Morphology, Rheology and Crystallization in Relation to the Viscosity Ratio of Polystyrene/Polypropylene Polymer Blends

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Compounding Procedure
2.3. Determination of Phase Morphology
2.4. Melting and Crystallization Behavior
2.5. Rheological Characterization
3. Results and Discussion
3.1. Morphological Characterization
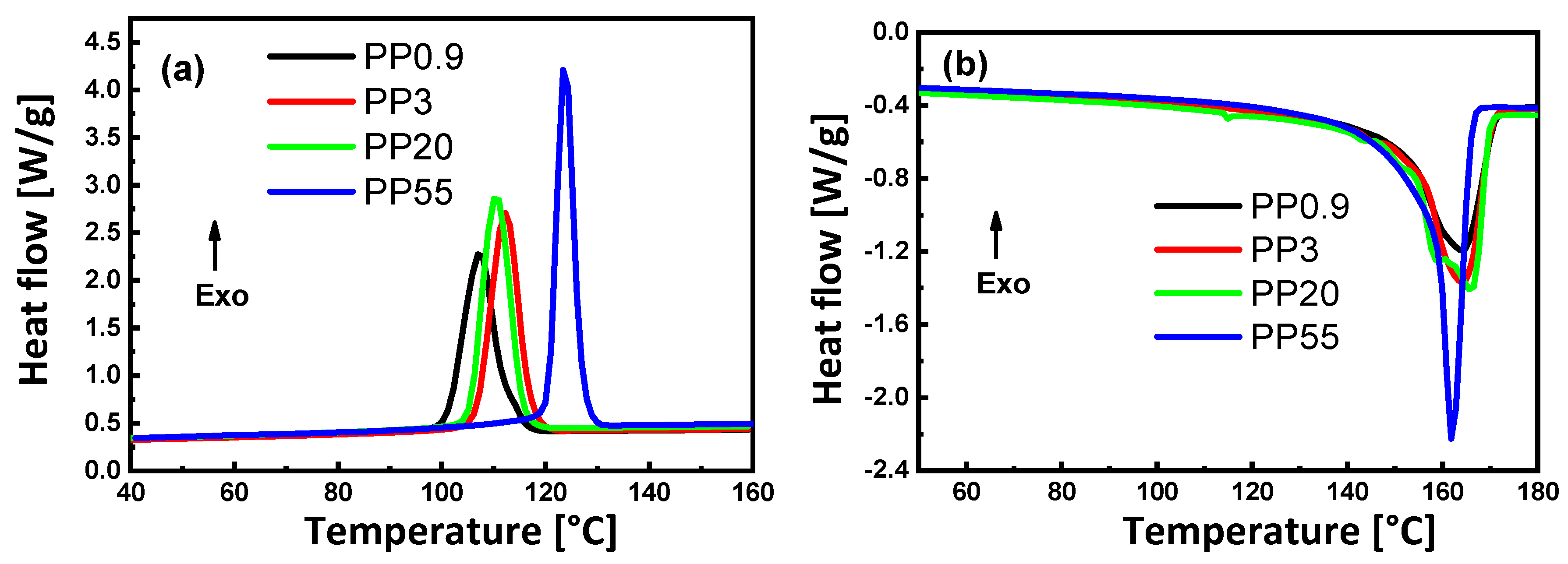
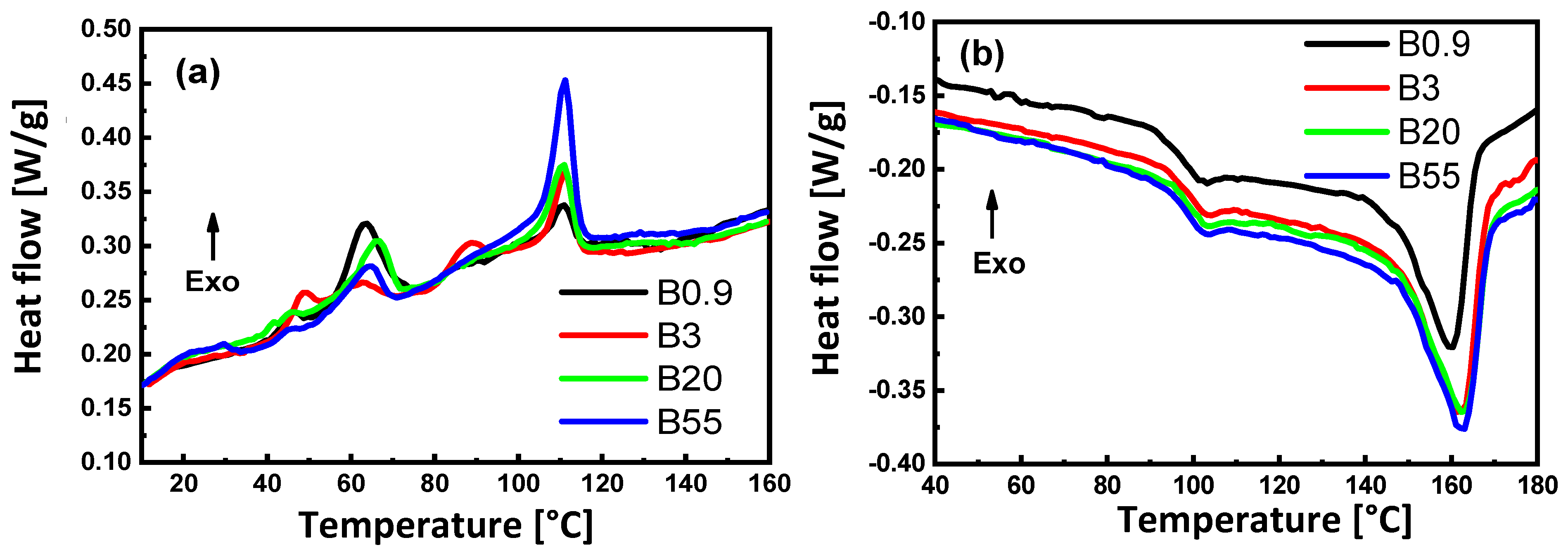
3.2. Melting and Crystallization Behavior
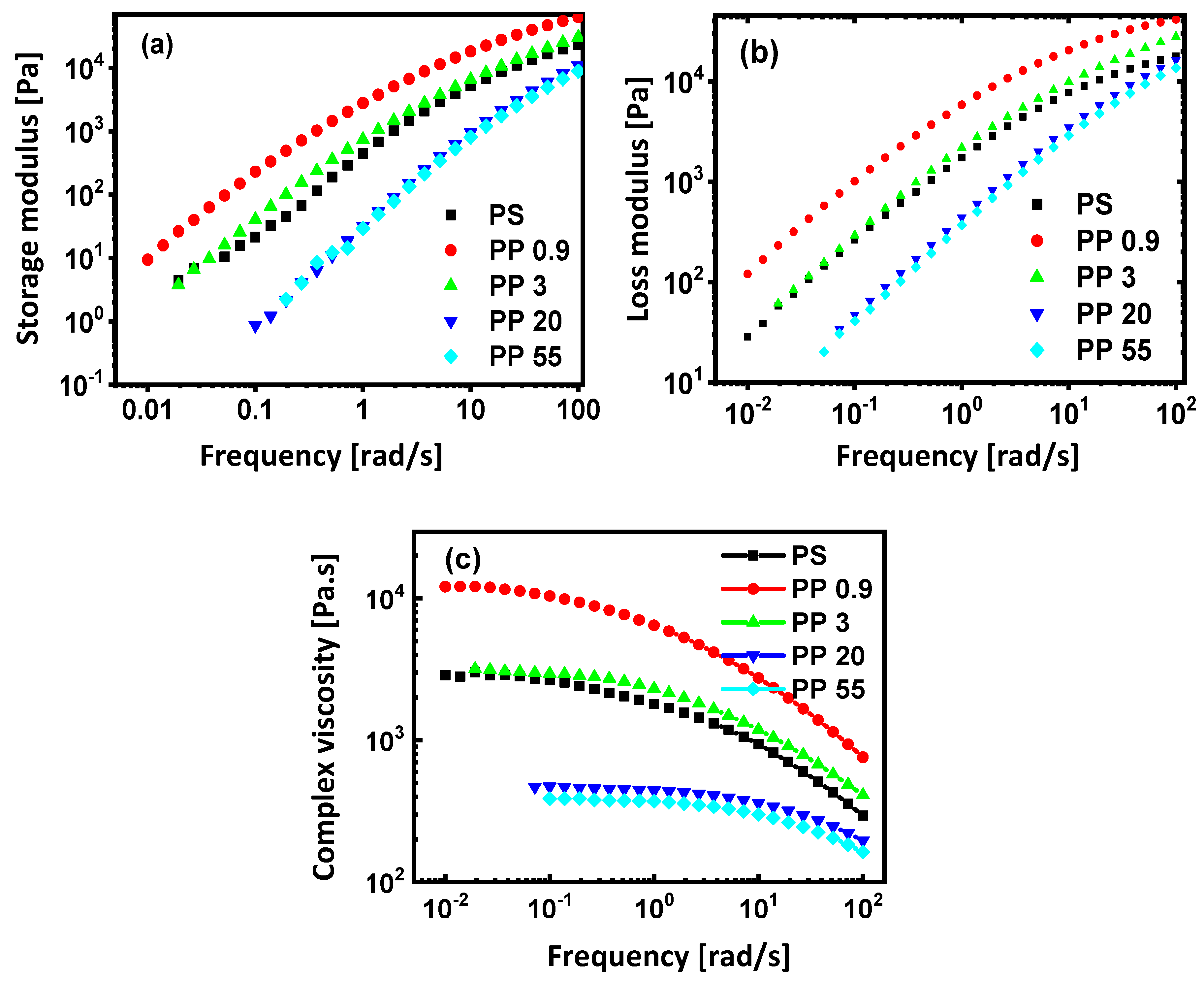
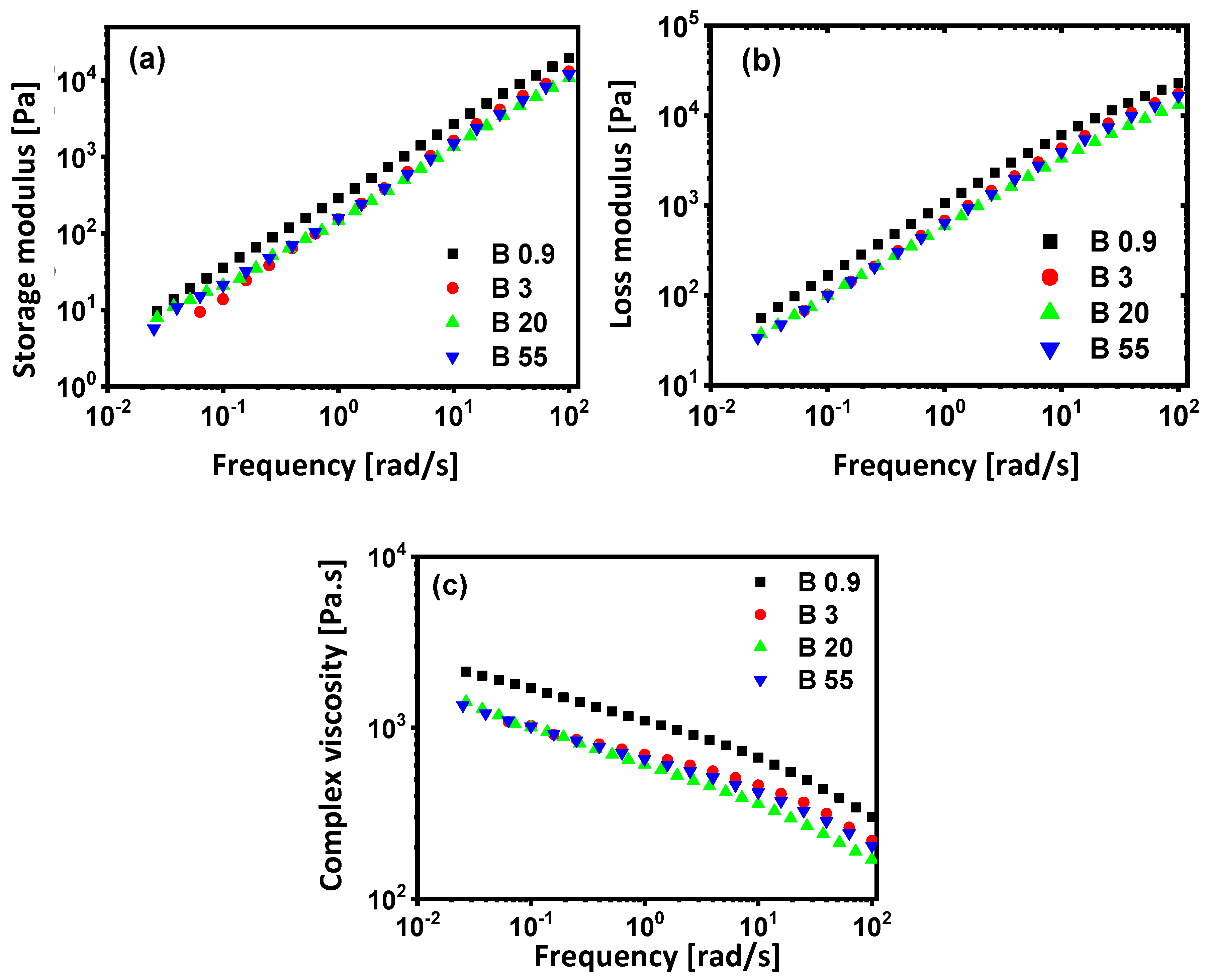
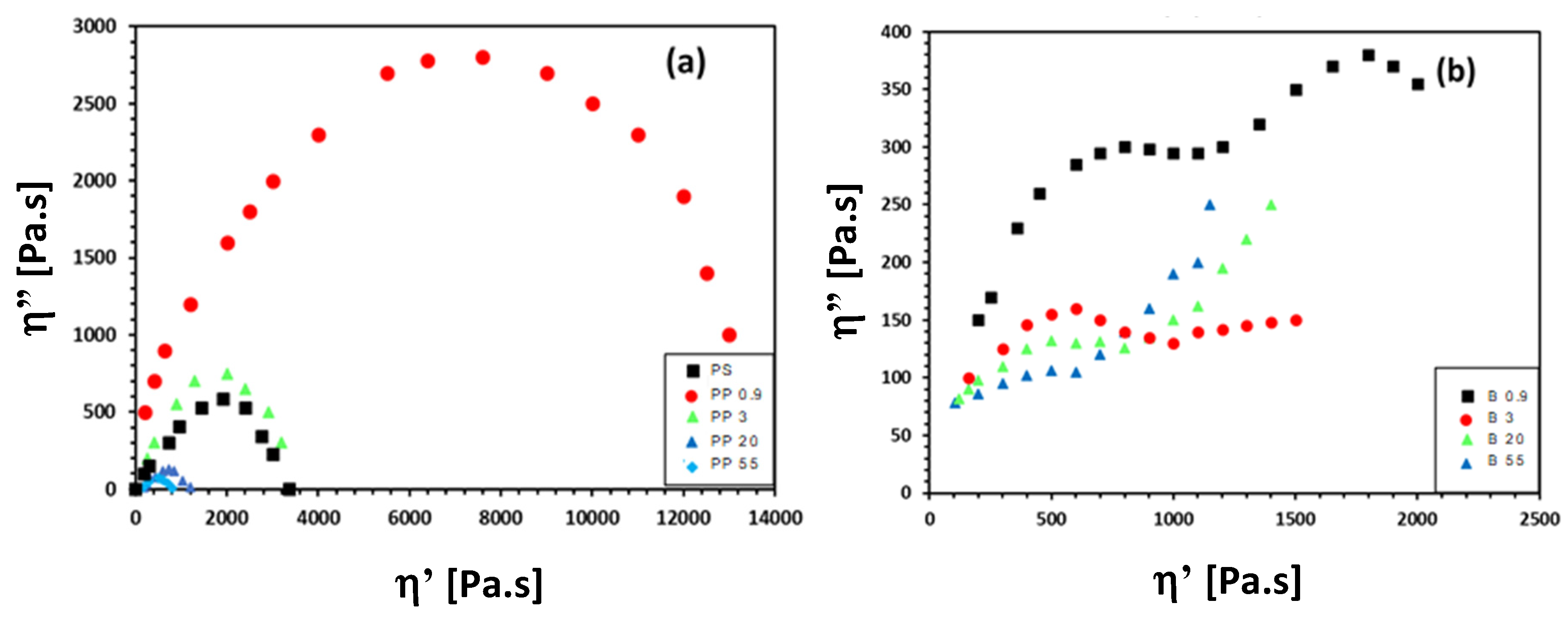
3.3. Rheological Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Brostow, W.; Grguric, T.H.; Meija, O.O.; Pietkiewicz, D.; Rek, V. Polypropylene polystyrene blends with a compatibilizer. E-Polymers 2008, 8. [Google Scholar] [CrossRef]
- Horak, Z.; Kolarik, J.; Sipek, M.; Hynek, V.; Vecerka, F. Gas permeability and mechanical properties of polystyrene–polypropylene blends. J. Appl. Polym. Sci. 1998, 69, 2615–2623. [Google Scholar] [CrossRef]
- Slouf, M.; Radonjic, G.; Hlavata, D.; Sikora, A. Compatibilized iPP/aPS blends: The effect of the viscosity ratio of the components on the blends morphology. J. Appl. Poylm. Sci. 2006, 101, 2236–2249. [Google Scholar] [CrossRef]
- Fujiyama, M. Structure and properties of injection moldings of polypropylene/polystyrene blends. J. Appl. Polym. Sci. 1997, 63, 1015–1027. [Google Scholar] [CrossRef]
- Gao, J.; Fu, X.T.; Ding, M.M.; Fu, Q. Studies of partial compatibility of PP and PS. Chin. J. Polym. Sci. 2010, 28, 647–656. [Google Scholar] [CrossRef]
- Bourry, D.; Favis, B.D. Cocontinuity and phase inversion in HDPE/PS blends: Influence of interfacial modification and elasticity. J. Polym. Sci. B 1998, 36, 1889–1899. [Google Scholar] [CrossRef]
- Hammani, S.; Barhoum, A.; Nagarajan, S.; Bechelany, M. Toner waste powder (twp) as a filler for polymer blends (ldpe/hips) for enhanced electrical conductivity. Materials 2019, 12, 3062. [Google Scholar] [CrossRef] [PubMed]
- Fenni, S.E.; Wang, J.; Haddaoui, N.; Favis, B.D.; Müller, A.J.; Cavallo, D. Crystallization and self-nucleation of PLA, PBS and PCL in their immiscible binary and ternary blends. Thermochim. Acta 2019, 677, 117–130. [Google Scholar] [CrossRef]
- Sarazin, P.; Li, G.; Orts, W.J.; Favis, B.D. Binary and ternary blends of polylactide, polycaprolactone and thermoplastic starch. Polymer 2008, 49, 599–609. [Google Scholar] [CrossRef]
- Habibi, M.; Najafi, S.K.; Ghasemi, I. Rheological and mechanical properties of composites made from wood flour and recycled LDPE/HDPE blend. Iran Polym. J. 2017, 26, 949–956. [Google Scholar] [CrossRef]
- Krisztina, A.; Minya, V.; Schausberger, A. Characterization of phase morphology of polymer melts (PP/PE blends) via rheology. J. Appl. Polym. Sci. 2007, 105, 2294–2298. [Google Scholar]
- Kwon, M.K.; Cho, K.S. Analysis of the Palierne model by relaxation time spectrum. Korea-Aust. Rheol. J. 2016, 28, 23–31. [Google Scholar] [CrossRef]
- Wang, L.; Feng, L.; Gu, X.; Zhang, C. Influences of the viscosity ratio and processing conditions on the formation of highly orientated ribbons in polymer blends by tape extrusion. Ind. Eng. Chem. Res. 2015, 54, 11080–11086. [Google Scholar] [CrossRef]
- Choi, G.D.; Jo, W.H.; Kim, H.G. The effect of the viscosity ratio of dispersed phase to matrix on the rheological, morphological, and mechanical properties of polymer blends containing a LCP. J. Appl. Polym. Sci. 1996, 59, 443–452. [Google Scholar] [CrossRef]
- Karingamanna, J.; Sabu, T.; Kuruvilla, J. Effect of blend ratio on the dynamic mechanical and thermal degradation behavior of polymer–polymer composites from low density polyethylene and polyethylene terephthalate. Iran. Polym. J. 2016, 25, 373–384. [Google Scholar]
- Utracki, L.A.; Shi, Z.H. Development of polymer blend morphology during compounding in a twin-screw extruder. Part I: Droplet dispersion and coalescence—A review. Polym. Eng. Sci. 1992, 32, 1824–1833. [Google Scholar] [CrossRef]
- Rizvi, A.; Park, C.B.; Favis, B.D. Tuning viscoelastic and crystallization properties of polypropylene containing in-situ generated high aspect ratio polyethylene terephthalate fibrils. Polymer 2015, 68, 83–91. [Google Scholar] [CrossRef]
- Pa, D.; Chen, L.; He, H.; Deng, K.; Qin, Z. Deformation of dispersed polystyrene droplets in immiscible polypropylene/polystyrene blend fibers under uniaxial elongational flow. Fibers Polym. 2016, 17, 1343–1351. [Google Scholar]
- Kong, M. Elongation thinning and morphology deformation of nanoparticle-filled polypropylene/polystyrene blends in elongational flow. J. Rheol. 2018, 62, 11–23. [Google Scholar] [CrossRef]
- Amoabeng, D.; Roell, D.; Clouse, K.M.; Young, B.A.; Velankar, S.S. A composition-morphology map for particle-filled blends of immiscible thermoplastic polymers. Polymer 2017, 119, 212–223. [Google Scholar] [CrossRef]
- Feigl, K.; Kaufmann, S.F.M.; Fischer, P.; Windhab, E.J. A numerical procedure for calculating droplet deformation in dispersing flows and experimental verification. Chem. Eng. Sci. 2003, 58, 2351–2363. [Google Scholar] [CrossRef]
- Pan, D.; He, H.; Ren, H.; Xu, H.; Wang, S.; Chen, L.; Qin, Z. Study on the interfacial tension of immiscible polystyrene/polypropylene blend with deformed drop retraction method. J. Thermoplast. Comp. 2019, 32, 205–215. [Google Scholar] [CrossRef]
- Willemse, R.C.; De Boer, A.P.; van Dam, J.; Gotsis, A.D. Co-continuous morphologies in polymer blends: The influence of interfacial tension. Polymer 1999, 40, 827–834. [Google Scholar] [CrossRef]
- Yi, X.; Xu, L.; Wang, Y.L.; Zhong, G.J.; Ji, X.; Li, Z.M. Morphology and properties of isotactic polypropylene/poly(ethylene terephthalate) in situ microfibrillar reinforced blends: Influence of viscosity ratio. Eur. Polym. J. 2010, 46, 719–730. [Google Scholar] [CrossRef]
- Birudugadda, A.K.; Madhav, C.V.; Bhukya, R. Evaluation and characterization of polystyrene blending with polypropylene by using compatibilizers. In Proceedings of the 8th International Conference on Chemical, Ecology and Environmental Science, Dubai, UAE, 12–13 January 2016; pp. 67–71. [Google Scholar]
- Omonov, T.S.; Harrats, C.; Moldenaers, P.; Groeninckx, G. Phase continuity detection and phase inversion phenomena in immiscible polypropylene/polystyrene blends with different viscosity ratios. Polymer 2007, 48, 5917–5927. [Google Scholar] [CrossRef]
- Li, J.; Zhao, J.; Yuan, X.; Sheng, J. Compositional dependence of static shear viscosity of immiscible pp/ps blends. Macromol. Sci. B 2007, 46, 651–665. [Google Scholar] [CrossRef]
- Han, D.C.; Kim, Y.W.; Chen, S.J. The effect of mixing on the modes of dispersion and rheological properties of two phase polymer blends in extrusion. J. Appl. Polym. Sci. 1975, 19, 2831–2843. [Google Scholar] [CrossRef]
- Everaert, V.; Aerts, L.; Groeninckx, G. Phase morphology development in immiscible PP/(PS/PPE) blends influence of the melt-viscosity ratio and blend composition. Polymer 1999, 40, 6627–6644. [Google Scholar] [CrossRef]
- Fortelny, I.; Michalkova, D.; Mikesova, J. Effect of the mixing conditions on the phase structure of PP/PS blends. J. Appl. Polym. Sci. 1996, 59, 155–164. [Google Scholar] [CrossRef]
- Michell, R.M.; Muller, A.J. Confined crystallization of polymeric materials. Prog. Polym. Sci. 2016, 54–55, 183–213. [Google Scholar] [CrossRef]
- Piorkowska, E.; Rutledge, G.C. Handbook of Polymer Crystallization; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Chen, C.; Xu, R.; Chen, X.; Xie, J.; Zhang, F.; Yang, Y.; Lei, C. Influence of cocrystallization behavior on structure and properties of HDPE/LLDPE microporous membrane. J. Polym. Res. 2016, 23, 46. [Google Scholar] [CrossRef]
- Gao, Q.; Scheinbeim, J. Crystallization studies of polymer blends of Nylon-11/Poly(vinylidene fluoride). Polym. J. 2003, 35, 345–352. [Google Scholar] [CrossRef]
- Weng, M.; Qiu, Z. Unusual fractional crystallization behavior of novel crystalline/crystalline polymer blends of poly(ethylene suberate) and poly(ethylene oxide) with similar melting points. Macromolecules 2014, 47, 8351–8358. [Google Scholar] [CrossRef]
- Naga, N.; Takagi, Y.; Urakami, S.; Noguchi, K. Crystallization and crystalline structure of syndiotactic polypropylene and syndiotactic-poly(1-Butene) blend. Open J. Polym. Chem. 2014, 4, 102–114. [Google Scholar] [CrossRef][Green Version]
- Wang, J.; Cheung, M.K.; Mi, Y. Miscibility and morphology in crystalline/amorphous blends of poly(caprolactone)/poly(4-vinylphenol) as studied by DSC, FTIR and 13C solid state NMR. Polymer 2002, 43, 1357–1364. [Google Scholar] [CrossRef]
- Topkanlo, H.A.; Ahmadi, Z.; Taromi, F.A. An in-depth study on crystallization kinetics of PET/PLA blends. Iran. Polym. J. 2018, 27, 13–22. [Google Scholar] [CrossRef]
- Xiang, F.; Shi, Y.; Li, X.; Huang, T.; Chen, C.; Peng, Y.; Wang, Y. Cocontinuous morphology of immiscible high density polyethylene/polyamide 6 blend induced by multiwalled carbon nanotubes network. Eur. Polym. J. 2012, 48, 350–361. [Google Scholar] [CrossRef]
- Tol, R.T.; Mathot, V.B.F.; Groeninckx, G. Confined crystallization phenomena in immiscible polymer blends with dispersed micro- and nanometer sized PA6 droplets, part 3: Crystallization kinetics and crystallinity of micro- and nanometer sized PA6 droplets crystallizing at high supercoolings. Polymer 2005, 46, 2955–2965. [Google Scholar] [CrossRef]
- Choudhur, N.R.; Chaki, T.K.; Dutta, A.; Bhowmick, A.K. Thermal, X-ray and dynamic mechanical properties of thermoplastic elastomeric natural rubber-polyethylene blends. Polymer 1989, 30, 2047–2053. [Google Scholar] [CrossRef]
- Thirtha, V.; Lehman, R.; Nosker, T. Glass transition phenomena in melt-processed polystyrene/polypropylene blends. Polym. Eng. Sci. 2005, 45, 1187–1193. [Google Scholar] [CrossRef]
- Barhoum, A.; Van Lokeren, L.; Rahier, H.; Dufresne, A.; Van Assche, G. Roles of in situ surface modification in controlling the growth and crystallization of CaCO3 nanoparticles, and their dispersion in polymeric materials. J. Mater. Sci. 2015, 50, 7908–7918. [Google Scholar] [CrossRef]
- Barhoum, A.; Garcia-Betancourt, M.L. Physicochemical characterization of nanomaterials: Size, morphology, optical, magnetic, and electrical properties. In Handbook of Nanoparticles and Architectural Nanostructured Materials; Barhoum, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Lopez-Barron, C.R.; Robledo-Ortiz, J.R.; Rodrigue, D.; Gonzalez-Nunez, R. Film processability, morphology, and properties of polyamide-6/low density polyethylene blends. J. Plast. Film Sheet. 2007, 23, 149–169. [Google Scholar] [CrossRef]
- Zare, Y.; Park, S.P.; Rhee, K.Y. Analysis of complex viscosity and shear thinning behavior in poly (lactic acid)/poly (ethylene oxide)/carbon nanotubes biosensor based on Carreau–Yasuda model. Results Phys. 2019, 13, 102245. [Google Scholar] [CrossRef]
- Salehiyan, R.; Song, H.Y.; Choi, W.J.; Hyun, K. Characterization of effects of silica nanoparticles on (80/20) pp/ps blends via nonlinear rheological properties from Fourier transform rheology. Macromolecules 2015, 48, 4669–4679. [Google Scholar] [CrossRef]
- Pan, S.; Hufenus, R.; Qin, Z.; Chen, L.; Gooneie, A. Tuning gradient microstructures in immiscible polymer blends by viscosity ratio. J. Appl. Polym. Sci. 2019, 136, 48165. [Google Scholar] [CrossRef]
- Vananroye, A.; Cardinaels, R.; Van Puyvelde, P.; Moldenaers, P. Effect of confinement and viscosity ratio on the dynamics of single droplets during transient shear flow. J. Rheol. 2008, 52, 1459–1475. [Google Scholar] [CrossRef]
- Jordhamo, G.M.; Manson, J.A.; Sperling, L.H. Phase continuity and inversion in polymer blends and simultaneous interpenetrating networks. Polym. Eng. Sci. 1986, 26, 517–524. [Google Scholar] [CrossRef]
- Chen, T.H.; Su, A.C. Morphology of poly(p-phenylene sulfide)polyethylene blends. Polymer 1993, 34, 4826–4831. [Google Scholar] [CrossRef]
- Fortelny, I.; Kovar, J. Droplet size of the minor component in the mixing of melts of immiscible polymers. Eur. Polym. J. 1989, 25, 317–319. [Google Scholar] [CrossRef]
- Tessier, R.; Lafranche, E.; Krawczak, P. Development of novel melt-compounded starch-grafted polypropylene-polypropylene-grafted maleic anhydride/organocaly ternary hybrids. Express Polym. Lett. 2012, 6, 937–952. [Google Scholar] [CrossRef]
- Sundararj, U.; Macosko, C.W. Drop breaking and coalescence in polymer blends: The effects of concentration and compatibilization. Macromolecules 1995, 28, 2647–2657. [Google Scholar] [CrossRef]
- Miroshnikov, Y.P.; Williams, H.L. Morphology of extruded polypropylene-polystyrene blends. Polym. Sci. USSR 1982, 24, 1811–1825. [Google Scholar] [CrossRef]
- Favis, B.D.; Therrien, D. Factors influencing structure formation and phase size in an immiscible polymer blend of polycarbonate and polypropylene prepared by twin-screw extrusion. Polymer 1991, 32, 1474–1481. [Google Scholar] [CrossRef]
- Berger, W.; Kammer, H.W. Structure formation in polymer blends as a result of phase separation and deformation processes. Makromol. Chem. Macromol. Symp. 1987, 12, 145–163. [Google Scholar] [CrossRef]
- Lyngaae-Jorgensen, J.; Andersen, F.E.; Alle, N. Domain Stability during Capillary Flow of Well Dispersed Two Phase Polymer Blends. Polystyrene/Polymethylmethacrylate Blends in Polymer Alloys III; Springer: Boston, MA, USA, 1983. [Google Scholar]
- Halimatudahliana Ismail, H.; Nasir, M. The effect of various compatibilizers on mechanical properties of polystyrene/polypropylene blend. Polymer Test. 2002, 21, 163–170. [Google Scholar] [CrossRef]
- Hammani, S.; Moulai-Mostefa, N.; Benyahia, L.; Tassin, J.F. Effects of shear during the cooling on the rheology and morphology of immiscible polymer blends. IOP Conf. Ser. Mater. Sci. Eng. 2014, 64, 012056. [Google Scholar] [CrossRef]
- Hammani, S.; Moulai-Mostefa, N.; Benyahia, L.; Tassin, J.F. Effects of composition and extrusion parameters on the morphological development and rheological properties of PP/PC blends. Co-continuity investigation. J. Polym. Res. 2012, 19, 9940. [Google Scholar] [CrossRef]
- Chapleau, N.; Favis, B.D. Droplet/fibre transitions in immiscible polymer blends generated during melt processing. J. Mater. Sci. 1995, 30, 142–150. [Google Scholar] [CrossRef]
- Taylor, G.I. The formation of emulsions in definable fields of flow. Proc. R. Soc. 1934, A146, 501–523. [Google Scholar]
- Grace, H.P. Dispersion phenomena in high viscosity immiscible fluid systems and application of static mixers as dispersion devices in such systems. Chem. Eng. Comm. 1982, 14, 225–277. [Google Scholar] [CrossRef]
- Gonzalez-Nunez, R.; Padilla, H.; De Kee, D.; Favis, B.D. Barrier properties of polyamide-6/high density polyethylene blends. Polym. Bull. 2001, 46, 323–330. [Google Scholar] [CrossRef]
- Kim, S.; Fang, Y.L.; Lafleur, P.G.; Carreau, P. Dynamics and criteria for bubble instabilities in a single layer film blowing extrusion. Polym. Eng. Sci. 2004, 44, 283–302. [Google Scholar] [CrossRef]
- Zhang, X.M.; Elkoun, S.; Ajji, A.; Hunneault, M.A. Oriented structure and anisotropy properties of polymer blown films: HDPE, LLDPE and LDPE. Polymer 2004, 45, 217–229. [Google Scholar] [CrossRef]
- Michell, R.M.; Blaszczyk-Lezak, I.; Mijangos, C.; Muller, A.J. Confinement effects on polymer crystallization: From droplets to alumina nanopores. Polymer 2013, 54, 4059–4077. [Google Scholar] [CrossRef]
- Guseinova, N.T.; Kurbanov, M.A. Study of the thermal properties of polyethylene of different MFI and molecular weight. Int. Polym. Sci. Technol. 2006, 34, 8–10. [Google Scholar] [CrossRef]
- Morrow, D.R.; Newman, B.A. Crystallization of low-molecular-weight polypropylene fractions. J. Appl. Phys. 1968, 39, 4944. [Google Scholar] [CrossRef]
- Jin, Y.; Hiltner, A.; Baer, E. Effect of a sorbitol nucleating agent on fractionated crystallization of polypropylene droplets. J. Polym. Sci. B 2007, 45, 1788–1797. [Google Scholar] [CrossRef]
- Thirtha, V.M.; Lehman, R.L.; Nosker, T.J. Morphological effects on glass transitions in immiscible polymer blends. Mater. Res. Soc. Symp. Proc. E 2005, 856, 12. [Google Scholar] [CrossRef]
- Perez, I.S.B.; Manrich, S.; Manrich, S. The effect of different block copolymers on post consumer HDPE/HIPS Blends: Phase morphology and thermal properties. Polim.-Cienc. Tecnol. 2008, 18, 207–214. [Google Scholar] [CrossRef]
- Huang, L.; Yuan, Q.; Jiang, W.; An, L.; Jiang, S.; Li, R.K.Y. Mechanical and thermal properties of glass bead–filled nylon-6. J. Appl. Polym. Sci. 2004, 94, 1885–1890. [Google Scholar] [CrossRef]
- Wu, D.; Zhang, Y.; Zhang, M.; Zhou, W. Phase behavior and its viscoelastic response of polylactide/poly(e-caprolactone) blend. Eur. Polym. J. 2008, 44, 2171–2183. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, H.; Song, Y.; Zheng, Q. Rheological behaviors of randomly crosslinked low density polyethylene and its gel network. Polymer 2012, 53, 3035–3042. [Google Scholar] [CrossRef]
- Sadiku-Agboola, O.; Sadiku, E.R.; Adegbola, A.T.; Biotidara, O.F. Rheological Properties of polymers: Structure and morphology of molten polymer blends. Mater. Sci. Appl. 2011, 2, 30–41. [Google Scholar] [CrossRef]
- Bochmann, E.S.; Neumann, D.; Gryczke, A.; Wagner, K.G. Micro-scale prediction method for API-solubility in polymeric matrices and process model for forming amorphous solid dispersion by hot-melt extrusion. Eur. J. Pharm. Biopharm. 2016, 107, 40–48. [Google Scholar] [CrossRef]
- Komuro, R.; Sukumaran, S.K.; Sugimoto, M.; Koyama, K. Slip at the interface between immiscible polymer melts I: Method to measure slip. Rheol. Acta 2014, 53, 23–30. [Google Scholar] [CrossRef]
- Stadler, F.J.; Piel, C.; Kaschta, J.; Rulhoff, S.; Kaminsky, W.; Munstdt, H. Dependence of the zero shear-rate viscosity and the viscosity function of linear highdensity polyethylenes on the mass-average molar mass and polydispersity. Rheol. Acta 2006, 45, 755–764. [Google Scholar] [CrossRef]
- Bousmina, M. Effect of interfacial tension on linear viscoelastic behavior of immiscible polymer blends. Short Comm. Rheol. Acta 1999, 38, 251–254. [Google Scholar] [CrossRef]
- Wu, T.; Tong, Y.; Qiu, F.; Yuan, D.; Zhang, G.; Qu, J. Morphology, rheology property, and crystallization behavior of PLLA/OMMT nanocomposites prepared by an innovative eccentric rotor extruder. Polym. Adv. Technol. 2018, 29, 41–51. [Google Scholar] [CrossRef]
- Graebling, D.; Muller, R.; Palierne, J.F. Linear viscoelastic behavior of some incompatible polymer blends in the melt. interpretation of data with a model of emulsion of viscoelastic liquids. Macromolecules 1993, 26, 320–329. [Google Scholar] [CrossRef]
- Luna, C.B.; Siqueira, D.D.; Araujo, E.M.; Wellen, R.M. Tailoring PS/PPrecycled blends compatibilized with SEBS. Evaluation of rheological, mechanical, thermomechanical and morphological characters. Mater. Res. Express 2019, 6, 075316. [Google Scholar] [CrossRef]
- Li, H.; Zhang, X.M.; Zhu, S.Y.; Chen, W.X.; Feng, L.F. Preparation of polypropylene and polystyrene with NCO and –NH2 functional groups and their applications in polypropylene/polystyrene blends. Polym. Eng. Sci. 2015, 55, 614–623. [Google Scholar] [CrossRef]
- Nascimento, W.A.; Agrawal, P.; Mélo, T.J.A. Effect of copolymers containing glycidyl methacrylate functional groups on the rheological, mechanical, and morphological properties of poly(ethylene terephthalate). Polym. Eng. Sci. 2019, 59, 683–693. [Google Scholar] [CrossRef]
- Walha, F.; Lamnawar, K.; Maazouz, A.; Jaziri, M. Rheological, morphological and mechanical studies of sustainably sourced polymer blends based on poly(lactic acid) and polyamide 11. Polymers 2016, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Komalan, C.; George, K.E.; Kumar, P.A.S.; Varughese, K.T.; Thomas, S. Dynamic mechanical analysis of binary and ternary polymer blends based on nylon copolymer/EPDM rubber and EPM grafted maleic anhydride compatibilizer. Express Polym. Lett. 2007, 1, 641–653. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Grade | Sample Code | Density at 23 °C 1 (g/cm3) | Melt Flow Index at 200 °C 1 (g/10 min) | Zero-Shear Viscosity 2 η0 (Pa·s) at 200 °C | Supplier |
|---|---|---|---|---|---|
| PS | PS | 1.050 | / | 3150 | BASF S.A. |
| 100GA01 | PP0.9 | 0.923 | 0.9 | 13349 | Ineos |
| ISPLEN PP-040G1E | PP3 | 0.905 | 3.0 | 3199 | Repsol YPF |
| ISPLEN PP080G2M | PP20 | 0.905 | 20.0 | 466 | Repsol YPF |
| ISPLEN PP099K2M | PP55 | 0.905 | 55.0 | 389 | Repsol YPF |
| Blend | PS + PP0.9 (80/20) | PS + PP3 (80/20) | PS + PP20 (80/20) | PS + PP55 (80/20) |
|---|---|---|---|---|
| Code | B0.9 | B3 | B20 | B55 |
| K1 at 200 °C | 4.23 | 1.01 | 0.15 | 0.12 |
| Blend | dn (µm) | dw (µm) | PDI | dmin (µm) | dmax (µm) | Sd |
|---|---|---|---|---|---|---|
| B09 | 2.80 | 3.07 | 1.10 | 1.0 | 6.0 | 0.43 |
| B3 | 6.38 | 7.04 | 1.10 | 2.5 | 12.3 | 0.19 |
| B20 | 9.03 | 9.82 | 1.09 | 2.5 | 15.5 | 0.13 |
| B50 | 10.3 | 11.1 | 1.098 | 3.0 | 17.0 | 0.12 |
| Polymer | Tc (°C) | Tg (°C) | Tm(Onset) (°C) | Tm (°C) | |
|---|---|---|---|---|---|
| PS | - | 95.5 | - | - | - |
| PP0.9 | 110.5 | N/D 1 | 153.9 | 168.9 | 28.5 |
| PP3 | 112.3 | N/D 1 | 154.6 | 168.7 | 35.1 |
| PP20 | 112.8 | N/D 1 | 157.9 | 170.9 | 34.1 |
| PP55 | 124.6 | N/D 1 | 149.9 | 165.1 | 44.8 |
| Blend | Tc (°C) | Tg (°C) | Tm (Onset) (°C) | Tm (°C) | Relative Loss Crystallinity (%) | |||
|---|---|---|---|---|---|---|---|---|
| Peak A | Peak B | Peak C | ||||||
| B0.9 | 110.0 | 63.5 | 45.0 | 98.7 | 145.5 | 160.0 | 24.7 | 13.33 |
| B3.0 | 111.0 | 88.0 | 48.4 | 98.6 | 149.5 | 161.8 | 25.8 | 26.49 |
| B20 | 110.7 | 66.1 | 42.0 | 98.0 | 149.6 | 162.8 | 28.2 | 17.30 |
| B55 | 111.1 | 64.2 | - | 99.9 | 151.8 | 163.1 | 19.3 | 56.91 |
| Polymer | n | A | 1/λ (s−1) | τ* (Pa) | ||
|---|---|---|---|---|---|---|
| PS | 3150 ± 86 | 0.517 ± 0.039 | 0.534 ± 0.075 | 0.457 ± 0.097 | 1.93 | 6092 |
| PP0.9 | 13,349 ± 186 | 1.020 ± 0.291 | 0.564 ± 0.037 | 0.419 ± 0.039 | 0.98 | 13,087 |
| PP3 | 3199 ± 19 | 0.508 ± 0.079 | 0.736 ± 0.033 | 0.495 ± 0.023 | 1.97 | 6297 |
| PP20 | 466 ± 3 | 0.140 ± 0.043 | 0.699 ± 0.034 | 0.975 ± 0.120 | 7.14 | 3329 |
| PP55 | 389 ± 2 | 0.114 ± 0.029 | 0.670 ± 0.031 | 0.933 ± 0.085 | 8.77 | 3412 |
| G′ (Pa) | Melt Elasticity (Pa·s) | Melt-Elasticity Factor | G″ (Pa) | Melt Dissipation (Pa·s) | Melt Dissipation Factor | Loss Factor tan δ | |
|---|---|---|---|---|---|---|---|
| PS | 1.5 × 104 | 250 | - | 1.6 × 104 | 266 | - | |
| PP0.9 | 5.1 × 104 | 850 | - | 3.2 × 104 | 530 | - | 0.62 |
| PP3 | 2.0 × 104 | 333 | - | 2.3 × 104 | 380 | - | 1.15 |
| PP20 | 0.8 × 104 | 133 | - | 1.2 × 104 | 200 | - | 1.50 |
| PP55 | 0.5 × 104 | 83 | - | 0.9 × 104 | 150 | - | 1.80 |
| B0.9 | 1.4 × 104 | 233 | 3.4 | 1.8 × 104 | 300 | 0.56 | 1.29 |
| B3 | 0.9 × 104 | 150 | 1.3 | 1.4 × 104 | 233 | 0.44 | 1.56 |
| B20 | 0.8 × 104 | 133 | 0.53 | 1.3 × 104 | 216 | 0.40 | 1.63 |
| B55 | 0.7 × 104 | 116 | 0.33 | 1.2 × 104 | 200 | 0.36 | 1.72 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hammani, S.; Moulai-Mostefa, N.; Samyn, P.; Bechelany, M.; Dufresne, A.; Barhoum, A. Morphology, Rheology and Crystallization in Relation to the Viscosity Ratio of Polystyrene/Polypropylene Polymer Blends. Materials 2020, 13, 926. https://doi.org/10.3390/ma13040926
Hammani S, Moulai-Mostefa N, Samyn P, Bechelany M, Dufresne A, Barhoum A. Morphology, Rheology and Crystallization in Relation to the Viscosity Ratio of Polystyrene/Polypropylene Polymer Blends. Materials. 2020; 13(4):926. https://doi.org/10.3390/ma13040926
Chicago/Turabian StyleHammani, Salim, Nadji Moulai-Mostefa, Pieter Samyn, Mikhael Bechelany, Alain Dufresne, and Ahmed Barhoum. 2020. "Morphology, Rheology and Crystallization in Relation to the Viscosity Ratio of Polystyrene/Polypropylene Polymer Blends" Materials 13, no. 4: 926. https://doi.org/10.3390/ma13040926
APA StyleHammani, S., Moulai-Mostefa, N., Samyn, P., Bechelany, M., Dufresne, A., & Barhoum, A. (2020). Morphology, Rheology and Crystallization in Relation to the Viscosity Ratio of Polystyrene/Polypropylene Polymer Blends. Materials, 13(4), 926. https://doi.org/10.3390/ma13040926