Hole Transfer in Open Carbynes



Abstract
1. Introduction
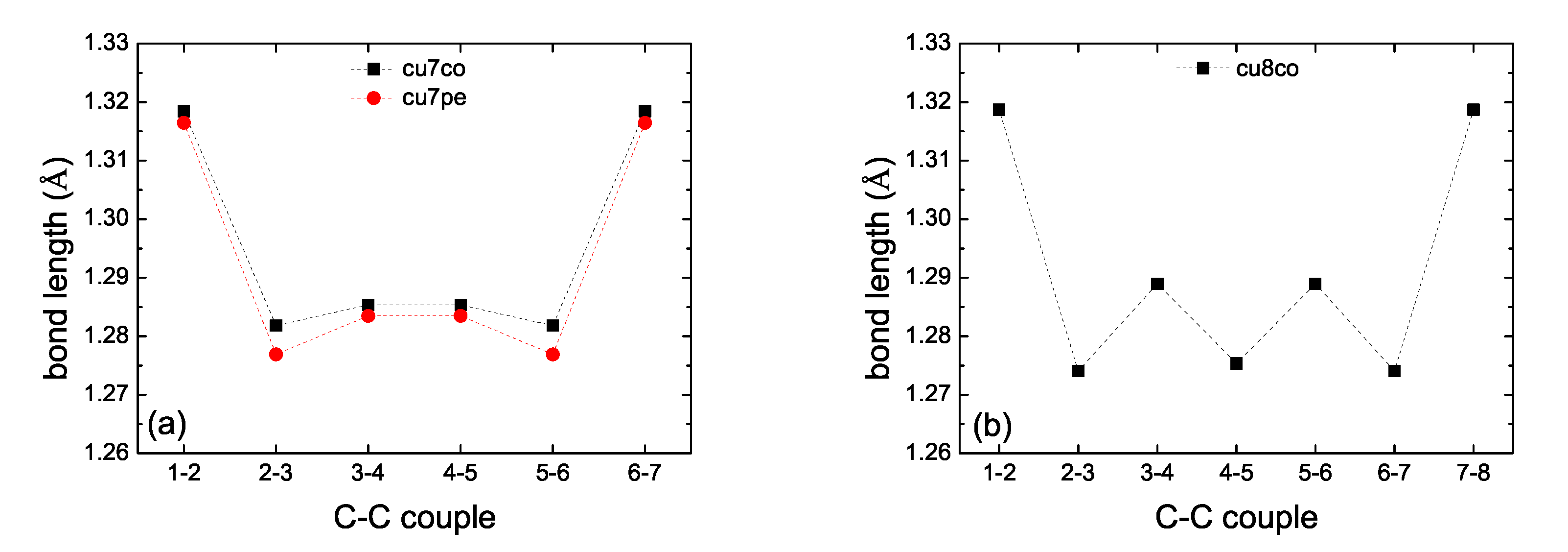
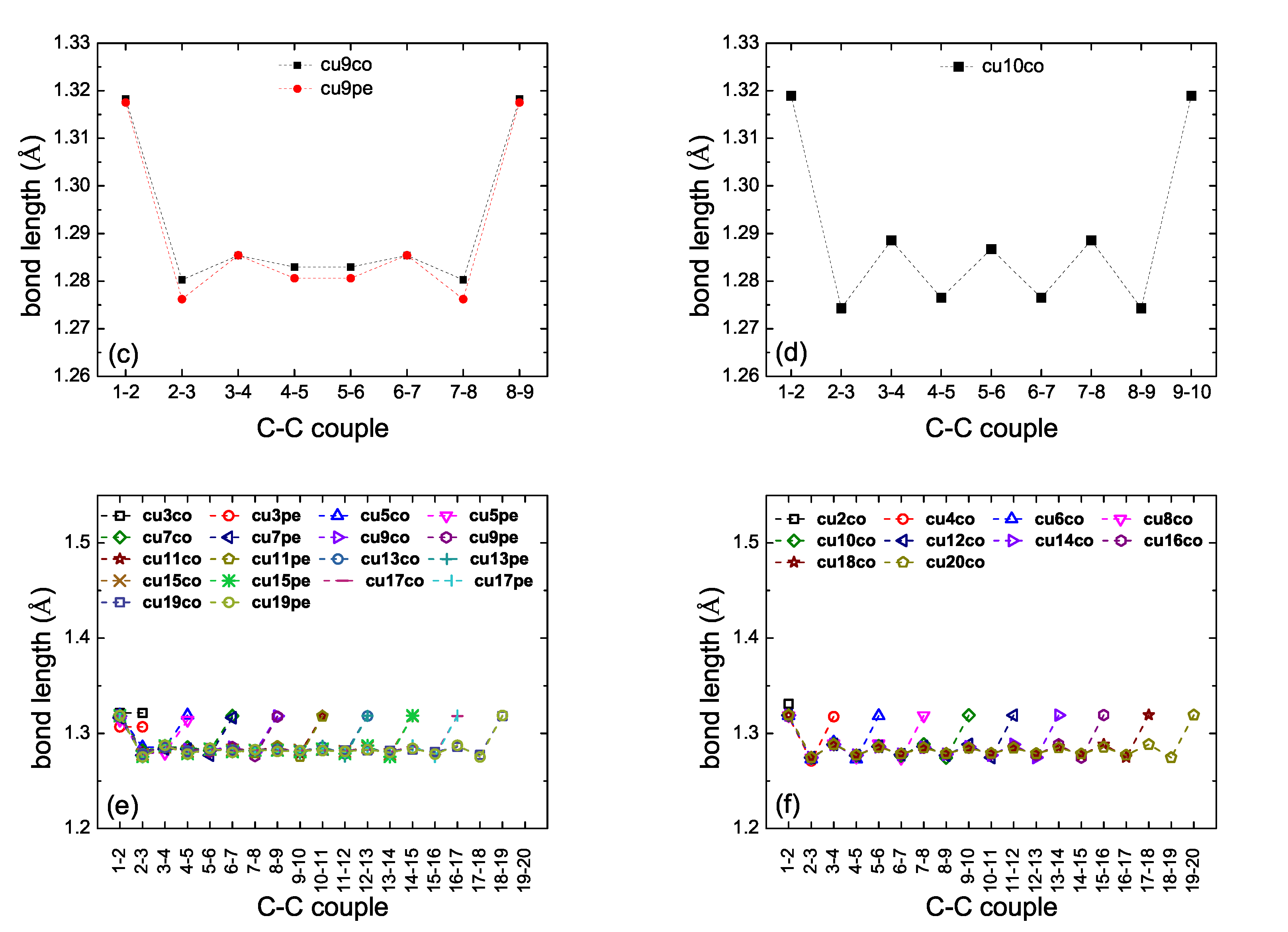
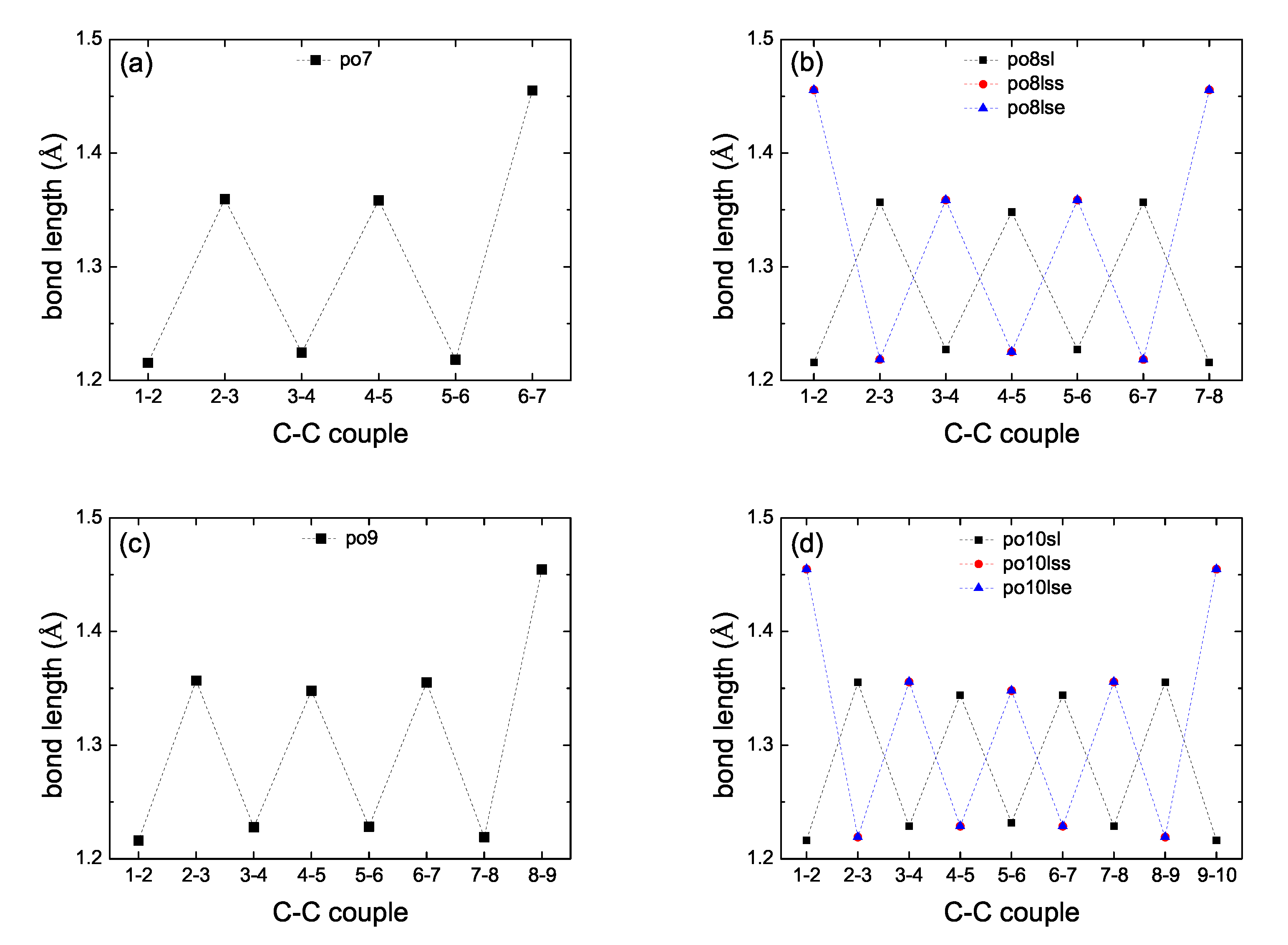
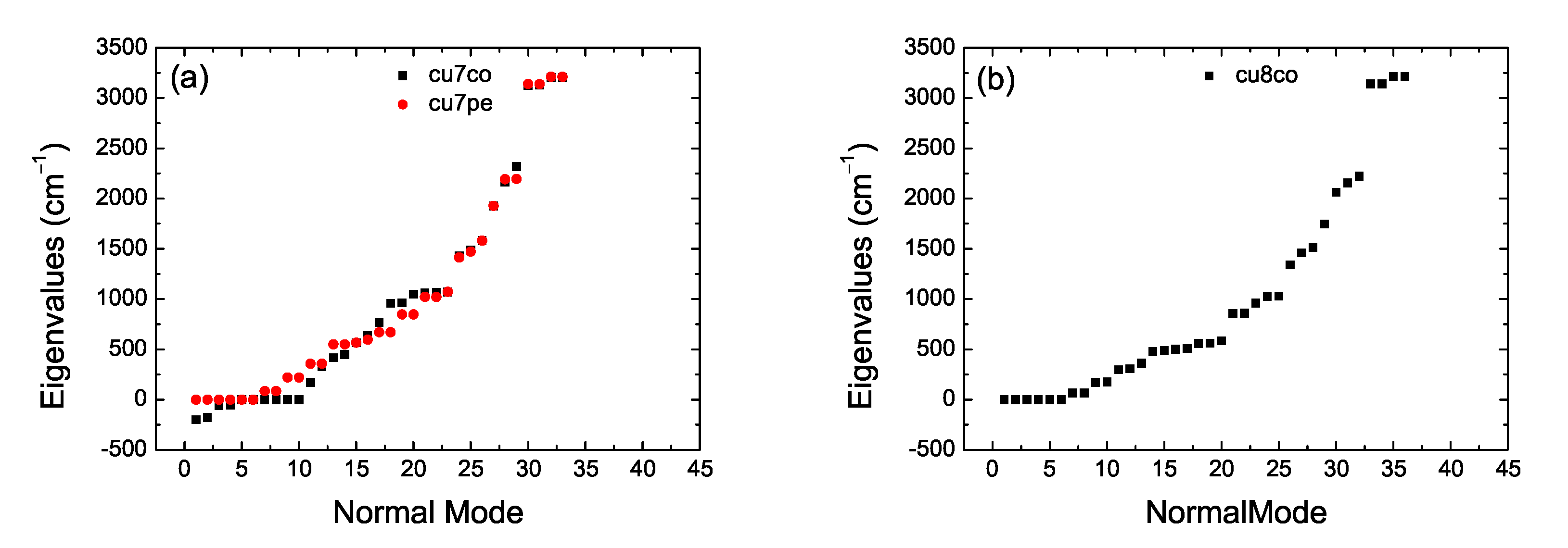
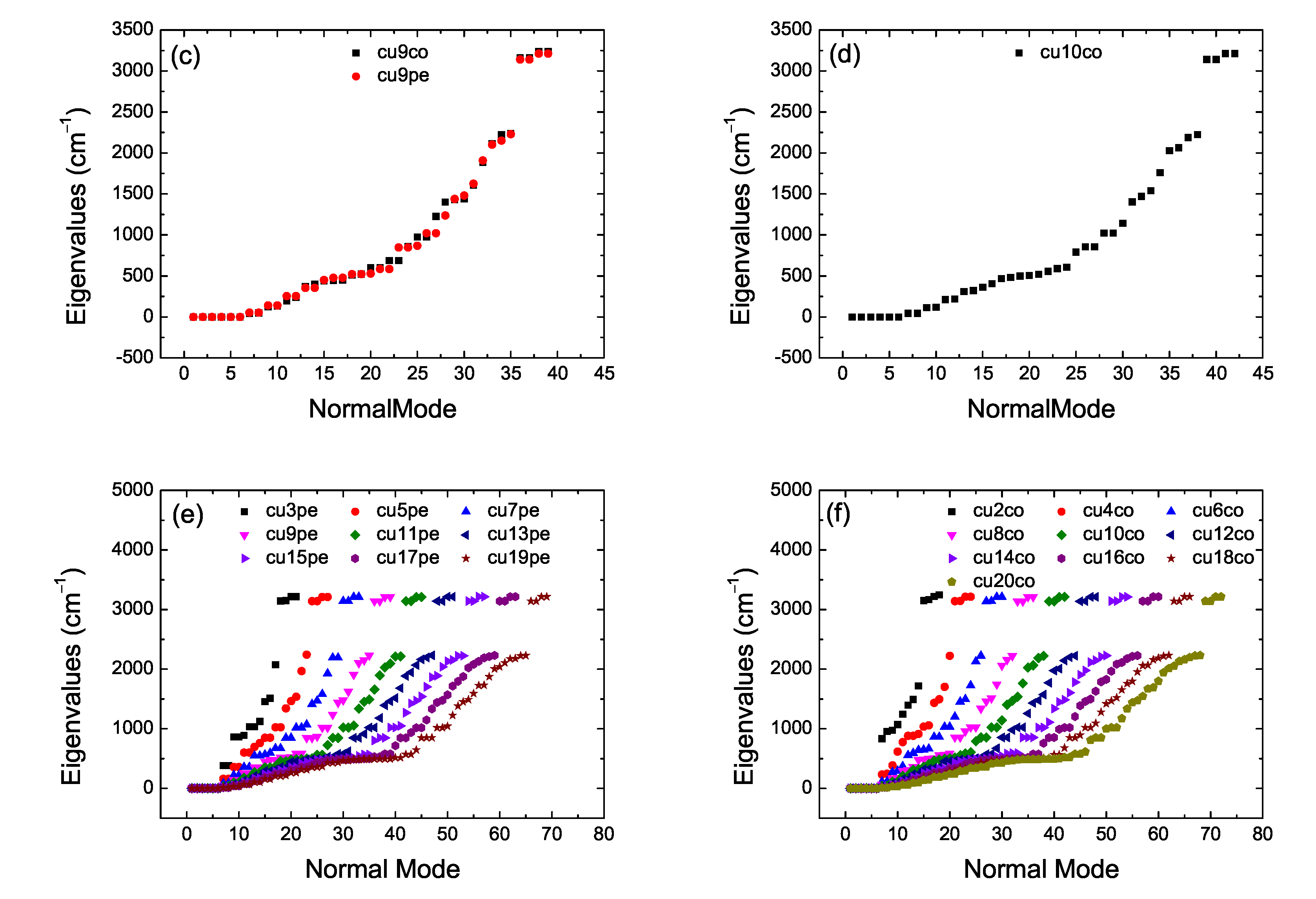
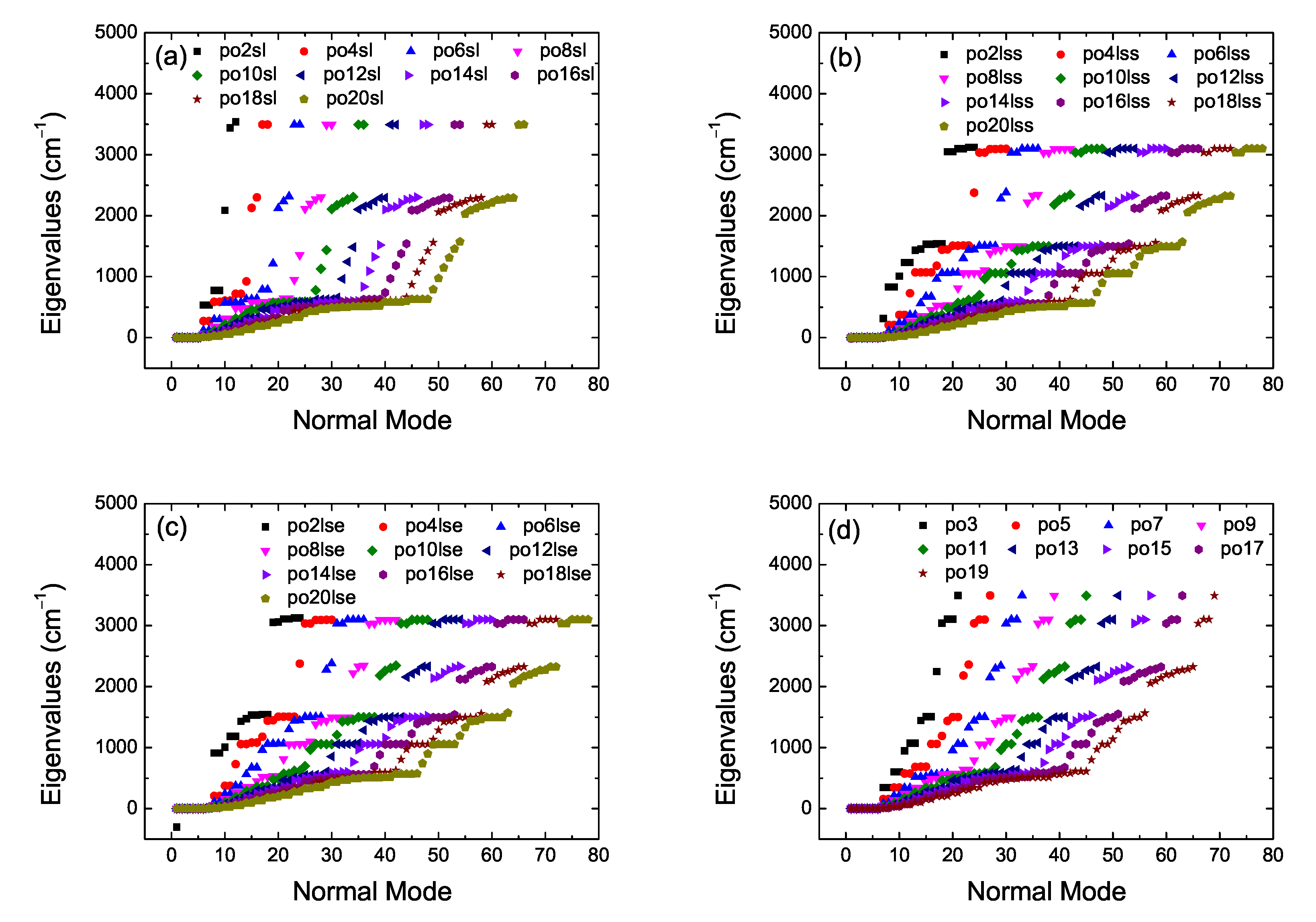
2. Bond Lengths-Structures-Vibrational Analysis
3. Tight-Binding Wire Model Variants
4. Real-Time Time-Dependent Density Functional Theory
5. Results
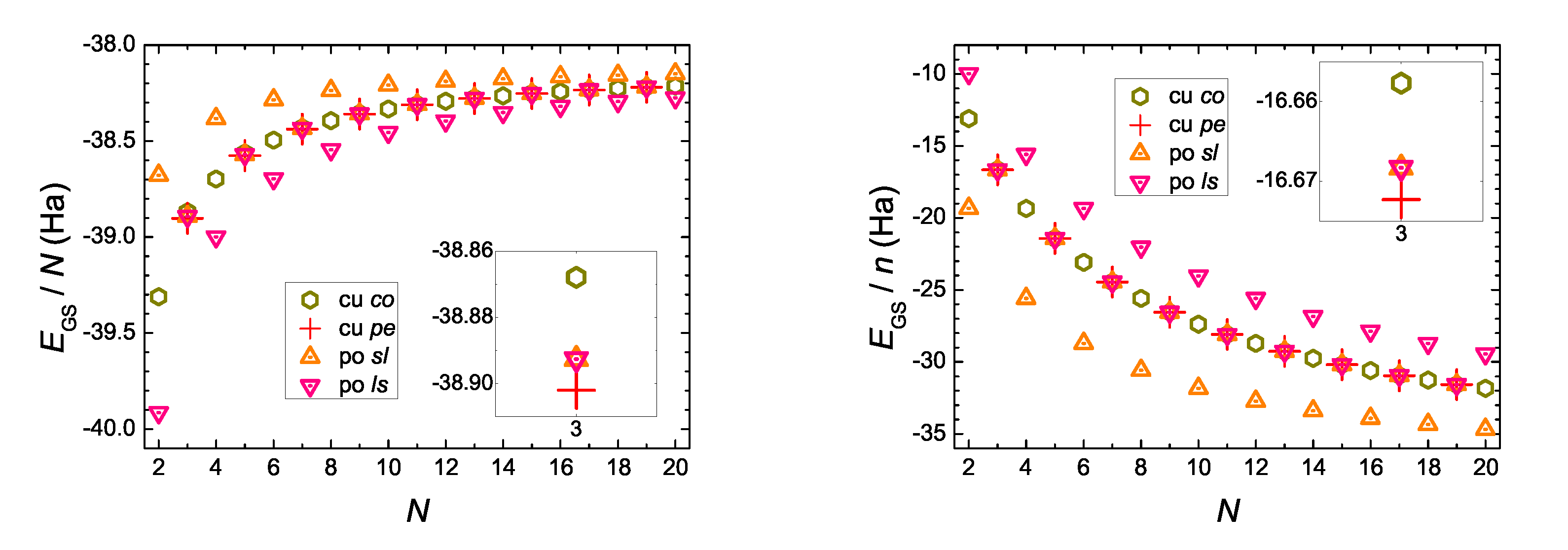
5.1. DFT Ground-State Energy
5.2. CDFT “Ground-State” Energy with a Hole at the First Site
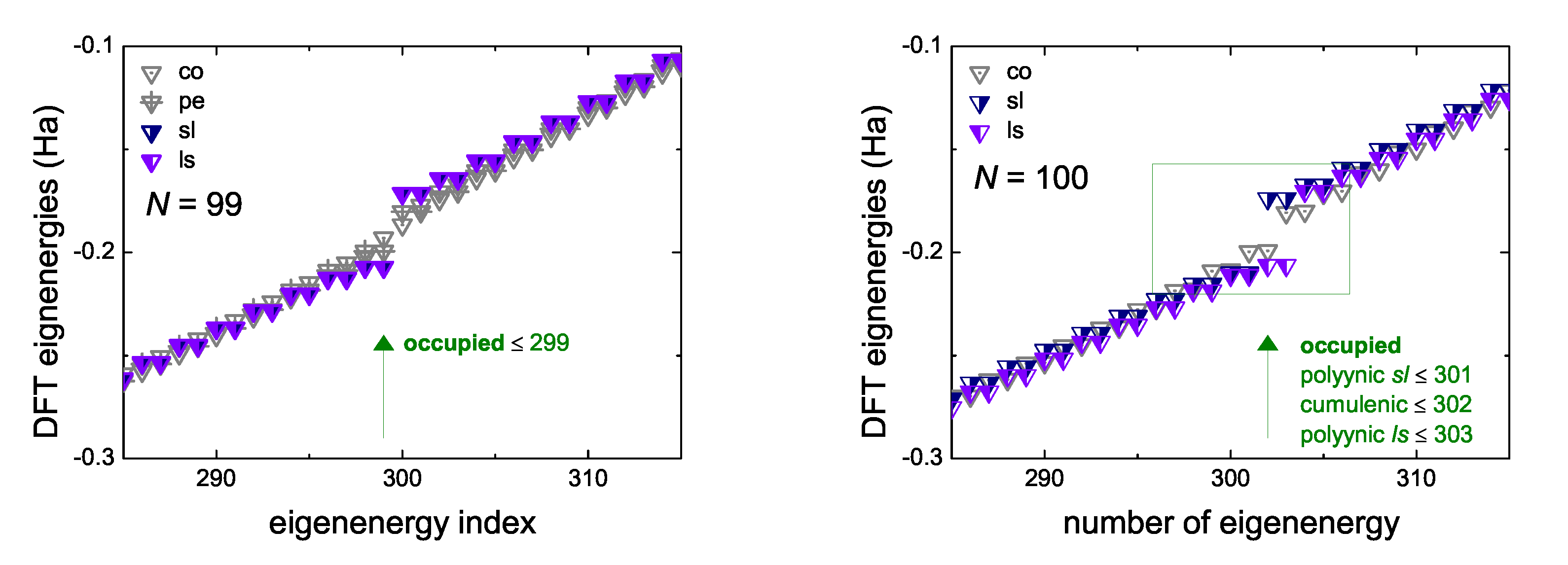
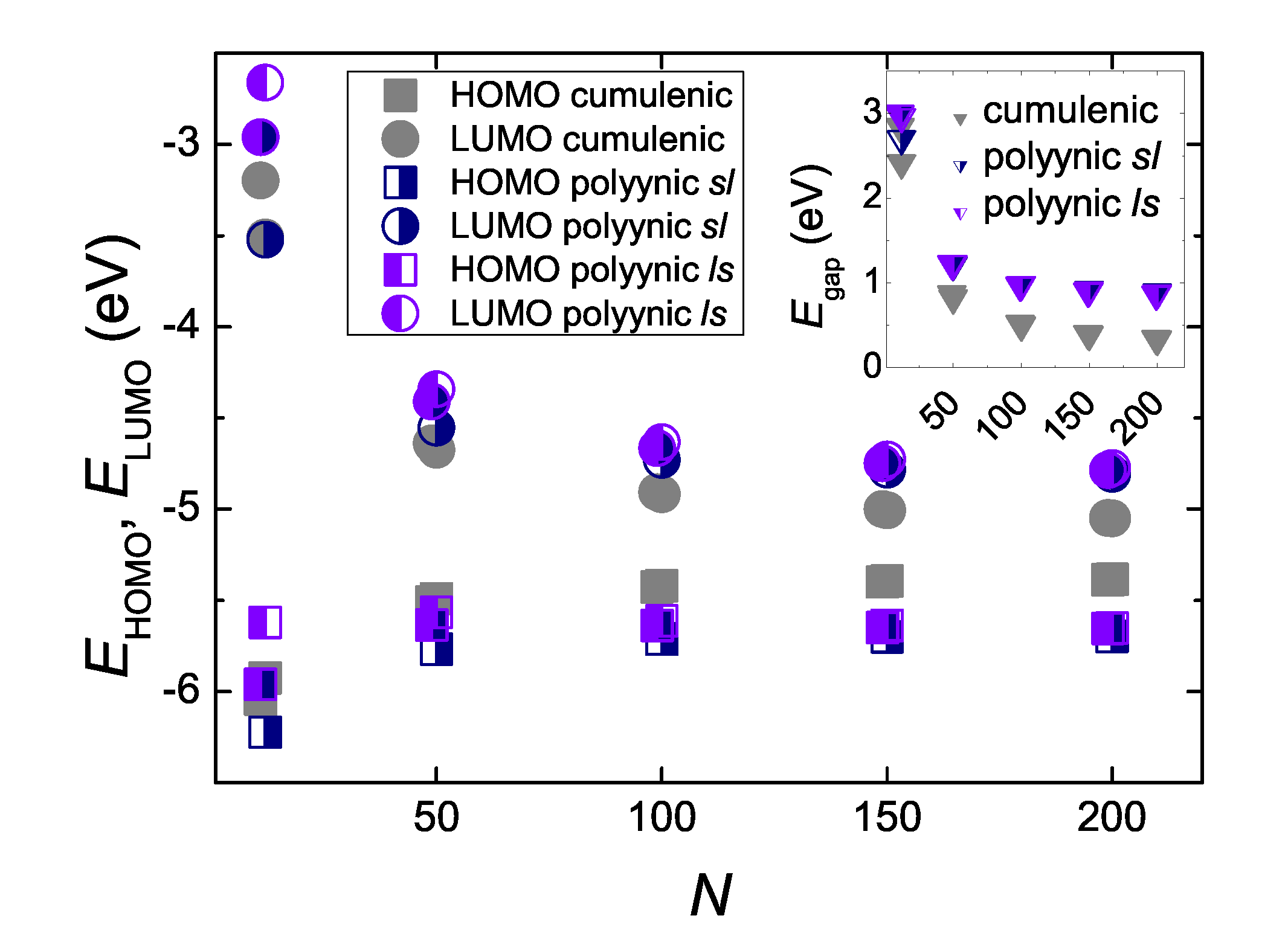
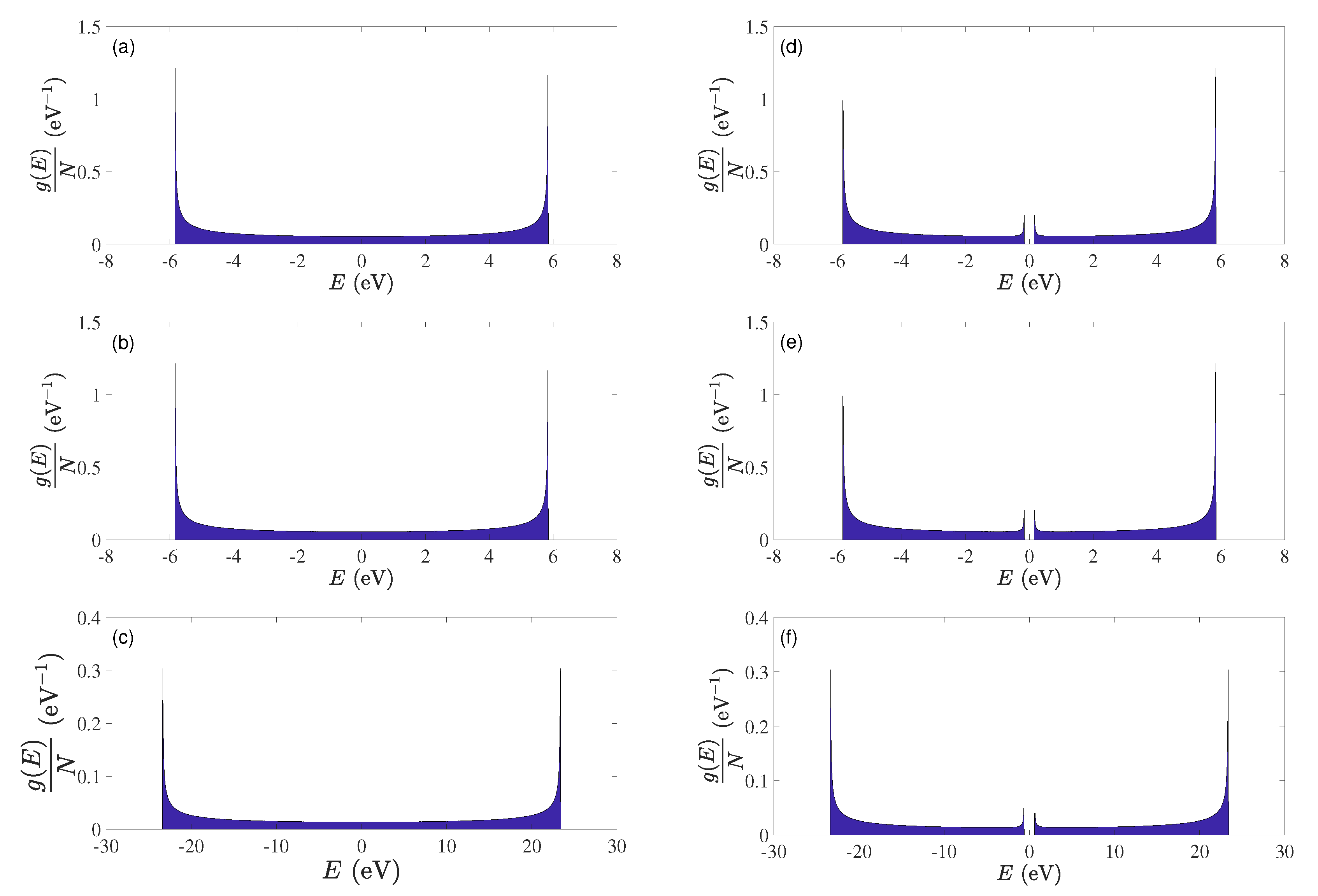
5.3. Eigenenergies, Density of States, and Energy Gap
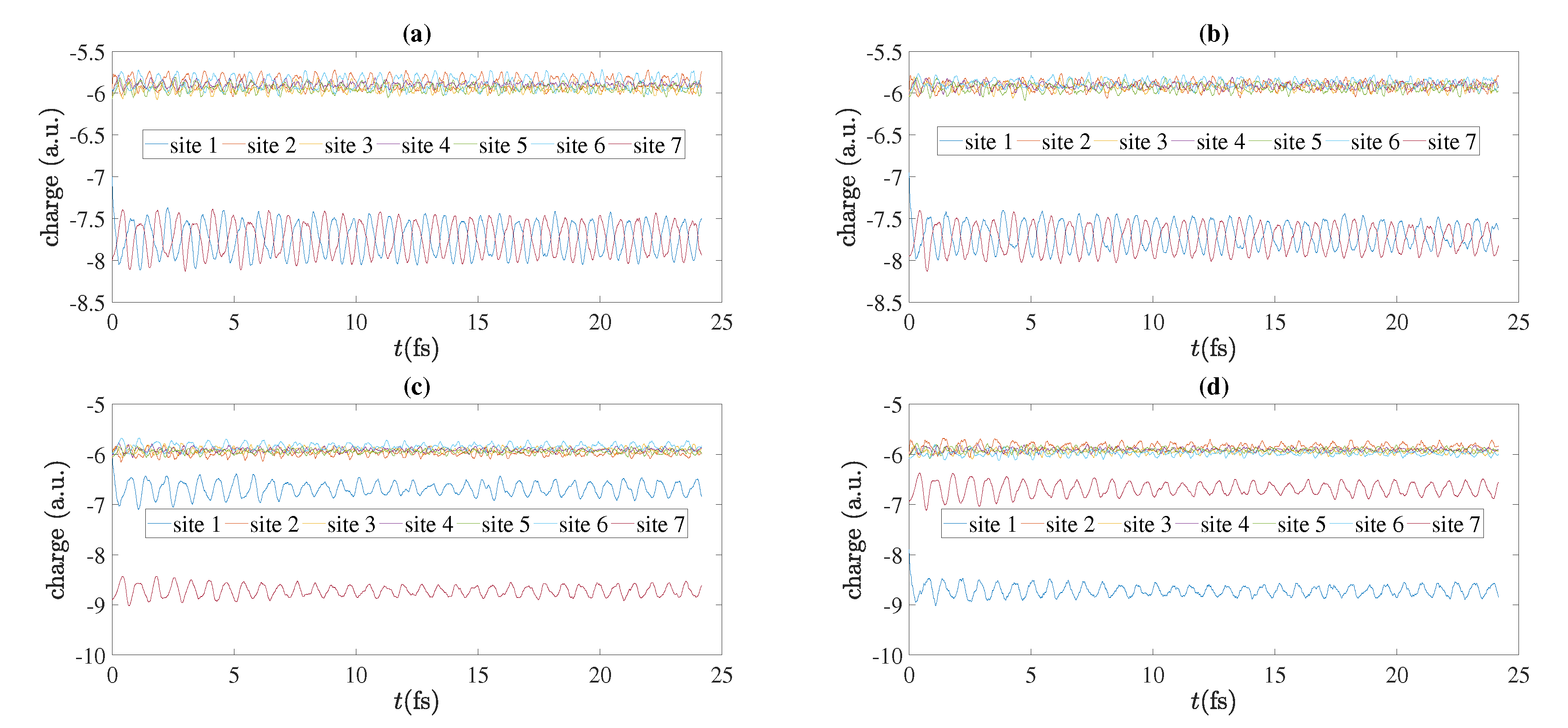
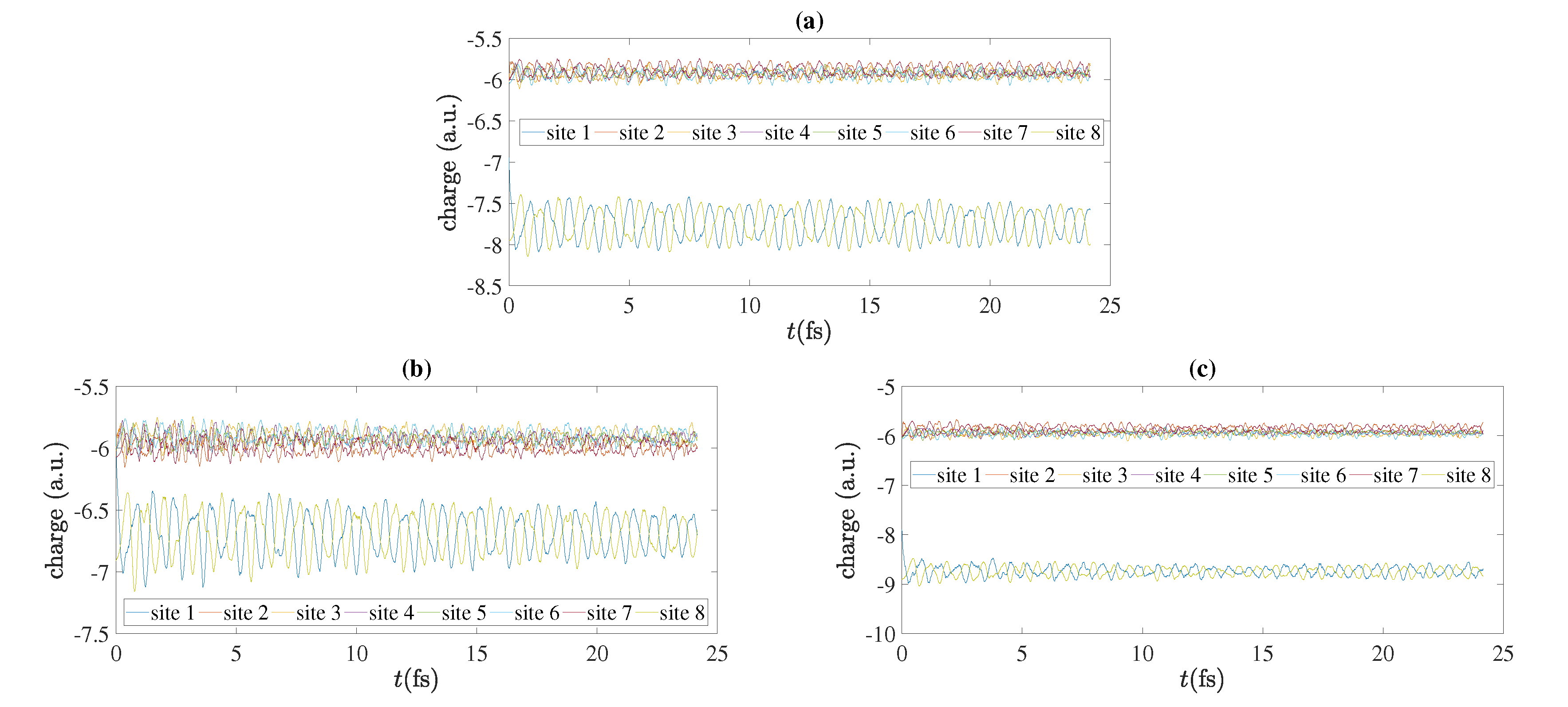
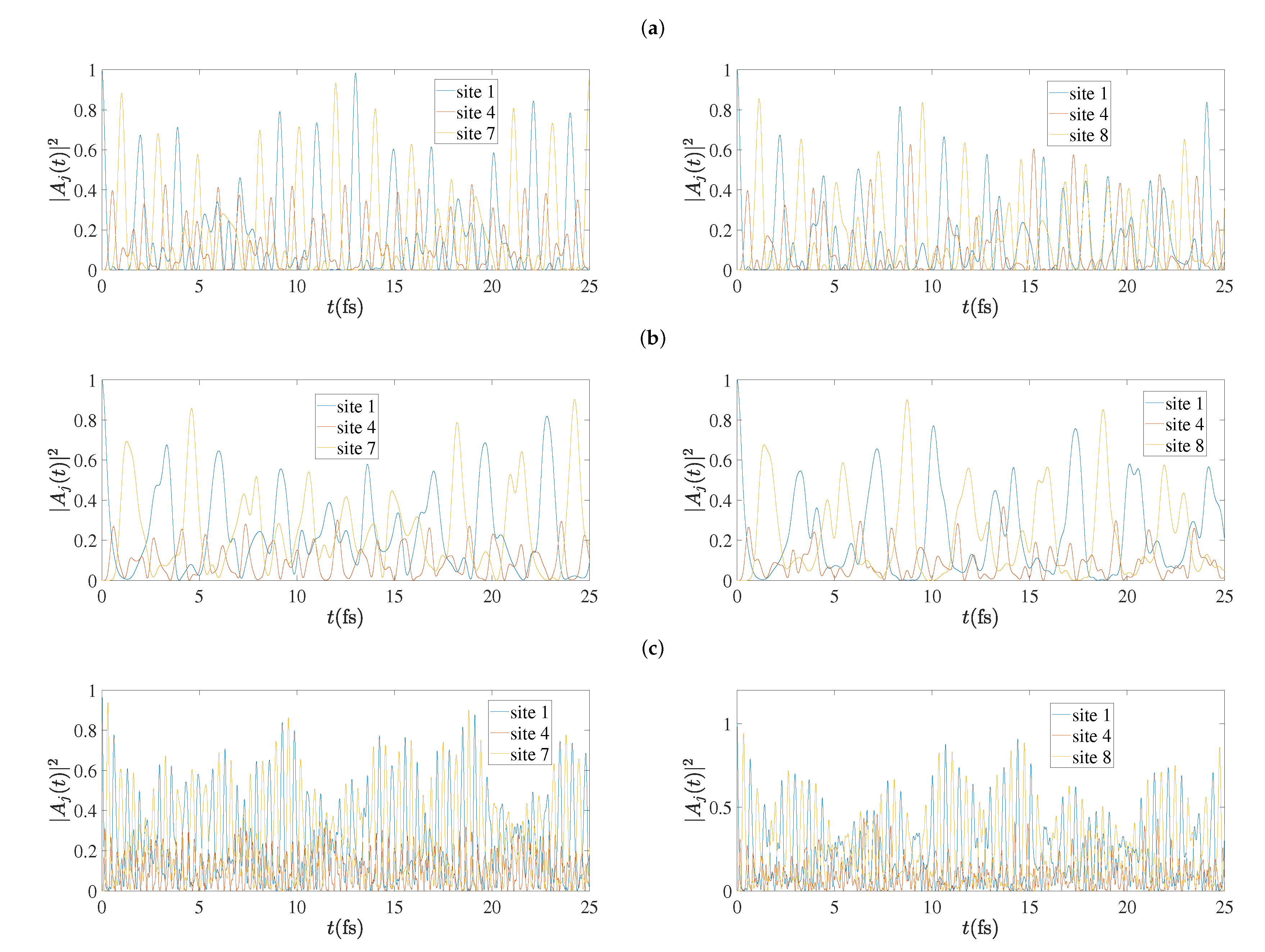
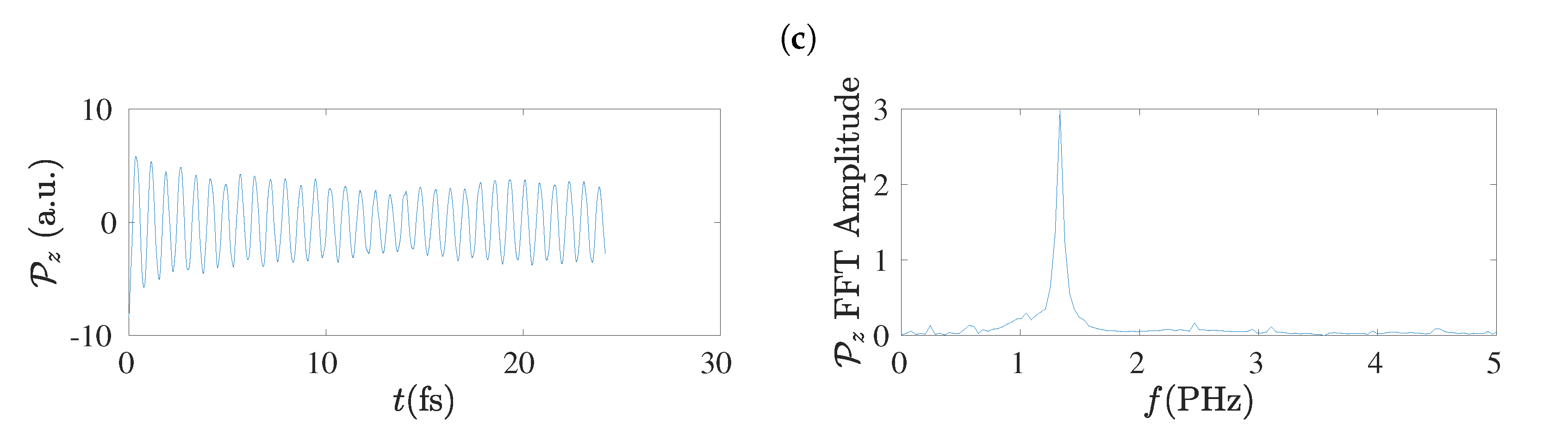
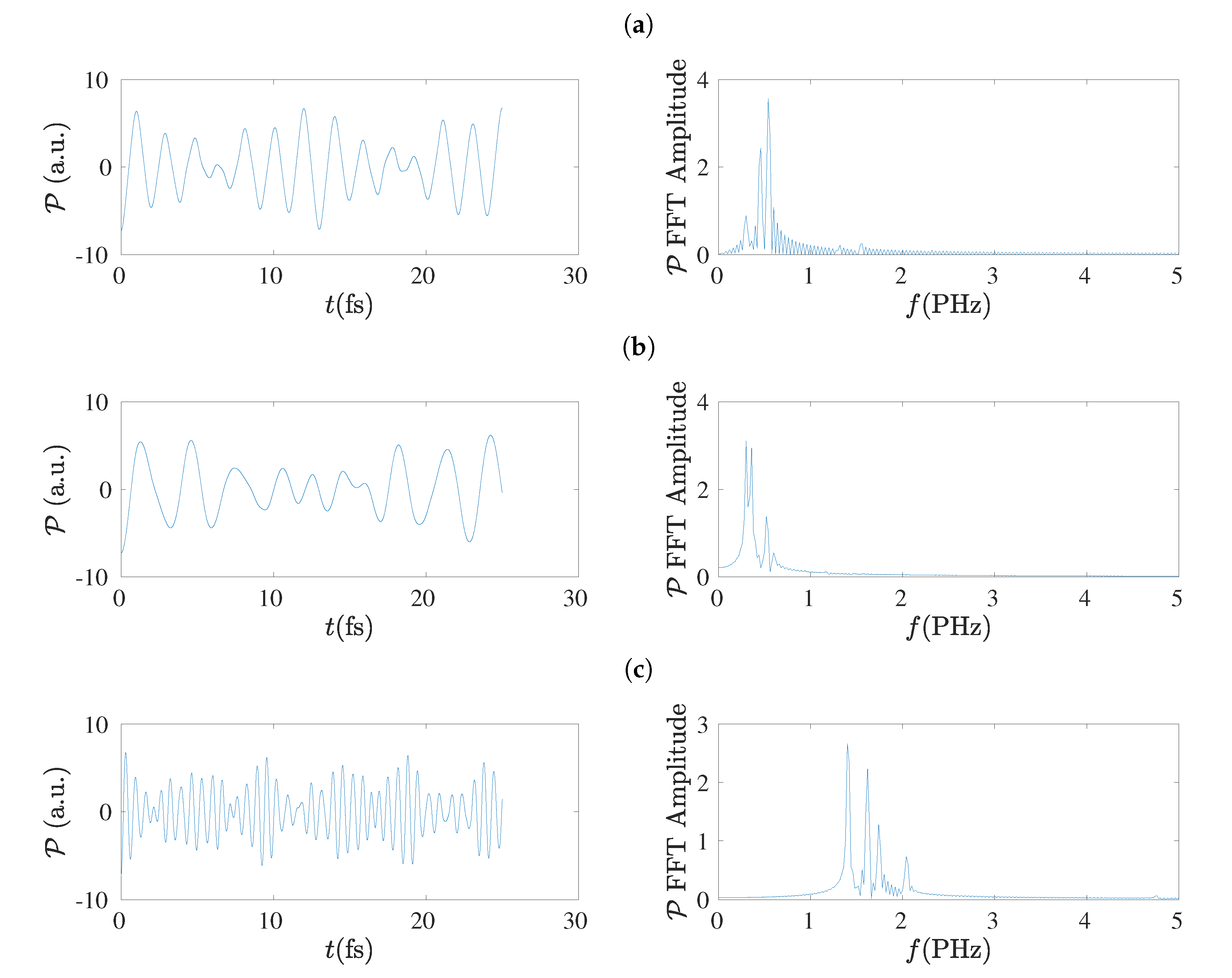
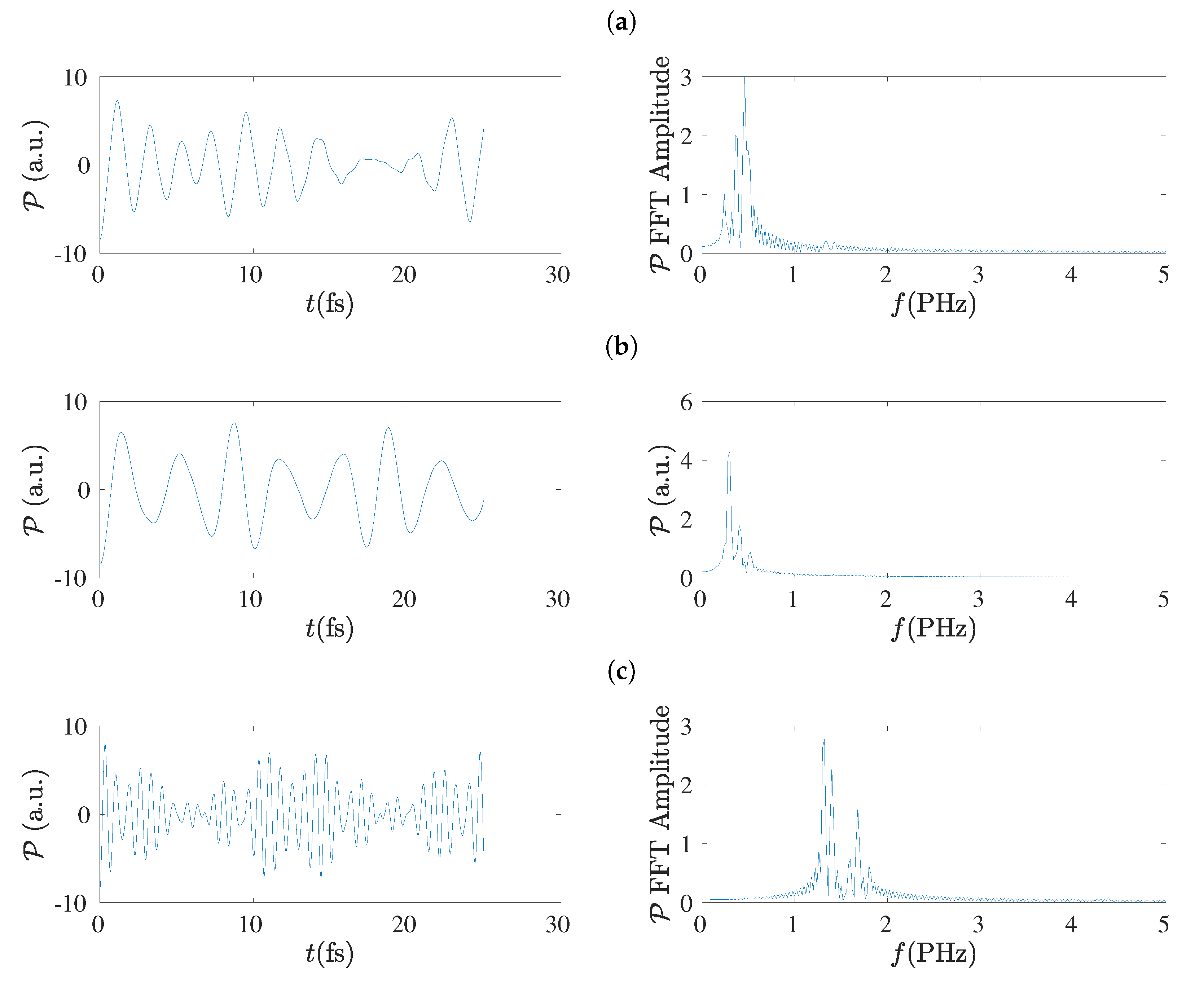
5.4. Charge Oscillations
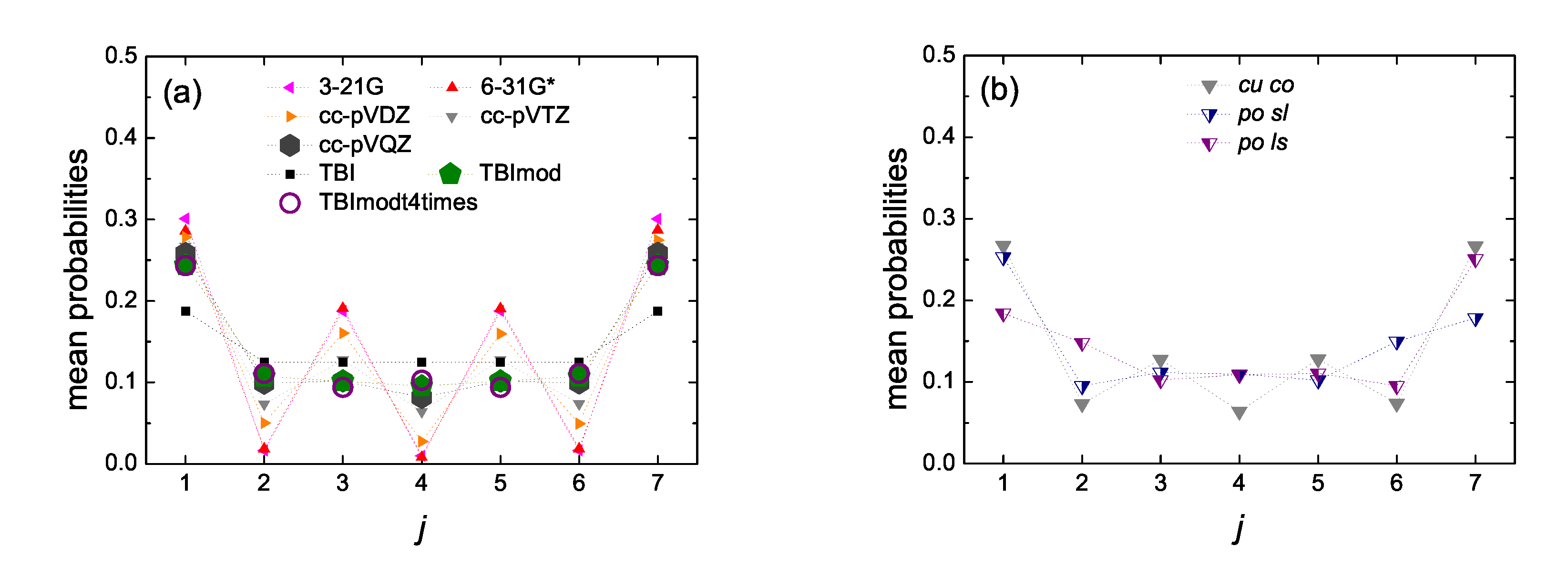
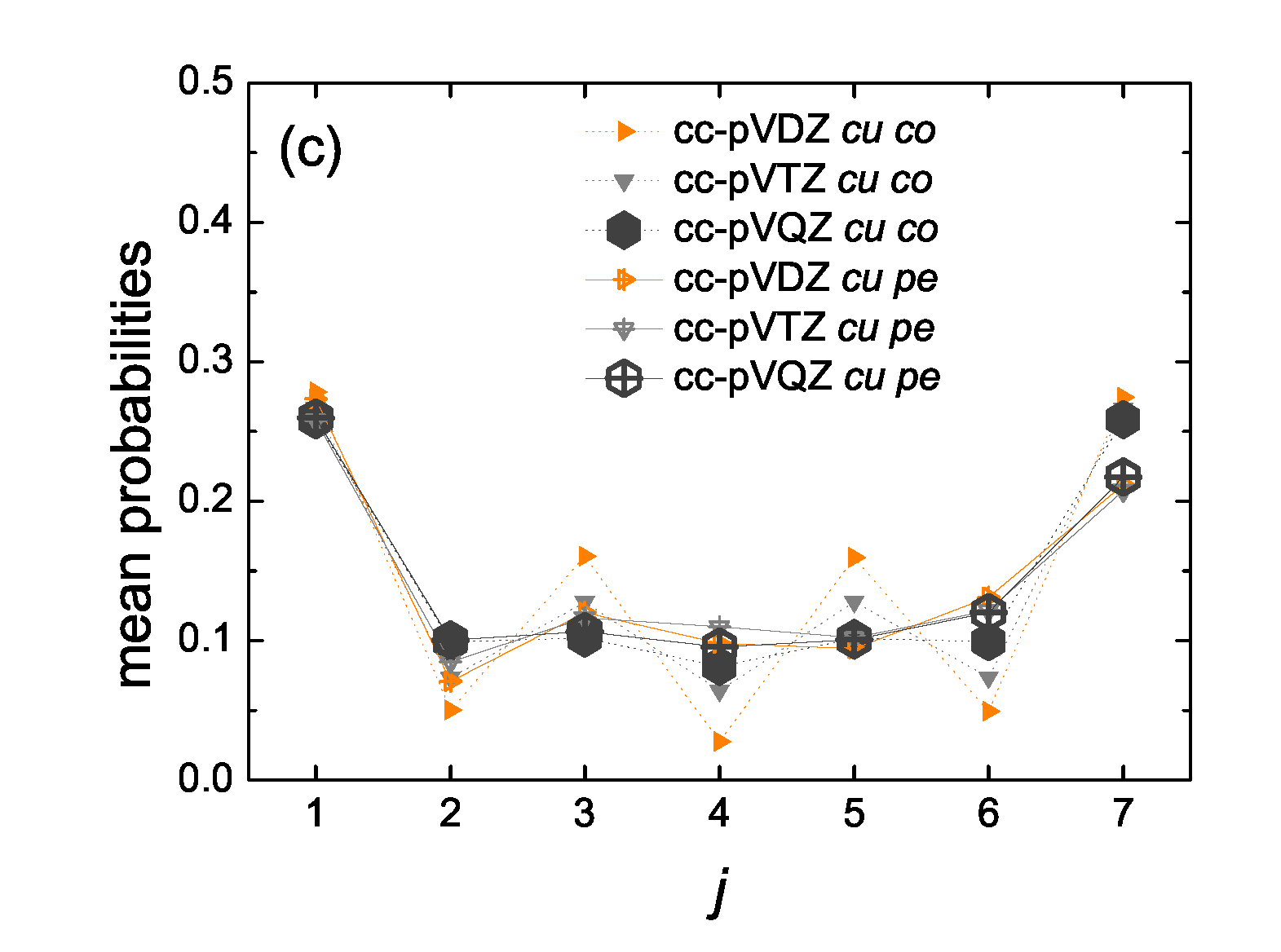
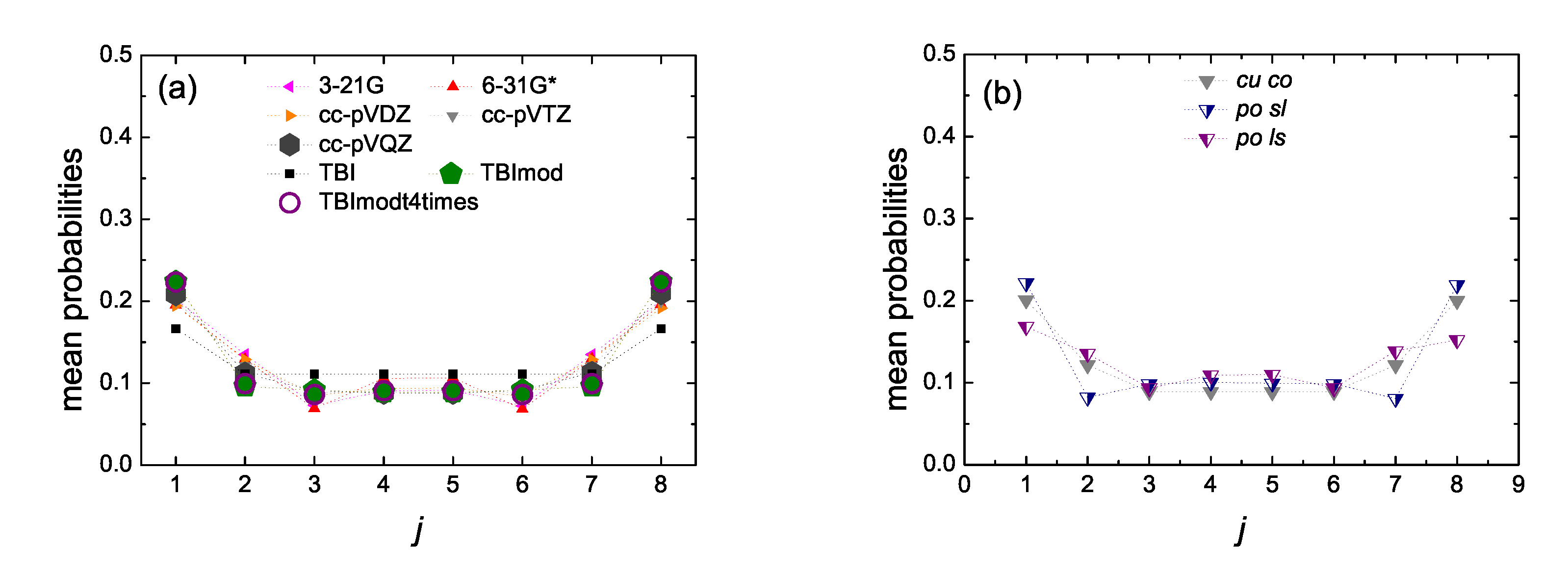
5.5. Mean over Time Probabilities
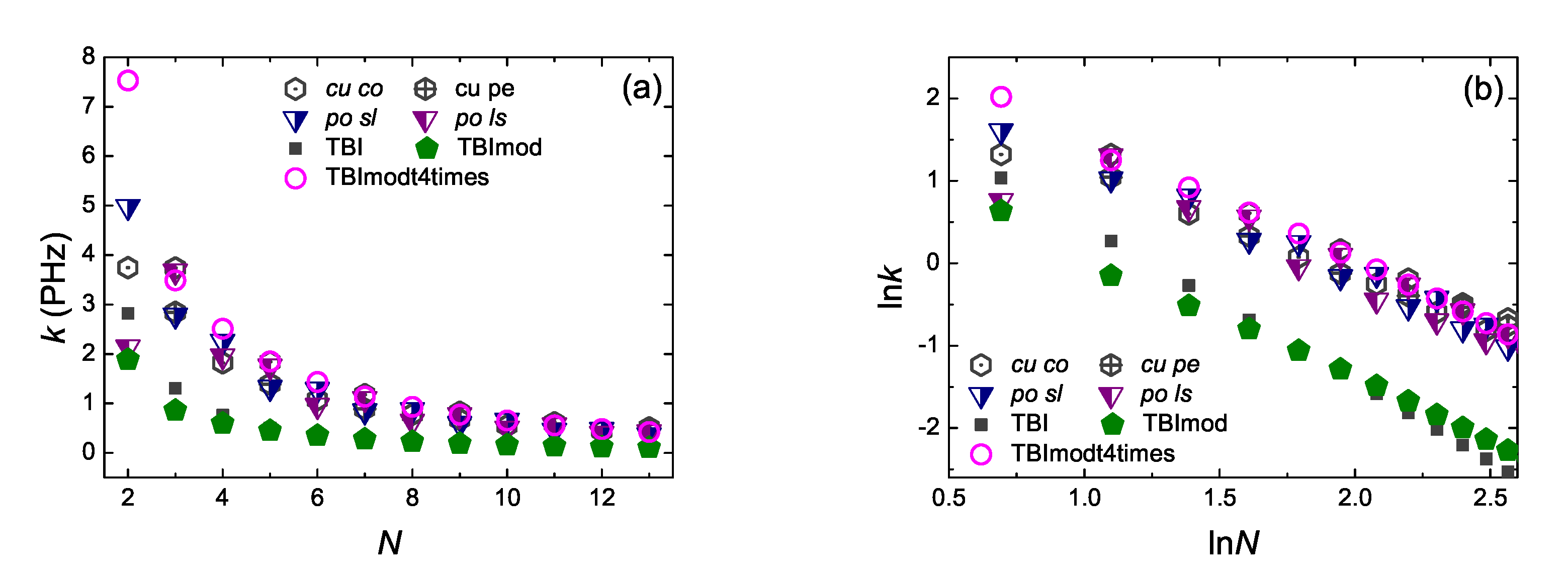
5.6. Coherent Transfer Rates
5.7. Electric Dipole Moment
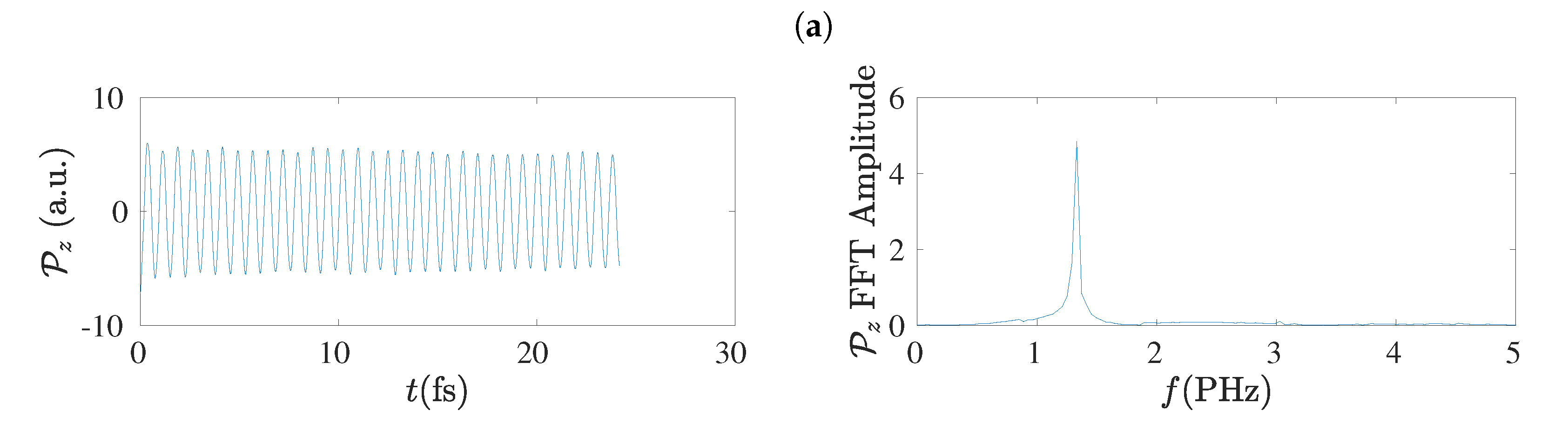
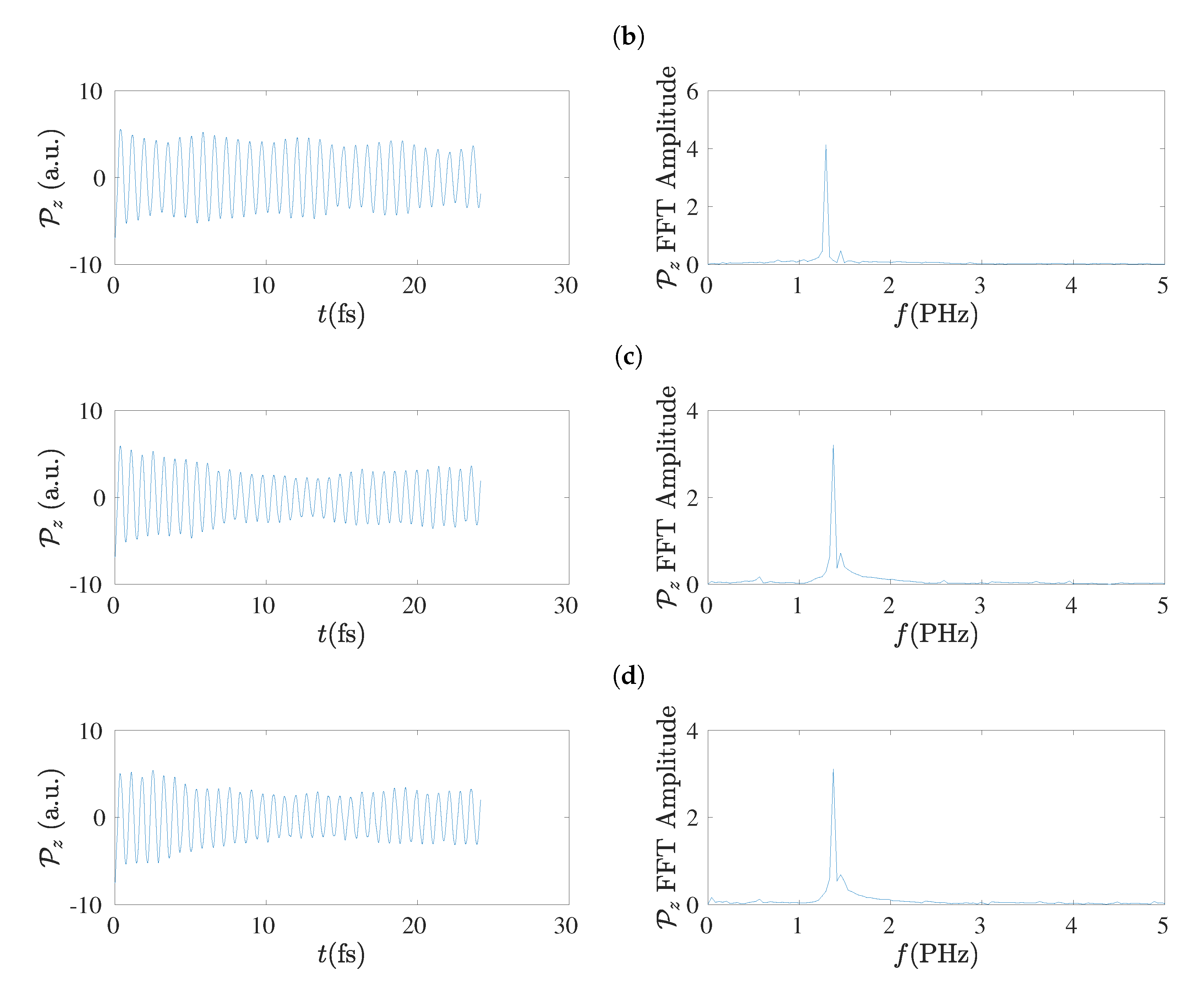
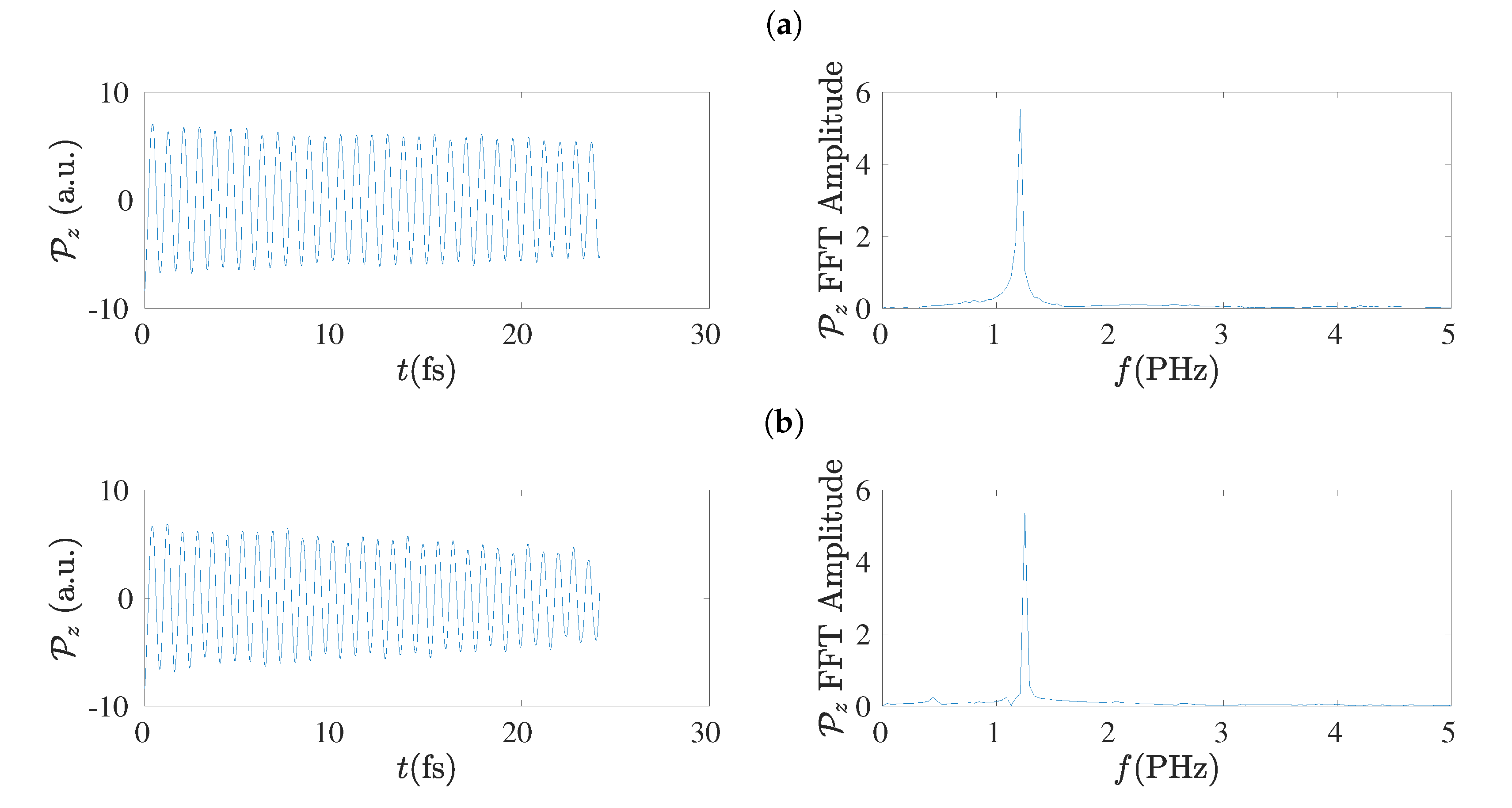
5.8. Frequency Content
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BLA | bond length alternation |
| CDFT | Constrained Density Functional Theory |
| DFT | Density Functional Theory |
| DOS | density of states |
| FFT | Fast Fourier Transform |
| RT-TDDFT | Real-Time Time-Dependent Density Functional Theory |
| TB | Tight Binding |
| TBI | TB wire model |
| TBImod | a crude modification of TB wire model |
| TBImodt4times | TBImod with four times greater transfer parameters |
| TDDFT | time-dependent DFT |
| TDKS | time-dependent Kohn-Sham |
References
- Cretu, O.; Botello-Mendez, A.R.; Janowska, I.; Pham-Huu, C.; Charlier, J.C.; Banhart, F. Electrical Transport Measured in Atomic Carbon Chains. Nano Lett. 2013, 13, 3487–3493. [Google Scholar] [CrossRef]
- La Torre, A.; Ben Romdhane, F.; Baaziz, W.; Janowska, I.; Pham-Huu, C.; Begin-Colin, S.; Pourroy, G.; Banhart, F. Formation and characterization of carbon–metal nano-contacts. Carbon 2014, 77, 906–911. [Google Scholar] [CrossRef]
- La Torre, A.; Botello-Mendez, A.; Baaziz, W.; Charlier, J.C.; Banhart, F. Strain-induced metal-semiconductor transition observed in atomic carbon chains. Nat. Commun. 2015, 6, 6636. [Google Scholar] [CrossRef] [PubMed]
- Banhart, F. Chains of carbon atoms: A vision or a new nanomaterial? Beilstein J. Nanotechnol. 2015, 6, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Lambropoulos, K.; Simserides, C. Electronic structure and charge transport properties of atomic carbon wires. Phys. Chem. Chem. Phys. 2017, 19, 26890–26897. [Google Scholar] [CrossRef] [PubMed]
- Milani, A.; Tommasini, M.; Zerbi, G. Carbynes phonons: A tight binding force field. J. Chem. Phys. 2008, 128, 064501. [Google Scholar] [CrossRef]
- Milani, A.; Tommasini, M.; Barbieri, V.; Lucotti, A.; Russo, V.; Cataldo, F.; Casari, C.S. Semiconductor-to-Metal Transition in Carbon-Atom Wires Driven by sp2 Conjugated End Groups. J. Phys. Chem. C 2017, 121, 10562–10570. [Google Scholar] [CrossRef]
- Milani, A.; Tommasini, M.; Del Zoppo, M.; Castiglioni, C.; Zerbi, G. Carbon nanowires: Phonon and π-electron confinement. Phys. Rev. B 2006, 74, 153418. [Google Scholar] [CrossRef]
- Milani, A.; Barbieri, V.; Facibeni, A.; Russo, V.; Bassi, A.L.; Lucotti, A.; Tommasini, M.; Tzirakis, M.D.; Diederich, F.; Casari, C.S. Structure modulated charge transfer in carbon atomic wires. Sci. Rep. 2019, 9, 1648. [Google Scholar] [CrossRef]
- Kawai, K.; Majima, T. Hole Transfer Kinetics of DNA. Acc. Chem. Res. 2013, 46, 2616–2625. [Google Scholar] [CrossRef]
- Lewis, F.D.; Wu, T.; Zhang, Y.; Letsinger, R.L.; Greenfield, S.R.; Wasielewski, M.R. Distance-Dependent Electron Transfer in DNA Hairpins. Science 1997, 277, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Fiebig, T.; Schiemann, O.; Barton, J.K.; Zewail, A.H. Femtosecond direct observation of charge transfer between bases in DNA. Proc. Natl. Acad. Sci. USA 2000, 97, 14052–14055. [Google Scholar] [CrossRef]
- Takada, T.; Kawai, K.; Fujitsuka, M.; Majima, T. Direct observation of hole transfer through double-helical DNA over 100 A. Proc. Natl. Acad. Sci. USA 2004, 101, 14002–14006. [Google Scholar] [CrossRef] [PubMed]
- Fujitsuka, M.; Majima, T. Charge transfer dynamics in DNA revealed by time-resolved spectroscopy. Chem. Sci. 2017, 8, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Mickley Conron, S.M.; Thazhathveetil, A.K.; Wasielewski, M.R.; Burin, A.L.; Lewis, F.D. Direct Measurement of the Dynamics of Hole Hopping in Extended DNA G-Tracts. An Unbiased Random Walk. J. Am. Chem. Soc. 2010, 132, 14388–14390. [Google Scholar] [CrossRef] [PubMed]
- Vura-Weis, J.; Wasielewski, M.R.; Thazhathveetil, A.K.; Lewis, F.D. Efficient Charge Transport in DNA Diblock Oligomers. J. Am. Chem. Soc. 2009, 131, 9722–9727. [Google Scholar] [CrossRef]
- Simserides, C.; Morphis, A.; Lambropoulos, K. Hole Transfer in Cumulenic and Polyynic Carbynes. J. Phys. Chem. C 2020. [Google Scholar] [CrossRef]
- Liu, M.; Artyukhov, V.I.; Lee, H.; Xu, F.; Yakobson, B.I. Carbyne from First Principles: Chain of C Atoms, a Nanorod or a Nanorope. ACS Nano 2013, 7, 10075–10082. [Google Scholar] [CrossRef]
- Fox, M.A.; Whitesell, J.K. Organic Chemistry; Jones and Bartlett: Boston, MA, USA; London, UK, 1994. [Google Scholar]
- Atkins, P.; de Paula, J. Physical Chemistry, 8th ed.; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Levine, I.N. Physical Chemistry, 6th ed.; McGraw-Hill: New York, NY, USA, 2009; p. 10020. [Google Scholar]
- Cahangirov, S.; Topsakal, M.; Ciraci, S. Long-range interactions in carbon atomic chains. Phys. Rev. B 2010, 82, 195444. [Google Scholar] [CrossRef]
- Wendinger, D.; Tykwinski, R.R. Odd [n]Cumulenes (n = 3, 5, 7, 9): Synthesis, Characterization, and Reactivity. Acc. Chem. Res. 2017, 50, 1468–1479. [Google Scholar] [CrossRef]
- Valiev, M.; Bylaska, E.J.; Govind, N.; Kowalski, K.; Straatsma, T.P.; Van Dam, H.J.J.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.L.; et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian~16 Revision C.01; Gaussian Inc.: Wallingford, UK, 2016. [Google Scholar]
- Ochterski, J.W. Vibrational Analysis in Gaussian; Gaussian Inc.: Wallingford, UK, 2018. [Google Scholar]
- Peierls, R.E. Quantum Theory of Solids; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Milani, A.; Tommasini, M.; Zerbi, G. Connection among Raman wavenumbers, bond length alternation and energy gap in polyynes. J. Raman Spectrosc. 2009, 40, 1931–1934. [Google Scholar] [CrossRef]
- Artyukhov, V.I.; Liu, M.; Yakobson, B.I. Mechanically Induced Metal-Insulator Transition in Carbyne. Nano Lett. 2014, 8, 4224–4229. [Google Scholar] [CrossRef] [PubMed]
- Slater, J.C.; Koster, G.F. Simplified LCAO Method for the Periodic Potential Problem. Phys. Rev. 1954, 94, 1498–1524. [Google Scholar] [CrossRef]
- Harrison, W.A. Electronic Structure and the Properties of Solids: The Physics of the Chemical Bond, 2nd ed.; Dover: New York, NY, USA, 1989. [Google Scholar]
- Harrison, W.A. Elementary Electronic Structure; World Scientific: River Edge, NJ, USA, 1999. [Google Scholar]
- Simserides, C. A systematic study of electron or hole transfer along DNA dimers, trimers and polymers. Chem. Phys. 2014, 440, 31–41. [Google Scholar] [CrossRef]
- Lambropoulos, K.; Chatzieleftheriou, M.; Morphis, A.; Kaklamanis, K.; Theodorakou, M.; Simserides, C. Unbiased charge oscillations in B-DNA: Monomer polymers and dimer polymers. Phys. Rev. E 2015, 92, 032725. [Google Scholar] [CrossRef]
- Lambropoulos, K.; Chatzieleftheriou, M.; Morphis, A.; Kaklamanis, K.; Lopp, R.; Theodorakou, M.; Tassi, M.; Simserides, C. Electronic structure and carrier transfer in B-DNA monomer polymers and dimer polymers: Stationary and time-dependent aspects of a wire model versus an extended ladder model. Phys. Rev. E 2016, 94, 062403. [Google Scholar] [CrossRef]
- Lambropoulos, K.; Vantaraki, C.; Bilia, P.; Mantela, M.; Simserides, C. Periodic polymers with increasing repetition unit: Energy structure and carrier transfer. Phys. Rev. E 2018, 98, 032412. [Google Scholar] [CrossRef]
- Mantela, M.; Lambropoulos, K.; Theodorakou, M.; Simserides, C. Quasi-Periodic and Fractal Polymers: Energy Structure and Carrier Transfer. Materials 2019, 12, 2177. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997. [Google Scholar] [CrossRef]
- Lopata, K.; Govind, N. Modeling Fast Electron Dynamics with Real-Time Time-Dependent Density Functional Theory: Application to Small Molecules and Chromophores. J. Chem. Theory Comput. 2011, 7, 1344. [Google Scholar] [CrossRef]
- Becke, A. Density-functional thermochemistry. III.The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51. [Google Scholar] [CrossRef]
- Binkley, J.S.; Pople, J.A.; Hehre, W.J. Self-consistent molecular orbital methods. 21. Small split-valence basis sets for first-row elements. J. Am. Chem. Soc. 1980, 102, 939–947. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Löwdin, P.O. On the Non-Orthogonality Problem Connected with the Use of Atomic Wave Functions in the Theory of Molecules and Crystals. J. Chem. Phys. 1950, 18, 365. [Google Scholar] [CrossRef]



























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| d | d | d | d | d | |||||
|---|---|---|---|---|---|---|---|---|---|
| - | 154 [19] | - | 146 [19] | C−C | 154 [20,21] | benzene | 140 [19] | polyynic long | 130.1 [22] |
| - | 150 [19] | - | 143 [19] | C=C | 134 [20,21] | alkene | 134 [19] | cumulenic | 128.2 [22] |
| - | 147 [19] | - | 137 [19] | C≡C | 120 [20,21] | alkyne | 120 [19] | polyynic short | 126.5 [22] |
| Type | N | n | TM | RM | VM |
|---|---|---|---|---|---|
| cumulenes | even, odd | 3 | 3 | ||
| polyynes sl | even | 3 | 2 | ||
| polyynes sl | odd | 3 | 3 | ||
| polyynes ls | even | 3 | 3 | ||
| polyynes ls | odd | 3 | 3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simserides, C.; Morphis, A.; Lambropoulos, K. Hole Transfer in Open Carbynes. Materials 2020, 13, 3979. https://doi.org/10.3390/ma13183979
Simserides C, Morphis A, Lambropoulos K. Hole Transfer in Open Carbynes. Materials. 2020; 13(18):3979. https://doi.org/10.3390/ma13183979
Chicago/Turabian StyleSimserides, Constantinos, Andreas Morphis, and Konstantinos Lambropoulos. 2020. "Hole Transfer in Open Carbynes" Materials 13, no. 18: 3979. https://doi.org/10.3390/ma13183979
APA StyleSimserides, C., Morphis, A., & Lambropoulos, K. (2020). Hole Transfer in Open Carbynes. Materials, 13(18), 3979. https://doi.org/10.3390/ma13183979