Anti-biofilm Fe3O4@C18-[1,3,4]thiadiazolo[3,2-a]pyrimidin-4-ium-2-thiolate Derivative Core-shell Nanocoatings

 ,
,  ,
,  ,
,  ,
, 


Abstract
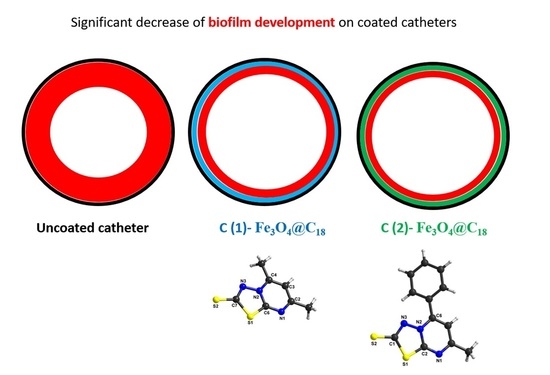
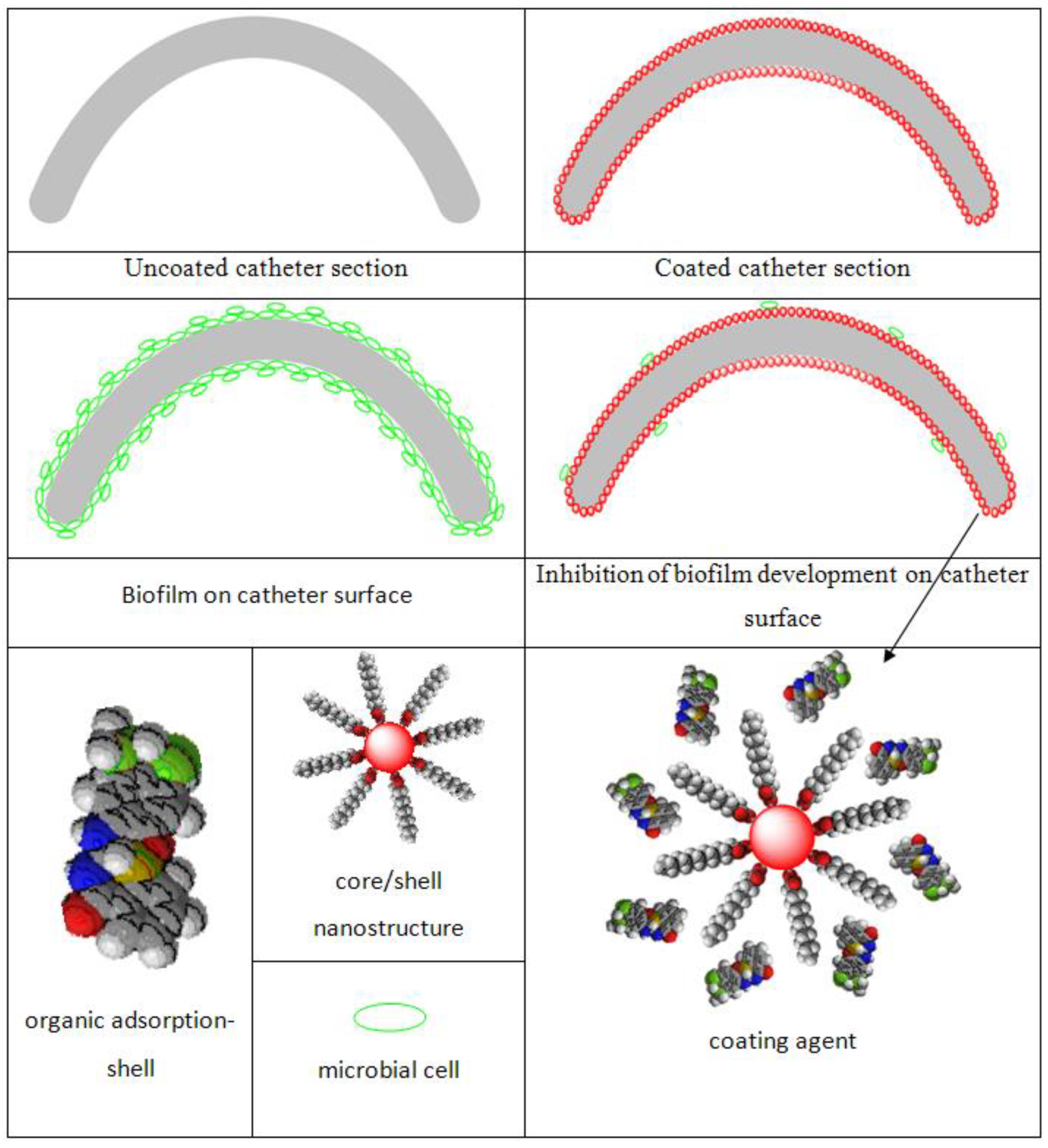
1. Introduction
2. Experimental
2.1. General Information
2.2. Synthesis and Spectral Data for [1,3,4]thiadiazolo[3,2-a]pyrimidine Derivatives (Adsorption-Shell)
2.3. X-ray Crystallography
2.4. Synthesis and Characterization of Core@Shell Nanostructure
2.5. The Core@Shell@Adsorption-Shell Nanostructure Deposition on Catheter Samples
2.6. Screening of the Antibiofilm Activity
2.7. Cytotoxicity Assay
3. Results and Discussion
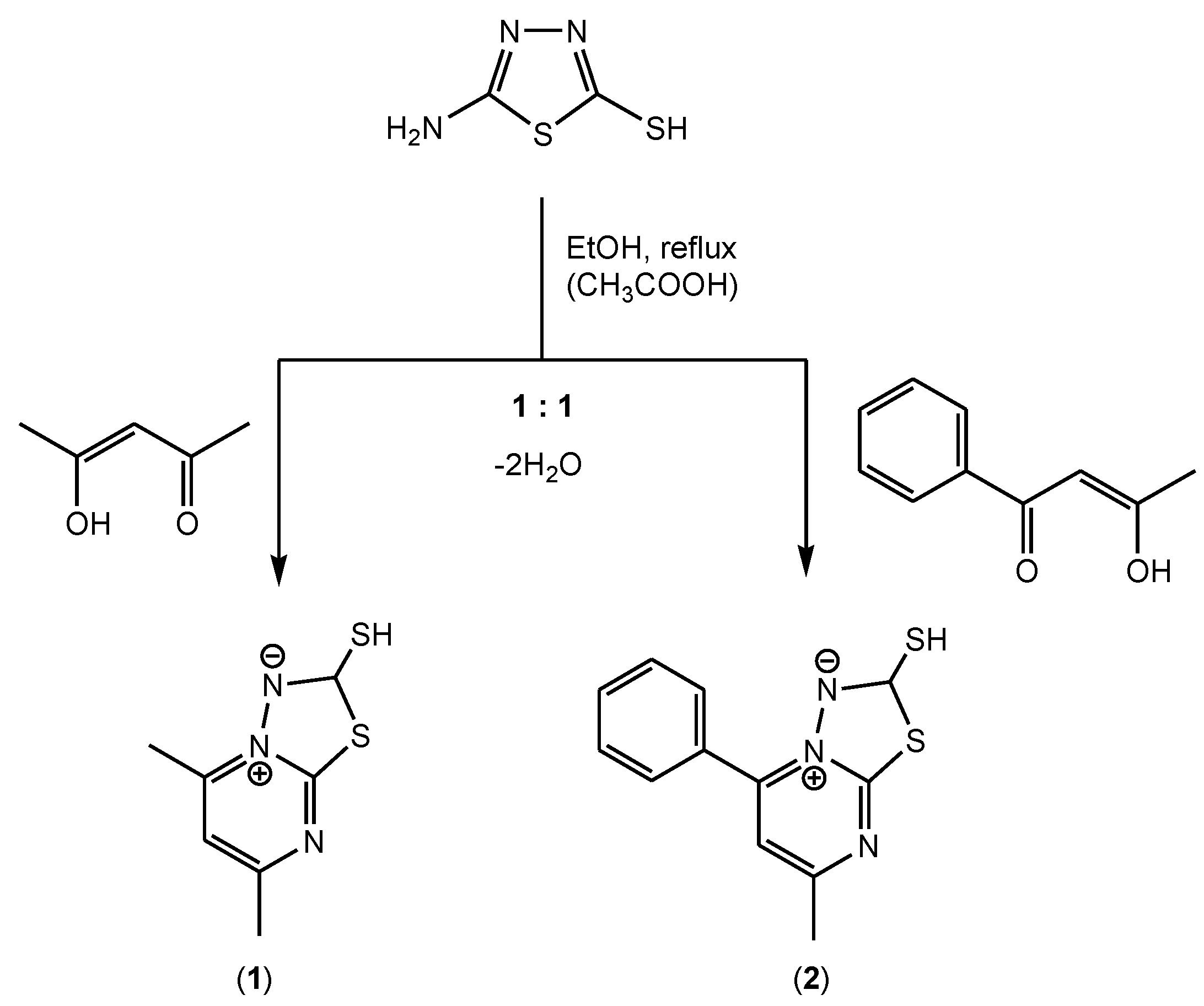
3.1. Synthesis and Characterization of [1,3,4]thiadiazolo[3,2-a]pyrimidine Derivatives
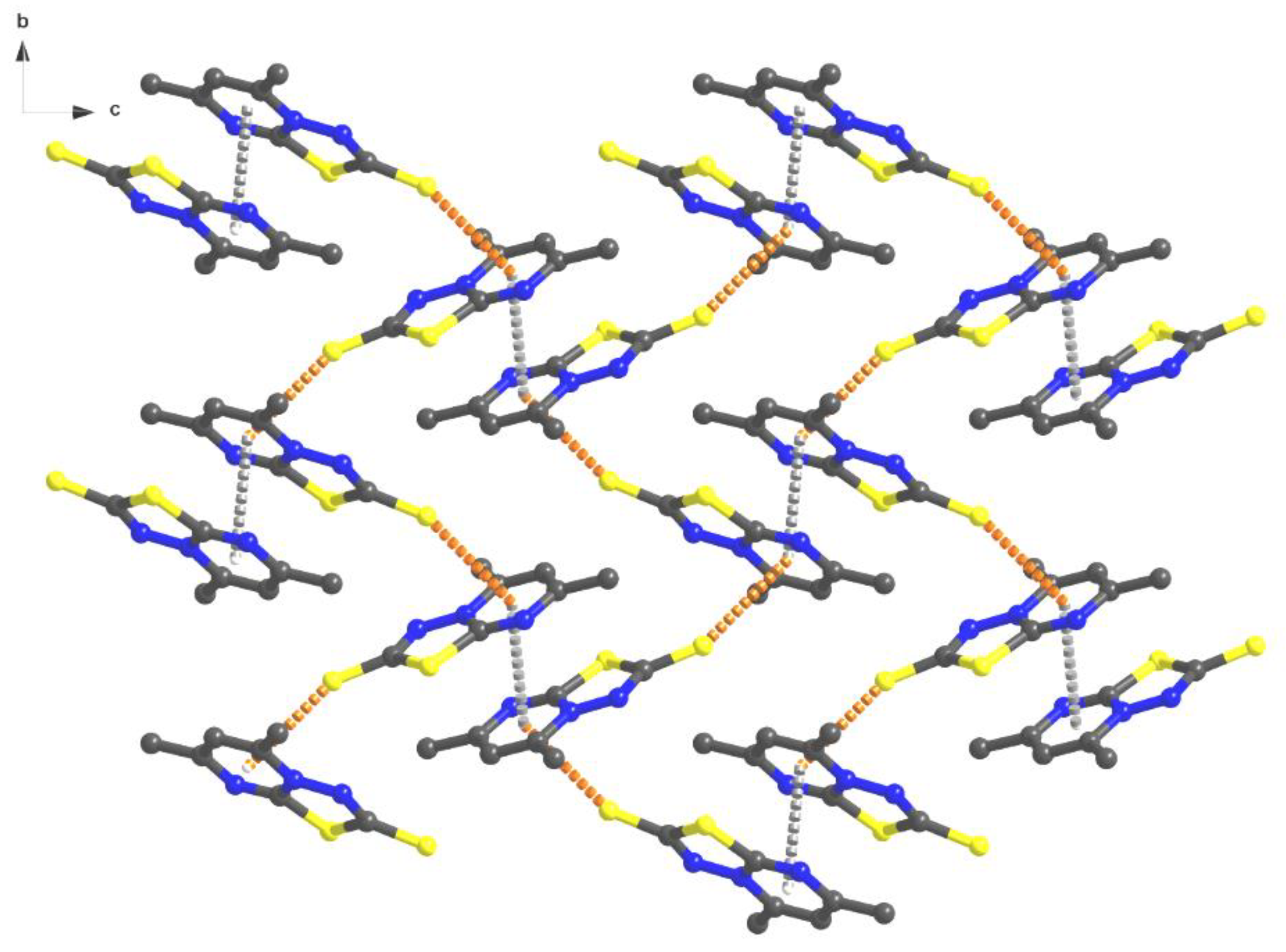
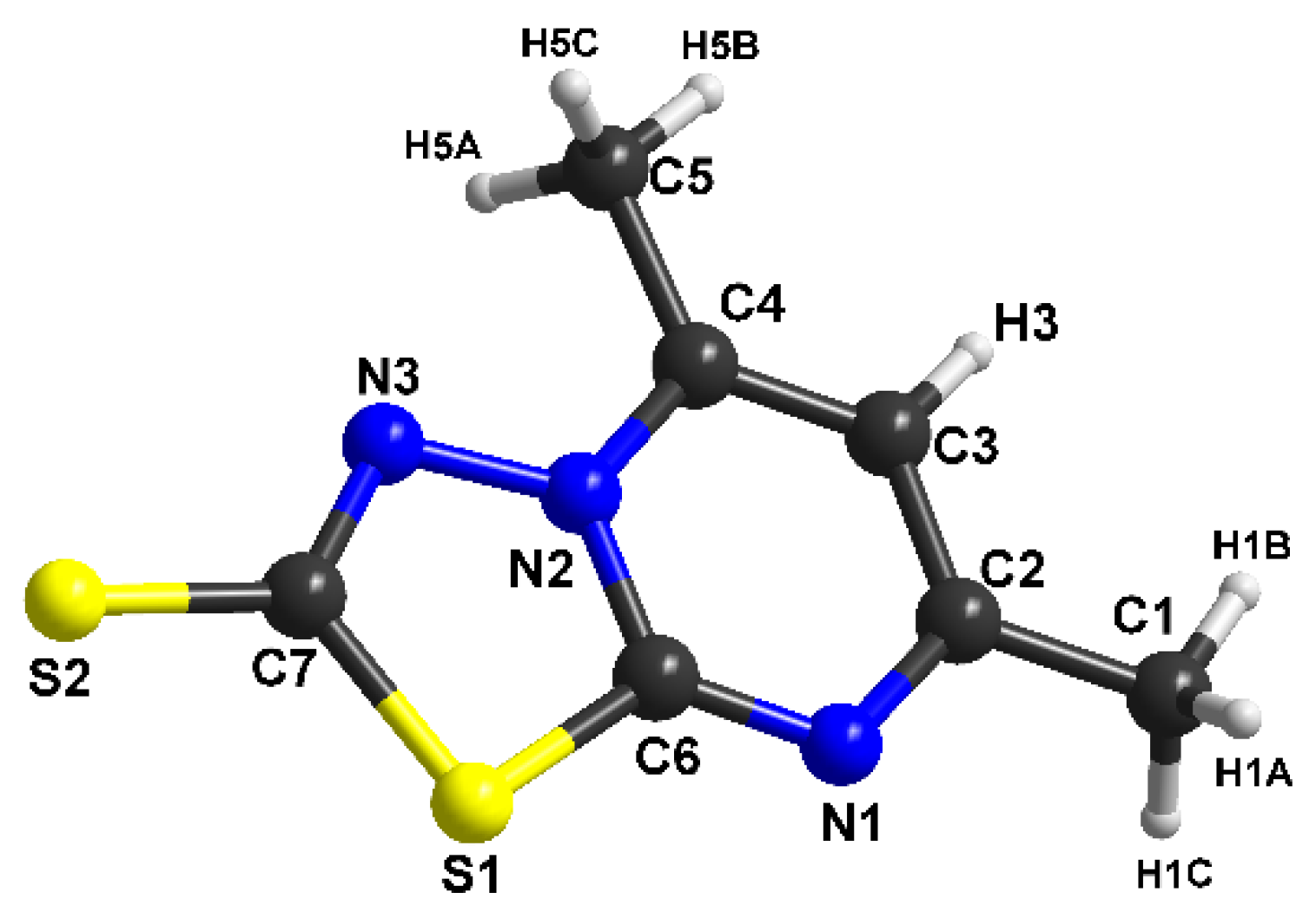
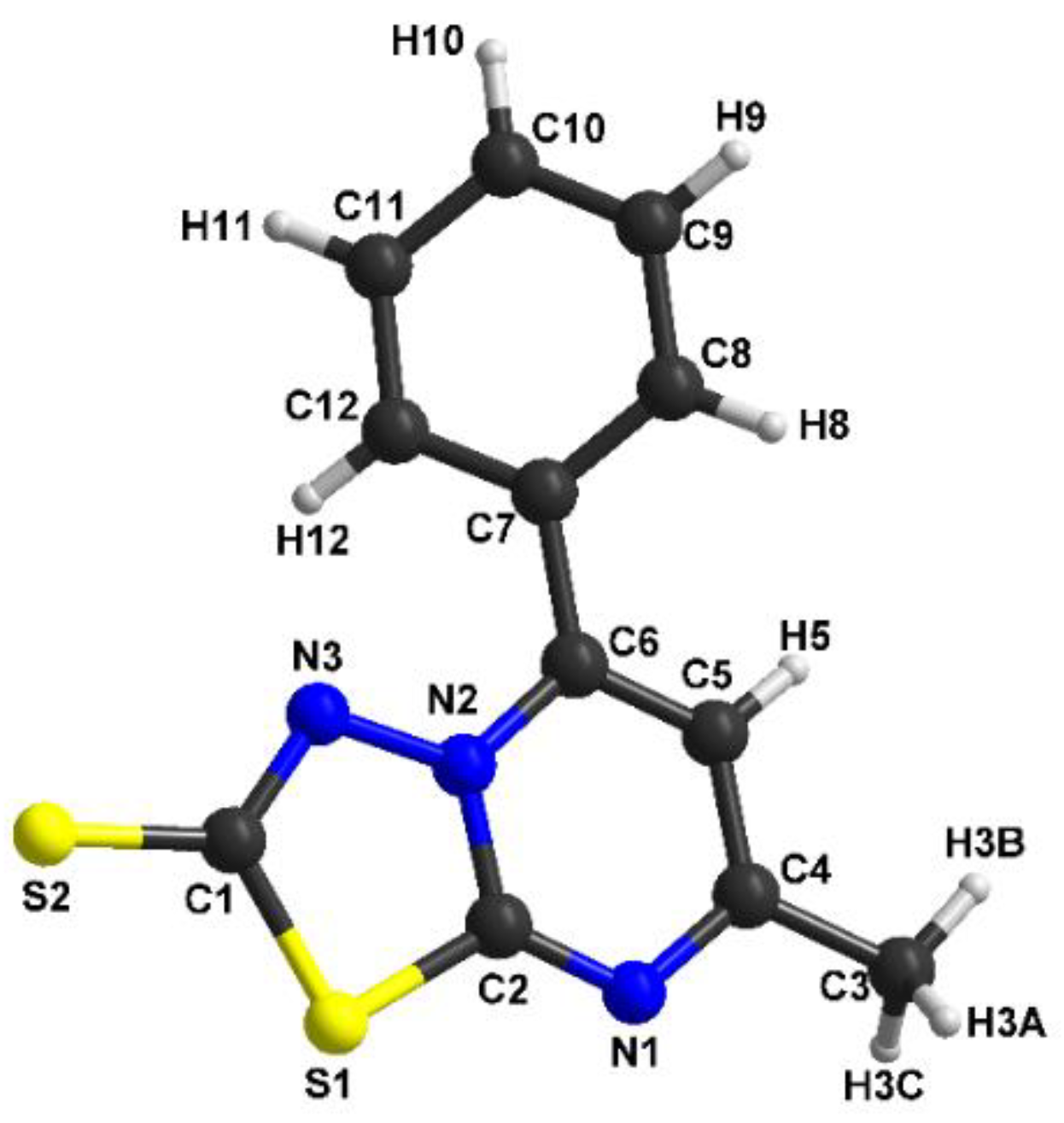
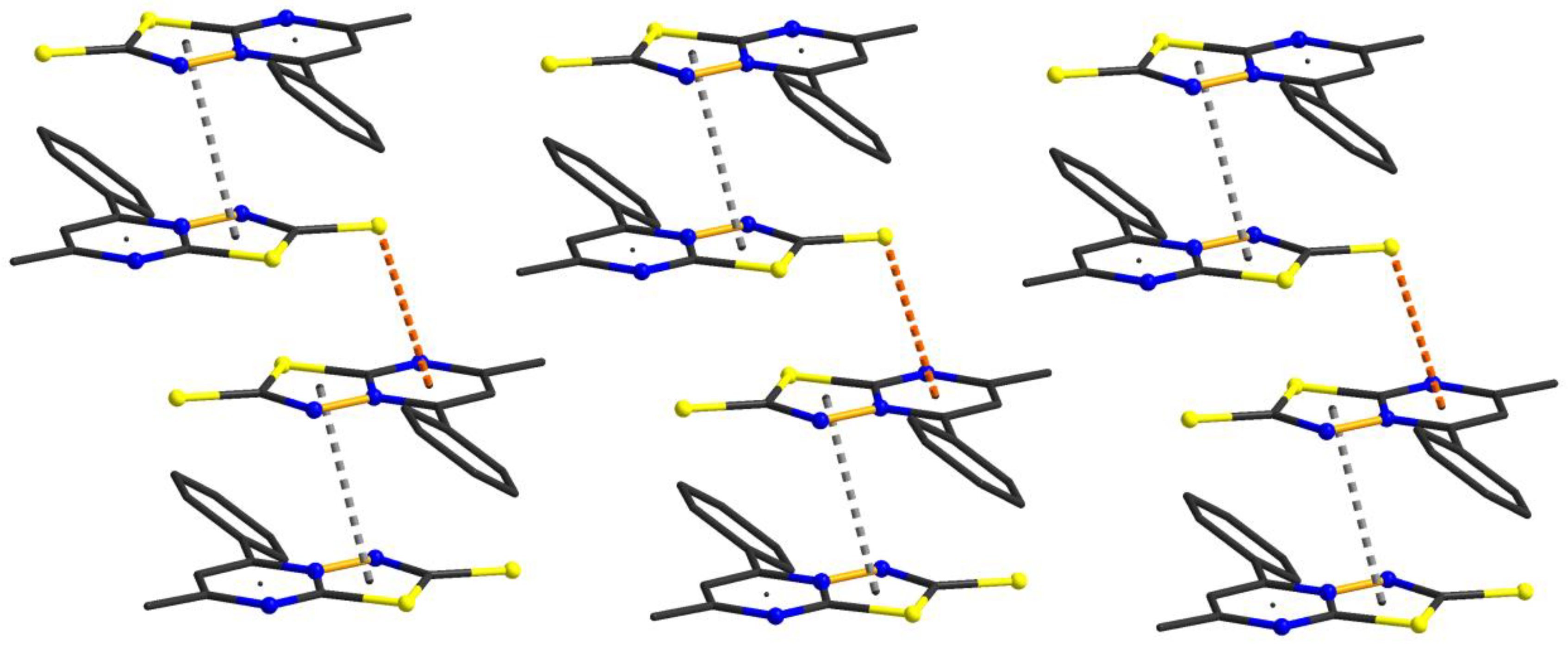
3.2. Structural Description of [1,3,4]thiadiazolo[3,2-a]pyrimidine Derivatives
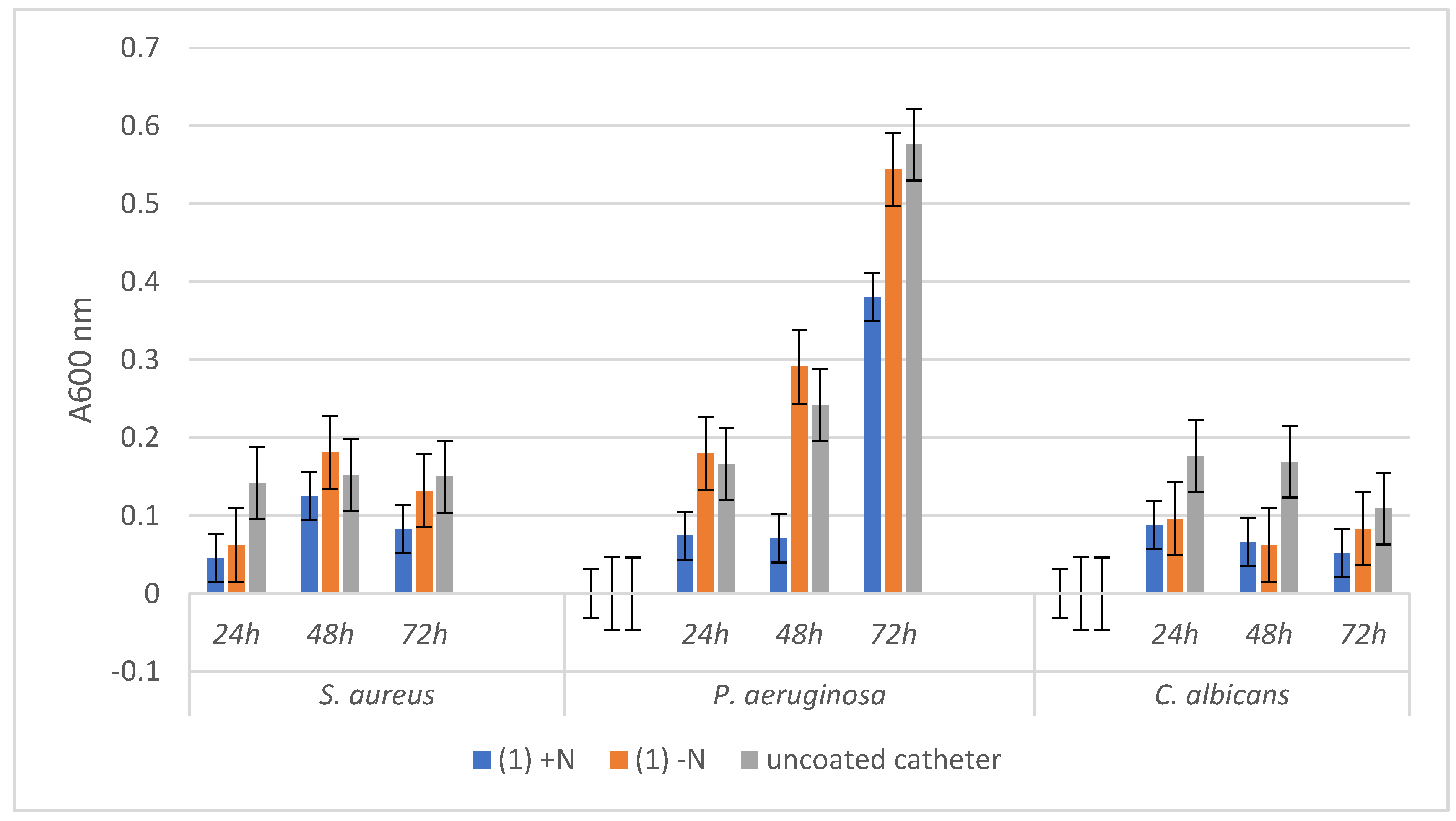
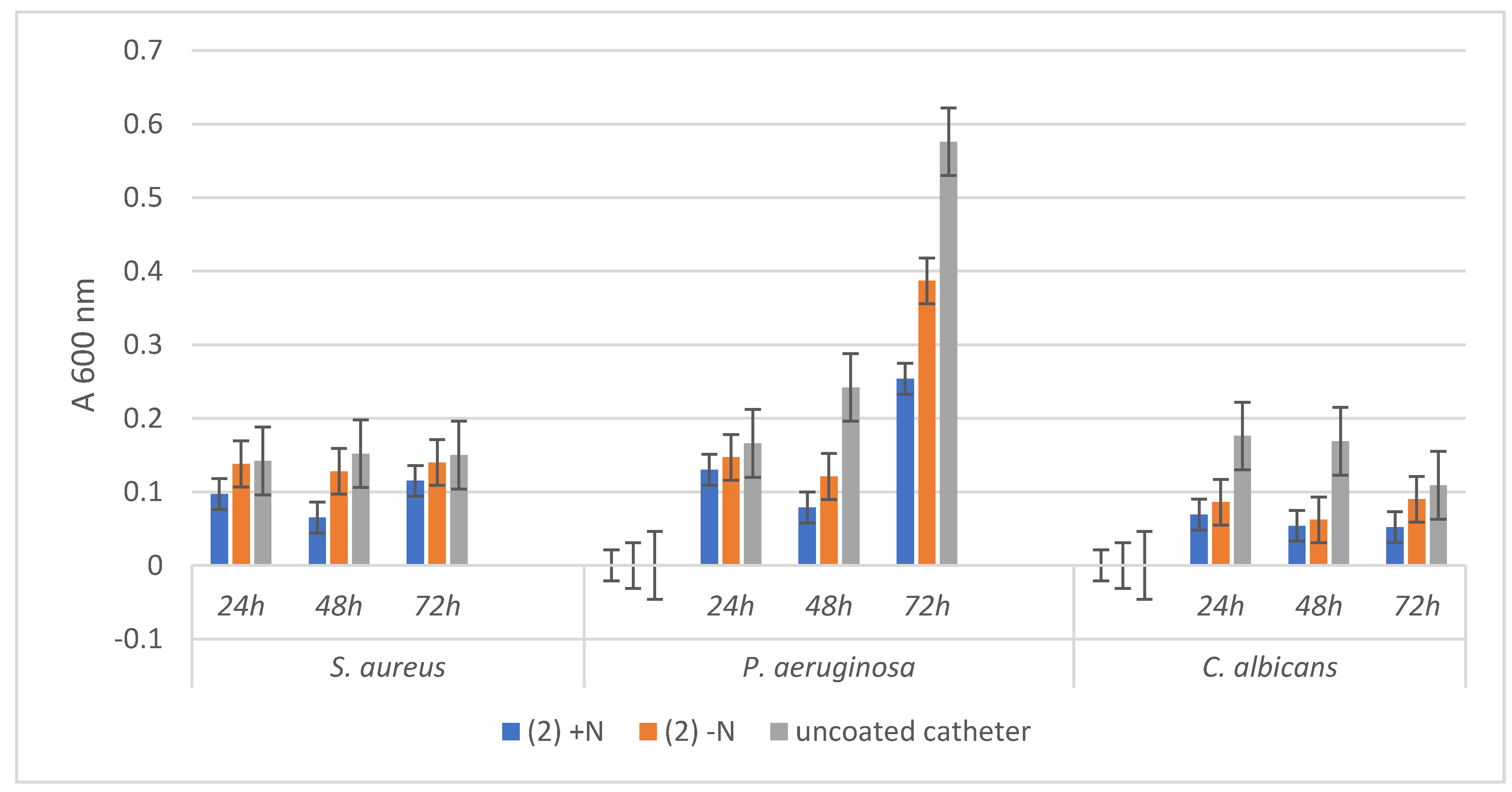
3.3. Antimicrobial Activity
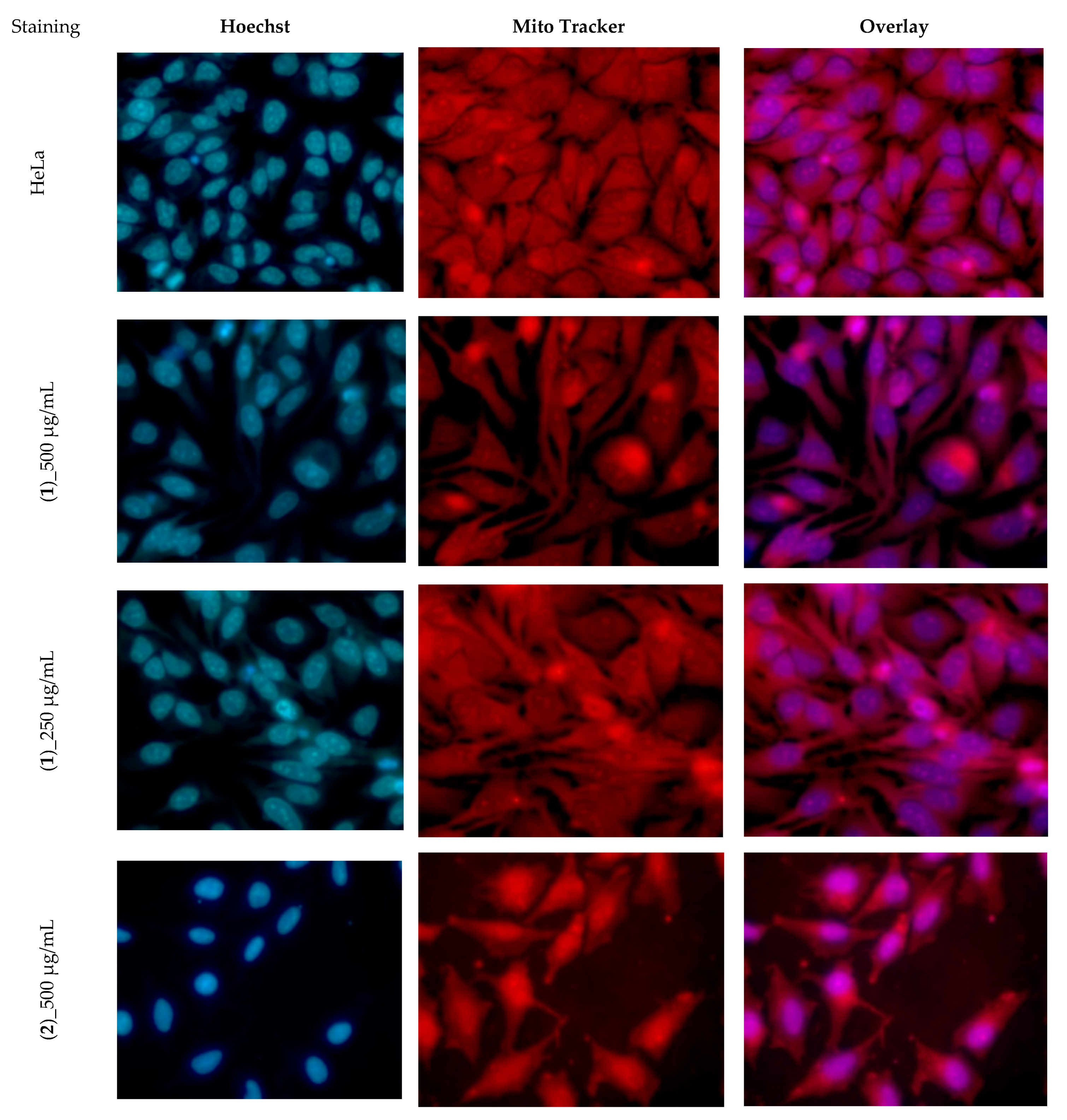
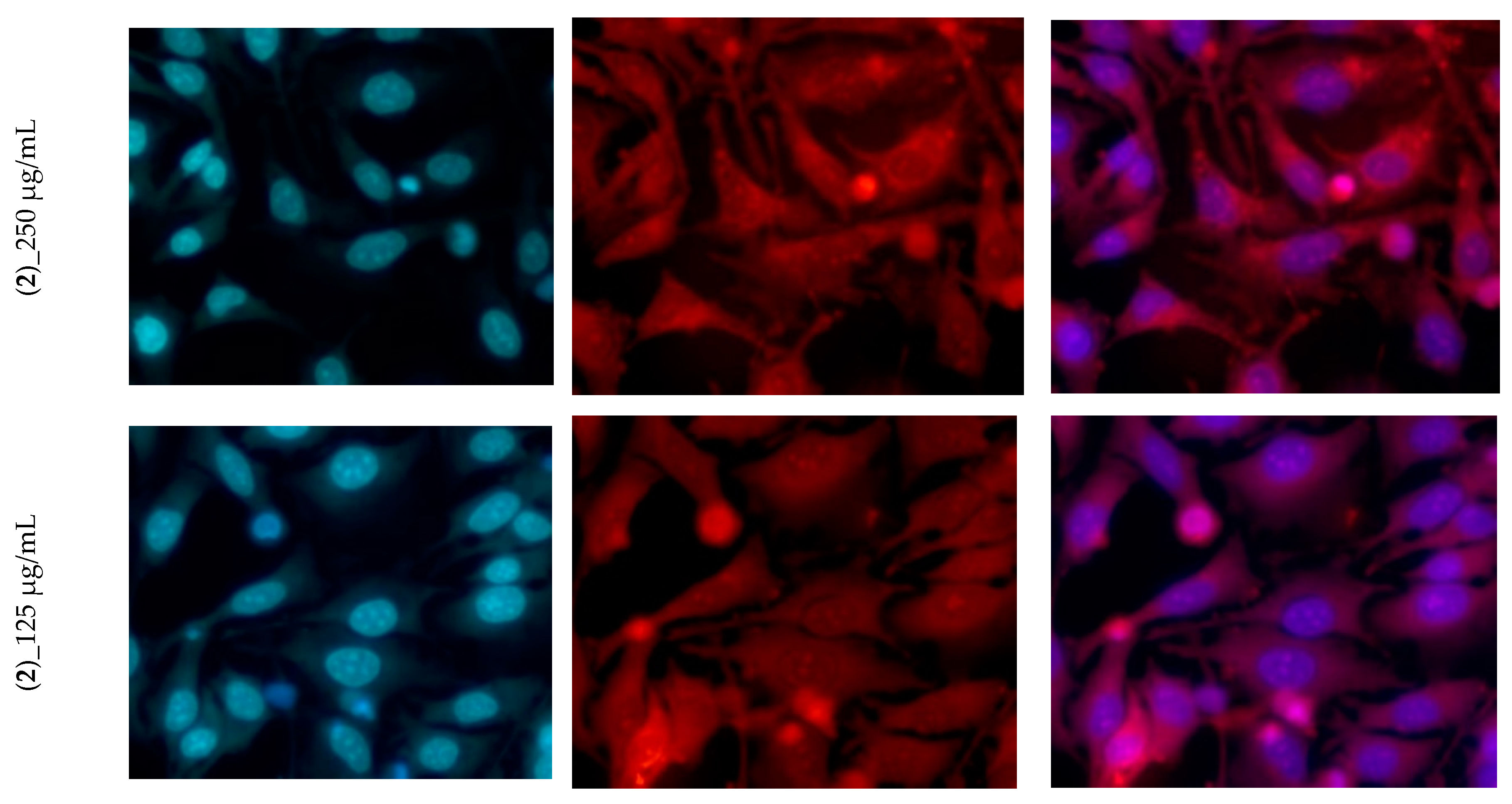
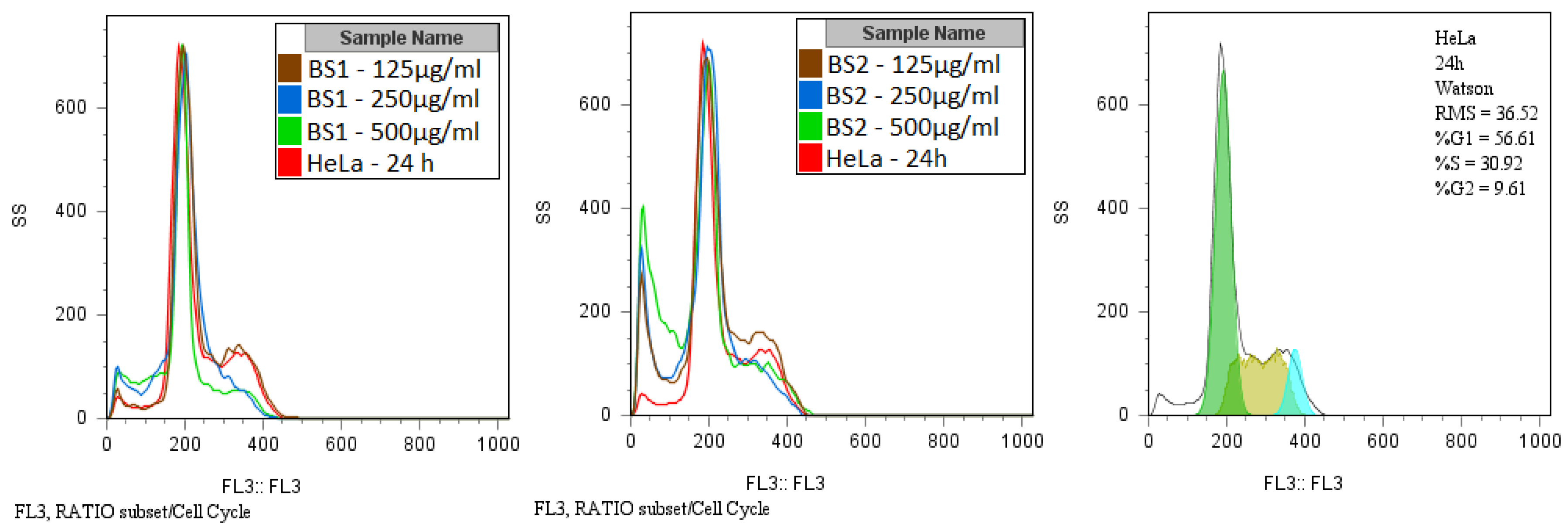
3.4. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Datta, P.; Gupta, V. Next-generation strategy for treating drug resistant bacteria: Antibiotic hybrids. Indian, J. Med. Res. 2019, 149, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Podgoreanu, P.; Negrea, S.M.; Buia, R.; Delcaru, C.; Trusca, S.B.; Lazar, V.; Chifiriuc, M.C. Alternative strategies for fighting multidrug resistant bacterial infections. Biointerface Res. Appl. Chem. 2019, 9, 3834–3841. [Google Scholar] [CrossRef]
- Ducu, R.; Gheorghe, I.; Chifiriuc, M.C.; Mihăescu, G.; Sârbu, I. Prevalence of vancomycin resistance phenotypes among Enterococcus species isolated from clinical samples in a Romanian hospital. Biointerface Res. Appl. Chem. 2019, 9, 4699–4704. [Google Scholar] [CrossRef]
- Vrancianu, C.O.; Popa, L.I.; Bleotu, C.; Chifiriuc, M.C. Targeting plasmids to limit acquisition and transmission of antimicrobial resistance. Front. Microbiol. 2020, 11, 761. [Google Scholar] [CrossRef] [PubMed]
- Bahramian, G.; Golestan, L.; Khosravi-Darani, K. Antimicrobial and antioxidant effect of nanoliposomes containing zataria multiflora boiss essential oil on the rainbow trout fillets during refrigeration. Biointerface Res. Appl. Chem. 2018, 8, 3505–3513. [Google Scholar]
- Dos Santos, L.D.R.; Dos Santos, A.E.S.; Cerávolo, I.P.; Figueiredo, F.J.B.; Dias-Souza, M.V. Antibiofilm activity of black tea leaf extract, its cytotoxicity and interference on the activity of antimicrobial drugs. Biointerface Res. Appl. Chem. 2018, 8, 3565–3569. [Google Scholar]
- Kasimanickam, R.; Ranjan, A.; Asokan, G.V.; Kasimanickam, V.R.; Kastelic, J. Prevention and treatment of biofilms by hybrid- and nanotechnologies. Int. J. Nanomed. 2013, 8, 2809–2819. [Google Scholar] [CrossRef] [PubMed]
- Domalaon, R.; Idowu, T.; Zhanel, G.G.; Schweizer, F. Antibiotic hybrids: The next generation of agents and adjuvants against gram-negative pathogens? Clin. Microbiol. Rev. 2018, 31, e00077-17. [Google Scholar] [CrossRef]
- Khanmohammadi, S.; Karimian, R.; Hajibonabi, F.; Mostafidi, E.; Tanomand, A.; Edjlali, L.; Azami, Z.K.; Mehrabani, M.G.; Kafil, H.S. Polythiophene/TiO2 and polythiophene/ZrO2 nanocomposites: Physical and antimicrobial properties against common infections. Biointerface Res. Appl. Chem. 2018, 8, 3457–3462. [Google Scholar]
- Theuretzbacher, U. Dual-mechanism antibiotics. Nat. Microbiol. 2020, 5, 984–985. [Google Scholar] [CrossRef]
- Del Olmo, J.A.; Ruiz-Rubio, L.; Pérez-Álvarez, L.; Saez-Martinez, V.; Vilas-Vilela, J.L. Antibacterial coatings for improving the performance of biomaterials. Coatings 2020, 10, 139. [Google Scholar] [CrossRef]
- Pircalabioru, G.G.; Chifiriuc, M.C. Nanoparticulate drug-delivery systems for fighting microbial biofilms: From bench to bedside. Futur. Microbiol. 2020, 15, 679–698. [Google Scholar] [CrossRef]
- Toledo, L.D.A.S.D.; Rosseto, H.C.; Bruschi, M.L. Iron oxide magnetic nanoparticles as antimicrobials for therapeutics. Pharm. Dev. Technol. 2017, 23, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, G.R.; López-Abarrategui, C.; Gómez, I.D.L.S.; Dias, S.C.; Otero-González, A.J.; Franco, O.L. Antimicrobial magnetic nanoparticles based-therapies for controlling infectious diseases. Int. J. Pharm. 2019, 555, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Gabrielyan, L.; Hovhannisyan, A.; Gevorgyan, V.; Ananyan, M.; Trchounian, A. Antibacterial effects of iron oxide (Fe3O4) nanoparticles: Distinguishing concentration-dependent effects with different bacterial cells growth and membrane-associated mechanisms. Appl. Microbiol. Biotechnol. 2019, 103, 2773–2782. [Google Scholar] [CrossRef] [PubMed]
- Sandhya, J.; Kalaiselvam, S. Biogenic synthesis of magnetic iron oxide nanoparticles using inedible borassus flabellifer seed coat: Characterization, antimicrobial, antioxidant activity and in vitro cytotoxicity analysis. Mater. Res. Express 2020, 7, 015045. [Google Scholar] [CrossRef]
- Limban, C.; Missir, A.V.; Caproiu, M.T.; Grumezescu, A.; Chifiriuc, M.-C.; Bleotu, C.; Marutescu, L.G.; Papacocea, M.T.; Nuță, D.C. Novel hybrid formulations based on thiourea derivatives and Core@Shell Fe3O4@C18 nanostructures for the development of antifungal strategies. Nanomaterials 2018, 8, 47. [Google Scholar] [CrossRef]
- Xiong, X.; Huang, Y.; Lin, C.; Liu, X.Y.; Lin, Y. Recent advances in nanoparticulate biomimetic catalysts for combating bacteria and biofilms. Nanoscale 2019, 11, 22206–22215. [Google Scholar] [CrossRef]
- Shi, Z.; Jin, L.; He, C.; Li, Y.; Jiang, C.; Wang, H.; Zhang, J.; Wang, J.; He, C.; Zhao, C. Hemocompatible magnetic particles with broad-spectrum bacteria capture capability for blood purification. J. Colloid Interface Sci. 2020, 576, 1–9. [Google Scholar] [CrossRef]
- Armijo, L.M.; Wawrzyniec, S.J.; Kopciuch, M.; Brandt, Y.I.; Rivera, A.C.; Withers, N.J.; Cook, N.C.; Huber, D.L.; Monson, T.C.; Smyth, H.D.; et al. Antibacterial activity of iron oxide, iron nitride, and tobramycin conjugated nanoparticles against Pseudomonas aeruginosa biofilms. J. Nanobiotechnology 2020, 18, 1–27. [Google Scholar] [CrossRef]
- Andersen, M.J.; Flores-Mireles, A.L. Urinary catheter coating modifications: The Race against catheter-associated infections. Coatings 2019, 10, 23. [Google Scholar] [CrossRef]
- Patrinoiu, G.; Calderon-Moreno, J.M.; Chifiriuc, M.C.; Saviuc, C.; Birjega, R.; Carp, O. Tunable ZnO spheres with high anti-biofilm and antibacterial activity via a simple green hydrothermal route. J. Colloid Interf. Sci. 2016, 462, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Rumyantceva, V.; Rumyantceva, V.; Koshel, E.I.; Vinogradov, V.V. Biocide-conjugated magnetite nanoparticles as an advanced platform for biofilm treatment. Ther. Deliv. 2019, 10, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Bilcu, M.; Grumezescu, A.; Grumezescu, A.; Popescu, R.C.; Mogosanu, G.D.; Hristu, R.; Stanciu, S.G.; Mihăilescu, D.F.; Lazar, V.; Bezirtzoglou, E.; et al. Efficiency of vanilla, patchouli and ylang ylang essential oils stabilized by iron oxide@C14 nanostructures against bacterial adherence and biofilms formed by Staphylococcus aureus and Klebsiella pneumoniae clinical strains. Molecules 2014, 19, 17943–17956. [Google Scholar] [CrossRef]
- Hu, Y.; Li, C.-Y.; Wang, X.-M.; Yang, Y.-H.; Zhu, H.-L. 1,3,4-Thiadiazole: Synthesis, reactions, and applications in medicinal, agricultural, and materials chemistry. Chem. Rev. 2014, 114, 5572–5610. [Google Scholar] [CrossRef]
- Kaur, G.; Singh, R. Thiadiazole analogs as potential pharmacological agents: A brief review. Int. J. Pharm. Pharm. Sci. 2014, 6, 35–46. [Google Scholar]
- Haider, S.; Alam, M.S.; Hamid, H. 1,3,4-Thiadiazoles: A potent multi targeted pharmacological scaffold. Eur. J. Med. Chem. 2015, 92, 156–177. [Google Scholar] [CrossRef]
- Mehta, D.; Taya, P.; Neetu. A review on the various biological activities of thiadiazole. Int. J. Pharm. Pharm. Sci. 2015, 7, 39–47. [Google Scholar]
- Husain, A.; Rashid, M.; Shaharyar, M.; Siddiqui, A.A.; Mishra, R. Benzimidazole clubbed with triazolo-thiadiazoles and triazolo-thiadiazines: New anticancer agents. Eur. J. Med. Chem. 2013, 62, 785–798. [Google Scholar] [CrossRef]
- Ramaprasad, G.C.; Kalluraya, B.; Kumar, B.S. Microwave-assisted synthesis of triazolothiadiazole analogs as anticancer agents. Med. Chem. Res. 2014, 23, 3644–3651. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, J.; Zhang, B.; Lu, T.; Chen, Y. Discovery of [1,2,4] triazolo [3,4-b] [1,3,4] thiadiazole derivatives as novel, potent and selective c-Met kinase inhibitors: Synthesis, SAR study, and biological activity. Eur. J. Med. Chem. 2018, 150, 809–816. [Google Scholar] [CrossRef]
- Sahu, J.K.; Ganguly, S.; Yasir, M. Synthesis, SAR and molecular docking studies of certain new derivatives of 1,2,4-Triazolo [3,4-b] [1,3,4] thiadiazole as potent antimicrobial agents. Anti-Infect. Agents 2018, 16, 40–48. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Akilandeswari, G.; Anusha, R.; Al-Shaalan, N.H.; Alkmali, O.M.; El-Emam, A.A.; Percino, J.M.; Thamotharan, S. Insights into the nature of weak noncovalent interactions in 3-(4-fluorophenyl)-6-(2-fluorophenyl)-1,2,4-triazolo[3,4-b] [1,3,4] thiadiazole, a potential bioactive agent: X-ray, QTAIM and molecular docking analysis. J. Mol. Struct. 2019, 1183, 331–341. [Google Scholar] [CrossRef]
- Settypalli, T.; Rao, C.V.; Kerru, N.; Nallapaneni, H.K.; Chintha, V.R.; Daggupati, T.; Yeguvapalli, S.; Wudayagiri, R. Design, synthesis, neuroprotective and antibacterial activities of 1,2,4-triazolo [3,4-b] 1,3,4-thiadiazole linked thieno [2,3-d] pyrimidine derivatives and in silico docking studies. ChemistySelect 2019, 4, 1627–1634. [Google Scholar] [CrossRef]
- Kamal, A.; Ponnampalli, S.; Vishnuvardhan, M.V.P.S.; Rao, M.P.N.; Mullagiri, K.; Nayak, V.L.; Chandrakant, B. Synthesis of imidazothiadiazole–benzimidazole conjugates as mitochondrial apoptosis inducers. MedChemComm 2014, 5, 1644–1650. [Google Scholar] [CrossRef]
- Ramprasad, J.; Nayak, N.; Dalimba, U.; Yogeeswari, P.; Sriram, D.; Peethambar, S.; Achur, R.; Dalimba, U. Synthesis and biological evaluation of new imidazo [2,1-b] [1,3,4] thiadiazole-benzimidazole derivatives. Eur. J. Med. Chem. 2015, 95, 49–63. [Google Scholar] [CrossRef]
- Cristina, A.; Leonte, D.; Vlase, L.; Bencze, L.C.; Imre, S.; Marc, G.; Apan, B.; Mogosan, C.; Zaharia, V. Heterocycles 48. Synthesis, characterization and biological evaluation of imidazo [2,1-b] [1,3,4] thiadiazole derivatives as anti-inflammatory agents. Molecules 2018, 23, 2425. [Google Scholar] [CrossRef] [PubMed]
- Tahtaci, H.; Karacık, H.; Ece, A.; Er, M.; Şeker, M.G. Design, synthesis, SAR and molecular modeling studies of novel imidazo [2,1-b] [1,3,4] thiadiazole derivatives as highly potent antimicrobial agents. Mol. Inform. 2017, 37, 1700083. [Google Scholar] [CrossRef] [PubMed]
- Cascioferro, S.; Parrino, B.; Petri, G.L.; Cusimano, M.G.; Schillaci, D.; Di Sarno, V.; Musella, S.; Giovannetti, E.; Cirrincione, G.; Diana, P. 2,6-Disubstituted imidazo[2,1-b] [1,3,4] thiadiazole derivatives as potent staphylococcal biofilm inhibitors. Eur. J. Med. Chem. 2019, 167, 200–210. [Google Scholar] [CrossRef]
- Coburn, R.A.; Glennon, R.A. Mesoionic purinone analogs IV: Synthesis and In Vitro antibacterial properties of mesoionic thiazolo [3,2-a] pyrimidin-5,7-diones and mesoionic 1,3,4-thiadiazolo[3,2-a]pyrimidin-5,7-diones. J. Pharm. Sci. 1973, 62, 1785–1789. [Google Scholar] [CrossRef]
- Coburn, R.A.; Glennon, R.A.; Chmielewicz, Z.F. Mesoionic purinone analogs. 7. In vitro antibacterial activity of mesoionic 1,3,4-thiadiazolo [3,2-a] pyrimidine-5,7-diones. J. Med. Chem. 1974, 17, 1025–1027. [Google Scholar] [CrossRef] [PubMed]
- Mahran, M.A.; El-Obaid, A.M.A.; Badria, F.A. Heterocyclic systems containing pyrimidine nucleus as potential antimicrobial and antitumor agents. Alexandria, J. Pharm. Sci. 1998, 12, 39–44. [Google Scholar]
- Deibel, M.R.; Bodnar, A.L.; Yem, A.W.; Wolfe, C.L.; Heckaman, C.L.; Bohanon, M.J.; Mathews, W.R.; Sweeney, M.T.; Zurenko, G.E.; Marotti, K.R.; et al. Immobilization of a novel antibacterial agent on solid phase and subsequent isolation of EF-Tu. Bioconjugate Chem. 2004, 15, 333–343. [Google Scholar] [CrossRef] [PubMed]
- El-Gohary, N.S.; Shaaban, M.I. Synthesis, antimicrobial, antiquorum-sensing, antitumor and cytotoxic activities of new series of fused [1,3,4] thiadiazoles. Eur. J. Med. Chem. 2013, 63, 185–195. [Google Scholar] [CrossRef]
- El-Gohary, N.S.; Shaaban, M.I. Antimicrobial and antiquorum-sensing studies. Part 2: Synthesis, antimicrobial, antiquorum-sensing and cytotoxic activities of new series of fused [1,3,4] thiadiazole and [1,3] benzothiazole derivatives. Med. Chem. Res. 2013, 23, 287–299. [Google Scholar] [CrossRef]
- El-Gohary, N.S.; Shaaban, M.I. Antimicrobial and antiquorum-sensing studies. Part 3: Synthesis and biological evaluation of new series of [1,3,4] thiadiazoles and fused [1,3,4] thiadiazoles. Arch. der Pharm. 2015, 348, 283–297. [Google Scholar] [CrossRef]
- El-Ashmawy, M.; El-Sherbeny, M.; El-Sayed, N.S. Synthesis, in vitro antitumor activity and DNA-binding affinity of novel thiadiazolopyrimidine and thiadiazoloquinazoline derivatives. Mansoura, J. Pharm. Sci. 2010, 26, 60–68. [Google Scholar]
- El-Sayed, N.S.; El-Bendary, E.R.; El-Ashry, S.M.; El-Kerdawy, M.M. Synthesis and antitumor activity of new sulfonamide derivatives of thiadiazolo [3,2-a] pyrimidines. Eur. J. Med. Chem. 2011, 46, 3714–3720. [Google Scholar] [CrossRef]
- Taher, A.T.; Georgey, H.H.; El-Subbagh, H.I. Novel 1,3,4-heterodiazole analogues: Synthesis and in-vitro antitumor activity. Eur. J. Med. Chem. 2012, 47, 445–451. [Google Scholar] [CrossRef]
- Hamama, W.S.; Gouda, M.A.; Badr, M.H.; Zoorob, H.H. Synthesis of some new fused and binary 1,3,4-thiadiazoles as potential antitumor and antioxidant agents. J. Heterocycl. Chem. 2013, 50, 787–794. [Google Scholar] [CrossRef]
- Rahman, D.E.A.; Mohamed, K.O. Synthesis of novel 1,3,4-thiadiazole analogues with expected anticancer activity. Der Pharma Chemica 2014, 6, 323–335. [Google Scholar]
- Baburajeev, C.P.; Mohan, C.D.; Rangappa, S.; Mason, D.J.; Fuchs, J.E.; Bender, A.; Barash, U.; Vlodavsky, I.; Basappa; Rangappa, K.S. Identification of novel class of triazolo-thiadiazoles as potent inhibitors of human heparanase and their anticancer activity. BMC Cancer 2017, 17, 235. [Google Scholar] [CrossRef] [PubMed]
- Chifiriuc, M.C.; Grumezescu, A.M.; Andronescu, E.; Ficai, A.; Cotar, A.I.; Grumezescu, V.; Bezirtzoglou, E.; Lazar, V.; Radulescu, R. Water dispersible magnetite nanoparticles influence the efficacy of antibiotics against planktonic and biofilm embedded Enterococcus faecalis cells. Anaerobe 2013, 22, 14–19. [Google Scholar] [CrossRef]
- Grumezescu, A.; Gestal, M.C.; Holban, A.; Grumezescu, V.; Vasile, B.S.; Mogoantă, L.; Iordache, F.; Bleotu, C.; Mogosanu, G.D. Biocompatible Fe3O4 increases the efficacy of amoxicillin delivery against gram-positive and gram-negative bacteria. Molecules 2014, 19, 5013–5027. [Google Scholar] [CrossRef]
- Abdel-Wahab, B.F.; Mohamed, H.A. Synthetic profiles to different thiadiazolopyrimidines. Phosphorus, Sulfur, Silicon Relat. Elem. 2014, 189, 1780–1793. [Google Scholar] [CrossRef]
- Olar, R.; Badea, M.; Marinescu, D.; Lazăr, V.; Chifiriuc, C.; Chifiriuc, M.-C. New complexes of Ni (II) and Cu (II) with Schiff bases functionalised with 1,3,4-thiadiazole: Spectral, magnetic, biological and thermal characterisation. J. Therm. Anal. Calorim. 2009, 97, 721–727. [Google Scholar] [CrossRef]
- Stoe & Cie, X-Area. 2002. Available online: https://www.stoe.com/product/software-x-area/ (accessed on 14 October 2020).
- Sheldrick, G.M.S. A Program. for the Solution of Crystal; Structures; University of Gottingen: Gottingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M.S. A Program. for Crystal Structures Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Voicu, G.; Andronescu, E.; Grumezescu, A.; Huang, K.-S.; Ficai, A.; Yang, C.-H.; Bleotu, C.; Chifiriuc, M.-C. Antitumor activity of magnetite nanoparticles: Influence of hydrocarbonated chain of saturated aliphatic monocarboxylic acids. Curr. Org. Chem. 2013, 17, 831–840. [Google Scholar] [CrossRef]
- Stecoza, C.E.; Caproiu, M.T.; Draghici, C.; Chifiriuc, M.C.; Dracea, N.O. Synthesis, characterization and antimicrobial activity evaluation of some new derivatives of 6,11-dihydrodibenzo[b,e]thiepin 5,5-dioxide. Rev. Chim. 2009, 6, 137–141. [Google Scholar]
- Anghel, I.; Grumezescu, A.M.; Holban, A.M.; Ficai, A.; Anghel, A.G.; Chifiriuc, M.C. Biohybrid nanostructured iron oxide nanoparticles and Satureja hortensis to prevent fungal biofilm development. Int. J. Mol. Sci. 2013, 14, 18110–18123. [Google Scholar] [CrossRef]
- Grumezescu, A.; Saviuc, C.; Chifiriuc, M.-C.; Hristu, R.; Mihaiescu, D.E.; Balaure, P.; Stanciu, G.; Lazar, V. Inhibitory activity of Fe3O4 Fe3O4/Oleic Acid/Usnic Acid-Core/Shell/Extra-Shell nanofluid on S. aureus biofilm development. IEEE Trans. NanoBioscience 2011, 10, 269–274. [Google Scholar] [CrossRef]
- Anghel, I.; Limban, C.; Grumezescu, A.; Anghel, A.G.; Bleotu, C.; Chifiriuc, M.-C. In vitro evaluation of anti-pathogenic surface coating nanofluid, obtained by combining Fe3O4/C12 nanostructures and 2-((4-ethylphenoxy)methyl)-N-(substituted-phenylcarbamothioyl)-benzamides. Nanoscale Res. Lett. 2012, 7, 513. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Crennell, K.M. Cambridge Crystallographic Data Centre. 6. Preparation and computer typesetting of the molecular structures and dimensions bibliographic volumes. J. Chem. Inf. Model. 1982, 22, 129–139. [Google Scholar] [CrossRef]
- Valley, C.C.; Cembran, A.; Perlmutter, J.D.; Lewis, A.K.; Labello, N.P.; Gao, J.; Sachs, J.N. The methionine-aromatic motif plays a unique role in stabilizing protein structure. J. Biol. Chem. 2012, 287, 34979–34991. [Google Scholar] [CrossRef]
- Iwaoka, M.; Isozumi, N. Hypervalent nonbonded interactions of a divalent sulfur atom. Implications in protein architecture and the functions. Molecules 2012, 17, 7266–7283. [Google Scholar] [CrossRef]
- Reid, K.; Lindley, P.; Thornton, J. Sulphur-aromatic interactions in proteins. FEBS Lett. 1985, 190, 209–213. [Google Scholar] [CrossRef]
- Riobé, F.; Avarvari, N. Electroactive oxazoline ligands. Co-ord. Chem. Rev. 2010, 254, 1523–1533. [Google Scholar] [CrossRef]
- Lorcy, D.; Bellec, N.; Fourmigué, M.; Avarvari, N. Tetrathiafulvalene-based group XV ligands: Synthesis, coordination chemistry and radical cation salts. Co-ord. Chem. Rev. 2009, 253, 1398–1438. [Google Scholar] [CrossRef]
- Pop, F.; Auban-Senzier, P.; Canadell, E.; Rikken, G.L.J.A.; Avarvari, N. Electrical magnetochiral anisotropy in a bulk chiral molecular conductor. Nat. Commun. 2014, 5, 3757. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | (1) | (2) |
|---|---|---|
| Chemical formula | C7H7N3S2 | C12H9N3S2 |
| M (g mol−1) | 197.28 | 259.34 |
| Temperature, (K) | 293 | 293 |
| Wavelength, (Å) | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P21/c |
| a (Å) | 7.3084(9) | 7.4623(11 |
| b (Å) | 8.4839(11) | 17.2931(17) |
| c (Å) | 14.3377(16) | 9.8647(13) |
| α (°) | 90 | 90 |
| β (°) | 96.638(9) | 111.000(10) |
| γ (°) | 90 | 90 |
| V (Å3) | 883.03(19) | 1188.5(3) |
| Z | 4 | 4 |
| Dc (g cm-3) | 1.484 | 1.449 |
| μ (mm−1) | 0.547 | 0.426 |
| F(000) | 408 | 536 |
| Goodness-of-fit on F2 | 1.041 | 1.079 |
| Final R1, wR2 (I > 2σ(I)) | 0.0399, 0.0923 | 0.0714, 0.1998 |
| R1, wR2 (all data) | 0.0576, 0.0987 | 0.0822, 0.2061 |
| Largest diff. peak and hole (eÅ−3) | −0.219, 0.223 | −0.783, 0.722 |
| Parameter | (1) | (2) | |
|---|---|---|---|
| C1 C2 | 1.492(3) | C1 N3 | 1.337(6) |
| C2 N1 | 1.346(2) | C1 S2 | 1.684(5) |
| C2 C3 | 1.384(3) | C1 S1 | 1.761(5) |
| C3 C4 | 1.373(2) | C2 N1 | 1.314(7) |
| C3 H3 | 0.9300 | C2 N2 | 1.374(6) |
| C4 N2 | 1.364(2) | C2 S1 | 1.715(5) |
| C4 C5 | 1.479(3) | C3 C4 | 1.484(8) |
| C5 H5A | 0.9600 | C3 H3A | 0.9600 |
| C5 H5B | 0.9600 | C3 H3B | 0.9600 |
| C5 H5C | 0.9600 | C3 H3C | 0.9600 |
| C6 N1 | 1.315(2) | C4 N1 | 1.339(7) |
| C6 N2 | 1.366(2) | C4 C5 | 1.391(8) |
| C6 S1 | 1.7216(18) | - | - |
| C7 N3 | 1.324(2) | - | - |
| C7 S2 | 1.6781(18) | - | - |
| C7 S1 | 1.767(2) | - | - |
| N2 N3 | 1.3786(18) | - | - |
| N1 C6 S1 | 126.71(15) | C7 C12 H12 | 120.5 |
| N2 C6 S1 | 108.88(12) | C2 N1 C4 | 116.6(5) |
| N3 C7 S2 | 125.48(15) | C6 N2 C2 | 119.4(4) |
| N3 C7 S1 | 113.70(13) | C6 N2 N3 | 123.0(4) |
| S2 C7 S1 | 120.82(11) | C2 N2 N3 | 117.6(4) |
| C6 N1 C2 | 116.34(16) | C1 N3 N2 | 109.5(4) |
| C4 N2 C6 | 120.54(15) | C2 S1 C1 | 90.1(2) |
| C4 N2 N3 | 121.57(15) | C7 C12 H12 | 120.5 |
| C6 N2 N3 | 117.85(14) | C2 N1 C4 | 116.6(5) |
| C7 N3 N2 | 109.71(15) | N3 C1 S2 | 125.8(4) |
| C6 S1 C7 | 89.84(9) | N3 C1 S1 | 113.6(4) |
| - | - | S2 C1 S1 | 120.6(3) |
| Compound | Concentration (µg/mL) | G0/G1 (%) | S (%) | G2/M (%) |
|---|---|---|---|---|
| (1) | 500 | 52.90 | 20.71 | 4.52 |
| 250 | 55.92 | 24.11 | 2.27 | |
| 125 | 53.73 | 31.18 | 9.58 | |
| (2) | 500 | 42.80 | 19.99 | 2.52 |
| 250 | 52.22 | 21.48 | 2.41 | |
| 125 | 50.54 | 26.43 | 9.86 | |
| HeLa | - | 56.61 | 30.92 | 9.61 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olar, R.; Badea, M.; Maxim, C.; Grumezescu, A.M.; Bleotu, C.; Măruţescu, L.; Chifiriuc, M.C. Anti-biofilm Fe3O4@C18-[1,3,4]thiadiazolo[3,2-a]pyrimidin-4-ium-2-thiolate Derivative Core-shell Nanocoatings. Materials 2020, 13, 4640. https://doi.org/10.3390/ma13204640
Olar R, Badea M, Maxim C, Grumezescu AM, Bleotu C, Măruţescu L, Chifiriuc MC. Anti-biofilm Fe3O4@C18-[1,3,4]thiadiazolo[3,2-a]pyrimidin-4-ium-2-thiolate Derivative Core-shell Nanocoatings. Materials. 2020; 13(20):4640. https://doi.org/10.3390/ma13204640
Chicago/Turabian StyleOlar, Rodica, Mihaela Badea, Cătălin Maxim, Alexandru Mihai Grumezescu, Coralia Bleotu, Luminiţa Măruţescu, and Mariana Carmen Chifiriuc. 2020. "Anti-biofilm Fe3O4@C18-[1,3,4]thiadiazolo[3,2-a]pyrimidin-4-ium-2-thiolate Derivative Core-shell Nanocoatings" Materials 13, no. 20: 4640. https://doi.org/10.3390/ma13204640
APA StyleOlar, R., Badea, M., Maxim, C., Grumezescu, A. M., Bleotu, C., Măruţescu, L., & Chifiriuc, M. C. (2020). Anti-biofilm Fe3O4@C18-[1,3,4]thiadiazolo[3,2-a]pyrimidin-4-ium-2-thiolate Derivative Core-shell Nanocoatings. Materials, 13(20), 4640. https://doi.org/10.3390/ma13204640