Exploration of Free Energy Surface and Thermal Effects on Relative Population and Infrared Spectrum of the Be6B11− Fluxional Cluster

 ,
, 

Abstract
1. Introduction
2. Theoretical Methods and Computational Details
2.1. Global Minimum Search
2.2. Thermochemistry Properties
2.3. Boltzmann Population
2.4. IR Spectra
2.5. Computational Details
3. Results and Discussion
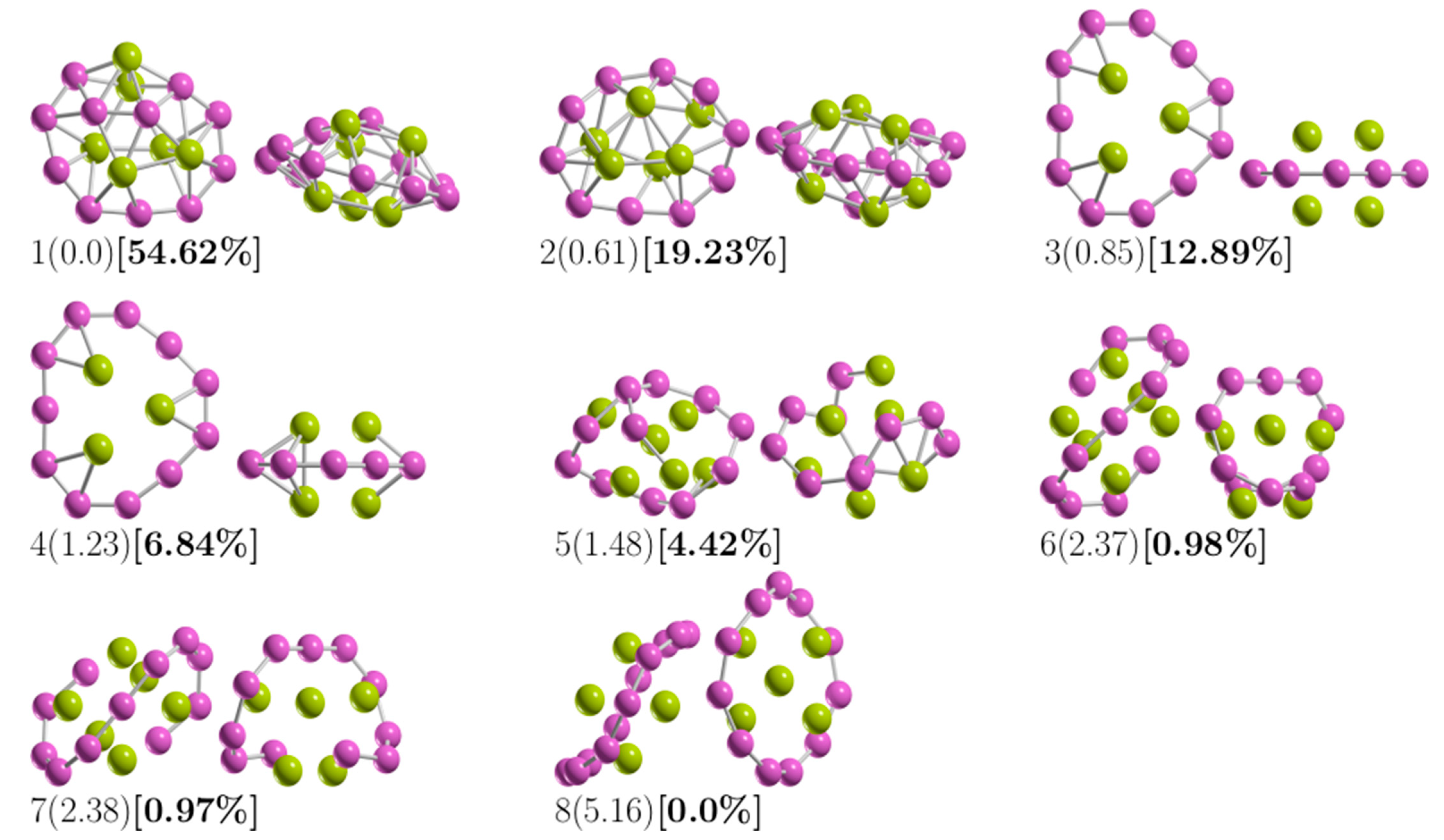
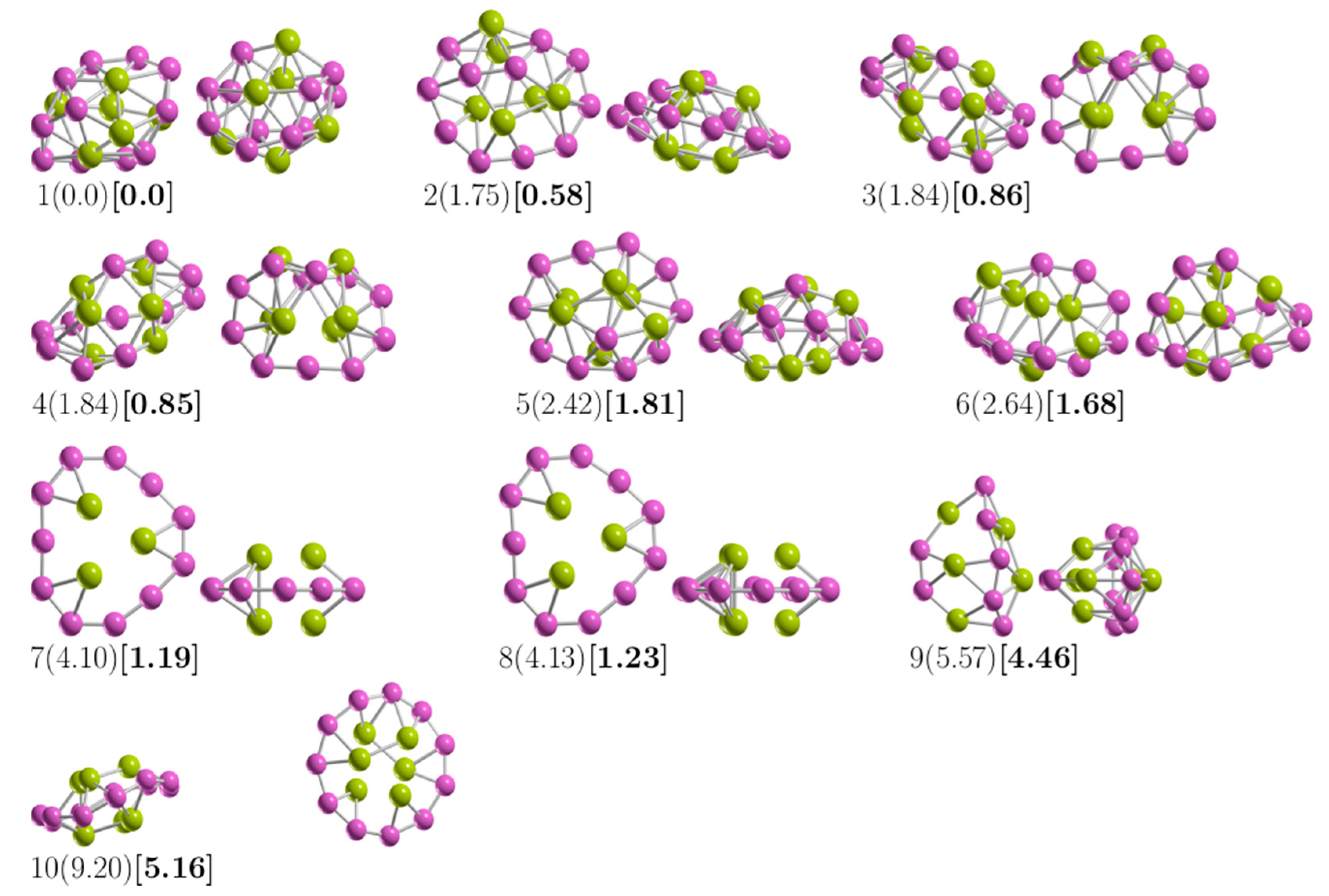
3.1. The Lowest-Energy Structures and Energetics
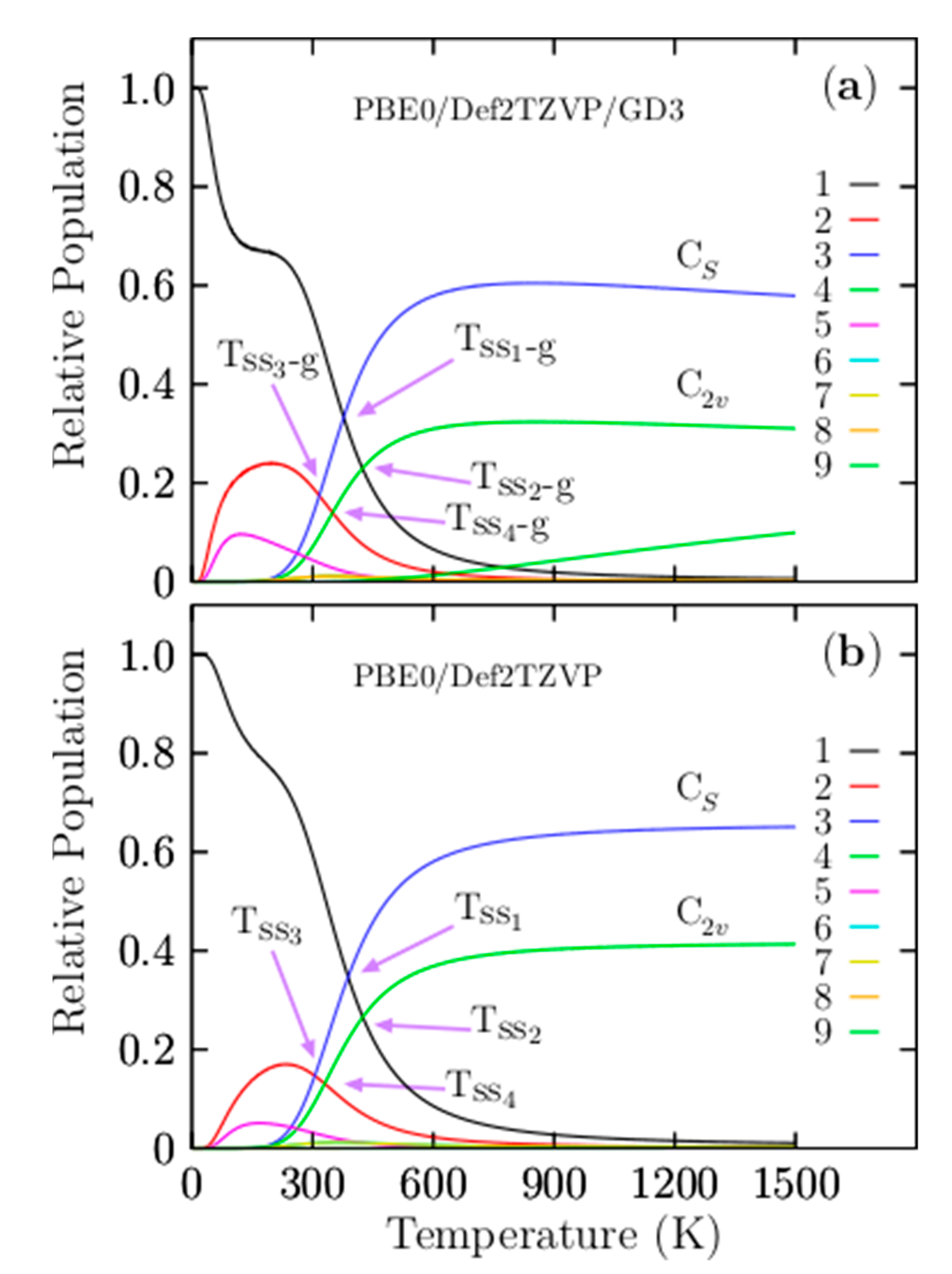
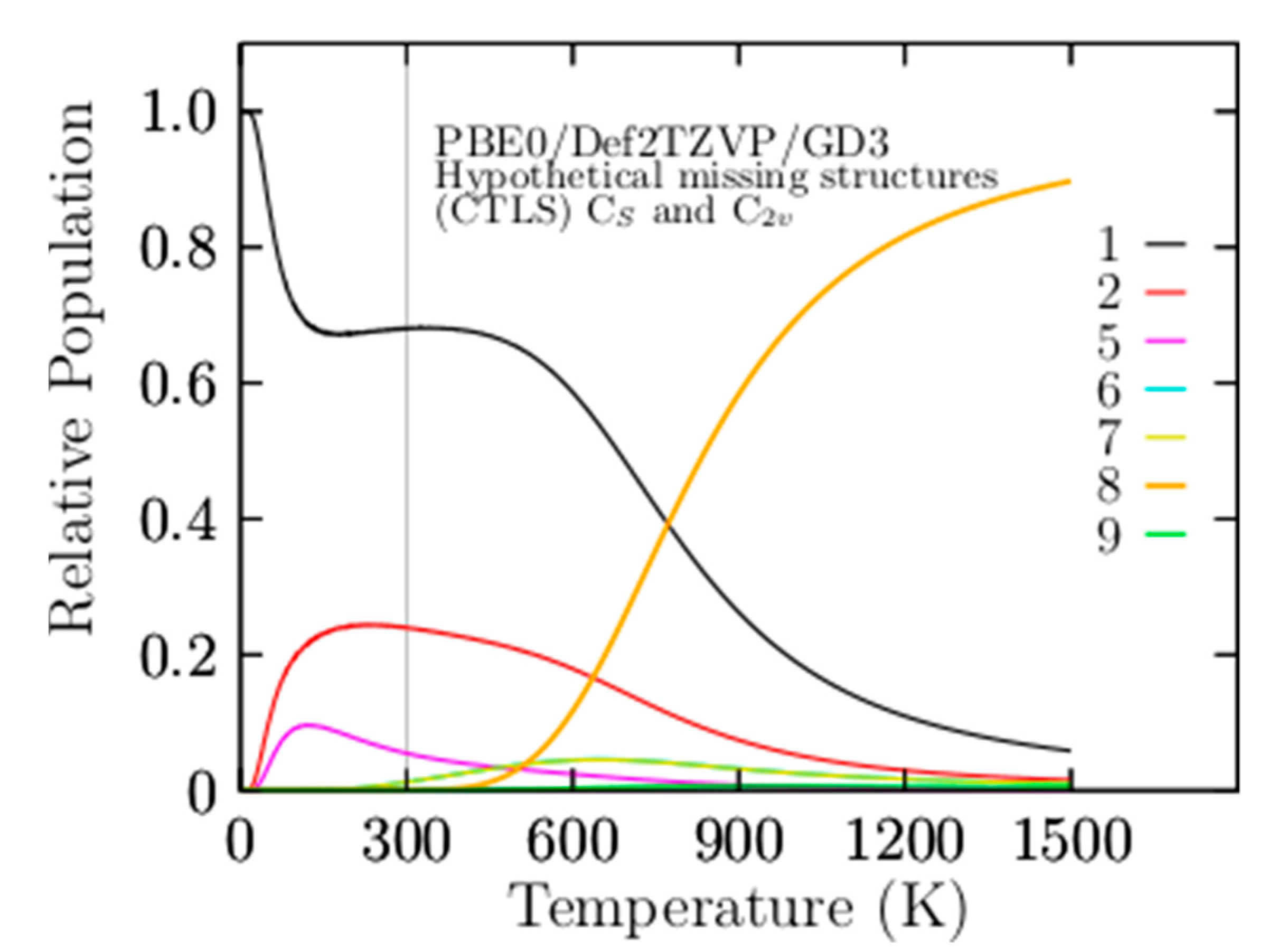
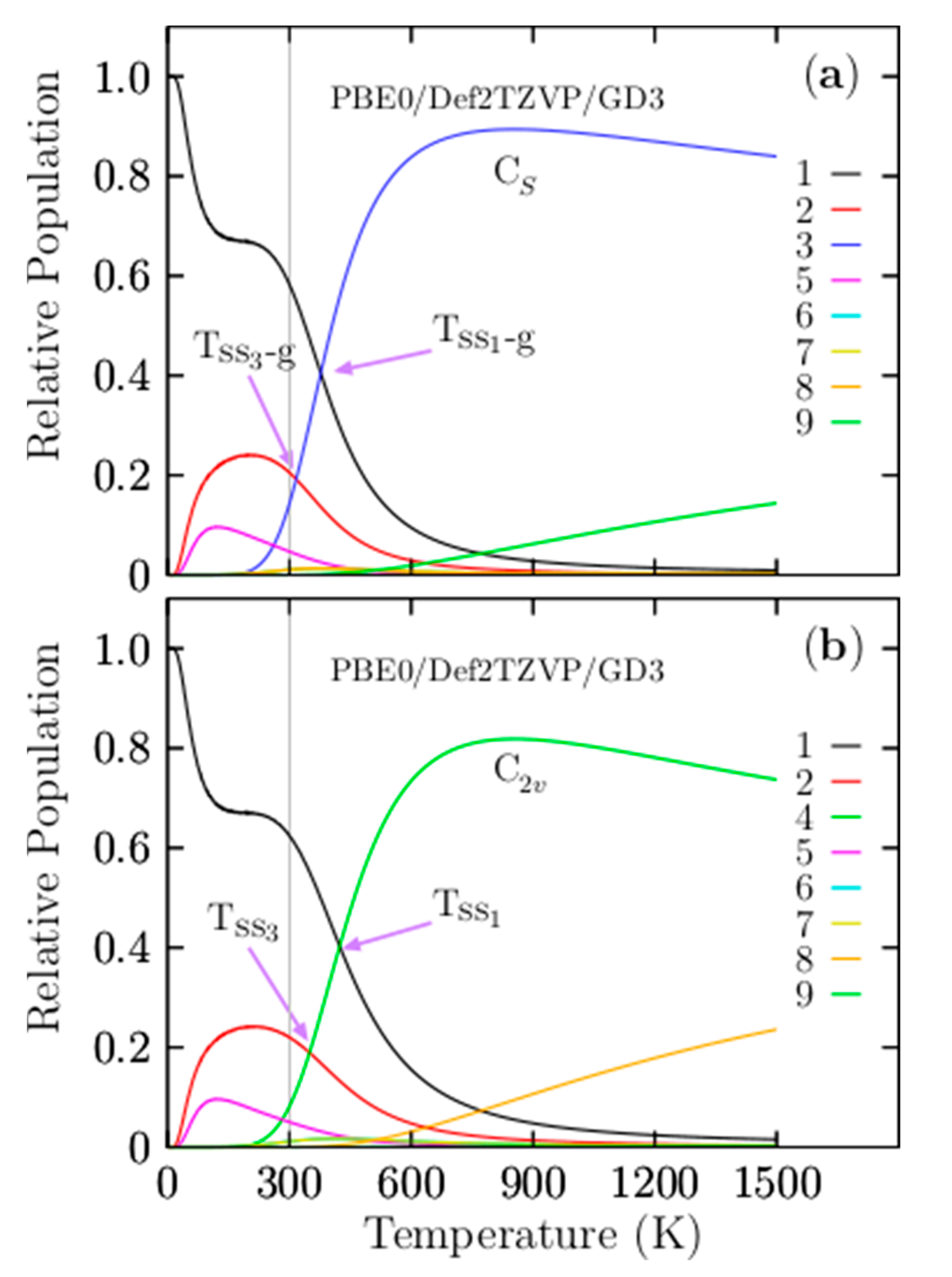
3.2. Relative Population
3.3. Molecular Dynamics
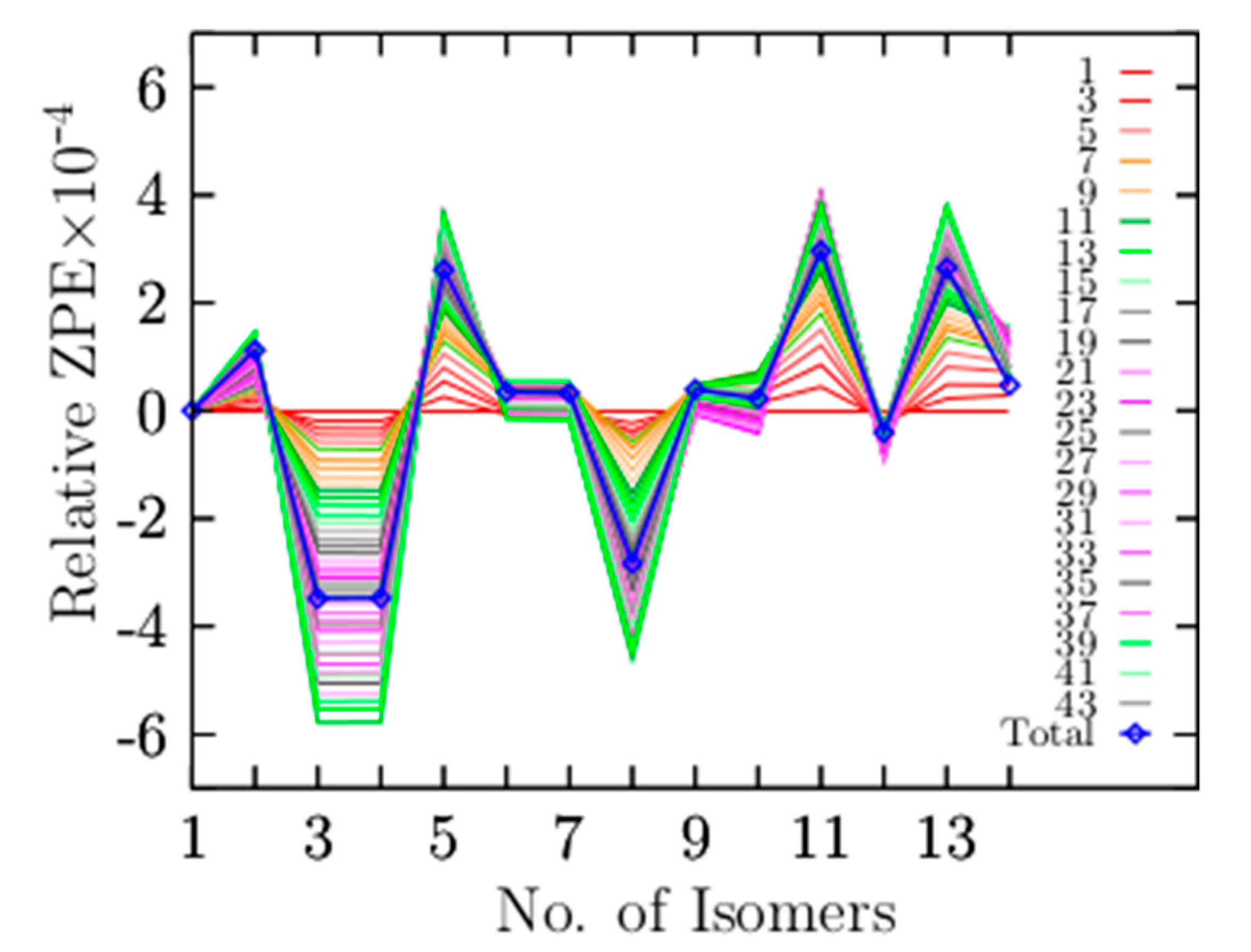
3.4. Contributions of the Vibrational Modes to the ZPE
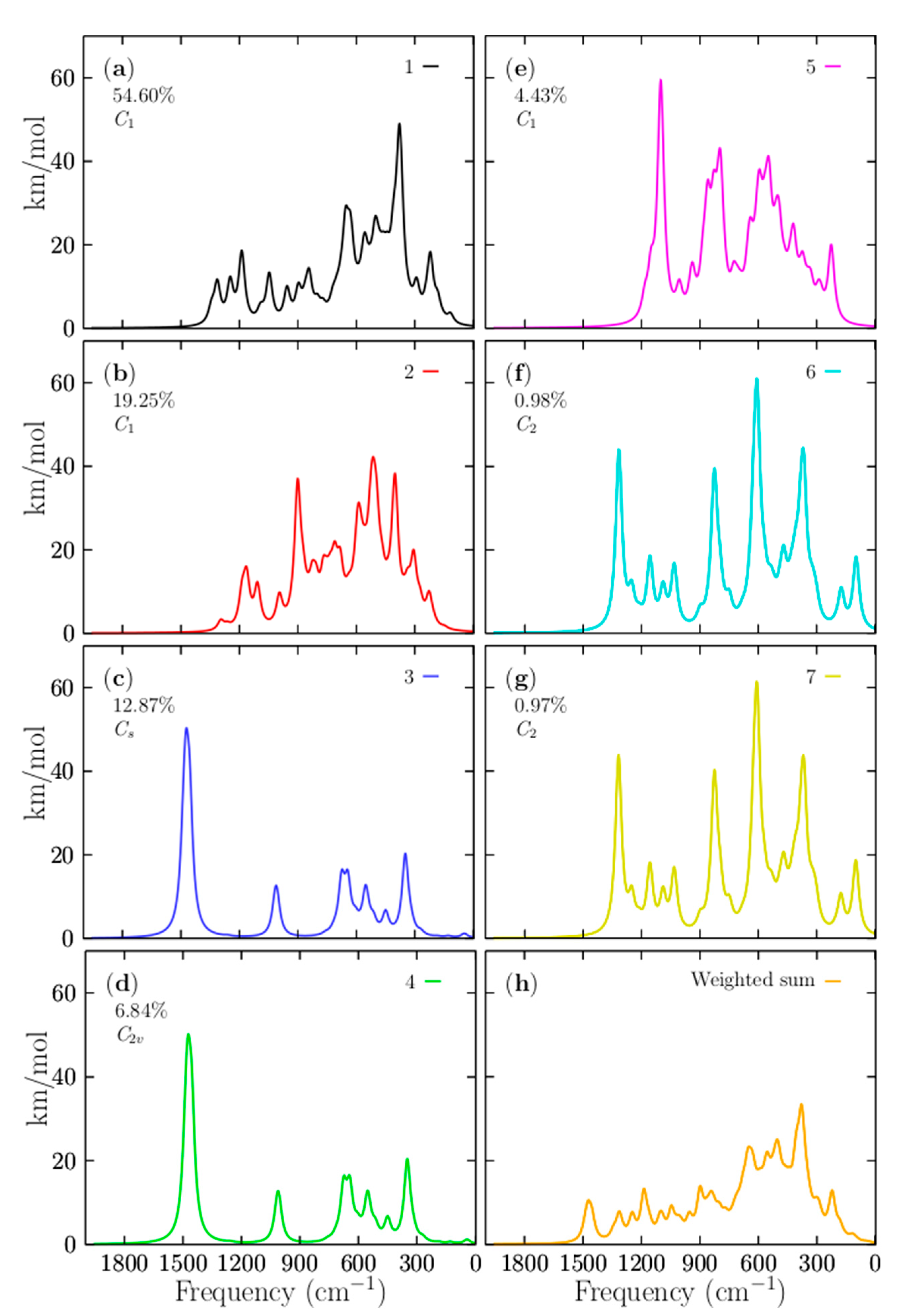
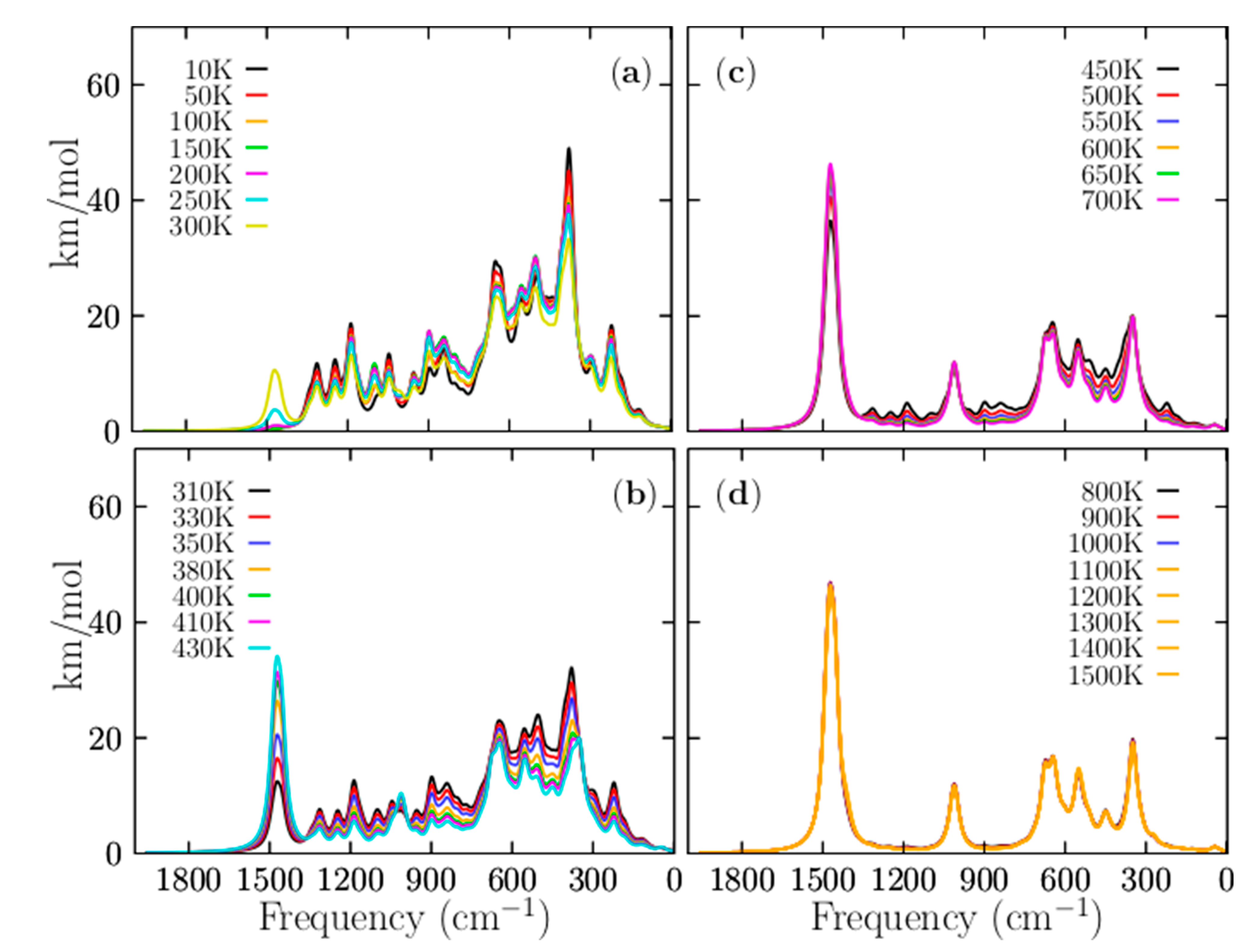
3.5. Infrared Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | Density Functional Theory |
| CCSD(T) | Coupled-cluster single-double and perturbative triple |
| ZPE | Zero point energy |
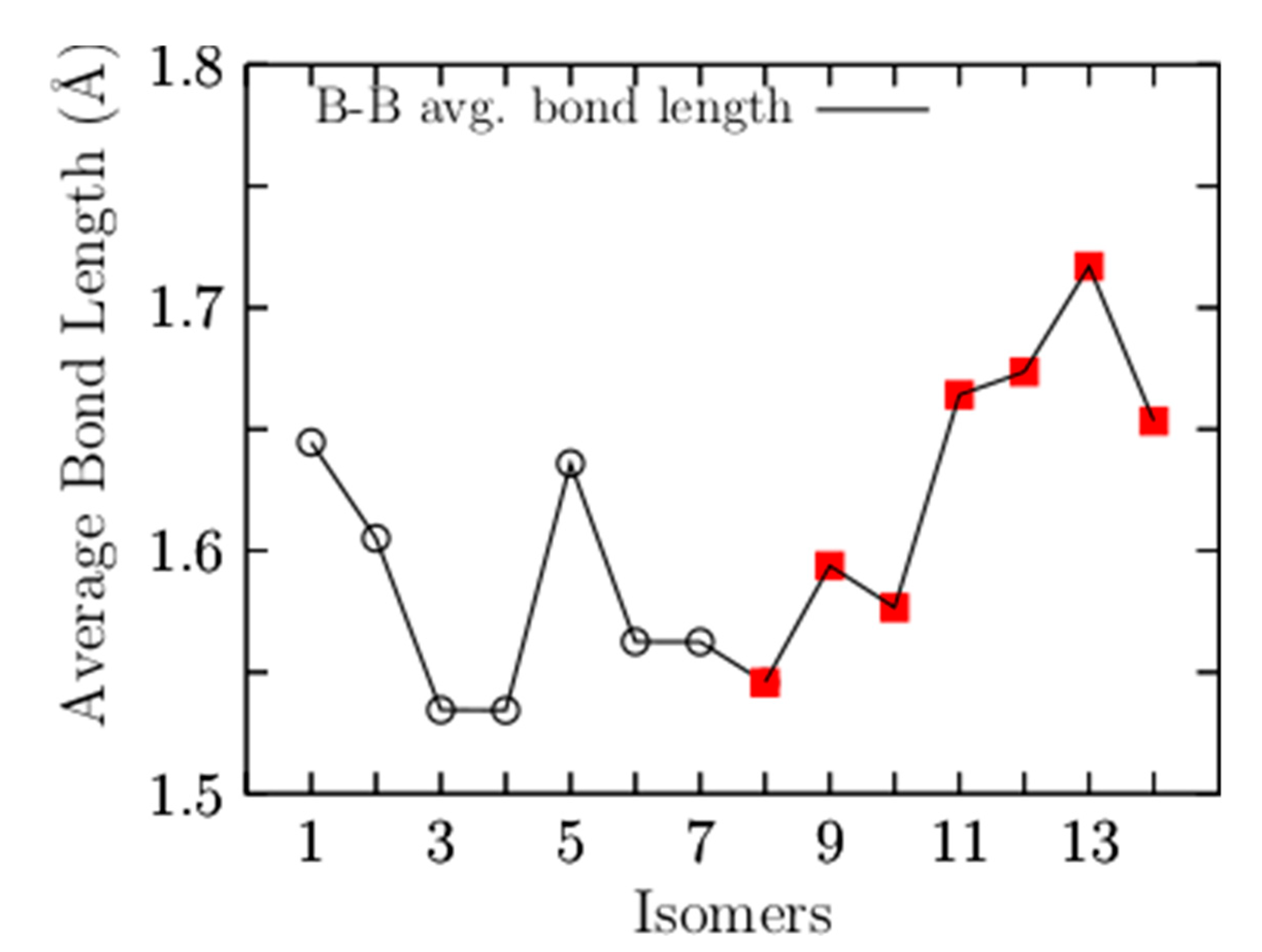
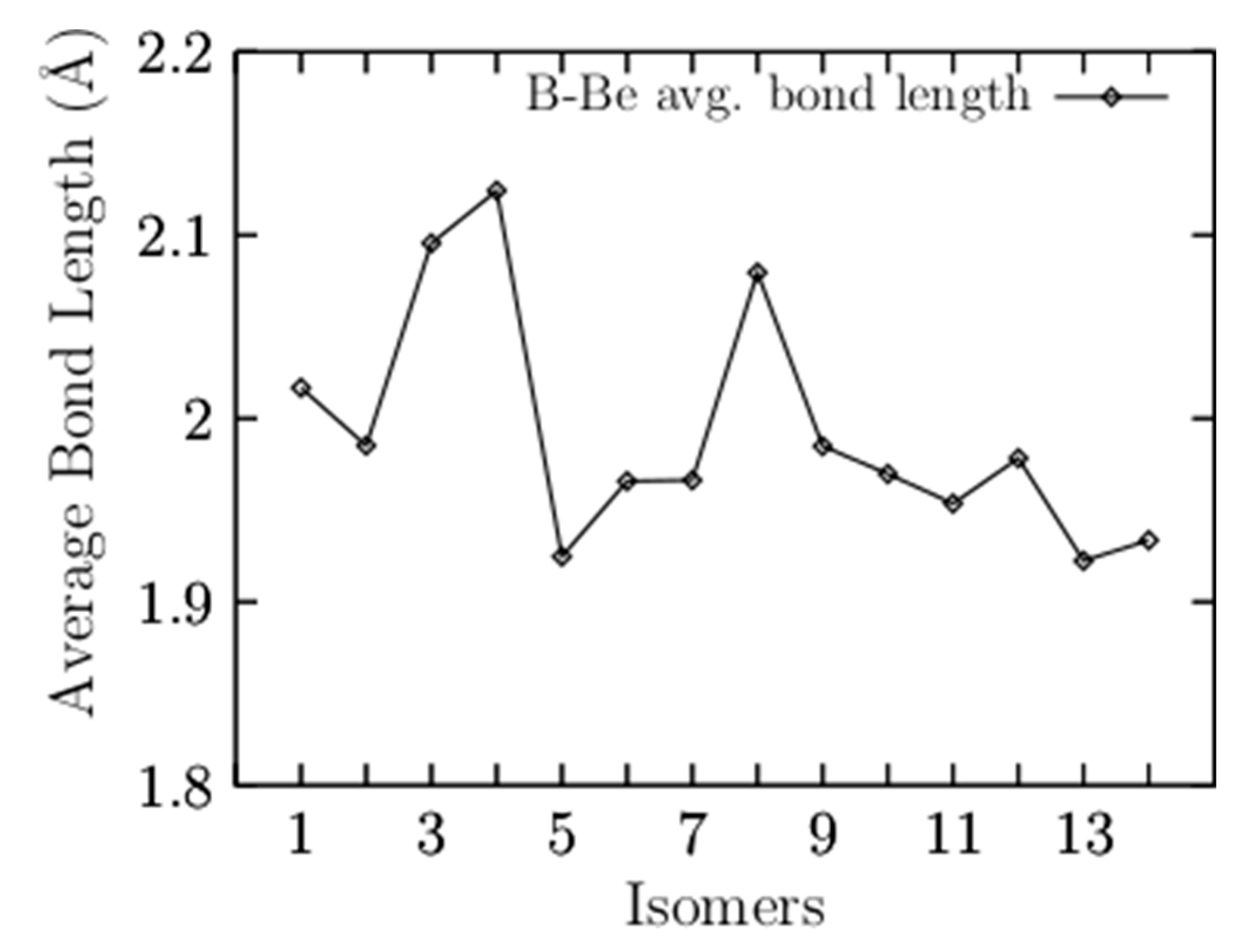
Appendix A. Average Be–B Bond Length

Appendix B. Energetic Ordering According to CCSD(T) Energies

References
- Jalife, S.; Liu, L.; Pan, S.; Cabellos, J.L.; Osorio, E.; Lu, C.; Heine, T.; Donald, K.J.; Merino, G. Dynamical behavior of boron clusters. Nanoscale 2016, 8, 17639–17644. [Google Scholar] [CrossRef]
- Zhai, H.J.; Zhao, Y.F.; Li, W.L.; Chen, Q.; Bai, H.; Hu, H.S.; Piazza, Z.A.; Tian, W.J.; Lu, H.G.; Wu, Y.B.; et al. Observation of an all-boron fullerene. Nat. Chem. 2014, 6, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Kang, D.; Chen, B.; Kuang, X.; Ding, K.; Lu, C. Tuning of Structure Evolution and Electronic Properties through Palladium-Doped Boron Clusters: PdB16 as a Motif for Boron-Based Nanotubes. J. Phys. Chem. A 2020, 1, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Jian, T.; Chen, X.; Li, S.D.; Boldyrev, A.I.; Li, J.; Wang, L.S. Probing the structures and bonding of size-selected boron and doped-boron clusters. Chem. Soc. Rev. 2019, 48, 3550–3591. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.T.; Li, W.L.; Chen, W.J.; Li, J.; Wang, L.S. La3 B1− 4: An inverse triple-decker lanthanide boron cluster. Chem. Commun. 2019, 55, 7864–7867. [Google Scholar] [CrossRef]
- Martínez-Guajardo, G.; Cabellos, J.L.; Díaz-Celaya, A.; Pan, S.; Islas, R.; Chattaraj, P.K.; Heine, T.; Merino, G. Dynamical behavior of Borospherene: A Nanobubble. Sci. Rep. 2015, 22, 11287–11297. [Google Scholar] [CrossRef]
- Kang, D.; Sun, W.; Shi, H.; Lu, C.; Kuang, X.; Chen, B.; Xia, X.; Maroulis, G. Probing the structure and electronic properties of beryllium doped boron clusters: A planar BeB16 cluster motif for metallo-borophene. Sci. Rep. 2019, 9, 14367–14387. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Du, X.; Wang, J.J.; Lu, C.; Chen, H. Probing the Structural Evolution and Stabilities of Medium-Sized MoBn0/– Clusters. J. Phys. Chem. C 2018, 122, 20000–20005. [Google Scholar] [CrossRef]
- Grande-Aztatzi, R.; Martínez-Alanis, P.R.; Cabellos, J.L.; Osorio, E.; Martínez, A.; Merino, G. Structural evolution of small gold clusters doped by one and two boron atoms. J. Comput. Chem. 2014, 35, 2288–2296. [Google Scholar] [CrossRef]
- Dong, X.; Jalife, S.; Vásquez-Espinal, A.; Ravell, E.; Pan, S.; Cabellos, J.L.; Liang, W.Y.; Cui, Z.H.; Merino, G. Li2 B12 and Li3 B12: Prediction of the Smallest Tubular and Cage-like Boron Structures. Angew. Chem. Int. Ed. 2018, 57, 4627–4631. [Google Scholar] [CrossRef]
- Brothers, P.J. Boron complexes of porphyrins and related polypyrrole ligands: Unexpected chemistry for both boron and the porphyrin. Chem. Commun. 2008, 2090–2102. [Google Scholar] [CrossRef] [PubMed]
- Axtell, J.C.; Saleh, L.M.A.; Qian, E.A.; Wixtrom, A.I.; Spokoyny, A.M. Synthesis and Applications of Perfunctionalized Boron Clusters. Inorg. Chem. 2018, 57, 2333–2350. [Google Scholar] [CrossRef] [PubMed]
- Piazza, Z.A.; Hu, H.S.; Li, W.L.; Zhao, Y.F.; Li, J.; Wang, L.S. From planar boron clusters to borophenes and metalloborophenes. Nat. Rev. Chem. 2017, 1, 0071. [Google Scholar] [CrossRef]
- Mannix, A.J.; Zhang, Z.; Guisinger, N.P.; Yakobson, B.I.; Hersam, M.C. Borophene as a prototype for synthetic 2D materials development. Nat. Nanotechnol. 2018, 13, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Vast, N.; Baroni, S.; Zerah, G.; Besson, J.M.; Polian, A.; Grimsditch, M.; Chervin, J.C. Lattice Dynamics of Icosahedral α-Boron under Pressure. Phys. Rev. Lett. 1997, 78, 693–696. [Google Scholar] [CrossRef]
- Fujimori, M.; Nakata, T.; Nakayama, T.; Nishibori, E.; Kimura, K.; Takata, M.; Sakata, M. Peculiar Covalent Bonds in α-Rhombohedral Boron. Phys. Rev. Lett. 1999, 82, 4452–4455. [Google Scholar] [CrossRef]
- Shi, H.; Kuang, X.; Lu, C. LiB13: A New Member of Tetrahedral-Typed B13 Ligand Half-Surround Cluster. Sci. Rep. 2020, 10, 1642–1651. [Google Scholar] [CrossRef]
- Piazza, Z.A.; Hu, H.S.; Li, W.L.; Zhao, Y.F.; Li, J.; Wang, L.S. Planar hexagonal B36 as a potential basis for extended single-atom layer boron sheets. Nat. Commun. 2014, 1, 3113. [Google Scholar] [CrossRef]
- Li, W.L.; Zhao, Y.F.; Hu, H.S.; Li, J.; Wang, L.S. [B30]: A Quasiplanar Chiral Boron Cluster. Angew. Chem. Int. Ed. 2014, 53, 5540–5545. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, T.T.; Li, H.R.; Zhao, X.Y.; Chen, W.J.; Zhai, H.J.; Li, S.D.; Wang, L.S. B31− and B32−: Chiral quasi-planar boron clusters. Nanoscale 2019, 11, 9698–9704. [Google Scholar] [CrossRef]
- Kiran, B.; Bulusu, S.; Zhai, H.J.; Yoo, S.; Zeng, X.C.; Wang, L.S. Planar-to-tubular structural transition in boron clusters: B20 as the embryo of single-walled boron nanotubes. Proc. Natl. Acad. Sci. USA 2005, 102, 961–964. [Google Scholar] [CrossRef]
- Chen, Q.; Li, W.L.; Zhao, Y.F.; Zhang, S.Y.; Hu, H.S.; Bai, H.; Li, H.R.; Tian, W.J.; Lu, H.G.; Zhai, H.J.; et al. Experimental and Theoretical Evidence of an Axially Chiral Borospherene. ACS Nano 2015, 9, 754–760. [Google Scholar] [CrossRef]
- Wang, Y.J.; Zhao, Y.F.; Li, W.L.; Jian, T.; Chen, Q.; You, X.R.; Ou, T.; Zhao, X.Y.; Zhai, H.J.; Li, S.D.; et al. Observation and characterization of the smallest borospherene, B28− and B28. J. Chem. Phys. 2016, 144, 064307. [Google Scholar] [CrossRef]
- Lv, J.; Wang, Y.; Zhang, L.; Lin, H.; Zhao, J.; Ma, Y. Stabilization of fullerene-like boron cages by transition metal encapsulation. Nanoscale 2015, 7, 10482–10489. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.Y.; Guo, J.C.; Li, P.F.; Zhai, H.J. Boron-Based Chiral Helix Be6 B10 and Be6 B11 Clusters: Structures, Chemical Bonding, and Formation Mechanism. Chem. Asian J. 2020, 15, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.C.; Feng, L.Y.; Wang, Y.J.; Jalife, S.; Vásquez-Espinal, A.; Cabellos, J.L.; Pan, S.; Merino, G.; Zhai, H.J. Coaxial Triple-Layered versus Helical Be6B11− Clusters: Dual Structural Fluxionality and Multifold Aromaticity. Angew. Chem. Int. Ed. 2017, 56, 10174–10177. [Google Scholar] [CrossRef] [PubMed]
- Mannix, A.J.; Zhou, X.F.; Kiraly, B.; Wood, J.D.; Alducin, D.; Myers, B.D.; Liu, X.; Fisher, B.L.; Santiago, U.; Guest, J.R.; et al. Synthesis of borophenes: Anisotropic, two-dimensional boron polymorphs. Science 2015, 350, 1513–1516. [Google Scholar] [CrossRef]
- Jimenez-Halla, J.; Islas, R.; Heine, T.; Merino, G. B19−: An Aromatic Wankel Motor. Angew. Chem. Int. Ed. 2010, 49, 5668–5671. [Google Scholar] [CrossRef]
- Barney, W.K.; Sehmel, G.A.; Seymour, W.E. The Use of Boron Carbide for Reactor Control. Nucl. Sci. Eng. 1958, 4, 439–448. [Google Scholar] [CrossRef]
- Leśnikowski, Z.J. Challenges and Opportunities for the Application of Boron Clusters in Drug Design. J. Med. Chem. 2016, 59, 7738–7758. [Google Scholar] [CrossRef]
- Ali, F.; S Hosmane, N.; Zhu, Y. Boron Chemistry for Medical Applications. Molecules 2020, 25, 828. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Wang, L.; Jiang, Y.; Liu, Q.; Huang, C. Hexagonal boron nitride nanoplates as emerging biological nanovectors and their potential applications in biomedicine. J. Mater. Chem. B 2016, 4, 6103–6110. [Google Scholar] [CrossRef] [PubMed]
- Oña, O.B.; Torres-Vega, J.J.; Torre, A.; Lain, L.; Alcoba, D.R.; Vásquez-Espinal, A.; Tiznado, W. Chemical bonding analysis in boron clusters by means of localized orbitals according to the electron localization function topology. Theor. Chem. Acc. 2015, 134, 28–37. [Google Scholar] [CrossRef]
- Alexandrova, A.N.; Boldyrev, A.I.; Zhai, H.J.; Wang, L.S. All-boron aromatic clusters as potential new inorganic ligands and building blocks in chemistry. Coord. Chem. Rev. 2006, 250, 2811–2866. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Comprehensive analysis of chemical bonding in boron clusters. J. Comput. Chem. 2007, 28, 251–268. [Google Scholar] [CrossRef] [PubMed]
- Poater, J.; Duran, M.; Solà, M.; Silvi, B. Theoretical Evaluation of Electron Delocalization in Aromatic Molecules by Means of Atoms in Molecules (AIM) and Electron Localization Function (ELF) Topological Approaches. Chem. Rev. 2005, 105, 3911–3947. [Google Scholar] [CrossRef]
- Mandado, M.; González-Moa, M.J.; Mosquera, R.A. QTAIM n-center delocalization indices as descriptors of aromaticity in mono and poly heterocycles. J. Comput. Chem. 2007, 28, 127–136. [Google Scholar] [CrossRef]
- Pan, S.; Barroso, J.; Jalife, S.; Heine, T.; Asmis, K.R.; Merino, G. Fluxional Boron Clusters: From Theory to Reality. Acc. Chem. Res. 2019, 52, 2732–2744. [Google Scholar] [CrossRef]
- Wang, Y.J.; Feng, L.Y.; Guo, J.C.; Zhai, H.J. Dynamic Mg2 B8 Cluster: A Nanoscale Compass. Chem. Asian J. 2017, 12, 2899–2903. [Google Scholar] [CrossRef]
- Zhai, H.; Alexandrova, A.N. Fluxionality of Catalytic Clusters: When It Matters and How to Address It. ACS Catal. 2017, 7, 1905–1911. [Google Scholar] [CrossRef]
- Merino, G.; Heine, T. And Yet It Rotates: The Starter for a Molecular. Wankel Motor. Angew. Chem. Int. Ed. 2012, 51, 10226–10227. [Google Scholar] [CrossRef] [PubMed]
- Romanescu, C.; Galeev, T.R.; Li, W.L.; Boldyrev, A.I.; Wang, L.S. Transition-Metal-Centered Monocyclic Boron Wheel Clusters (M c Bn): A New Class of Aromatic Borometallic Compounds. Acc. Chem. Res. 2013, 46, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.Y.; Barroso, J.; Jalife, S.; Orozco-Ic, M.; Zarate, X.; Dong, X.; Cui, Z.H.; Merino, G. B10M2 (M = Rh, Ir): Finally a stable boron-based icosahedral cluster. Chem. Commun. 2019, 55, 7490–7493. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.T.; Li, W.L.; Bai, H.; Chen, W.J.; Dong, X.R.; Li, J.; Wang, L.S. ReB8 and ReB9−: New Members of the ransition-Metal-Centered Borometallic Molecular Wheel Family. J. Phys. Chem. A 2019, 123, 5317–5324. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Jalife, S.; Vásquez-Espinal, A.; Barroso, J.; Orozco-Ic, M.; Ravell, E.; Cabellos, J.L.; Liang, W.Y.; Cui, Z.H.; Merino, G. Li2 B24: The simplest combination for a three-ring boron tube. Nanoscale 2019, 11, 2143–2147. [Google Scholar] [CrossRef] [PubMed]
- Popov, I.A.; Jian, T.; Lopez, G.V.; Boldyrev, A.I.; Wang, L.S. Cobalt-centred boron molecular drums with the highest coordination number in the CoB16 cluster. Nat. Commun. 2015, 6, 8654. [Google Scholar] [CrossRef]
- Van Duong, L.; Tho Nguyen, M. Silicon doped boron clusters: How to make stable ribbons? Phys. Chem. Chem. Phys. 2017, 19, 14913–14918. [Google Scholar] [CrossRef]
- Cervantes-Navarro, F.; Martínez-Guajardo, G.; Osorio, E.; Moreno, D.; Tiznado, W.; Islas, R.; Donald, K.J.; Merino, G. Stop rotating! One substitution halts the B19 motor. Chem. Commun. 2014, 50, 10680–10682. [Google Scholar] [CrossRef]
- Ferrari, P.; Vanbuel, J.; Hansen, K.; Lievens, P.; Janssens, E.; Fielicke, A. Effect of radiative cooling on the size-dependent stability of small boron clusters. Phys. Rev. A 2018, 98, 012501. [Google Scholar] [CrossRef]
- Martínez-Guajardo, G.; Sergeeva, A.P.; Boldyrev, A.I.; Heine, T.; Ugalde, J.M.; Merino, G. Unravelling phenomenon of internal rotation in B13+ through chemical bonding analysis. Chem. Commun. 2011, 47, 6242–6244. [Google Scholar] [CrossRef]
- Moreno, D.; Pan, S.; Zeonjuk, L.L.; Islas, R.; Osorio, E.; Martínez-Guajardo, G.; Chattaraj, P.K.; Heine, T.; Merino, G. B18: A quasi-planar bowl member of the Wankel motor family. Chem. Commun. 2014, 50, 8140–8143. [Google Scholar] [CrossRef] [PubMed]
- Tai, T.B.; Ceulemans, A.; Nguyen, M.T. Disk Aromaticity of the Planar and Fluxional Anionic Boron Clusters B20-/2-. Chem. A Eur. J. 2012, 18, 4510–4512. [Google Scholar] [CrossRef] [PubMed]
- Fagiani, M.R.; Song, X.; Petkov, P.; Debnath, S.; Gewinner, S.; Schöllkopf, W.; Heine, T.; Fielicke, A.; Asmis, K.R. Structure and Fluxionality of B13+ Probed by Infrared. Photodissociation Spectroscopy. Angew. Chem. Int. Ed. 2017, 56, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jia, D.; Wang, Y.J.; Zhai, H.J.; Man, Y.; Li, S.D. A universal mechanism of the planar boron rotors B11−, B13+, B15+, and B19−: Inner wheels rotating in pseudo-rotating outer bearings. Nanoscale 2017, 9, 1443–1448. [Google Scholar] [CrossRef]
- Wang, Y.J.; Zhao, X.Y.; Chen, Q.; Zhai, H.J.; Li, S.D. B11−: A moving subnanoscale tank tread. Nanoscal 2015, 7, 16054–16060. [Google Scholar] [CrossRef]
- Wang, Y.J.; Guo, J.C.; Zhai, H.J. Why nanoscale tank treads move? Structures, chemical bonding, and molecular dynamics of a doped boron cluster B10C. Nanoscale 2017, 9, 9310–9316. [Google Scholar] [CrossRef]
- Yañez, O.; Inostroza, D.; Usuga-Acevedo, B.; Vásquez-Espinal, A.; Pino-Rios, R.; Tabilo-Sepulveda, M.; Garza, J.; Barroso, J.; Merino, G.; Tiznado, W. valuation of restricted probabilistic cellular automata on the exploration of the potential energy surface of Be6 B11. Theor. Chem. Acc. 2020, 139, 139–147. [Google Scholar] [CrossRef]
- Baletto, F.; Ferrando, R. Structural properties of nanoclusters: Energetic, thermodynamic, and kinetic effects. Rev. Mod. Phys. 2005, 77, 371–423. [Google Scholar] [CrossRef]
- Grigoryan, V.G.; Springborg, M. Temperature and isomeric effects in nanoclusters. Phys. Chem. Chem. Phys. 2019, 21, 5646–5654. [Google Scholar] [CrossRef]
- Calvo, F.; Doye, J.P.K.; Wales, D.J. Quantum partition functions from classical distributions: Application to rare-gas clusters. J. Chem. Phys. 2001, 114, 7312–7329. [Google Scholar] [CrossRef]
- Bréchignac, C.; Cahuzac, P.; de Frutos, M.; Kebaïli, N.; Sarfati, A.; Akulin, V. Experimental Evidence for the Entropy Effect in Coulombic Cluster Fission. Phys. Rev. Lett. 1996, 77, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.; Ferrando, R.; Palmer, R. Experimental determination of the energy difference between competing isomers of deposited, size-selected gold nanoclusters. Nat. Commun. 2018, 9, 1323–1334. [Google Scholar] [CrossRef]
- Dzib, E.; Cabellos, J.L.; Ortíz-Chi, F.; Pan, S.; Galano, A.; Merino, G. Eyringpy: A program for computing rate constants in the gas phase and in solution. Int. J. Quantum Chem. 2019, 119, e25686. [Google Scholar] [CrossRef]
- Vargas-Caamal, A.; Cabellos, J.L.; Ortiz-Chi, F.; Rzepa, H.S.; Restrepo, A.; Merino, G. How Many Water Molecules Does it Take to Dissociate HCl? Chem. A Eur. J. 2016, 22, 2812–2818. [Google Scholar] [CrossRef] [PubMed]
- Shkrebtii, A.; Heron, J.; Cabellos, J.; Witkowski, N.; Pluchery, O.; Mendoza, B.; Borensztein, Y. Temperature Dependent Optical Response of Si(100): Theory vs. Experiment. MRS Proc. 2011, 1370. [Google Scholar] [CrossRef]
- Goldsmith, B.R.; Florian, J.; Liu, J.X.; Gruene, P.; Lyon, J.T.; Rayner, D.M.; Fielicke, A.; Scheffler, M.; Ghiringhelli, L.M. Two-to-three dimensional transition in neutral gold clusters: The crucial role of van der Waals interactions and temperature. Phys. Rev. Mater. 2019, 3, 016002. [Google Scholar] [CrossRef]
- Ghiringhelli, L.M.; Gruene, P.; Lyon, J.T.; Rayner, D.M.; Meijer, G.; Fielicke, A.; Scheffler, M. Not so loosely bound rare gas atoms: Finite-temperature vibrational fingerprints of neutral gold-cluster complexes. New J. Phys. 2013, 15, 083003. [Google Scholar] [CrossRef]
- Schebarchov, D.; Baletto, F.; Wales, D.J. Structure, thermodynamics, and rearrangement mechanisms in gold clusters—insights from the energy landscapes framework. Nanoscale 2018, 10, 2004–2016. [Google Scholar] [CrossRef]
- Mendoza-Wilson, A.M.; Balandrán-Quintana, R.R.; Cabellos, J.L. Thermochemical behavior of sorghum procyanidin trimers with C4-C8 and C4-C6 interflavan bonds in the reaction with superoxide anion radical and H2O2-forming NADH-oxidase flavoenzyme. Comput. Theor. Chem. 2020, 1186, 112912. [Google Scholar] [CrossRef]
- Seitsonen, A.P.; Laasonen, K.; Nieminen, R.M.; Klein, M.L. Structure of CAl12. J. Chem. Phys. 1995, 103, 8075–8080. [Google Scholar] [CrossRef][Green Version]
- Chandrachud, P.; Joshi, K.; Kanhere, D.G. Thermodynamics of carbon-doped Al and Ga clusters: Ab initio molecular dynamics simulations. Phys. Rev. B 2007, 76, 235423. [Google Scholar] [CrossRef]
- Doye, J.P.K.; Calvo, F. Entropic effects on the structure of Lennard-Jones clusters. J. Chem. Phys. 2002, 116, 8307–8317. [Google Scholar] [CrossRef]
- Li, Z.H.; Jasper, A.W.; Truhlar, D.G. Structures, Rugged Energetic Landscapes, and Nanothermodynamics of Aln (2, <n< 65) Particles. J. Am. Chem. Soc. 2007, 129, 14899–14910. [Google Scholar] [CrossRef]
- Darby, S.; Mortimer-Jones, T.V.; Johnston, R.L.; Roberts, C. Theoretical study of Cu–Au nanoalloy clusters using a genetic algorithm. J. Chem. Phys. 2002, 116, 1536–1550. [Google Scholar] [CrossRef]
- Doye, J.P.K.; Wales, D.J. Global minima for transition metal clusters described by Sutton-Chen potentials. New J. Chem. 1998, 22, 733–744. [Google Scholar] [CrossRef]
- Ohno, K.; Maeda, S. Global Reaction Route Mapping on Potential Energy Surfaces of Formaldehyde, Formic Acid, and Their Metal-Substituted Analogues. J. Phys. Chem. A 2006, 110, 8933–8941. [Google Scholar] [CrossRef] [PubMed]
- Strøm, B.A.; Simon, J.M.; Schnell, S.K.; Kjelstrup, S.; He, J.; Bedeaux, D. Size and shape effects on the thermodynamic properties of nanoscale volumes of water. Phys. Chem. Chem. Phys. 2017, 19, 9016–9027. [Google Scholar] [CrossRef]
- Hill, T.L. Extension of Nanothermodynamics to Include a One-Dimensional Surface Excess. Nano Lett. 2001, 1, 159–160. [Google Scholar] [CrossRef]
- Gibbs, J. Thermodynamics; Scientific Papers; Dover Publications: New York, NY, USA, 1961. [Google Scholar]
- Li, Z.H.; Truhlar, D.G. Nanothermodynamics of metal nanoparticles. Chem. Sci. 2014, 5, 2605–2624. [Google Scholar] [CrossRef]
- Hill, T.L. Thermodynamics of Small Systems. J. Chem. Phys. 1962, 36, 3182–3197. [Google Scholar] [CrossRef]
- Calvo, F. Thermodynamics of nanoalloys. Phys. Chem. Chem. Phys. 2015, 17, 27922–27939. [Google Scholar] [CrossRef] [PubMed]
- Bixon, M.; Jortner, J. Energetic and thermodynamic size effects in molecular clusters. J. Chem. Phys. 1989, 91, 1631–1642. [Google Scholar] [CrossRef]
- Kristensen, W.D.; Jensen, E.J.; Cotterill, R.M.J. Thermodynamics of small clusters of atoms: A molecular dynamics simulation. J. Chem. Phys. 1974, 60, 4161–4169. [Google Scholar] [CrossRef]
- Wales, D.J. Structure, Dynamics, and Thermodynamics of Clusters: Tales from Topographic Potential Surfaces. Science 1996, 271, 925–929. [Google Scholar] [CrossRef]
- Jena, P.; Khanna, S.; Rao, B. Physics and Chemistry of Finite Systems: From Clusters to Crystals; Number v. 2 in NATO ASI Series: Advanced Science Institutes Series: Series C, Mathematical and Physical Sciences; Kluwer Academic Publishers: Richmond, VA, USA, 1992. [Google Scholar]
- Fox, H.; Horsfield, A.P.; Gillan, M.J. Density functional calculations of surface free energies. J. Chem. Phys. 2006, 124, 134709. [Google Scholar] [CrossRef] [PubMed]
- Beret, E.C.; Ghiringhelli, L.M.; Scheffler, M. Free gold clusters: Beyond the static, monostructure description. Faraday Discuss 2011, 152, 153–167. [Google Scholar] [CrossRef]
- Lv, Z.L.; Xu, K.; Cheng, Y.; Chen, X.R.; Cai, L.C. Ab initio investigation of the lower energy candidate structures for (H2O)5+ water cluster. J. Chem. Phys. 2014, 141, 054309. [Google Scholar] [CrossRef]
- Malloum, A.; Fifen, J.J.; Dhaouadi, Z.; Engo, S.G.N.; Jaidane, N.E. Structures and relative stabilities of ammonia clusters at different temperatures: DFT vs. ab initio. Phys. Chem. Chem. Phys. 2015, 17, 29226–29242. [Google Scholar] [CrossRef]
- Malloum, A.; Fifen, J.J.; Dhaouadi, Z.; Nana Engo, S.G.; Jaidane, N.E. Structures and spectroscopy of medium size protonated ammonia clusters at different temperatures, H+(NH3)10–16. J. Chem. Phys. 2017, 146, 044305. [Google Scholar] [CrossRef] [PubMed]
- Malloum, A.; Fifen, J.J.; Conradie, J. Structures and infrared spectroscopy of large sized protonated ammonia clusters. J. Chem. Phys. 2018, 149, 244301. [Google Scholar] [CrossRef]
- Fifen, J.J.; Agmon, N. Structure and Spectroscopy of Hydrated Sodium Ions at Different Temperatures and the Cluster Stability Rules. J. Chem. Theory Comput. 2016, 12, 1656–1673. [Google Scholar] [CrossRef] [PubMed]
- Gruene, P.; Rayner, D.M.; Redlich, B.; van der Meer, A.F.G.; Lyon, J.T.; Meijer, G.; Fielicke, A. Structures of Neutral Au7, Au19, and Au20 Clusters in the Gas Phase. Science 2008, 321, 674–676. [Google Scholar] [CrossRef] [PubMed]
- Fielicke, A.; von Helden, G.; Meijer, G.; Pedersen, D.B.; Simard, B.; Rayner, D.M. Gold Cluster Carbonyls Saturated Adsorption of CO on Gold Cluster Cations, Vibrational Spectroscopy, and Implications for Their Structures. J. Am. Chem. Soc. 2005, 127, 8416–8423. [Google Scholar] [CrossRef]
- Fielicke, A.; Kirilyuk, A.; Ratsch, C.; Behler, J.; Scheffler, M.; von Helden, G.; Meijer, G. Structure Determination of Isolated Metal Clusters via Far-Infrared Spectroscopy. Phys. Rev. Lett. 2004, 93, 023401. [Google Scholar] [CrossRef]
- Sieber, C.; Buttet, J.; Harbich, W.; Félix, C.; Mitrić, R.; Bonačić Koutecký, V. Isomer-specific spectroscopy of metal clusters trapped in a matrix: Ag9. Phys. Rev. A 2004, 70, 041201. [Google Scholar] [CrossRef]
- Ji, M.; Gu, X.; Li, X.; Gong, X.; Li, J.; Wang, L.S. Experimental and Theoretical Investigation of the Electronic and Geometrical Structures of the Au32 Cluster. Angew. Chem. Int. Ed. 2005, 44, 7119–7123. [Google Scholar] [CrossRef]
- Wille, L.T.; Vennik, J. Computational complexity of the ground-state determination of atomic clusters. J. Phys. A Math. Gen. 1985, 18, L419–L422. [Google Scholar] [CrossRef]
- Xu, S.G.; Zhao, Y.J.; Liao, J.H.; Yang, X.B. Understanding the stable boron clusters: A bond model and first-principles calculations based on high-throughput screening. J. Chem. Phys. 2015, 142, 214307. [Google Scholar] [CrossRef]
- Rossi, G.; Ferrando, R. Searching for low-energy structures of nanoparticles: A comparison of different methods and algorithms. J. Phys. Condens. Matter 2009, 21, 084208. [Google Scholar] [CrossRef]
- Cheng, L.; Feng, Y.; Yang, J.; Yang, J. Funnel hopping: Searching the cluster potential energy surface over the funnels. J. Chem. Phys. 2009, 130, 214112. [Google Scholar] [CrossRef]
- Kirkpatrick, S.; Gelatt, C.D.; Vecchi, M.P. Optimization by Simulated Annealing. Science 1983, 220, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- Xiang, Y.; Gong, X.G. Efficiency of generalized simulated annealing. Phys. Rev. E 2000, 62, 4473–4476. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Gubian, S.; Suomela, B.; Hoeng, J. Generalized Simulated Annealing for Global Optimization: The GenSA Package for R. R J. 2013, 5, 13–29. [Google Scholar] [CrossRef]
- Vlachos, D.; Schmidt, L.; Aris, R. Comparison of small metal clusters: Ni, Pd, Pt, Cu, Ag, Au. Z. Phys. D At. Mol. Clst. 1993, 26, 156–158. [Google Scholar] [CrossRef]
- Granville, V.; Krivanek, M.; Rasson, J.P. Simulated annealing: A proof of convergence. IEEE Trans. Pattern Anal. Mach. Intell. 1994, 16, 652–656. [Google Scholar] [CrossRef]
- Saunders, M. Stochastic search for isomers on a quantum mechanical surface. J. Comput. Chem. 2004, 25, 621–626. [Google Scholar] [CrossRef]
- Saunders, M. Stochastic exploration of molecular mechanics energy surfaces. Hunting for the global minimum. J. Am. Chem. Soc. 1987, 109, 3150–3152. [Google Scholar] [CrossRef]
- Hsu, P.J.; Lai, S.K. Structures of bimetallic clusters. J. Chem. Phys. 2006, 124, 044711-0. [Google Scholar] [CrossRef]
- Qin, W.; Lu, W.C.; Zhao, L.Z.; Zang, Q.J.; Wang, C.Z.; Ho, K.M. Stabilities and fragmentation energies of Si n clusters (n = 2–33). J. Phys. Condens. Matter 2009, 21, 455501. [Google Scholar] [CrossRef]
- Goldberg, D.E. Genetic Algorithms in Search, Optimization and Machine Learning, 1st ed.; Addison-Wesley Longman Publishing Co., Inc.: Boston, MA, USA, 1989. [Google Scholar]
- Alexandrova, A.N.; Boldyrev, A.I. Search for the Li0, +1, −1 (n = 5–7) Lowest-Energy Structures Using the ab Initio Gradient Embedded Genetic Algorithm (GEGA). Elucidation of the Chemical Bonding in the Lithium Clusters. J. Chem. Theory Comput. 2005, 1, 566–580. [Google Scholar] [CrossRef]
- Alexandrova, A.N.; Boldyrev, A.I.; Fu, Y.J.; Yang, X.; Wang, X.B.; Wang, L.S. Structure of the Nax Clx (x = 1, 4) clusters via ab-initio genetic algorithm and photoelectron spectroscopy. J. Chem. Phys. 2004, 121, 5709–5719. [Google Scholar] [CrossRef]
- Alexandrova, A.N. H·(H2O)n Clusters: Microsolvation of the Hydrogen Atom via Molecular ab Initio Gradient Embedded Genetic Algorithm (GEGA). J. Phys. Chem. A 2010, 114, 12591–12599. [Google Scholar] [CrossRef] [PubMed]
- Harding, D.; Mackenzie, S.R.; Walsh, T.R. Structural Isomers and Reactivity for Rh6 and Rh6+. J. Phys. Chem. B 2006, 110, 18272–18277. [Google Scholar] [CrossRef] [PubMed]
- Wales, D.J.; Doye, J.P.K. Global Optimization by Basin-Hopping and the Lowest Energy Structures of Lennard-Jones Clusters Containing up to 110 Atoms. J. Phys. Chem. A 1997, 101, 5111–5116. [Google Scholar] [CrossRef]
- Mondal, S.; Cabellos, J.L.; Pan, S.; Osorio, E.; Torres-Vega, J.J.; Tiznado, W.; Restrepo, A.; Merino, G. 10-pi-Electron arenes a la carte: Structure and bonding of the [E-(Cn H)-E]n-6 (E = Ca, Sr, Ba; n = 6–8) complexes. Phys. Chem. Chem. Phys. 2016, 18, 11909–11918. [Google Scholar] [CrossRef] [PubMed]
- Ravell, E.; Jalife, S.; Barroso, J.; Orozco-Ic, M.; Hernandez-Juarez, G.; Ortiz-Chi, F.; Pan, S.; Cabellos, J.L.; Merino, G. Structure and Bonding in CE5—(E=Al-Tl) Clusters: Planar Tetracoordinate Carbon versus Pentacoordinate Carbon. Chem. Asian J. 2018, 13, 1467–1473. [Google Scholar] [CrossRef]
- Pan, S.; Moreno, D.; Cabellos, J.L.; Romero, J.; Reyes, A.; Merino, G.; Chattaraj, P.K. In Quest of Strong Be-Ng Bonds among the Neutral Ng-Be Complexes. J. Phys. Chem. A 2014, 118, 487–494. [Google Scholar] [CrossRef]
- Cui, Z.H.; Ding, Y.H.; Cabellos, J.L.; Osorio, E.; Islas, R.; Restrepo, A.; Merino, G. Planar tetracoordinate carbons with a double bond in CAl3E clusters. Phys. Chem. Chem. Phys. 2015, 17, 8769–8775. [Google Scholar] [CrossRef]
- Vargas-Caamal, A.; Pan, S.; Ortiz-Chi, F.; Cabellos, J.L.; Boto, R.A.; Contreras-Garcia, J.; Restrepo, A.; Chattaraj, P.K.; Merino, G. How strong are the metallocene–metallocene interactions? Cases of ferrocene, ruthenocene, and osmocene. Phys. Chem. Chem. Phys. 2016, 18, 550–556. [Google Scholar] [CrossRef]
- Cui, Z.H.; Vassilev-Galindo, V.; Luis Cabellos, J.; Osorio, E.; Orozco, M.; Pan, S.; Ding, Y.H.; Merino, G. Planar pentacoordinate carbon atoms embedded in a metallocene framework. Chem. Commun. 2017, 53, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Caamal, A.; Ortiz-Chi, F.; Moreno, D.; Restrepo, A.; Merino, G.; Cabellos, J.L. The rich and complex potential energy surface of the ethanol dimer. Theor. Chem. Acc. 2015, 134, 16. [Google Scholar] [CrossRef]
- Flórez, E.; Acelas, N.; Ibargüen, C.; Mondal, S.; Cabellos, J.L.; Merino, G.; Restrepo, A. Microsolvation of NO3−: Structural exploration and bonding analysis. RSC Adv. 2016, 6, 71913–71923. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Wallingford, CT, USA, 2009. [Google Scholar]
- McQuarrie, D.A. Statistical Mechanics; Chemistry Series; Harper & Row: Manhattan, NY, USA, 1975. [Google Scholar]
- Hill, T. An Introduction to Statistical Thermodynamics; Addison-Wesley Series in Chemistry; Dover Publications: New York, NY, USA, 1986. [Google Scholar]
- Teague, S.J. Implications of protein flexibility for drug discovery. Nat. Rev. Drug Discov. 2003, 2, 527–541. [Google Scholar] [CrossRef]
- An, W.; Bulusu, S.; Gao, Y.; Zeng, X.C. Relative stability of planar versus double-ring tubular isomers of neutral and anionic boron cluster B20 and B20−. J. Chem. Phys. 2006, 124, 154310. [Google Scholar] [CrossRef]
- Shortle, D. Propensities, probabilities, and the Boltzmann hypothesis. Comput. Theor. Chem. 2003, 12, 1298–1302. [Google Scholar] [CrossRef]
- Kubicki, J.; Watts, H. Quantum Mechanical Modeling of the Vibrational Spectra of Minerals with a Focus on Clays. Minerals 2019, 9, 141. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Pan, L.L.; Li, J.; Wang, L.S. Low-lying isomers of the B9− boron cluster: The planar molecular wheel versus three-dimensional structures. J. Chem. Phys. 2008, 129, 024302. [Google Scholar] [CrossRef] [PubMed]
- Shoji, Y.; Matsuo, T.; Hashizume, D.; Gutmann, M.J.; Fueno, H.; Tanaka, K.; Tamao, K. Boron-Boron σ-Bond Formation by Two-Electron Reduction of a H-Bridged Dimer of Monoborane. J. Am. Chem. Soc. 2011, 133, 11058–11061. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.J.; Alexandrova, A.N.; Birch, K.A.; Boldyrev, A.I.; Wang, L.S. Hepta- and Octacoordinate Boron in Molecular Wheels of Eight- and Nine-Atom Boron Clusters: Observation and Confirmation. Angew. Chem. Int. Ed. 2003, 42, 6004–6008. [Google Scholar] [CrossRef] [PubMed]
- Moezzi, A.; Olmstead, M.M.; Power, P.P. Boron-boron double bonding in the species [B2R4]2-: Synthesis and structure of [(Et2O)Li2Mes2BB(Mes)Ph], a diborane(4) dianion analog of a substituted ethylene. J. Am. Chem. Soc. 1992, 114, 2715–2717. [Google Scholar] [CrossRef]
- Zhou, M.; Tsumori, N.; Li, Z.; Fan, K.; Andrews, L.; Xu, Q. OCBBCO: A Neutral Molecule with Some Boron-Boron Triple Bond Character. J. Am. Chem. Soc. 2002, 124, 12936–12937. [Google Scholar] [CrossRef]
- Feixas, F.; Matito, E.; Poater, J.; Sola, M. Understanding Conjugation and Hyperconjugation from Electronic Delocalization Measures. J. Phys. Chem. A 2011, 115, 13104–13113. [Google Scholar] [CrossRef]
- Szabo, A.; Kovacs, A.; Frenking, G. Theoretical Studies of Inorganic Compounds. 341) Energy Decomposition Analysis of E-E Bonding in Planar and Perpendicular X2E-EX2 (E= B, Al, Ga, In, Tl; X = H, F, Cl, Br, I). Z. Anorg. Allg. Chem. 2005, 631, 1803–1809. [Google Scholar] [CrossRef]
- Geudtner, G.; Calaminici, P.; Carmona-Espíndola, J.; del Campo, J.M.; Domínguez-Soria, V.D.; Moreno, R.F.; Gamboa, G.U.; Goursot, A.; Köster, A.M.; Reveles, J.U.; et al. deMon2k. Wires Comput. Mol. Sci. 2012, 2, 548–555. [Google Scholar] [CrossRef]
- Li, M.; Wang, J.; Fu, B.; Hou, Q. A molecular dynamics study of melting and dissociation of tungsten nanoparticles. AIP Adv. 2015, 5, 127131. [Google Scholar] [CrossRef]
- Spickermann, C. Entropies of Condensed Phases and Complex Systems: A First Principles Approach; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contributions | Partition Function |
|---|---|
| Translational | |
| Rotational linear | |
| Rotational nonlinear | |
| Vibrational | |
| Electronic |
| Contributions | Internal Energy | Entropy |
|---|---|---|
| Translational | ||
| Rotational linear | ||
| Rotational nonlinear | ||
| Vibrational | ||
| - | ||
| Electronic | ||
| Be6B11− | Level | i1 | i2 | i3 | i4 | i5 | i6 | i6 | i8 |
|---|---|---|---|---|---|---|---|---|---|
| 0.0 | 1.75 | 1.84 | 1.84 | 4.10 | 4.13 | 2.64 | 2.42 | ||
| 0.0 | 0.58 | 0.85 | 0.86 | 1.19 | 1.23 | 1.68 | 1.81 | ||
| 0.0 | −1.48 | 0.89 | 0.88 | −0.63 | −0.25 | 4.14 | −0.87 | ||
| Be6B11− | 0.0 | −0.29 | 1.51 | 1.52 | 2.41 | 2.42 | 5.0 | −0.08 | |
| 0.0 | 0.87 | 2.50 | 2.50 | 5.32 | 5.32 | 5.96 | 0.52 | ||
| Point-Group Symmetry | C1 | C1 | C2 | C2 | CS | C2v | C1 | C1 | |
| Electronic ground state | 1A | 1A | 1A | 1A | 1A′ | 1A1 | 1A | 1A | |
| Frequencies (cm−1) | 230 | 119 | 102 | 100 | 46 | 43 | 161 | 151 |
| PBE0-D3/def2-TZVP | PBE0/def2-TZVP | |
|---|---|---|
| 1 | (377)/(33) | (388)/(34.5) |
| 2 | (424)/(22.9) | (444)/(22.8) |
| 3 | (316.7)/(14) | (305.4)/(14.7) |
| 4 | (349)/(17.6) | (346.6)/(12.2) |
| 5 | (258)/(5.7) | (246)/(4.2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buelna-Garcia, C.E.; Cabellos, J.L.; Quiroz-Castillo, J.M.; Martinez-Guajardo, G.; Castillo-Quevedo, C.; de-Leon-Flores, A.; Anzueto-Sanchez, G.; Martin-del-Campo-Solis, M.F. Exploration of Free Energy Surface and Thermal Effects on Relative Population and Infrared Spectrum of the Be6B11− Fluxional Cluster. Materials 2021, 14, 112. https://doi.org/10.3390/ma14010112
Buelna-Garcia CE, Cabellos JL, Quiroz-Castillo JM, Martinez-Guajardo G, Castillo-Quevedo C, de-Leon-Flores A, Anzueto-Sanchez G, Martin-del-Campo-Solis MF. Exploration of Free Energy Surface and Thermal Effects on Relative Population and Infrared Spectrum of the Be6B11− Fluxional Cluster. Materials. 2021; 14(1):112. https://doi.org/10.3390/ma14010112
Chicago/Turabian StyleBuelna-Garcia, Carlos Emiliano, José Luis Cabellos, Jesus Manuel Quiroz-Castillo, Gerardo Martinez-Guajardo, Cesar Castillo-Quevedo, Aned de-Leon-Flores, Gilberto Anzueto-Sanchez, and Martha Fabiola Martin-del-Campo-Solis. 2021. "Exploration of Free Energy Surface and Thermal Effects on Relative Population and Infrared Spectrum of the Be6B11− Fluxional Cluster" Materials 14, no. 1: 112. https://doi.org/10.3390/ma14010112
APA StyleBuelna-Garcia, C. E., Cabellos, J. L., Quiroz-Castillo, J. M., Martinez-Guajardo, G., Castillo-Quevedo, C., de-Leon-Flores, A., Anzueto-Sanchez, G., & Martin-del-Campo-Solis, M. F. (2021). Exploration of Free Energy Surface and Thermal Effects on Relative Population and Infrared Spectrum of the Be6B11− Fluxional Cluster. Materials, 14(1), 112. https://doi.org/10.3390/ma14010112