Abstract
The stability, physical properties, and electronic structures of Cr(NCN)2 were studied using density functional theory with explicit electronic correlation (GGA+U). The calculated results indicate that Cr(NCN)2 is a ferromagnetic and half-metal, both thermodynamically and elastically stable. A comparative study on the electronic structures of Cr(NCN)2 and CrO2 shows that the Cr atoms in both compounds are in one crystallographically equivalent site, with an ideal 4+ valence state. In CrO2, the Cr atoms at the corner and center sites have different magnetic moments and orbital occupancies, moreover, there is a large difference between the intra- (12.1 meV) and inter-chain (31.2 meV) magnetic couplings, which is significantly weakened by C atoms in Cr(NCN)2.
1. Introduction
Recently, high-valence chromium oxides have attracted much attention because of their unusual physical properties and complicated microscopic mechanism [1,2,3,4,5,6,7,8]. For example, CaCrO3 as a rare metallic antiferromagnet was reported to have a Bose–Einstein condensate at TN [2,5]. The quasi-one-dimensional Hollandite-type structure K2Cr8O16 undergoes an unconventional metal–insulator transition while maintaining the ferromagnetic state [3,8]. CrO2, as a simplest Cr4+ compound, is a half-metal ferromagnet (TC = 390 K) and has been widely used in magnetic recording media. It has been widely studied both in experiment and theory; however, the origin of its half-metal ferromagnetism remains controversial and unclear. Early theoretical studies suggest a self-doping double exchange mechanism for describing the material’s intertwined metallicity and ferromagnetism [9,10]. Shim et al. indeed observed two different Cr ions coexistence in CrO2 using 53Cr nuclear magnetic resonance (NMR), supporting the self-doping and double exchange mechanism [11]. Latterly, Takeda et al. also observed the presence of two Cr sites using the orbital-resolved NMR method, however, they revealed that two Cr ions at the corner and body center sites have the same valence state, but different orbital occupancies [12]. A local orbital order takes place with breaking of the local symmetry. They ascribed it to the negative charge transfer between chromium and oxygen ions. Nevertheless, a realistic low-energy model derived from the first principles calculations presents that the direct exchange interactions and the magnetic polarization of the oxygen 2p band play a very important role in the stability of the ferromagnetic ground state of CrO2 instead of double exchange [13,14,15]. Thus, a clear and uniform microscopic model is desired to elucidate it.
Here, we predicted a new Cr4+-based compound Cr(NCN)2 by first principles theory. It has a tetragonal structure with a space group of P42/mnm, similar t-o CrO2. NCN2−as a pseudochalcogen umligand, has the same oxidation state as O2−, but different structure and electronegative. Experimental reports indicate that NCN2−-based 3d transition-metal compounds display rich physical and chemical properties, showing the similarity and difference to corresponding oxides [16,17,18,19]. For instance, Cr2NCN3 has the same crystal structure as Cr2O3 with the R–3c space group; however, it is a rare ferromagnetic semiconductor, quite different from antiferromagnetic Cr2O3 [17].
As in CrO2, Cr atoms in Cr(NCN)2 are octahedrally coordinated by nitrogen, forming edge-sharing octahedral ribbons along the c axis. However, the octahedra on adjacent ribbons are connected by NCN2−, leading to a larger distance of 5.906 Å, not like those in CrO2 that share an apex O2− with a shorter distance of 3.450 Å. The large inter-chain distance induces a weak magnetic interaction, and as a result Cr(NCN)2, provides a simpler theoretical model for disclosing the mechanism behind the ferromagnetic and half-metal property for CrO2, even for other Cr4+-based magnetic compounds. Therefore, in this paper, besides predicting the crystal structure, we also calculated the electronic and magnetic structure through the use of density functional theory with explicit electronic correlation (GGA+U). In our study, both GGA and GGA+U calculations present a ferromagnetic half-metal for Cr(NCN)2. The electronic structures show the Cr atoms in both Cr(NCN)2 and CrO2 are in one crystallographically equivalent site, with an ideal 4+ valence state. In CrO2, the Cr atoms at the corner and center sites have the different magnetic moments and orbital occupancies, moreover, there is a large difference between the intra- (12.1 meV) and inter-chain (31.2 meV) magnetic couplings, which is significantly weakened by C atoms in Cr(NCN)2.
2. Methods
As an AB2-type compound, two kinds of initial crystal structures were chosen for Cr(NCN)2, that is, tetragonal P42/mnm from CrO2 [20] and orthorhombic Pnnm from the high-pressure CrO2 and M(NCNH)2 (M = Fe, Co, Ni) [21,22]. This is because, as an analog of O2− ligand, NCN2−-based 3d transition metal compounds have been proven to own a similar crystal structure as corresponding oxides [16,17,18,19]. The initial structures were optimized by letting all lattice parameters and the positions of Cr, C, and N relax simultaneously until self-consistency was achieved.
The calculations were based on density functional theory [22,23] in which the ground state properties of a many-electron system are determined by an electron density with three spatial coordinates instead of N electrons with 3N spatial coordinates many-body problem, and performed using the plane–wave pseudopotential Vienna ab initio Simulation Package [24,25,26], which is a well-tested code and has been successfully used to calculate a great variety of materials [17,18,19,27,28,29,30]. The generalized gradient approximation [31] was used for the exchange-correlation functional, which was formulated by Perdew, Burke, and Ernzerhof (GGA-PBE). The projector-augmented wave (PAW) method was employed with a cutoff energy of 800 eV, which was proposed by Blöchl [32] and implemented by Kresse and Joubert [33]. A uniform mesh grid with an actual spacing of 0.031 Å−1 was used to sample the complete Brillouin zone (k-point grid of 4 × 4 × 10). Brillouin zone integrations were performed with the Methfessel–Paxton method for structure optimization and the tetrahedron method with Blöchl’s correction for electronic structure [34]. The PAW pseudopotentials are 2p63d54s1 for Cr, 2s22p2 for C, 2s22p3 for N, and 2s22p4 for O. Electron–electron Coulomb interactions in combination with the self-interaction correction were considered for the Cr atom in the rotationally invariant method (GGA+U) with an effective Hubbard parameter Ueff = U − J [35,36], which is set as 3.0 eV, obtained from the previous theoretical studies on the oxides based on Cr4+ [6,37].
Density-functional perturbation theory (DFPT) [38] in Vienna ab initio Simulation Package combined with the analysis program PHONOPY [39] was used to calculate the phonon frequencies of Cr(NCN)2. The 2 × 2 × 2 supercell including 112 atoms was used to calculate the force constants. A total of 101 k-points was used to sample each segment of band paths in order to obtain phonon dispersion relations.
3. Results and Discussion
The calculated results show that both initial structures (P42/mnm and Pnnm) are stable in the same ground state, that is, P42/mnm. Thus, Cr(NCN)2 was predicted to be a tetragonal crystal with the same space group as CrO2 in ambient condition (Figure 1), and Table S1 in the Supporting Information shows all of the theoretical structure parameters. This is quite similar as the scenario in Cr3+ ion-based compound Cr2(NCN)3, which has the same crystal structure as Cr2O3. The calculated lattice parameters are a = 8.04 Å, c = 3.1318 Å, V = 202.445 Å, and Z = 2. The Cr4+ ion in the Wyckoff position 2a (0 0 0) is coordinated by six nitrogen atoms, leading to a slightly flattened octahedral coordination with four Cr–N bonds at 2.023 Å and two shorter Cr–N bonds at 1.934 Å. The average Cr–N bond length is 1.993 Å, close to the sum of Cr4+ and N3− effective ionic radii [40] of 2.01 Å. Edge-sharing CrN6 octahedra form a single chain along the z-axis with a Cr–Cr distance of 3.132 Å (Figure 1). C atoms is in Wyckoff position 4 g (0.2238 0.2238 0) and N atoms occupy in two different positions, that is, N1 4 g (0.1126 0.1126 0) and N2 4 g (0.3299 0.3299 0), forming a strictly linear [N-C-N]2− unit (∠N-C-N = 180°). The bond lengths between C and N are 1.264 Å (C-N1) and 1.206 Å (C-N2), indicating the asymmetrical structure of NCN2−. The theoretical predicted Cr(NCN)2 is different from other symmetrical 3d transition-metal carbondiimides MNCN (M = Mn, Fe, Co, Ni) and Cr2NCN3, in which two C–N bonds are the same, around 1.23 Å. This asymmetric structure results from the coordination of N atoms, N1 is connected by two Cr atoms and one C, but N2 is connected by one Cr and one C. Until now, this asymmetrical structure has only been reported in d10 nonmagnetic cyanamides Ag2NCN [41], HgNCN (II) [42], CdNCN (R3m) [43], and PbNCN [44]. For example, in Ag2NCN, there is a single bond C–N 1.270 Å and a triple bond C≡N 1.187 Å. In Cr(NCN)2, the edge-sharing CrN6 chains are connected by the asymmetrical [N–C≡N]2–, as a result of a large distance between CrN6 chains of about 5.906 Å. Therefore, Cr(NCN)2 would be the first transition-metal cyanamide with partially filled 3d orbitals.
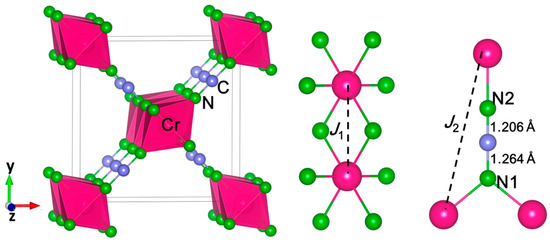
Figure 1.
Crystal structures of Cr(NCN)2 (left) and the coordination environments of the Cr4+ ion (middle) and the NCN2– ions (right). The Cr, N, and C are in pink, green, and gray, respectively.
The possible reaction routes were summarized in Equations (S1) and (S2) (see Supporting Information), based on successfully synthesized compounds Cr2NCN3 and MNCN (M = Mn, Fe, Co, Ni) [16,17]. The calculated reaction energies are −7.46 eV and −4.74 eV, respectively, indicating that the proposed reactions are possible candidates. The formation enthalpy, which is calculated from the direct reaction (Equation (S3)) with elements, is −13.54 eV, suggesting that Cr(NCN)2 is thermodynamically stable relative to the elements. Figure 2 shows the phonon dispersion curves of Cr(NCN)2 in the ferromagnetism, which is the most stable magnetic state and will be discussed below. It has no imaginary modes and hence is dynamically stable.
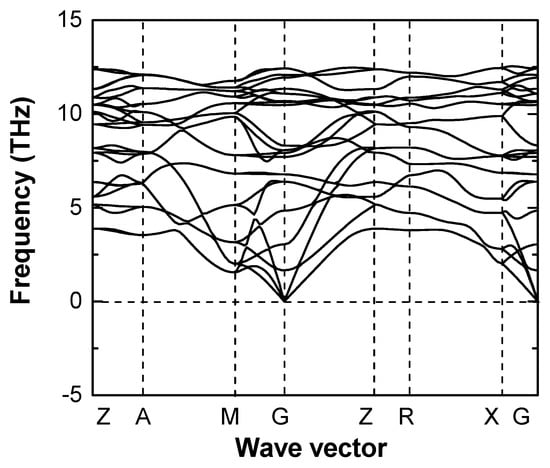
Figure 2.
Calculated phonon dispersion relations of Cr(NCN)2 in ferromagnetic state. Imaginary phonon frequencies are shown by negative values.
To achieve quantum-chemical insight into the conductivity and magnetic properties, we performed electronic-structure calculations for Cr(NCN)2 and CrO2 in P42/mnm structure. Considering the structure as shown in Figure 1, two effective magnetic interactions were expected for Cr(NCN)2, the intra-chain J1 (two neighbors) and inter-chain J2 (eight neighbors) between the “nearest-neighbor” Cr ions as corresponding to the Cr–N–Cr along CrN6 octahedral chain and Cr–N–C≡N–Cr superexchange path between chains, respectively (see Figure 1). Thus, we considered a ferromagnetic (FM) and two anti-ferromagnetic structures (AFM1 and AFM2), shown in Figure S1 in the Supporting Information, needed to identify the most probable ground state magnetic structure and estimate the magnitudes of J1 and J2. As described in CuNCN [18,19], based on the Heisenberg spin Hamiltonian,
the total energies per unit cell (two formula units) of FM, AFM1, and AFM2 can be written as
where N is the number of unpaired electrons, N = 2 for Cr4+ ion. Thus, we can extract J1 and J2 from
The total energies (Table 1) as calculated from GGA and GGA+U evidence that a ferromagnetic structure is most stable for both Cr(NCN)2 and CrO2, with the result of CrO2 consistent with previous experimental and theoretical reports [12,13]. The effective exchange parameters of J1 and J2 listed in Table 1 were estimated from the total energies. In the GGA context, the calculated total saturation magnetic moment Mtot is 2.01 μB f. u. and the spin saturation moment for Cr4+ ion MCr is 2.18 μB in Cr(NCN)2, larger than the S = 1 scenario. A small negative spin moment about −0.06 μB was found for N3−. When including the Coulomb interaction (GGA+U), the Mtot increases to 2.26 μB f. u., and there present two Cr spin moments of 2.63 μB and 2.62 μB at the corner Cr1 (0 0 0) and body-center Cr2 (0.5 0.5 0.5) sites, respectively, with a small difference about 0.01 μB. Accordingly, the spin moments of N3− diverge to four different values, with an average of −0.11 μB. The similar values of J1 and J2 calculated from the GGA+U calculation suggest that the couplings of the intra- and inter-chain are comparative.

Table 1.
Total energies (Etotal) (meV) of the ferromagnetic (FM), anti-ferromagnetic (AFM)1, and AFM2 states per formula unit relative to that of the FM state for Cr(NCN)2 and CrO2; the effective exchange coupling constants (C) (meV); the calculated saturated magnetic moment (M) of Cr, N, and O ions; and the total magnetic moment per formula unit (μB), as coming from the GGA and density functional theory with explicit electronic correlation (GGA+U) (U = 3 eV) calculations.
For CrO2, GGA and GGA+U both present different spin saturation moments for Cr1 and Cr2, that is, 2.11 μB and 2.05 μB (GGA), and 2.38 and 2.31 (GGA+U), respectively, with the difference of 0.06 and 0.07 μB, respectively. The apex and basal plane oxygens of the CrO6 octahedron also have different spin moments of −0.05 μB and −0.06 μB for GGA, and −0.13 μB and −0.12 μB for GGA+U, respectively. The total saturation magnetic moment increases to 2.09 μB in GGA+U from 1.97 in GGA. Obviously, Cr1 and Cr2 in CrO2 are nonequivalent in magnetism in spite of one crystallographic Cr site, nevertheless, this nonequivalence is not obvious in iso-structure Cr(NCN)2. The values of J1 and J2 calculated from the GGA+U calculation are 12.1 and 31.2 meV, respectively. This indicates that the coupling of the inter-chain is stronger than that of the intra-chain.
The scenario of two Cr sites (corner and center sites) with different spontaneous moments has been reported for CrO2 in experiments [9,10,11]. However, two different microscopic mechanisms are proposed: (1) a mixed valence state of Cr+4±δ resulting from a self-doping effect activates the double exchange mechanism, and thus induces the metallic ferromagnetism [9,10,11]; (2) the two Cr sites do not have different valence states, but have 3d orbital occupation numbers different from each other owing to the negative charge transfer between chromium and oxygen ions; as a result, a local orbital order takes place with breaking of the local symmetry, leading to the difference between the body-center and corner Cr sites [12]. In our calculation, Cr1 and Cr2 in both CrO2 and Cr(NCN)2 are in one crystallographically equivalent site, with ideal 4+ valence states. Clearly, the nonequivalent magnetic sites do not result from the mixed valence states. In order to disclose the physical mechanism, we perform electronic-structure calculations in the following.
The local density-of-states (DOS) within the FM states of Cr(NCN)2 and CrO2, as derived from GGA and GGA+U theory, are shown in Figure 3, with the band structure shown in Figure S2 in the Supporting Information. In the GGA description for Cr(NCN)2, there is a finite DOS on the Fermi level in majority spin with a strong Cr–N orbital mixing, while for minority spin, there is an energy gap of 0.62 eV between the highest occupied valence bands with N 2p character and the lowest-lying conduction bands of Cr 3d character, a p–d charge transfer insulating property. These suggest that Cr(NCN)2 is a half-metal compound, with 100% spin polarized thermally induced current at the Fermi level. Upon including an on-site Coulomb interaction (GGA+U), Cr(NCN)2 keeps the half-metallic character, with the p–d charge transfer energy gap increasing to 1.348 eV for the minority spin.
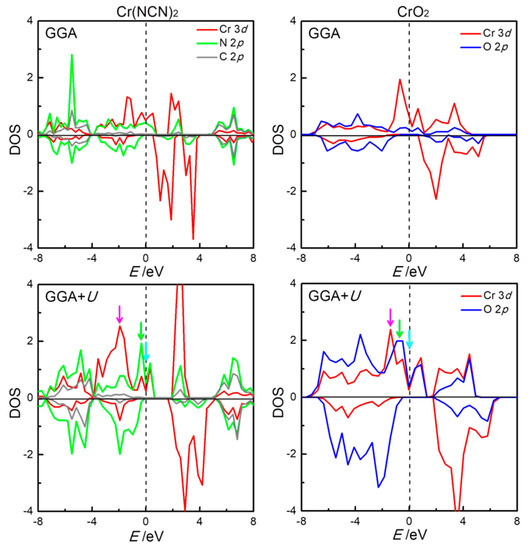
Figure 3.
The density-of-states (DOS) of Cr(NCN)2 (left) and CrO2 (right) projected to Cr 3d (red), N 2p (green), C 2p (grey), and O 2p (blue) orbitals on the basis of GGA (upper row) and density functional theory with explicit electronic correlation (GGA+U) (lower row) calculations. The energy zero indicates the Fermi energy level.
For CrO2, a half-metallic character is throughout for GGA and GGA+U calculations (Figure 3), in good agreement with experimental reports [12] and previous theoretical study [13]. It is a metallic property for majority spin with a strong Cr–O orbital mixing. For minority spin, the energy gaps of 1.348 and 2.254 eV from GGA and GGA+U theory are found between the highest occupied valence bands with O 2p character and the lowest-lying conduction bands of Cr 3d character, as a p–d charge transfer insulating property. In short, the electronic structures of Cr(NCN)2 from both GGA and GGA+U are similar to those of CrO2, both of which display the p–d charge transfer half-metal property.
To better understand what is going on with the 3d orbitals of Cr1 (0.5 0.5 0.5) and Cr2 (0 0 0) ions in Cr(NCN)2 and CrO2, we consider the transformation of the corresponding orbital-projected DOS in more detail. In both compounds, the distortion of CrN6/CrO6 results in a symmetry lowering Oh to D2h, that is, a contraction of the octahedron along one of its threefold axes. Therefore, the forms of three t2g orbitals in the global coordinate frame are , , and where the “+” and “−” signs stand for Cr1 and Cr2, respectively [13]. Sometimes these orbitals are denoted as , , and in the local coordinate frame, respectively [45]. The relevant orbital-projected DOS of Cr1 and Cr2 in Cr(NCN)2 and CrO2 from GGA+U calculations given in Figure 4 are performed on symmetry grounds; we projected densities in majority spin channel of the |1>, |2>, and |3> symmetry with respect to the crystal coordinate frame. We also inserted the numerical orbital populations obtained by integration up to the Fermi energy in Figure 4. In Cr(NCN)2, the projected DOS of |1>, |2>, and |3> of Cr1 and Cr2 are similar, and the orbital populations are quite close, reflecting almost the same spin moment of Cr1 and Cr2 discussed before. In CrO2, the projected DOS |1> and |3> of both Cr ions are similar with the close orbital population. However, the projected DOS of |2> of Cr1 and Cr2 are dissimilar, with the difference of 0.226 in the orbital population. Obviously, the different spin moments of Cr1 and Cr2 in CrO2 mainly result from |2>.
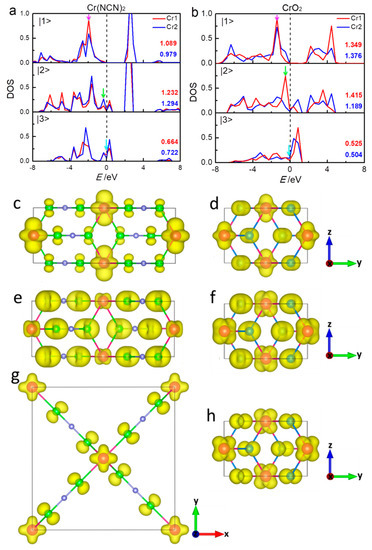
Figure 4.
The partial densities-of-states in the spin majority of the Cr 3d orbitals on the basis of GGA+U calculations for Cr(NCN)2 (a) and CrO2 (b). The values inserted are the populations of the five Cr 3d orbitals in the majority channels, as calculated by integration (up to Fermi level EF) of the partial densities-of-states. The energy zero indicates the Fermi energy level. The corresponding three-dimensional electron density contour plots (e/Å−3) in the regions of (–2.20)–(–1.60) eV (c), (–0.69)–(–0.40) eV (e), and (–0.10)–0.0 eV (g) for Cr(NCN)2 and (–1.57)–(–1.35) eV (d), (–0.64)–(–0.37) eV (f), and (–0.10)–0.0 (h) eV for CrO2 (energy regions shown in blue, green, and purple arrows in Figure 3 and Figure 4a,b).
For both compounds, |1> mainly locates below −1.0 eV and is localized with an energy gap of 1.9 eV for Cr(NCN)2 and 1.3 eV for CrO2 at the Fermi level. Moreover |1> is less hybridized with O 2p orbitals (Figure 3), suggesting a possible direct Cr–Cr interaction. |1> of Cr ions in Cr(NCN)2 have a smaller orbital occupation (1.089 and 0.979) than those in CrO2 (1.349 and 1.376). In both compounds, the electrons in |2> and |3> are itinerantly crossing the Fermi level with a strong hybridization with N/O 2p orbitals (Figure 3). The Fermi level is mainly occupied by |3> in Cr(NCN)2, while that of CrO2 is occupied by both |2> and |3>. Besides, |3> in Cr(NCN)2 (0.66 and 0.72) has a bigger orbital population than that in CrO2 (0.53 and 0.50). The total orbital populations of 3d orbitals of Cr1 and Cr2 are 2.985 and 2.995 in Cr(NCN)2, and 3.289 and 3.069 in CrO2, respectively, larger than 2.0 of ideal Cr4+ ion (3d2). That probably implies a large negative charge transfer from N/O 2p orbitals to Cr 3d orbitals. In short, the different spin moments of Cr1 and Cr2 in CrO2 mainly result from |2>. The conductivity of Cr(NCN)2 mainly results from |3>, however, that of CrO2 is attributed from both |2> and |3>.
In order to gain insight into the magnetic coupling between the Cr1 and Cr2 ions, we analyze the three-dimensional electronic-density contour plots near the Fermi level, according to orbital-projected DOS. Figure 4c,d show the density of electron distribution in the energy regions of (–2.20)–(–1.60) eV for Cr(NCN)2 and (–1.57)–(–1.35) eV for CrO2 (purple arrows in Figure 3 and in bottom panel of Figure 4b). In this region, for both compounds, the electrons are almost occupied in Cr atoms |1> orbitals, forming d–d direct superexchange in the edge-shared CrN6/CrO6 chain. This superexchange, where an electron is assumed to drift from one cation, exists in the compounds with edge-sharing or face-sharing octahedral chain or dimer. The d–d hopping is very important for early 3d meals such as Ti, V, and Cr [46]. As shown in Figure 4a,b, Cr4+ ion in both Cr(NCN)2 and CrO2 has |1> orbital only partially occupied for the up-spin state, that is, the integrated value in the full energy range is around 2, while the orbital occupation (integration below Fermi level) is only around 1, thus an intra-chain ferromagnetic interaction is expected [46,47]. The calculated effective exchange coupling constants of intra-chain J1 from GGA+U (Table 1) are 20.5 and 12.1 meV for Cr(NCN)2 and CrO2, respectively, indicating a stronger intra-chain coupling in Cr(NCN)2. Figure 4e,f shows the density of electron distribution in the energy regions of (–−0.69)–− (–0.40) eV and (–0.64)–(–0.37) eV for Cr(NCN)2 and CrO2, respectively (green arrows in Figure 3 and middle panel of Figure 4a,b). In this region, the electrons are mainly occupied by Cr |2> orbitals and O 2p orbitals, forming dpπ–dpπ correlation ferromagnetic superexchange between Cr1 and Cr2 sites. In CrO2, this interaction is strong, while it is cut off by C atoms in Cr(NCN)2. This is consistent with the results of the inter-chain coupling constants J2 from GGA+U calculations, that is, 21.5 meV for Cr(NCN)2 and 31.2 meV for CrO2. Figure 4g,f show the density of electron distribution in the energy regions near the Fermi level, that is, from −0.10–0.0 eV for Cr(NCN)2 and −0.10–0.0 eV for CrO2, respectively (blue arrows in Figure 3 and bottom panel of Figure 4a,b). In this region, the electrons are mainly occupied by Cr |3> orbitals and O 2p orbitals, also forming dpπ–dpπ correlation ferromagnetic superexchange between Cr1 and Cr2 sites. However, the electrons are more localized in Cr(NCN)2 than those in CrO2 because of the large size ligand of NCN2−. Obviously, there are two magnetic couplings, that is, direct d–d exchange and indirect dpπ–dpπ superexchange, dominating the ferromagnetic properties of Cr(NCN)2 and CrO2. In Cr(NCN)2, the strengths of the intra- and inter-chain couplings are comparative, however, in CrO2, there is a large difference between them, that is, 12.1 meV for intra-chain and 31.2 meV for inter-chain.
4. Conclusions
In summary, on the basis of the results of density functional theory with explicit electronic correlation, Cr(NCN)2 is a ferromagnetic and half-metal material, and stable both thermodynamically and elastically. It was predicted to be a tetragonal structure in the space group of P42/mnm with an asymmetrical [N–C≡N]2− ligand; as a result, it would be the first transition-metal cyanamide with partially filled 3d orbitals. A comparative study on the electronic structures of Cr(NCN)2 and CrO2 presents that the Cr atoms in both compounds are in one crystallographically equivalent site, however, in CrO2, the Cr atoms at the corner and center sites have different magnetic moments and orbital occupancies, and there is a large difference between the intra- (Cr atoms at the same site) and inter-chain (Cr atoms at the different sites) magnetic couplings. This difference is significantly weakened by C atoms in Cr(NCN)2. Thus, ferromagnetically half-metallic Cr(NCN)2 might be an interesting spintronic material, and we hope it could be synthesized in the future.
Supplementary Materials
The following are available online at https://www.mdpi.com/1996-1944/13/8/1805/s1. Table S1. The structure parameters of predicted Cr(NCN)2. Thermodynamically stability. Figure S1. Ordered spin arrangements of Cr(NCN)2 and CrO2 designed as (a) FM, (b) AFM1, and (c) AFM2 employed to extract the spin exchange parameters J1 and J2. Figure S2. Band structures near the Fermi energy for (a) Cr(NCN)2 and (b) CrO2 from GGA+U (U = 3 eV) calculations.
Author Contributions
Z.W. and S.Q. performed the experiments; all authors analyzed the data and discussed the results; J.S. analyzed the result; Z.W., H.X., and Z.H. wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundations of China (51601187 and 51971159), and the Natural Science Foundation of Fujian Province (No. 2016J05059).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Sakurai, H.; Kolodiazhnyi, T.; Michiue, Y.; Takayama-Muromachi, E.; Tanabe, Y.; Kikuchi, H. Unconventional colossal magnetoresistance in sodium chromium oxide with a mixed-valence state. Angew. Chem. Int. Ed. 2012, 124, 6757–6760. [Google Scholar] [CrossRef]
- Komarek, A.C.; Streltsov, S.V.; Isobe, M.; Möller, T.; Braden, M. CaCrO3: An anomalous antiferromagnetic metallic oxide. Phys. Rev. Lett. 2008, 101, 167204. [Google Scholar] [CrossRef] [PubMed]
- Toriyama, T.; Nakao, A.; Yamaki, Y.; Nakao, H.; Otha, Y. Peierls Mechanism of the metal-insulator transition in ferromagnetic hollandite K2Cr8O16. Phys. Rev. Lett. 2011, 107, 266402. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.S.; Ahn, K.H.; Jung, M.C.; Lee, K.W. Strain and spin-orbit coupling induced orbital-ordering in mott insulator BaCrO3. Phys. Rev. B 2014, 9, 205124. [Google Scholar] [CrossRef]
- Komarek, A.C.; Möller, T.; Isobe, M.; Drees, Y.; Braden, M. Magnetic order, transport and infrared optical properties in the ACrO3 system (A = Ca, Sr, and Pb). Phys. Rev. B 2011, 84, 125114. [Google Scholar] [CrossRef]
- Cheng, J.G.; Kweon, K.E.; Larregola, S.A.; Ding, Y.; Shirako, Y.; Marshall, L.G.; Li, Z.-Y.; Li, X.; António, M.; Suchomel, M.R.; et al. Charge disproportionation and the pressure-induced insulator-metal transition in cubic perovskite PbCrO3. Proc. Natl. Acad. Sci. USA 2015, 112, 1670–1674. [Google Scholar] [CrossRef]
- Xiao, W.; Tan, D.; Xiong, X.; Liu, J.; Xu, J. Large volume collapse observed in the phase transition in cubic PbCrO3 perovskite. Proc. Natl. Acad. Sci. USA 2010, 107, 14026–14029. [Google Scholar] [CrossRef]
- Hasegawa, K.; Isobe, M.; Yamauchi, T.; Ueda, H.; Ueda, Y. Discovery of ferromagnetic-half-metal-to-insulator transition in K2Cr8O16. Phys. Rev. Lett. 2009, 103, 146403. [Google Scholar] [CrossRef]
- Korotin, M.A.; Anisimov, V.I.; Khomskii, D.I.; Sawatzky, G.A. CrO2: A self-doped double exchange ferromagnet. Phys. Rev. Lett. 1998, 80, 4305–4308. [Google Scholar] [CrossRef]
- Schlottmann, P. Double-exchange mechanism for CrO2. Phys. Rev. B 2003, 67, 386–393. [Google Scholar] [CrossRef]
- Shim, J.H.; Lee, S.; Dho, J.; Kim, D.H. Coexistence of two different Cr Ions by self-doping in half-metallic CrO2 nanorods. Phys. Rev. Lett. 2007, 99, 057209. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Shimizu, Y.; Kobayashi, Y.; Itoh, M.; Jinno, T.; Isobe, M. Local electronic state in the half-metallic ferromagnet CrO2 investigated by site-selective 53Cr NMR measurements. Phys. Rev. B 2016, 93, 235129. [Google Scholar] [CrossRef]
- Solovyev, I.V.; Kashin, I.V.; Mazurenko, V.V. Mechanisms and origins of half-metallic ferromagnetism in CrO2. Phys. Rev. B 2015, 92, 144407. [Google Scholar] [CrossRef]
- Heffernan, K.H.; Yu, S.; Deckoff-Jones, S.; Zhang, X.; Talbayev, D. The role of spin fluctuations in the conductivity of CrO2. Phys. Rev. B 2016, 93, 165143. [Google Scholar] [CrossRef]
- Toropova, A.; Kotliar, G.; Savrasov, S.Y.; Oudovenko, V.S. Electronic structure and magnetic anisotropy of CrO2. Phys. Rev. B 2005, 71, 172403. [Google Scholar] [CrossRef]
- Boyko, T.D.; Green, R.J.; Dronskowski, R.; Moewes, A. Electronic band gap reduction in manganese carbodiimide: MnNCN. J. Phys. Chem. C 2013, 117, 12754–12761. [Google Scholar] [CrossRef]
- Tang, X.J.; Xiang, H.P.; Liu, X.H.; Speldrich, M.; Dronskowski, R. A ferromagnetic carbodiimide: Cr2(NCN)3. Angew. Chem. Int. Ed. 2010, 49, 4738–4742. [Google Scholar] [CrossRef]
- Liu, X.; Dronskowski, R.; Kremer, R.K.; Ahrens, M.; Lee, C.; Whangbo, M. Characterization of the Magnetic and Structural Properties of Copper Carbodiimide, CuNCN, by Neutron Diffraction and First-Principles Evaluations of Its Spin Exchange Interactions. J. Phys. Chem. C 2008, 112, 11013–11017. [Google Scholar] [CrossRef]
- Xiang, H.P.; Liu, X.H.; Dronskowski, R. Theoretical reinvestigation of the electronic structure of CuNCN: The influence of packing on the magnetic properties. J. Phys. Chem. C 2009, 113, 18891–18896. [Google Scholar] [CrossRef]
- Maddox, B.R. High-pressure structure of half-metallic CrO2. Phys. Rev. B 2006, 73, 144111. [Google Scholar] [CrossRef]
- Tang, X.J.; Houben, A.; Liu, X.H.; Stork, L.; Dronskowski, R. Crystal structure refinement of M(NCNH)2 (M = Fe, Co) based on combined neutron and X-ray diffraction Data. ZAAC 2011, 637, 1089–1091. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. B 1964, 136, 864–871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 1993, 48, 13115–13118. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Altmeyer, M.; Jeschke, H.O.; Hijano-Cubelos, O.; Martins, C.; Lechermann, F.; Koepernik, K.; Santander-Syro, A.F.; Rozenberg, M.; Valenti, R.; Gabay, M. Magnetism, spin texture, and in-gap states: Atomic specialization at the surface of oxygen-deficient SrTiO3. Phys. Rev. Lett. 2015, 116, 157203. [Google Scholar] [CrossRef]
- Beutier, G.; Collins, S.P.; Dimitrova, O.V.; Dmitrienko, V.E. Band Filling Control of the Dzyaloshinskii-Moriya Interaction in Weakly Ferromagnetic Insulators. Phys. Rev. Lett. 2017, 119, 167201. [Google Scholar] [CrossRef]
- Wen, X.D.; Martin, R.L.; Henderson, T.M.; Scuseria, G.E. Density functional theory studies of the electronic structure of solid state actinide oxides. Chem. Rev. 2013, 113, 1063–1096. [Google Scholar] [CrossRef]
- Zheng, S.; Huang, C.; Yu, T.; Xu, M.; Zhang, S.; Xu, H.; Liu, Y.; Kan, E.; Wang, Y.; Yang, G. High-Temperature Ferromagnetism in Fe3P Monolayer with Large Magnetic Anisotropy. J. Phys. Chem. Lett. 2019, 10, 2733–2738. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Peter, E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for brillouin-zone integrations. Phys Rev. B 1994, 49, 16223–16233. [Google Scholar] [CrossRef]
- Anisimov, V.I.; Poteryaev, A.I.; Korotin, M.A.; Anokhin, A.O.; Kotliar, G. First-principles calculations of the electronic structure and spectra of strongly correlated systems: Dynamical mean-field theory. J. Phys.-Condens. Matter 1997, 9, 7359–7367. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Bhobe, P.A.; Chainani, A.; Taguchi, M.; Eguchi, R.; Shin, S. Electronic structure of an antiferromagnetic metal: CaCrO3. Phys. Rev. B 2010, 83, 165132. [Google Scholar] [CrossRef]
- Liu, X.; Müller, P.; Kroll, P.; Dronskowski, R. Synthesis, structure determination, and quantum-chemical characterization of an alternate HgNCN polymorph. Inorg. Chem. 2002, 41, 4259–4265. [Google Scholar] [CrossRef]
- Baroni, S.; Gironcoli, S.; Corso, A.D.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515. [Google Scholar] [CrossRef]
- Togo, A.; Oba, F.; Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B 2008, 78, 134106. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Becker, M.; Nuss, J.; Jansen, M. ChemInform Abstract: Synthesis and characterization of sodium cyanamide. ChemInform 2010, 32. [Google Scholar] [CrossRef]
- Baldinozzi, G.; Malinowska, B.; Rakib, M.; Durand, G. Crystal structure and characterisation of cadmium cyanamide. J. Mater. Chem. A 2002, 12, 268–272. [Google Scholar] [CrossRef]
- Cooper, M.J. The structures of some inorganic cyanamides. II. The structure of lead cyanamide. Acta Crystallogr. 1964, 17, 1452–1456. [Google Scholar] [CrossRef]
- Yamasaki, A.; Chioncel, L.; Lichtenstein, A.I.; Andersen, O.K. Model hamiltonian parameters for half-metallic ferromagnets NiMnSb and CrO2. Phys. Rev. B 2006, 74, 024419. [Google Scholar] [CrossRef]
- Goodenough, J.B. Magnetism and the Chemical Bond; Interscience Publishers: New York, NY, USA, 1963; p. 180. [Google Scholar]
- Xiang, H.P.; Tang, Y.Y.; Zhang, S.Y.; He, Z. Intra-chain superexchange couplings in quasi-1D 3d transition-metal magnetic compounds. J. Phys.-Condens. Matter 2016, 28, 276003. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).