASH) hydroceramic system from 200 to 350 °C. Cem. Concr. Res. 2009, 39, 45–53. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Applied Conditions | wpc | wSF |
|---|---|---|---|
| S0_5 | 0.5 MPa, 165 °C | 100 | - |
| S0_12 | 1.2 MPa, 195 °C | ||
| S0_20 | 2.0 MPa, 220 °C | ||
| S30_5 | 0.5 MPa, 165 °C | 70 | 30 |
| S30_12 | 1.2 MPa, 195 °C | ||
| S30_20 | 2.0 MPa, 220 °C | ||
| S50_5 | 0.5 MPa, 165 °C | 50 | 50 |
| S50_12 | 1.2 MPa, 195 °C | ||
| S50_20 | 2.0 MPa, 220 °C |
| Oxide Composition (Mass%) | Specific Surface (m2 kg−1) | |||||||
|---|---|---|---|---|---|---|---|---|
| CaO | SiO2 | Al2O3 | Fe2O3 | MgO | SO3 | |||
| CEM I 52.5 R | 61.84 | 21.84 | 5.15 | 2.85 | 1.56 | 3.33 | Blaine | 560.9 ± 0.8 |
| SF | 0.50 | 97.10 | 0.21 | - | 0.40 | - | BET | 15,000 |
| Type of Used Cement | Mineral Composition (Mass%) | ||||||
|---|---|---|---|---|---|---|---|
| C3S | C2S | C3A (ort.) | C3A (cub.) | C4AF | Free Lime | MgO | |
| CEM I 52.5 R | 60.36 | 11.38 | 5.72 | 2.32 | 8.35 | 2.69 | 0.24 |
| Sample | S0_5 | S30_5 | S50_5 |
|---|---|---|---|
| Temperature Interval (°C) | Mass Loss (Mass%) | ||
| 165 °C, 0.5 MPa | |||
| r.t.–400 (C(-A)-S-H, C-A-H) | 5.7 | 15.0 | 12.9 |
| 400–480 (CH, α-C2SH) | 5.4 | ||
| 490–560 (jaffeite) | 1.1 | ||
| 560–1000 (carbonates) | 4.4 | 1.8 | 1.2 |
| Total mass loss (%) | 16.6 | 16.8 | 14.1 |
| Sample | S0_12 | S30_12 | S50_12 |
| 195 °C, 1.2 MPa | |||
| r.t.–400 (C(-A)-S-H, C-A-H) | 4.3 | 10.3 | 13.8 |
| 400–480 (CH, α-C2SH) | 4.4 | ||
| 490–560 (jaffeite) | 2.0 | ||
| 560–1000 (carbonates) | 4.5 | 2.3 | 1.4 |
| Total mass loss (%) | 15.2 | 12.6 | 15.2 |
| Sample | S0_20 | S30_20 | S50_20 |
| 220 °C, 2.0 MPa | |||
| r.t.–400 (C(-A)-S-H, C-A-H) | 4.3 | 8.0 | 12.8 |
| 400–480 (CH, α-C2SH) | 2.6 | ||
| 490–560 (jaffeite) | 2.1 | 1.9 | |
| 490–1000 (carbonates) | 4.4 | 2.8 | 1.8 |
| Total mass loss (%) | 13.4 | 12.7 | 14.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuzielová, E.; Slaný, M.; Žemlička, M.; Másilko, J.; Palou, M.T. Phase Composition of Silica Fume—Portland Cement Systems Formed under Hydrothermal Curing Evaluated by FTIR, XRD, and TGA. Materials 2021, 14, 2786. https://doi.org/10.3390/ma14112786
Kuzielová E, Slaný M, Žemlička M, Másilko J, Palou MT. Phase Composition of Silica Fume—Portland Cement Systems Formed under Hydrothermal Curing Evaluated by FTIR, XRD, and TGA. Materials. 2021; 14(11):2786. https://doi.org/10.3390/ma14112786
Chicago/Turabian StyleKuzielová, Eva, Michal Slaný, Matúš Žemlička, Jiří Másilko, and Martin Tchingnabé Palou. 2021. "Phase Composition of Silica Fume—Portland Cement Systems Formed under Hydrothermal Curing Evaluated by FTIR, XRD, and TGA" Materials 14, no. 11: 2786. https://doi.org/10.3390/ma14112786
APA StyleKuzielová, E., Slaný, M., Žemlička, M., Másilko, J., & Palou, M. T. (2021). Phase Composition of Silica Fume—Portland Cement Systems Formed under Hydrothermal Curing Evaluated by FTIR, XRD, and TGA. Materials, 14(11), 2786. https://doi.org/10.3390/ma14112786