Penta-C20: A Superhard Direct Band Gap Carbon Allotrope Composed of Carbon Pentagon



Abstract
1. Introduction
2. Calculation Methods
3. Results and Discussion
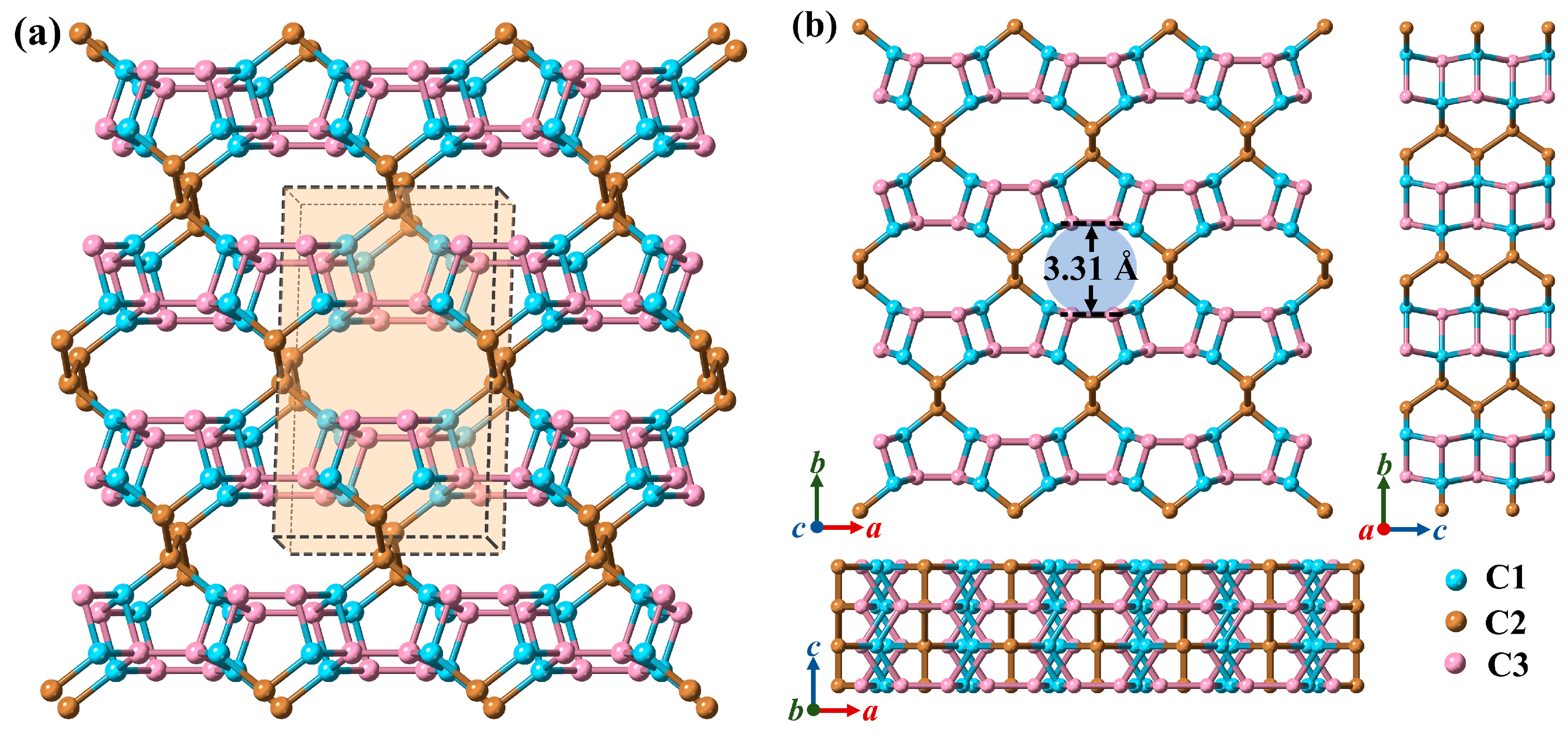
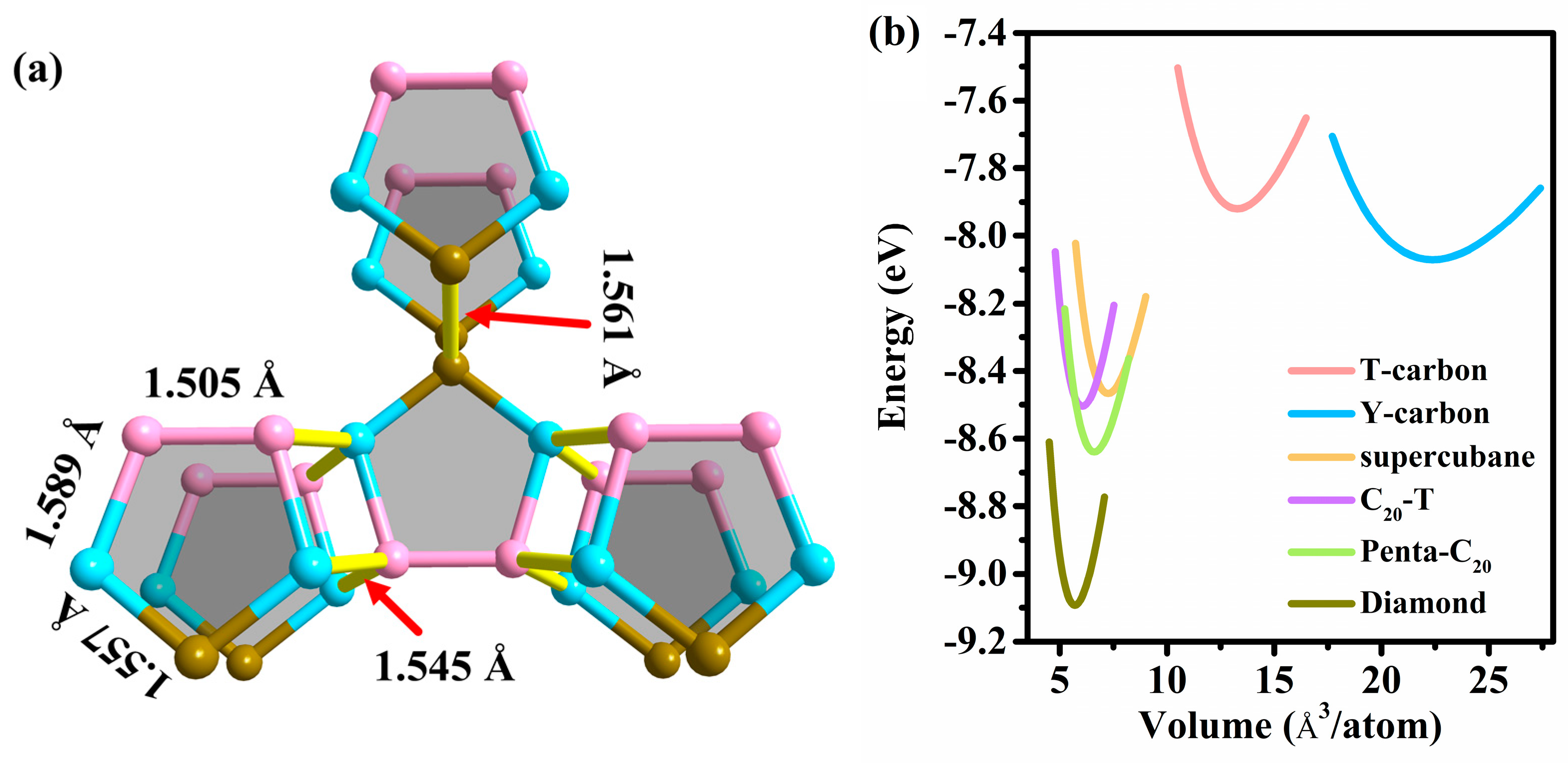
3.1. Structural Properties
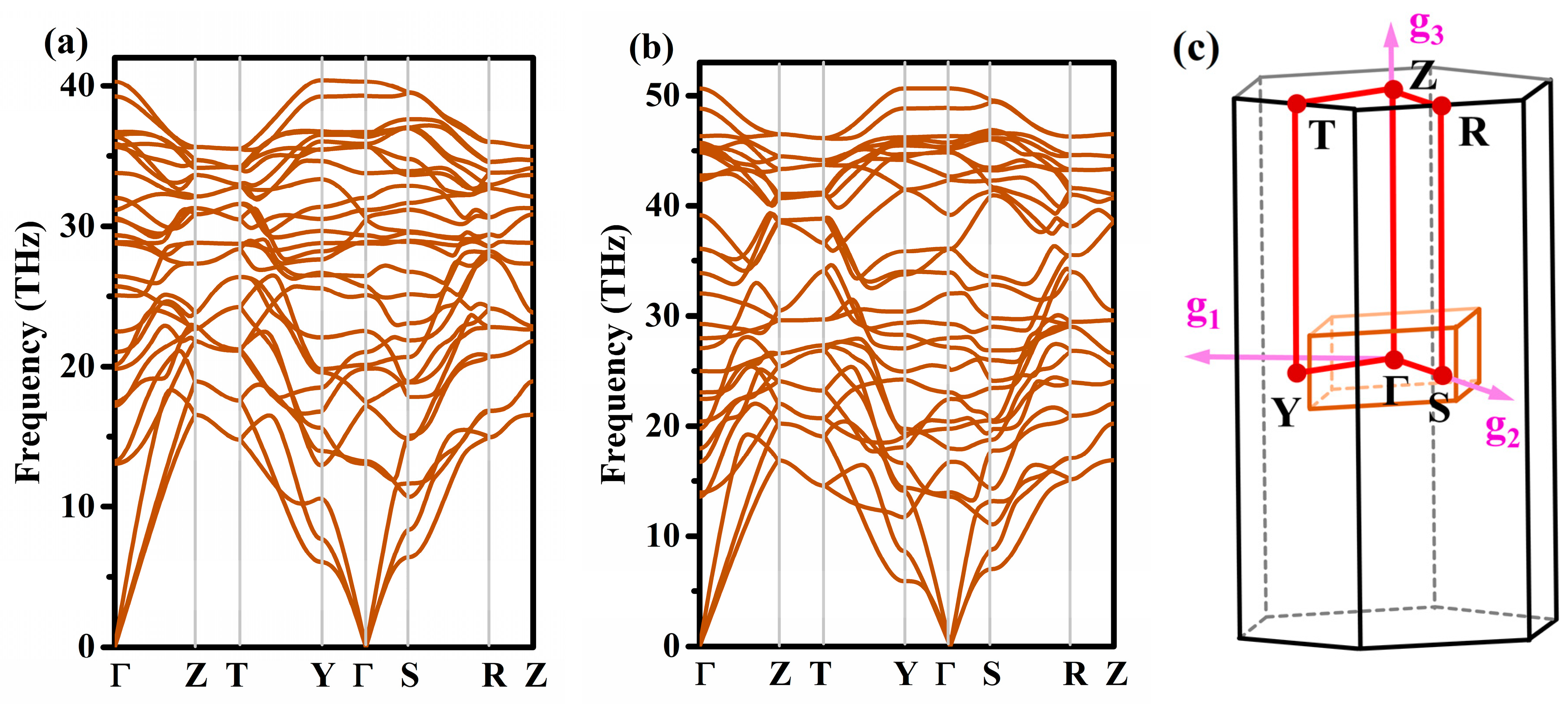
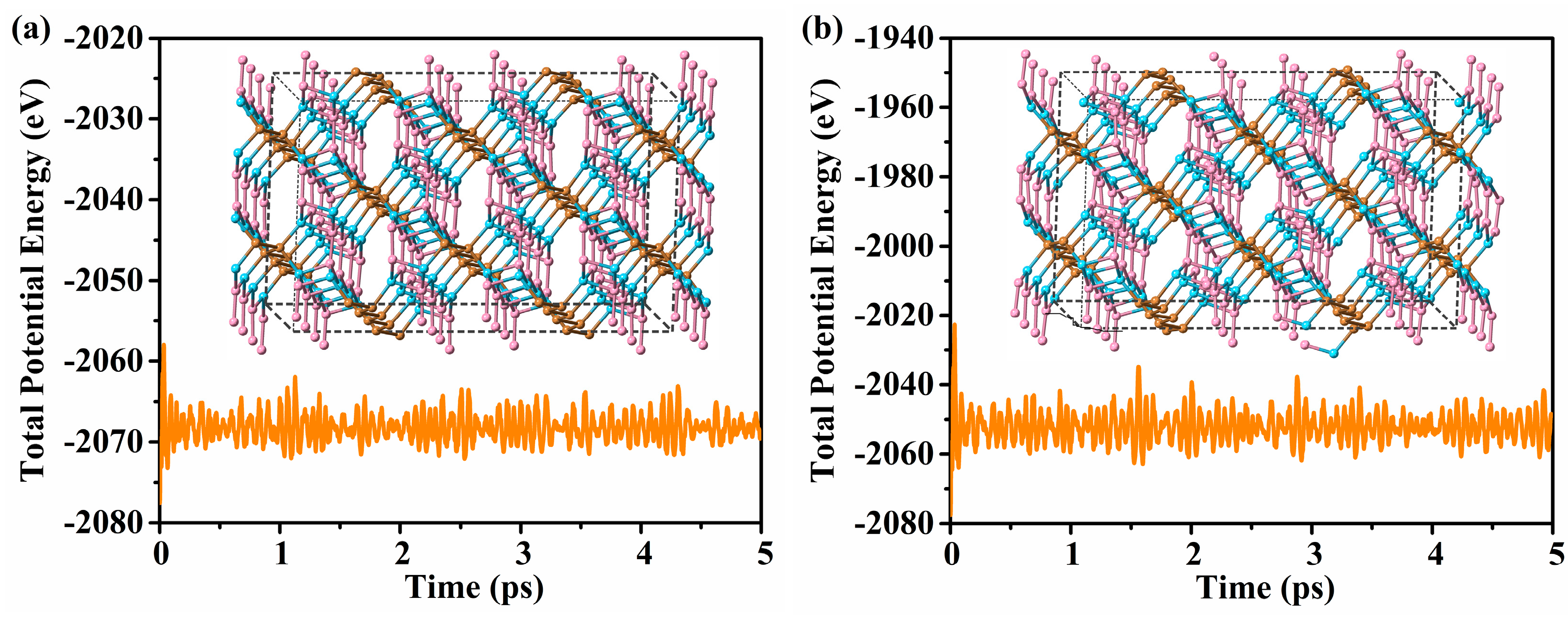
3.2. Stability
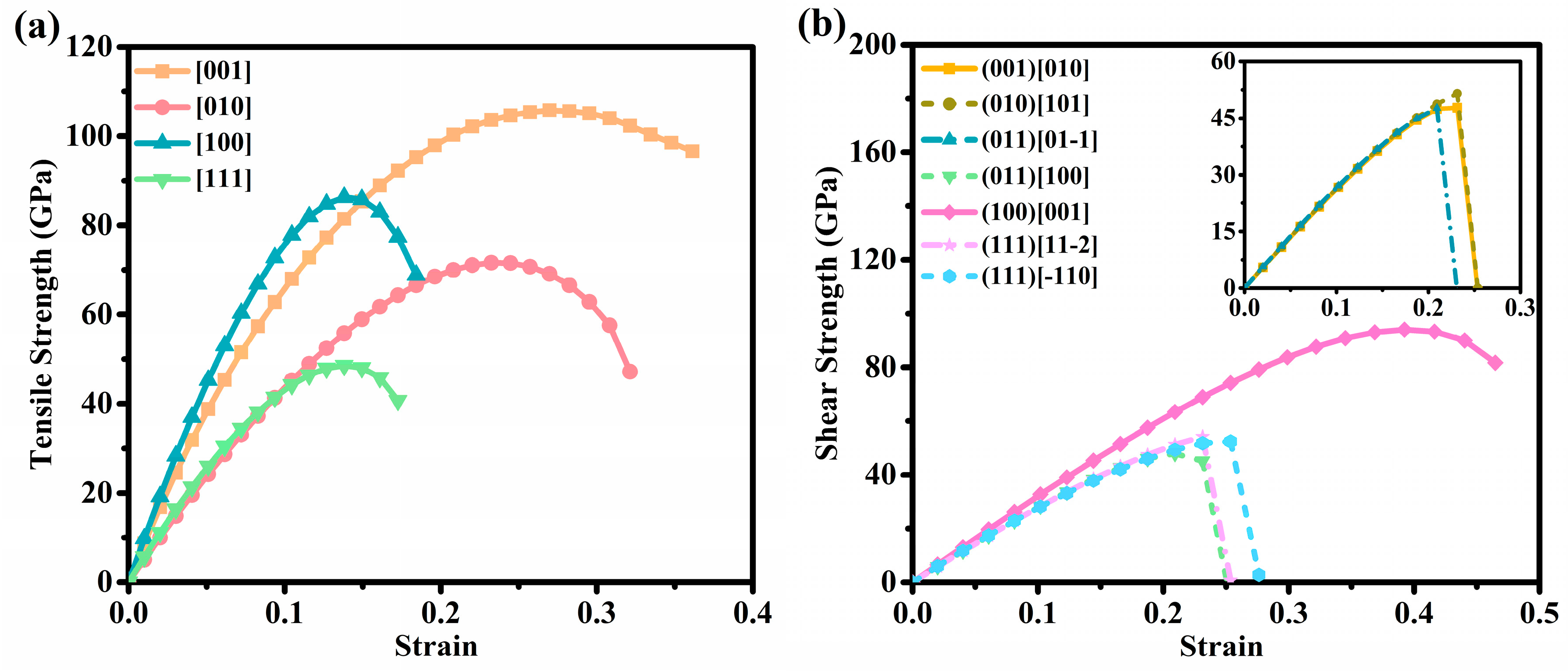
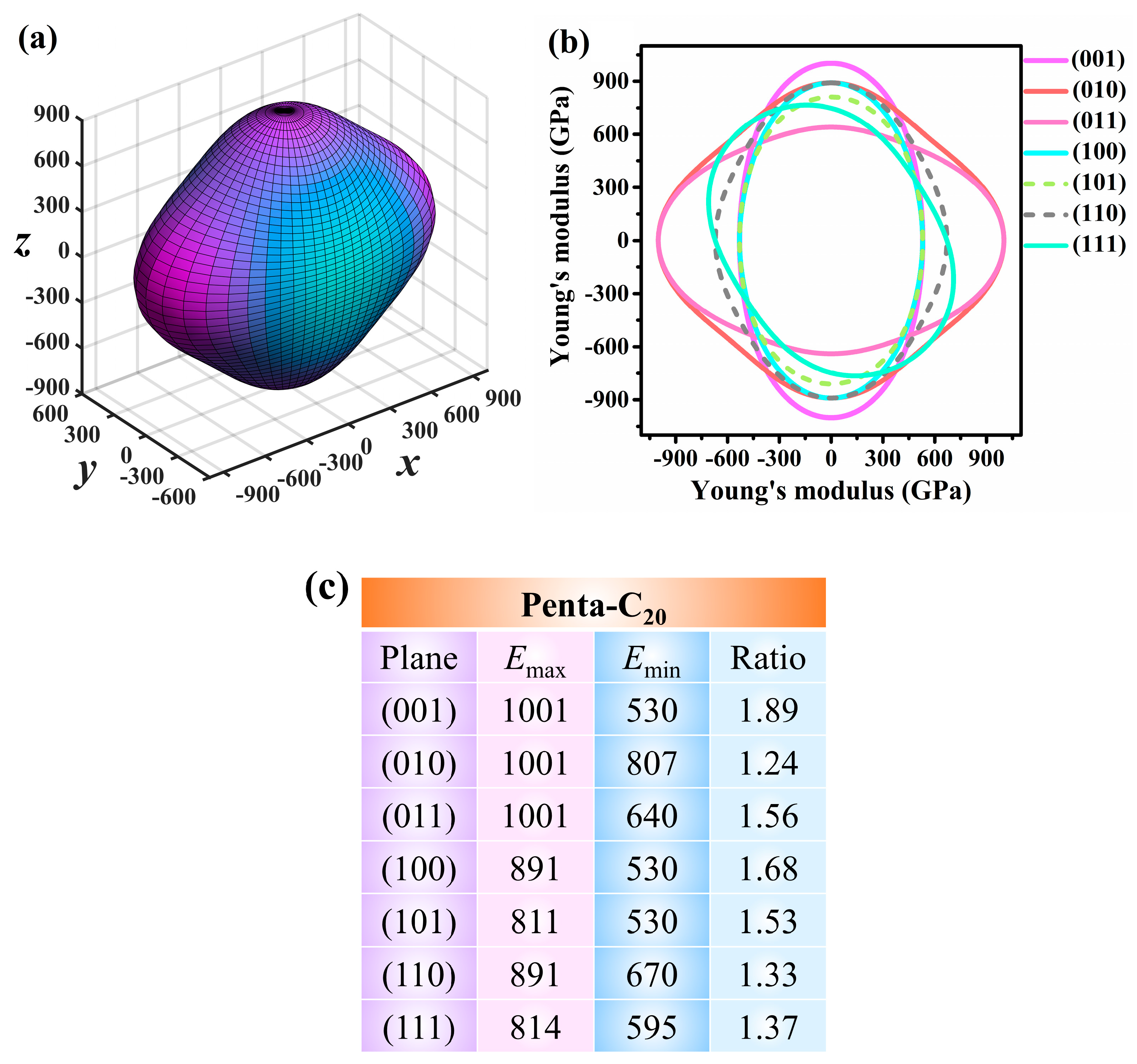
3.3. Mechanical Properties
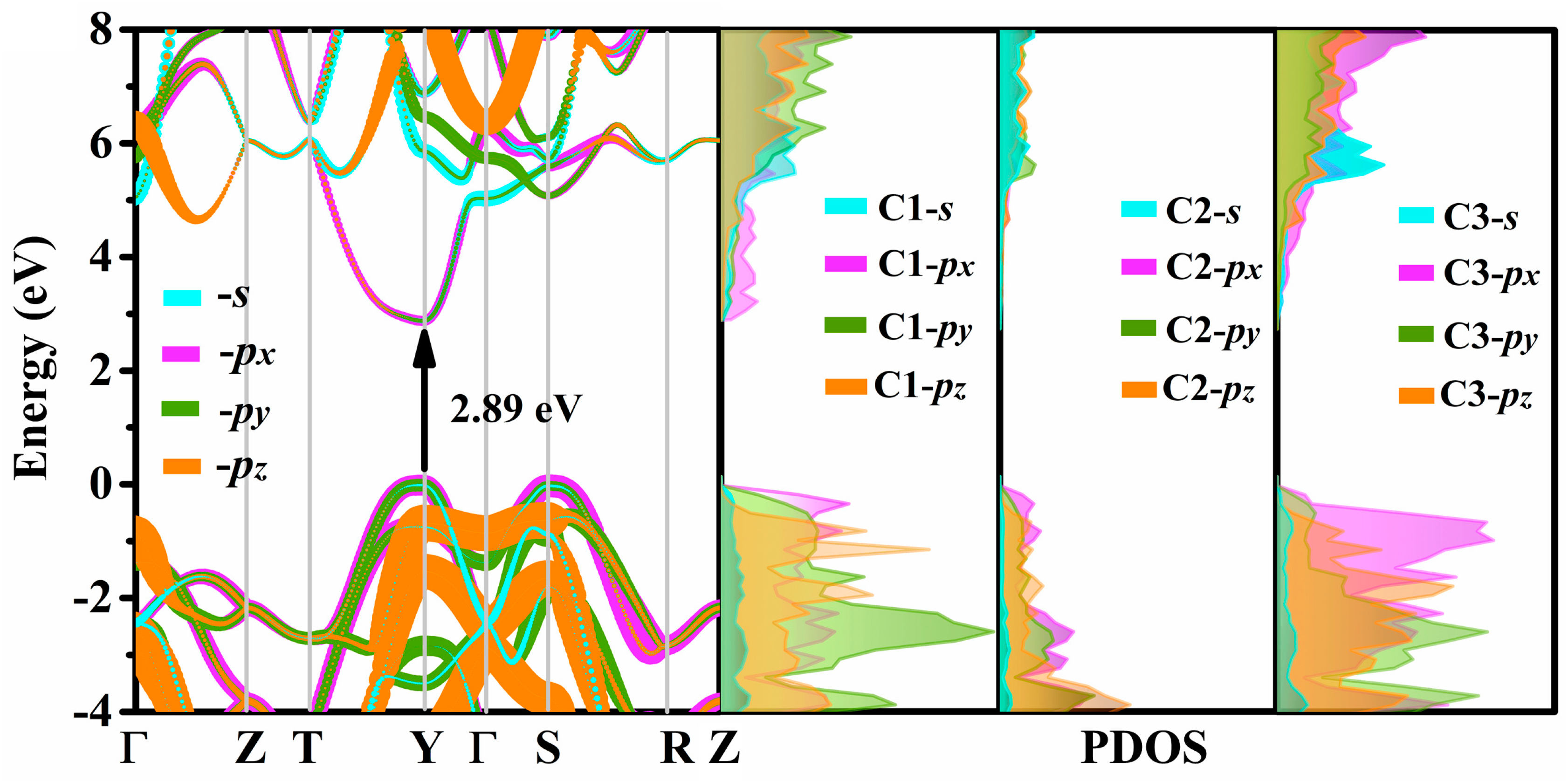
3.4. Electrical Properties
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kroto, H.W.; Heath, J.R.; O’Brien, S.C.; Curl, R.F.; Smalley, R.E. C60—A new form of carbon. Nature 1985, 318, 162–163. [Google Scholar] [CrossRef]
- Iijima, S. Synthesis of carbon nanotubes. Nature 1991, 354, 56–58. [Google Scholar] [CrossRef]
- Matsuoka, R.; Sakamoto, R.; Hoshiko, K.; Sasaki, S.; Masunaga, H.; Nagashio, K.; Nishihara, H. Crystalline graphdiyne nanosheets produced at a gas/liquid or liquid/liquid interface. J. Am. Chem. Soc. 2017, 139, 3145–3152. [Google Scholar] [CrossRef]
- Diederich, F.; Kivala, M. All-carbon scaffolds by rational design. Adv. Mater. 2010, 22, 803–812. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Neto, A.H.C.; Guinea, F.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef]
- Andrew, R.C.; Mapasha, R.E.; Ukpong, A.M.; Chetty, N. Mechanical properties of graphene and boronitrene. Phys. Rev. B 2012, 85, 125428. [Google Scholar] [CrossRef]
- Zhang, W.; Chai, C.; Fan, Q.; Weng, K.; Yang, Y. Theoretical investigations of Ge1−xSnx alloys (x = 0, 0.333, 0.667, 1) in P42/ncm phase. J. Mater. Sci. 2018, 53, 9611–9626. [Google Scholar] [CrossRef]
- Zhang, W.; Chai, C.; Song, Y.; Fan, Q.; Yang, Y. Structural, mechanical, anisotropic, and thermal properties of AlAs in oC12 and hP6 phases under pressure. Materials 2018, 11, 740. [Google Scholar] [CrossRef]
- Song, Y.; Chai, C.; Fan, Q.; Zhang, W.; Yang, Y. Physical properties of Si–Ge alloys in C2/m phase: A comprehensive investigation. J. Phys. Condens. Matter 2019, 31, 255703. [Google Scholar] [CrossRef]
- Fan, Q.; Zhang, W.; Song, Y.; Zhang, W.; Yun, S. P63/mmc-Ge and their Si-Ge alloys with a mouldable direct band gap. Semicond. Sci. Tech. 2020. In press. [Google Scholar] [CrossRef]
- Miao, J.; Chai, C.; Zhang, W.; Song, Y.; Yang, Y. First-Principles Study on Structural, Mechanical, Anisotropic, Electronic and Thermal Properties of III-Phosphides: XP (X = Al, Ga, or In) in the P6422 Phase. Materials 2020, 13, 686. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Duan, Z.; Song, Y.; Zhang, W.; Zhang, Q.; Yun, S. Electronic, Mechanical and Elastic Anisotropy Properties of X-Diamondyne (X = Si, Ge). Materials 2019, 12, 3589. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Wang, H.; Zhang, W.; Wei, M.; Song, Y.; Zhang, W.; Yun, S. Si–Ge alloys in C2/c phase with tunable direct band gaps: A comprehensive study. Curr. Appl. Phys. 2019, 19, 1325–1333. [Google Scholar] [CrossRef]
- Zhang, W.; Chai, C.; Song, Y.; Fan, Q.; Yang, Y. Six novel carbon and silicon allotropes with their potential application in photovoltaic field. J. Phys. Condens. Matter 2020. In press. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, X.; Fu, J.; Zhao, J. New metallic carbon: Three dimensionally carbon allotropes comprising ultrathin diamond nanostripes. Carbon 2018, 126, 601–610. [Google Scholar] [CrossRef]
- Fan, Q.; Xu, J.; Zhang, W.; Song, Y.; Yun, S. Physical properties of group 14 semiconductor alloys in orthorhombic phase. J. Appl. Phys. 2019, 126, 045709. [Google Scholar] [CrossRef]
- Li, D.; Tian, F.; Chu, B.; Duan, D.; Wei, S.; Lv, Y.; Zhang, H.; Wang, L.; Lu, N.; Liu, B.; et al. Cubic C 96: A novel carbon allotrope with a porous nanocube network. J. Mater. Chem. A 2015, 3, 10448–10452. [Google Scholar] [CrossRef]
- Umemoto, K.; Saito, S.; Berber, S.; Tománek, D. Carbon foam: Spanning the phase space between graphite and diamond. Phys. Rev. B 2001, 64, 193409. [Google Scholar] [CrossRef]
- Li, X.; Xing, M. Prediction of a novel carbon allotrope from frst-principle calculations: A potential superhard material in monoclinic symmetry. Mater. Chem. Phys. 2020, 104, 125504. [Google Scholar]
- Li, Q.; Ma, Y.; Oganov, A.R.; Wang, H.; Wang, H.; Xu, Y.; Cui, T.; Mao, H.K.; Zou, G. Superhard monoclinic polymorph of carbon. Phys. Rev. Lett. 2009, 102, 175506. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.T.; Chen, C.; Kawazoe, Y. Low-temperature phase transformation from graphite to orthorhombic carbon. Phys. Rev. Lett. 2011, 106, 075501. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Zhao, C.X.; Niu, C.Y.; Sun, Q.; Jia, Y. C 20–T carbon: A novel superhard sp3 carbon allotrope with large cavities. J. Phys. Condens. Matter 2016, 28, 475402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Wang, R.; Zhu, X.; Pan, A.; Han, C.X.; Li, X.; Zhao, D.; Ma, C.S.; Wang, W.; Su, H.B.; et al. Pseudo-topotactic conversion of carbon nanotubes to T-carbon nanowires under picosecond laser irradiation in methanol. Nat. Commun. 2017, 8, 683. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Cui, H.; Yang, G.W. Synthesis of body-centered cubic carbon nanocrystals. Cryst. Growth Des. 2008, 8, 581–586. [Google Scholar] [CrossRef]
- Fan, Q.; Wang, H.; Song, Y.; Zhang, W.; Yun, S. Five carbon allotropes from Squaroglitter structures. Comp. Mater. Sci. 2020, 178, 109634. [Google Scholar] [CrossRef]
- Hoffmann, R.; Kabanov, A.A.; Golov, A.A.; Proserpio, D.M. Homo citans and carbon allotropes: For an ethics of citation. Angew. Chem. Int. Ed. 2016, 55, 10962–10976. [Google Scholar] [CrossRef]
- Baburin, I.A.; Proserpio, D.M.; Saleev, V.A.; Shipilova, A.V. From zeolite nets to sp3 carbon allotropes: A topology-based multiscale theoretical study. Phys. Chem. Chem. Phys. 2015, 17, 1332–1338. [Google Scholar] [CrossRef]
- Zhang, W.; Chai, C.; Fan, Q.; Song, Y.; Yang, Y. Two novel superhard carbon allotropes with honeycomb structures. J. Appl. Phys. 2019, 126, 145704. [Google Scholar] [CrossRef]
- Laranjeira, J.; Marques, L.; Fortunato, N.M.; MelleFranco, M.; Strutyński, K.; Barroso, M. Three-dimensional C60 polymers with ordered binary-alloy-type structures. Carbon 2018, 137, 511–518. [Google Scholar] [CrossRef]
- Yang, X.; Yao, M.; Wu, X.; Liu, S.; Chen, S.; Yang, K.; Liu, R.; Cui, T.; Sundqvist, B.; Liu, B. Novel Superhard sp3 Carbon Allotrope from Cold-Compressed C 70 Peapods. Phys. Rev. Lett. 2017, 118, 245701. [Google Scholar] [CrossRef] [PubMed]
- Avery, P.; Wang, X.; Oses, C.; Gossett, E.; Proserpio, D.M.; Toher, C.; Curtarolo, S.; Zurek, E. Predicting superhard materials via a machine learning informed evolutionary structure search. NPJ Comput. Mater. 2019, 5, 89. [Google Scholar] [CrossRef]
- Zhang, W.; Chai, C.; Fan, Q.; Song, Y.; Yang, Y. PBCF-graphene: A 2Dsp2 hybridized honeycomb carbon allotrope with a direct band gap. ChemNanoMat 2020, 6, 139–147. [Google Scholar] [CrossRef]
- Shi, X.; He, C.; Pickard, C.J.; Tang, C.; Zhong, J. Stochastic generation of complex crystal structures combining group and graph theory with application to carbon. Phys. Rev. B 2018, 97, 014104. [Google Scholar] [CrossRef]
- He, C.; Shi, X.; Clark, S.J.; Li, J.; Pickard, C.J.; Ouyang, T.; Zhang, C.; Tang, C.; Zhong, J. Complex low energy tetrahedral polymorphs of group IV elements from first principles. Phys. Rev. Lett. 2018, 121, 175701. [Google Scholar] [CrossRef] [PubMed]
- Blatov, V.A.; Shevchenko, A.P.; Proserpio, D.M. Applied topological analysis of crystal structures with the program package ToposPro. Cryst. Growth Des. 2014, 14, 3576–3586. [Google Scholar] [CrossRef]
- Foster, M.D.; Treacy, M.M.J. A Database of Hypothetical Zeolite Structures. Available online: http://www.hypotheticalzeolites.net/DATABASE/DEEM/DEEM_PCOD/index.php (accessed on 7 April 2020).
- Kresse, G.; Furthmüller, J. Self-interaction correction to density functional approximation for many electron systems. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Density functional theory (DFT). Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Togo, A.; Oba, F.; Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B 2008, 78, 134106. [Google Scholar] [CrossRef]
- Gonze, X.; Vigneron, J.P. Density-functional approach to nonlinear-response coefficients of solids. Phys. Rev. B 1989, 39, 13120–13128. [Google Scholar] [CrossRef] [PubMed]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z Kristallogr 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Jo, J.Y.; Kim, B.G. Carbon allotropes with triple bond predicted by first-principle calculation: Triple bond modified diamond and T-carbon. Phys. Rev. B 2012, 86, 075151. [Google Scholar] [CrossRef]
- Sheng, X.L.; Yan, Q.B.; Ye, F.; Zheng, Q.R.; Su, G. T-carbon: A novel carbon allotrope. Phys. Rev. Lett. 2011, 106, 155703. [Google Scholar] [CrossRef]
- Niu, C.Y.; Wang, X.Q.; Wang, J.T. K6 carbon: A metallic carbon allotrope in sp3 bonding networks. J. Chem. Phys. 2014, 140, 054514. [Google Scholar] [CrossRef]
- Mouhat, F.; Couder, F.X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2004, 90, 224104. [Google Scholar] [CrossRef]
- Fu, W.; Zhang, Y.; Shang, J.; Zeng, L.; Cai, Y. Lattice thermal conductivity and bandgap engineering of a three-dimensional sp2-hybridized Dirac carbon material: HS-C48. Comput. Mater. Sci. 2018, 155, 293–297. [Google Scholar] [CrossRef]
- Nulakani, N.V.R.; Subramanian, V. Superprismane: A porous carbon allotrope. Chem. Phys. Lett. 2019, 715, 29–33. [Google Scholar] [CrossRef]
- Ma, J.L.; Song, D.L.; Wu, Y.L.; Fu, Z.F.; Zhou, J.P.; Liu, P.; Zhu, X.; Wei, Q. C72: A novel low energy and direct band gap carbon phase. Phys. Lett. A 2020, 126325. [Google Scholar] [CrossRef]
- Wei, Q.; Zhang, Q.; Zhang, M.G.; Yan, H.Y.; Guo, L.X.; Wei, B. A novel hybrid sp-sp2 metallic carbon allotrope. Front. Phys. 2018, 13, 136105. [Google Scholar] [CrossRef]
- Grimsditch, M.H.; Ramdas, A.K. Brillouin scattering in diamond. Phys. Rev. B 1975, 11, 3139. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. Lond. 1952, 65, 349. [Google Scholar] [CrossRef]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Lyakhov, A.O.; Oganov, A.R. Evolutionary search for superhard materials: Methodology and applications to forms of carbon and TiO2. Phys. Rev. B 2011, 84, 092103. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, B.; Zhao, Z. Microscopic theory of hardness and design of novel superhard crystals. Int. J. Refract. Met. Hard Mater. 2012, 33, 93–106. [Google Scholar] [CrossRef]
- Andrievski, R.A. Superhard materials based on nanostructured high-melting point compounds: Achievements and perspectives. Int. J. Refract. Met. Hard Mater. 2001, 19, 447–452. [Google Scholar] [CrossRef]
- Zhang, M.; Lu, M.; Du, Y.; Gao, L.; Lu, C.; Liu, H. Hardness of FeB4: Density functional theory investigation. J. Chem. Phys. 2014, 140, 174505. [Google Scholar] [CrossRef] [PubMed]
- Ting, T.C.T. On anisotropic elastic materials for which young’s modulus e(n) is independent of n or the shear modulus G(n,m) is independent of n and m. J. Elasticity 2005, 81, 271–292. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structures | Methods | Space Group | ρ (g/cm3) | a (Å) | b (Å) | c (Å) | Etot (eV) |
|---|---|---|---|---|---|---|---|
| Penta-C20 | PBE | Cmcm | 3.031 | 5.595 | 9.168 | 2.577 | −8.639 |
| T-carbon | PBE | Fd-3m | 1.503 | 7.516 | - | - | −7.921 |
| - | PBE [a] | Fd-3m | 1.503 | 7.517 | - | - | −7.922 |
| - | Exp. [b] | Fd-3m | - | 7.80 | - | - | - |
| Y-carbon | PBE | Fd-3m | 0.892 | 9.636 | - | - | −8.071 |
| - | PBE [c] | Fd-3m | 0.894 | 9.636 | - | - | −8.074 |
| TY-carbon | PBE | Fd-3m | 0.524 | 13.459 | - | - | −8.038 |
| - | PBE [c] | Fd-3m | 0.523 | 13.460 | - | - | −8.034 |
| C20-T | PBE | P213 | 3.293 | 4.948 | - | - | −8.505 |
| - | PBE [d] | P213 | 3.298 | 4.945 | - | - | |
| Diamond | PBE | Fd-3m | 3.518 | 3.567 | - | - | −9.093 |
| - | Exp. [e] | Fd-3m | 3.516 | 3.567 | - | - | - |
| Materials | C11 | C12 | C13 | C22 | C23 | C33 | C44 | C55 | C66 | B | G | B/G | HUSPEX | HTian | HExp |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Penta-C20 | 1020 | 76 | 97 | 539 | 59 | 905 | 289 | 332 | 299 | 313 | 327 | 0.96 | 76.23 | 58.30 | - |
| HS-C48 [a] | 656 | 137 | 266 | 151 | 94 | 777 | 112 | 92 | 323 | 287 | 178 | 1.61 | - | - | - |
| C96 [b] | 623 | 108 | - | - | - | - | 194 | - | - | 279 | 219 | 1.2 | - | - | - |
| Superprismane [c] | 306 | 136 | 185 | - | - | 525 | 226 | - | - | 238 | 150 | 1.59 | - | - | - |
| C72 [d] | 273 | 139 | - | - | - | - | 81 | - | - | 183 | 75 | 2.46 | - | - | - |
| K6-carbon [e] | 250 | 187 | - | - | - | - | 29 | - | - | 209 | 30 | 6.97 | - | - | - |
| T-carbon [e] | 203 | 136 | - | - | - | - | 70 | - | - | 159 | 52 | 3.08 | - | - | - |
| Diamond | 1053 | 119 | - | - | - | - | 566 | - | - | 431 | 524 | 0.82 | 89.77 | 96.73 | - |
| Diamond [f] | 1076 | 125 | - | - | - | - | 577 | - | - | 442 | 634 | 0.83 | 89.72 [g] | 93.6 [h] | 96 ± 5 [i] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Chai, C.; Fan, Q.; Song, Y.; Yang, Y. Penta-C20: A Superhard Direct Band Gap Carbon Allotrope Composed of Carbon Pentagon. Materials 2020, 13, 1926. https://doi.org/10.3390/ma13081926
Zhang W, Chai C, Fan Q, Song Y, Yang Y. Penta-C20: A Superhard Direct Band Gap Carbon Allotrope Composed of Carbon Pentagon. Materials. 2020; 13(8):1926. https://doi.org/10.3390/ma13081926
Chicago/Turabian StyleZhang, Wei, Changchun Chai, Qingyang Fan, Yanxing Song, and Yintang Yang. 2020. "Penta-C20: A Superhard Direct Band Gap Carbon Allotrope Composed of Carbon Pentagon" Materials 13, no. 8: 1926. https://doi.org/10.3390/ma13081926
APA StyleZhang, W., Chai, C., Fan, Q., Song, Y., & Yang, Y. (2020). Penta-C20: A Superhard Direct Band Gap Carbon Allotrope Composed of Carbon Pentagon. Materials, 13(8), 1926. https://doi.org/10.3390/ma13081926