1. Introduction
Hydrogen sulfide (H
2S) is a colorless, corrosive, water-soluble, highly toxic, and flammable acid gas with the characteristic foul odor of rotten eggs, which can be typically found in natural gas, petroleum, and biogas (a byproduct of anaerobic decomposition) [
1,
2,
3,
4]. Its separation has significant economic and environmental repercussions for the relevant industries. In this regard, different physicochemical methods have been developed and commercially adopted, such as biological treatment, chemical oxidation, chemical scrubbing, etc. Among these methods, the dry adsorption process has received the most attention for both large- and small-scale applications due to its superior performance in terms of H
2S removal efficiency even at low temperatures and pressures [
5,
6].
Obviously, for any adsorption application, it is desirable to design adsorbents with high selectivity towards the target molecules and high adsorption capacity [
7,
8]. Traditional adsorbents, such as natural and synthetic zeolites, have received considerable attention in recent years. Structurally, zeolites are microporous crystalline materials formed by a combination of SiO
4 and AlO
4ˉ or exclusively SiO
4 tetrahedra sharing a vertex, and they have been widely used in the petrochemical industry as catalytic materials and adsorbents as well as water softeners in detergents [
9].
Karge et al. [
10], who pioneered the studies of the properties of faujasite-type (FAU-type) zeolites, found that, on NaX zeolites, OHˉ and HSˉ groups are produced on the surface of the sorbent as a result of the dissociative adsorption of H
2S molecules, indicating that these materials can be considered as good candidates for H
2S removal. Moreover, there is a considerable amount of studies probing the Claus reaction between H
2S and SO
2 to produce elemental sulfur in cation-exchanged zeolites [
11]. For example, Liu et al. [
12] investigated a 4A zeolite synthesized from attapulgite to remove H
2S from different industrial gases. The results revealed a maximum H
2S capture capacity of 8.36 mg g
−1 at 50 °C. Nevertheless, it is common knowledge that, although most zeolites can effectively adsorb acid species, such as H
2S, they require energetically demanding regeneration processes (typically above 450 °C). A different method for the reactivation of these solids is chemical treatment with hydrogen peroxide (H
2O
2), which also poses risks as it can damage the pores of the sorbent.
Another group of solid materials that have been thoroughly examined for desulfurization processes are metal oxides. Of note is the work or Westmoreland et al. [
13], who reported that Fe, Zn, Mo, Mn, V, Ca, Sr, Ba, Co, Cu, and W-containing metal oxides demonstrated thermodynamic feasibility for high-temperature desulfurization. However, high working temperatures result in loss of stability of the metal oxides [
14,
15] while at lower temperatures they experience poor capture performance [
16]. To avoid these limitations, mixed metal Zn-based oxides came to prominence for low temperature H
2S removal [
17].
Generally, the major shortcoming for this class of materials is the irreversible structure transformation caused by the chemisorption of H
2S, preventing their reactivation [
18]. In addition, their strong interactions with H
2S can stimulate the formation of other environmental contaminants, as is the case of Fe
2O
3 used for H
2S capture
, which produces SO
2 by the exhaustive oxidation of H
2S and elemental sulfur [
19]. Metal oxides also exhibit poor porosity and low surface areas (SSAs) which can also afflict their adsorption capacity. One strategy to enhance their gas capture performances and decrease the operating temperatures is to support these solids on a porous matrix, such as stable mesoporous materials (i.e., MCM-41 and SBA−15). Thus, this method introduces new perspectives to consider in order to accomplish sufficient gas removal although, in most cases, increases the implementation costs [
20]. Another way is the use of metal hydroxide [
21] or oxohydroxide [
22] composites, which offer the opportunity of operating in lower temperatures in comparison to those of pure metal oxides, but with average adsorption capacities. It should be stressed that, unlike Fe oxides, where increased temperatures are needed to provide sufficient gas capture performance, techniques based on metal oxides are yet to be commercialized for desulfurization processes. These technical limitations can also occur in the case of other H
2S adsorbents such as activated carbons, amino silanes, and ionic liquids [
23,
24,
25].
That said, a need of developing holistic technologies toward the design of materials that meet the industrial requirements and conform with environmental laws and regulation standards as well as exhibit improved gas capture performance arises.
A class of inorganic-organic hybrid materials, known as metal-organic frameworks (MOFs), with a crystalline nature that bear an open and porous framework have shown decent performance in CO
2 capture over the last twenty years, and recently they have been probed in the adsorption of acid gases (i.e., H
2S), showing promising potential [
26]. They consist of metal ions or clusters of metal ions, inorganic secondary building units (ISBUs), linked together by organic linkers. The organic units which are negatively charged molecules, are usually ditopic or polytopic organic carboxylates which, when linked to metal ions, form nodes that bind the arms of the linkers, resulting in architecturally robust crystalline MOF structures [
27,
28].
MOFs can exhibit significant advantages in gas selectivity and separation due to their large surface area (typically ranging from 1000 to 10,000 m
2/g), diverse structure, molecular dimensions, and tunable functionality [
29]. For instance, the chemistry of their pores can be improved by different methods, such as metal ion exchange. The ability to vary their size and nature, without changing their underlying topology and tailorability in terms of chemical functionality, affords control over the structure and properties required for given applications. MOFs can operate through physical or weak chemical adsorption interactions, thereby requiring lower regeneration energy in comparison to other methods (e.g., aqueous amine solutions) due to their lower heat of adsorption and heat capacities [
30,
31]. Another advantage of MOFs is that they can cover the full pore size gap between zeolites (microporous) and mesoporous silicas due to their highly tunable pores (0–3 nm up to 9.8 nm).
For most of the twentieth century, the synthesis of crystalline structures was rather fortuitous and the construction of materials with atomic precision posed an insuperable challenge. However, in 1959, Yukio and his co-workers, in an attempt to build large structures from well-defined building units, studied the crystal structure of bis(adiponitrilo)copper(I) nitrate [Cu(NC–CH
2–CH
2–CH
2–CH
2–CN)
2]NO
3. Using two-dimensional Fourier methods, the authors demonstrated that bis(adiponitrilo)copper(I) nitrate is orthorhombic and consists of infinite three-dimensional networks of complex ions of
and nitrate ions. They also showed that the copper ions were linked together by rod-like links of adiponitrile, creating a framework that was based on the widely known dense structure of diamonds, except here, due to the length of the units, the structure was open. Nevertheless, in this particular case, the openness of the structure resulted in interpenetration, thus preventing the space from being accessible [
32].
The work of Yukio et al. [
32] was followed by numerous efforts that were reported in the literature, but the term “metal-organic framework” itself was introduced for the first time in 1994, in a pioneering study carried out by Yaghi et al. [
33]. For this work, Omar Yaghi earned the moniker “the father of MOFs”. Two additional key works in the development of MOFs were reported in 1999 [
34,
35]. Firstly, Williams et al. [
34] synthesized a highly porous open-framework metal coordination polymer, referred as Cu-BTC (also stands for HKUST−1 and MOF−199), which was comprised of copper-based clusters and benzene tricarboxylate linkers. Not to be outdone, a few months later, Yaghi et al. [
35] reported MOF-5, a structure comprised of zinc-based clusters and benzene dicarboxylate linkers. The authors showed that compared to conventionally used microporous inorganic materials such as zeolites, these robust materials exhibited considerably higher SSAs and pore volumes.
The ability to alter the metrics of MOF structures, by using an expanded version of the parent organic linker, without changing their underlying topology, gave rise to the isoreticular (having the same network topology) principle and its application in making MOFs with even larger pore apertures and lower densities (reaching up to 98 Å and 0.13 g cm
−3) [
35]. Similar to the ISBUs, these organic linkers, appropriately functionalized, can also redound in promoting the selectivity towards target molecules [
36,
37,
38].
Intense efforts have been carried out to exploit the properties of MOFs in a large number of applications and in diverse fields, such as gas storage and separation, liquid separation and purification, electrochemical energy storage, catalysis, and sensing. However, despite the recent momentum, MOFs have yet to find a commercial impact, and concerns in terms of their cost-effectiveness and stability remain. For example, the capture of acidic H
2S has proven to be a tough challenge, largely owing to the formation of a strong and occasionally irreversible bond. This bond can cleave other coordination bonds between the metal centers and the linkers, leading to a structural disintegration of the MOF. Moreover, in the presence of water, the formation of acidic species can accelerate their structural degradation. In particular, the H
2S/H
2O reaction can result in the formation of HSˉ and H
3Oˉ species [
39]. Consequently, it is important to regulate the host–guest binding interactions between MOFs and H
2S. As a rule, in order to achieve a reversible process, these interactions should transpire through non-covalent bonding between H
2S and the functionalized linkers, such as hydrogen bonds, or by means of donor-acceptor bonds with the uncoordinated metal sites within the structure of the MOF [
40]. To predict these basal interactions density functional theory (DFT) and grand canonical Monte Carlo simulations (GCMC) have been widely implemented by numerous works, as will be discussed throughout the text below. Although the topic of desulfurization using MOFs has been thoroughly reviewed in the past [
41,
42,
43,
44,
45,
46,
47,
48], the work presented herein is the first to collect and critically review all works found in the literature that focus exclusively on H
2S capture. As such, attention has been paid to the structural characteristics of these materials that provide significant advantages in H
2S selectivity and separation, but also to issues related to the reversibility of the process. Efforts have also been made in identifying promising areas for future research.
2. H2S Capture via Materials of the Institute Lavoisier (MILs)
Hamon et al. [
49] pioneered the investigation of H
2S adsorption in MOFs. In particular, the group probed several MIL-series MOFs (MIL stands for materials of the Institute Lavoisier), including the small-pore (SP) MIL-53(Al
3+, Cr
3+, Fe
3+) and MIL-47(V
4+) (approximately 11 Å), as well as, the large-pore (LP) MIL−100(Cr) and MIL−101(Cr) (25 and 30 Å cage diameter and 4.8 × 5.8 and 12.5 × 12.5 Å pore apertures respectively), as seen in
Figure 1, for H
2S capture at 30 °C. SP MILs solids, presenting the same topology but containing two different metal centers, are built from corner-sharing chains of either (M
3+O
4(OH)
2) or metal oxide (M
4+O
6) octahedra interconnected through terephthalate moieties (SSA ≈ 1000 m
2 g
−1) (
Figure 2). LP MILs solids consist of trimers of chromium octahedra linked with trimesate (MIL−100) or terephthalate (MIL−101) (SSA > 2000 m
2 g
−1). The isotherms observed for MIL-47, MIL−100(Cr), and MIL−101(Cr) MOFs were type-I-shaped, illustrating that they can be considered as rigid structures. In contrast, two step adsorption isotherms were shown for MIL-53(Al, Cr, Fe), which the authors hypothesized to be caused by the strong polarity-based interactions (hydrogen bond) between H
2S molecules and -OH groups of the inorganic chains, resulting in pore occlusion at low gas or vapor loading.
Soon thereafter, these findings were claimed to be confirmed by the same group using a combination of IR measurements and modeling [
50]. Regarding the breathing MOF (MIL-53(Cr)), isotherms were simulated to represent both the narrow and the large-pore regimes in the experimental adsorption isotherm. Even though the simulations provided a decent match with the experimental results, the analysis failed to examine the ability to predict the structural transformation. CH
4 adsorption tests at room temperature before and after high pressure H
2S treatment, followed by vacuum desorption (8 h at 120 °C), showed that all the small pore solids, except for MIL-53(Fe), exhibited fully recovery. The latter material completely lost its adsorption capacity, owing to the formation of iron sulfide (FeS) and the crystallization of terephthalic acid at the surface of the adsorption cell. Simply put, the structure of MIL-53(Fe) collapses and forms FeS. On the other hand, such reversibility was not observed in the mesoporous type materials, i.e., MIL−100(Cr) and MIL−101(Cr) (hydrogen bond between μ-O group and H
2S molecules). The Henry’s constants were determined to be 72.9 mmol g
−1 MPa
−1 and 61.3 mmol g
−1 MPa
−1 for the MIL−100(Cr) and MIL−101(Cr) MOFs, respectively, while the maximum adsorbed quantities at 2 MPa were immensely high: 16.7 and 38.4 mmol g
−1, respectively. However, their inability to regenerate hinders the use of these larger pore MIL-series (mesoporous) as potential candidates for practical applications. The authors also reported that MIL-53(Al, Cr) and MIL-47 exhibited saturation loadings of 11.8, 13.1, and 14.6 mmol g
−1, respectively, which are considerably high in comparison to those of other conventional materials for H
2S uptake (i.e., activated carbons: 1.8 mmol g
−1, 13X zeolites: 5.62 mmol g
−1).
H
2S capture by MIL-53(Al), in both powder and pellet form, was also studied by Heymans et al. [
51]. The authors conducted a thorough experimental and theoretical study of MOF-53(Al) as an adsorbent for the removal of H
2S and CO
2 from biogas. High-pressure pure gas isotherms of CO
2, H
2S and CH
4 gave evidence of H
2S steep uptake in comparison to those of the two other species (
Figure 3). This pure compound study also revealed that MOF-53(Al) was fully regenerable at relatively low temperature (200 °C), suggesting that physisorption occurred. Alongside the simulations, the interaction of H
2S with the framework as reported by the Hamon group was corroborated [
49,
50]. It was also reported that the powdered MOF-53(Al) exhibited higher H
2S adsorption capacity, presumably owing to its increased SSA and pore volume compared to the pellet-form MOF-53(Al).
MIL-68(Al) was investigated at high H
2S pressures up to 12 bar at room temperature by Yang et al. [
52] using experimental and modeling (Grand Canonical Monte Carlo-GCMC) approaches. The three-dimensional networks of these materials consist of two types of channels, triangular (6.0–6.4 Å) and hexagonal (16–17 Å) running along the c axis coordinate system (
Figure 4). It was reported that only a specific number of the hydroxyls were available for interaction with H
2S, as confirmed by the simulated isotherm occurring by blocking the triangular channels, in harmony with the experimental isotherm. Moreover, the adsorption enthalpy of −21.6 kJ mol
−1 was calculated experimentally. Bearing in mind the modeling as well as the experimental approaches, one may conclude that the triangular pores of MIL-68(Al) were blocked by some remaining organic or solvent molecules owing to the incomplete activation of the material. This partially activated material was shown to be fully regenerable for at least five consecutive cycles, without loss of capacity. Nevertheless, it is unclear from this work whether the MIL-68(Al) can resist corrosiveness of H
2S if fully activated. However, it is also worth noting that MIL-68(Al) exhibited a rigid structure, as confirmed by its type-I-adsorption isotherm, despite the fact that it is considered as a polymorph of MIL-53(Al), which in comparison shows framework flexibility. This behavior highlights the significant role of an MOF’s framework topology in addition to the choice of the secondary building units.
Vaesen et al. [
53] investigated the adsorption performance of the amino-functionalized titanium terephthalate MIL−125(Ti)-NH
2 towards its parent MIL−125(Ti) analogue, for the concurrent elimination of CO
2 and H
2S from biogas and natural gas using a joint experimental/modeling approach. Generally, Ti MOFs bear two intriguing characteristics, namely that they are H
2S resistant and they can be scaled up to the multi-gram scale under ambient pressure conditions. MIL−125(Ti)-NH
2 consists of tetrahedral (4.7 Å) and octahedral (10.7 Å) cages, accessible through triangular windows of 5–7 Å. The cage diameters of the parent material (6 Å for the tetrahedral cage and 12 Å the octahedral cage) are moderately bigger than those of the functionalized material (
Figure 5). The pure-component adsorption tests at 30 °C and low pressures demonstrated reduced H
2S adsorption capacities for both MOFs. However, the adsorption enthalpies calculations for each gas denoted that the MIL−125(Ti)-NH
2 can achieve selective H
2S removal over CH
4 (∼70). In practical terms, they verified that the gas adsorption performances following H
2S and H
2O exposures were not compromised. Afterwards, in situ IR in the ν(H
2S) range revealed that for NH
2-free MOF the adsorbate/adsorbent interaction was established with the μ
2-OH groups through μ
2-OH·S(H
2S) interactions (2570 cm
−1), while for MIL−125(Ti)-NH
2, two additional interactions occurred; when H
2S acts as an H donor HS-H·N (from amino radical functionalization) with a 2550 cm
−1 band, and when the adsorbate molecule acts as H acceptor H
2S·H (NH
2). These interactions were corroborated by GCMC simulations. Compared to other adsorbents, such as 13X zeolites (∼45 kJ mol
−1), these amine-functionalized MOFs exhibit lower H
2S adsorption enthalpies (∼30 kJ mol
−1), which result in a lower energy footprint for recycling the sorbent. Moreover, the relatively high CO
2/CH
4 selectivity (∼7) of MIL−125(Ti)-NH
2 allows one to envision a one-step process for the synchronous capture of CO
2 and H
2S. Yet still, on the grounds that these MOFs are unstable in water vapor at temperatures above 100 °C, its improvement (structural stability) is a topic for further consideration. Overall, this work corroborates the simulations carried out by Yang et al. [
52], highlighting the positive role of amino functional group in capturing H
2S molecules.
Motivated by Biswas et al. [
54], who studied the adsorption properties of CO
2 using functionalized MIL-47-X (X = –Cl, –Br, CH
3, –CF
3, –OCH
3), Sokhanvaran et al. [
55] conducted molecular simulations (GCMC) to investigate the adsorption and selectivity of H
2S, CO
2, and CH
4 molecules and their mixtures on the functionalized MIL-47-X (X = –OH and –OCH
3) and pristine MOF MIL-47. The three studied MIL-47 MOFs exhibited type-I shaped isotherms, clearly indicating a rigid microporous structure. Results showed that the gas adsorption on MIL-47-X at low pressures was superior compared to that of the MIL-47. Even though the uptake at saturation decreased by functionalization, the adsorption selectivities of the MIL-47-X were higher when evaluated against the pristine MOF. In particular, using H
2S/CH
4, H
2S/CO
2, CO
2/CH
4 mixtures, the materials studied showed selectivity in the order of MIL-47 < MIL-47-OH < MIL-47-OCH
3. The authors attributed this finding to the fact that the adsorption isotherms showed greater affinity for polar H
2S over non-polar CO
2 molecules for the materials under consideration, probably owing to the strong interaction of H
2S with -OH groups. In combination with the results of the adsorption isotherms, the higher calculated isosteric heat of H
2S for all MIL-47 solids compared to that of CO
2 implied the stronger interaction between the H
2S molecules and the atoms in the framework of MOFs. The simulated adsorption isotherm of H
2S on the MIL-47, obtained at 30 °C, showed a decent agreement with the experimental data of the Hamon group [
49].
In another recent study focusing on the MIL-family, Pourreza et al. [
56] investigated the adsorption capacity of H
2S on three different MOFs, namely MIL−101(Cr), MIL−101(Cr)-SO
3H and MIL−101(Cr)-SO
3Ag in a dynamic adsorption system. In particular, the MIL−101(Cr)-SO
3H, which demonstrated high SSA, was used as a matrix to load Ag(I) by ion exchange (
Figure 6). Results showed that the breakthrough time measured for the Ag-free MOFs was short while the curve was moderately steep, suggesting that these sorbents presented poor capacity in capturing H
2S (MIL−101(Cr)—24.32 mg g
−1/MIL−101(Cr)-SO
3H—28.67 mg g
−1). Conversely, the breakthrough time for MIL−101(Cr)-SO
3Ag was longer, illustrating enhanced H
2S adsorption capacity (96.75 mg g
−1). Thus, although the Ag-functionalized MOF exhibited lower SSA (1534 m
2 g
−1) compared for example to the MIL−101(Cr) (1725 m
2 g
−1), its adsorption capacity was almost four times higher, indicating that sulfur molecules binded preferentially on the Ag(I) ions. Moreover, by performing 5 adsorption-desorption cycles using the MIL−101(Cr)-SO
3Ag, the authors showed that the capacity of the sorbent remained almost unchanged (approximately 96 mg g
−1). It was also noticed that increasing temperature had an adverse effect on H
2S adsorption, suggesting that physical adsorption predominated. Namely, the adsorption capacity of H
2S on MIL−101(Cr)-SO
3Ag decreased by 25.56 mg g
−1 following a temperature increase from 20 to 60 °C, in agreement with the exothermic nature of physical adsorption. The authors also applied the density functional theory (DFT) with a view to probe the adsorption mechanism of H
2S on these solids and reported that the Ag-modified sorbent could double the value of the H
2S adsorption energy compared to that of MIL−101(Cr)-SO
3H. Simply put, the Ag atoms could improve the retention of H
2S in the pores of the sorbent in comparison to SO
3H-modified linker. Overall, the electrostatic forces and the amount of charge transfer terms, in conjunction with polarization, which holds a significant role in H
2S-containing adsorption processes, could provide enhanced adsorption properties for MIL−101(Cr)-SO
3Ag in terms of H
2S removal.
However, serious concerns should be raised in regard to the likely future commercialization of MIL−101(Cr) due to the use of HF as modulator during its synthesis. Structurally, the nanoporous chromium terephthalate MIL−101(Cr) is composed by terephthalic acid (benzene−1,4-dicarboxylic acid), chromium salt (Cr(NO3)3·9H2O) and hydrofluoric acid (HF), as a modulator, in an aqueous medium. Even though HF can significantly enhance the synthesis efficacy and crystal size of this solid, it is a highly toxic and corrosive liquid capable of afflicting the nervous system. In this context, numerous attempts have been made to prepare HF-free MIL−101(Cr) sorbents.
For example, Leng et al. [
57] prepared and tested a solvent-free MIL−101(Cr). Noorpoor et al. [
58] prepared MIL−101(Cr) using a series of modulators (HF included) and assessed the H
2O uptake and physical properties of products in liquids. The authors found that the sorbents synthesized by ethanoic acid exhibited enhanced SSA (i.e., 2927 m
2 g
−1) and H
2O uptake (130 g g
−1), which corresponds to 40% and 73% improvement, respectively, in comparison to the HF-containing ones. Hu et al. [
59] probed highly porous HCl-assisted MIL−101(Cr). Results showed improved SSA and pore volume when evaluated against HF-assisted samples. Ren et al. [
60] used methanoic acid as modulator and found relatively promising H
2 capture for practical applications (at 1 bar and 77K the H
2 uptake was 1.9 wt.%). Jiang et al. [
61] also tested HF-free MOFs and found that these samples prepared using monocarboxylic acid as modulator presented decent SSA ranging from 2600 to 2900 m
2 g
−1. The study also reported increased CO
2/N
2 selectivity, presumably since these sorbents have the ability to tailor their particle size in a controlled manner (i.e., 19 to 84 nm). Zhao et al. [
62] and Zhou et al. [
63] used a combination of weak alkaline acetates, such as lithium acetate and potassium acetate, and acetic acid to obtain MIL−101(Cr) crystals and reported that the samples exhibited high SSA (3200–3500 m
2 g
−1) and tolerable production yield (over 60%).
Specifically, for H
2S removal, Alivand et al. [
64] conducted a very interesting work, in which they synthesized HF-free MIL−101(Cr) crystals using different HNO
3 concentrations to study the adsorption capacity of polar and non-polar molecules (i.e., H
2S, CH
4, CO
2, and N
2) on MIL−101-HNO
3 at different temperatures (i.e., 0, 10, 20 °C) and up to high pressures (up to 3500 kPa). Adsorption equilibrium data were well-fitted with Langmuir, Langmuir–Freundlich, and dual-site Langmuir-Freundlich models. Results showed that the MIL−101-HNO
3−1 (HNO
3/H
2BDC=1) exhibited the highest SSA (3841 m
2 g
−1) and pore volume (1.72 cm
3 g
−1), this being the optimal concentration of HNO
3. Moreover, at ambient temperature, the adsorption capacity of H
2S, CO
2, and CH
4 on MIL−101-HNO
3−1 was improved significantly (i.e., H
2S: 21.7%, CO
2: 29.3% and CH
4: 12.3%) at 100 kPa in comparison to that at 3500 kPa (i.e., H
2S: 7.1%, CO
2: 13.7% and CH
4: 11.5%). The authors also used ideal absorbed solution theory (IAST) to predict the selectivity of H
2S/CH
4, CO
2/CH
4, and CO
2/N
2 on the MIL−101-HNO
3−1, and found it to be 12.6, 14.2, and 336.1. It is noteworthy that these IAST predicted selectivities displayed an increment of approximately 167%, 22%, and 14% at 10 kPa. Furthermore, the authors stated that the improved adsorption capacity and separation ability of HNO
3−1-assisted sample over MIL−101-HF−1, is due to additional electrostatic adsorptive sites and more open Cr
3+ metal sites, which were both a result of the increased SSA (3609 to 3841 m
2 g
−1) and pore volume (1.55 to 1.72 cm
3 g
−1). Finally, the authors extolled the recyclability, as well as moisture and thermal stability of MIL−101-HNO
3−1, signaling that these materials can be regarded as superior alternatives for MIL−101(Cr) prepared by HF, in industrial desulfurization processes.
Kooti et al. [
65] synthesized new hybrids consisting of nanoporous carbon (GCKP2) and MIL−101(Cr) in different ratios (i.e., ranging from 10 to 50%) and used them as adsorbents for H
2S removal. A series of characterization methods were carried out to gain insight into the physicochemical properties of these samples. Results showed adsorption capacities of 10, 6.2, 7.9 and 6 mmol g
−1 at approximately 90 kPa for MIL−101 samples containing 10%, 30%, and 50% of GCKP2, respectively. On the other hand, the amount of H
2S adsorbed on pristine MIL−101(Cr) was higher, namely 10 mmol g
−1. The authors claimed that the decreased H
2S adsorption capacities of GCKP2-containing materials were due to their higher packing density (i.e., 1.4 times higher than MIL−101). Furthermore, the authors also obtained H
2S adsorption equilibrium isotherms for the above samples at low pressures (up to 10 kPa) by means of Langmuir, Freundlich, and Toth models. The nonlinear behavior of the isotherms, which is most likely ascribed to the contribution of physisorption to the process, illustrates the heterogeneity of adsorption sites for hybrids that contain GCKP2.
In a recent work, Díaz-Ramírez et al. [
66] reported the partial functionalization of MIL−101(Cr) with fluorine using 2,3,5,6-tetrafluoro−1,4-benzenedicarboxylate (BDC-4F) providing MIL−101(Cr)-4F(1%). Cyclic voltammetry measurements showed that the acidity of the metal centers of the modified form of the MOF was improved. The authors conducted several adsorption tests, including H
2S adsorption at 30 °C and 15% of H
2S volume. Results demonstrated the highest H
2S capture for the MIL−101(Cr)-4F(1%) (36.9 mmol g
−1) in comparison to all other mesoporous MOF materials mentioned in the literature. However, H
2S exposure partially damaged the crystallinity in both structures.
Xu et al. [
67] performed a computational study (Monte Carlo simulation) to investigate the adsorption and separation of H
2S in the monohalogenated MIL-47-(V)-X (X = F, Cl, Br) and its parent analogue MIL-47(V). Results showed that both halogenated and initial materials exhibit a considerably high H
2S adsorption capacity, in comparison to that of CH
4 and N
2. It was also noted that the adsorption isotherms of all gases in halogenated and initial MOFs are of typical type I, suggesting a rigid structure. Functionalization with halogens could improve the adsorption of H
2S in unit volume of sorbents, notably in low-pressure range, following an order MIL-47(V) < MIL-47(V)-F < MIL-47(V)-Cl < MIL-47(V)-Br, according with the increasing polarizability of the linkers, insofar as the H
2S mass fraction follows an opposite order MIL-47(V) > MIL-47(V)-F > MIL-47(V)-Cl > MIL-47(V)-Br, due to the rising mass density of MOFs after halogenation. The authors found that the four sorbents exhibited increased selectivity toward H
2S molecules in H
2S/CH
4 and H
2S/N
2 mixtures, at low temperature, high pressure, and high H
2S mole fraction.
More recently, Zheng et al. [
68] probed the selective oxidation of H
2S using the classical amino-modified NH
2-MIL-53(Fe) and its parent analogue MIL-53(Fe), prepared by means of a simple hydrothermal method. Various characterization methods were carried out, such as x-ray diffraction (XRD), Brunauer–Emmett–Teller (BET), scanning electron microscopy (SEM), Fourier-transform infrared spectroscopy (FTIR), CO
2 temperature-programmed desorption (CO
2-TPD), and x-ray photoelectron spectroscopy (XPS), to provide an exegesis regarding the physicochemical properties of the solids. It was reported that the insertion of the amines decreased the activation energies for H
2S oxidation and enriched the solid surface with moderate basic sites, thus, increasing the desulfurization performance. Specifically, the amino-functionalized material presented almost 100% sulfur selectivity in temperatures ranging from 130–160 °C, catalytically outperforming the commercial Fe
2O
3, MIL-53(Fe) and activated carbon that was evaluated against.
To further investigate the selective oxidation of H
2S to sulfur, the same group prepared a porous MIL−100(Fe) with coordinatively unsaturated (CUS) Fe
2+/Fe
3+ sites (CUS-MIL−100(Fe)) [
69]. It was reported that the functionalized materials exhibited high desulfurization performance and 100% sulfur selectivity at a temperature range of 100–190 °C and space velocity of 6400 h
−1, outperforming both the commercial Fe
2O
3 and MIL−100(Fe). It is noteworthy that the H
2S conversion over MIL−100(Fe) and CUS-MIL−100(Fe) remained stable at 100% in a continuous run of 100 h, while sulfur selectivity remained higher than 93.9% and 95.1%, respectively, indicating that these catalysts may be regarded as ideal candidates for practical applications. This enhanced catalytic performance of the CUS-MIL−100(Fe) can most likely be ascribed to the ordered pore structure of the MOF, which provides a chemically stable environment for the active pore sites, thereby hampering the development of potential side reactions.
3. H2S Capture via HKUST−1 (Hong King University of Science and Technology)
Cu-BTC [Cu
3(TMA)
2(H
2O)
3]
n polymer, which also stands for MOF−199 or HKUST−1 (Hong King University of Science and Technology), consists of Cu nodes and organic linkers (benzene-tricarboxylate, BTC), with each Cu coordinated with four oxygen atoms and water molecules (
Figure 7) [
70]. The framework of this Cu-based material contains a bimodal pore size distribution, i.e., a large cage with 9 Å diameter and small pores with 3.5 Å diameter [
71,
72]. In addition, they exhibit a reasonable degree of thermal stability (up to 240 °C). In the recent past, various scientific works turned their attention to probing several aspects of H
2S adsorption using this hybrid material [
73,
74,
75,
76,
77,
78,
79,
80,
81,
82,
83,
84,
85,
86,
87,
88,
89].
Petit et al. [
73] first investigated HKUST−1 and its composites with graphene oxide (GO) (5 to 46 wt.% graphene oxide) for H
2S removal at ambient conditions using a flow of H
2S (i.e., 1000 ppm, 250 ml min
−1) diluted in moist air. The breakthrough capacities obtained denoted that the composite with 5 wt.% of GO exhibited the highest H
2S uptake (199 mg g
−1), while the capacities of HKUST−1 and GO were significantly lower (92 mg g
−1 and 9 mg g
−1, respectively). It was reported that H
2O, which is present due to bed pre-humidification, does not prevent the H
2S adsorption (2.7 mmol g
−1). Conversely, it promotes its retention by means of dissolution in the water film. However, further H
2S tests in absence of H
2O would help to better elucidate the role of humidity. Following the H
2S adsorption tests, the formation of CuS (black powder) was observed, illustrating the major disadvantage associated with the H
2S adsorption on HKUST−1. Along these lines, the authors proposed that a possible H
2S adsorption mechanism for HKUST−1 and the composite materials entails the substitution of H
2O molecules coordinated to the Cu(II) metal centers by H
2S. This mechanism hinted that the MOF structure is vulnerable to H
2S exposure and may collapse. These observations were confirmed by XRD and IR spectroscopy. The authors also observed a decrement in pH during H
2S adsorption, which is probably attributed to the acid nature of benzene-tricarboxylate linkers (no more coordinated to Cu). Overall, H
2S capture in the presence of open metal sites can afflict the stability of the MOF framework. Results of this study show that MOFs based on metal clusters with open metal sites particularly confine the employment of these solids for H
2S removal processes. However, the insertion of small amounts of GO can alleviate this limitation.
Pokhrel et at. [
74] investigated the H
2S adsorption on MOF−199 and MOF−199/GO. The authors argued that the GO did not favor the adsorption of H
2S and attributed the observed capacity of MOF−199/GO was due to the presence of well dispersed crystals of the MOF. It was also reported that both physisorption and reactive adsorption occurred, owing to the unsaturated Cu sites in the MOF structure, which interact with H
2S molecules. However, due to the fact that physisorption was the dominant capture mechanism, an increasing temperature led to favoring kinetics, but decreasing H
2S capture. In the presence of moisture, the stability of MOF−199 and MOF−199/GO composites exhibited gradual degradation, suggesting that the unsaturated atoms of Cu in the metal center of MOF−199, having a coordination number of 4, could bind with H
2O molecules via chemisorption.
In a more recent study by the same group [
75] in-situ grown MOF−199 on GO, was doped with polyethyleneimine (PEI), and hybridized with GO along with functionalized GO for H
2S capture. It was reported that, at ambient conditions, the pristine MOF−199 exhibited lower H
2S adsorption capacity (0.5 mmol g
−1) in comparison to that of the MOF−199/GO, which was pre-functionalized with low molecular weight PEI. In addition to the 80% improvement in H
2S capture, the sorbent showed considerably enhanced sorption kinetics. Furthermore, MOF−199/GO hybrid sorbent exhibited 27% increase in H
2S capacity (2.1 mmol g
−1) in comparison to that of the pristine material by rising the temperature (150 °C). These GO-containing hybrid materials which are amenable to functionalizing both the GO support and MOF crystallites may be considered as promising alternatives for the removal of H
2S.
Ebrahim et al. [
76] studied composites consisting of HKUST−1 and S and N doped graphite oxides. The authors reported that the HKUST−1 composites exhibited improved adsorption performance in comparison to the parent MOFs, presumably owing to the insertion of polar groups and development of microporosity by linkages between the S and N groups of the modified GO and Cu centers of HKUST−1. In the presence of moisture, S doped GO/HKUST−1 and N doped GO/HKUST−1 showed increased H
2S breakthrough capacities of 241 ± 6.44 mg g
−1 and 125 ± 2.17 mg g
−1, possibly due to the synergistic effect of the modified graphene phase. Synopsizing, H
2O present, the adsorption capacities were improved in comparison to those in dry conditions, due to the dissolution and dissociation of H
2S. The occurrence of acid-base reactions led to the formation of Cu/S salts instead of H
2S, thus preventing the direct attack of H
2S molecules on Cu centers of HKUST−1.
Zhang et al. [
77] studied the effects of temperature and pressure on the performance of H
2S adsorption in HKUST−1 using GCMC simulations. The interaction between adsorbate and adsorbent was further investigated through DFT calculations. The authors found that the H
2S adsorption capacity on HKUST−1 increases with increasing pressure. When the pressure was higher than 0.4 Mpa, the adsorption tended to equilibrium. At low pressures, the capacity of the sorbent in capturing H
2S molecules is strongly affected by the frameworks containing the binding sites of Cu dimers and benzene−1,3,5-tricarboxylic acid (trimeric acid). At high pressures, HKUST−1’s free volume also contributes to the adsorption performance of the MOF. Conversely, by increasing temperature, the H
2S adsorption capacity of HKUST−1 decreases. This behavior is anticipated as the increased kinetic energy of H
2S molecules compromised the effectiveness of the interactions between the framework atoms of HKUST−1 and guest H
2S molecules. Consequently, residence time of H
2S on the surface of HKUST−1 was shortened, resulting in poor adsorption performance. The interactions between H
2S and organic linkers were weak (>−14.469 kJ/mol). It was reported that the most stable adsorption configuration for H
2S to adsorb onto the organic linker, is the one with H
2S located in the same plane as the benzene ring with hydrogen atoms of H
2S nearby the oxygen atom of benzene−1,3,5-tricarboxylic acid. In addition, in the case that H
2S adsorbs onto the Cu-Cu bridge, the binding energies of the modes with hydrogen placed inward of the Cu dimer are typically smaller in comparison to that where hydrogen is outward. The smallest binding energies (<−50 kJ/mol) were observed when the adsorption occurred on the top of the copper ion, probably owing to the trend of forming a saturated six-coordinated configuration.
A quite interesting work was conducted by Watanabe et al. [
70] who tried to calculate the binding energies of several molecules (i.e., H
2S, H
2O, CO, NO, pyridine, C
2H
2, and NH
3) on HKUST−1 using plane wave periodic density functional theory (DFT). H
2S was found to exhibit a binding strength of 0.49 eV on Cu dimers, very close to that of H
2O, which has shown a large affinity for the metal center of HKUST−1 (
Figure 8). Employing GCMC simulations, at 27 °C and even at partial pressures less than 0.01 kPa, H
2S was shown to adsorb at approximately 50% of active Cu sites, illustrating that HKUST−1 is a promising material for removing H
2S from various industrial gases. It is worth mentioning that in the cases when two adsorbate molecules per dimer were adsorbed, the second molecule on a Cu dimer exhibited a lower binding energy. The effect of physisorption on the adsorption isotherm was not considered in this work.
Similar to the work discussed above [
70], several other simulation studies reported that H
2O molecules bind preferentially to the Cu atoms. Castillo et al. [
78] noticed that H
2O exhibits strong affinity for the metal centers in HKUST−1 compared to N
2, CO
2 or hydrocarbons. Kristof et al. [
79] also corroborated that the H
2O molecules interact more strongly with Cu atoms, whilst H
2S molecules are accommodated at the center of the cages of the framework. Supronowicz et al. [
80] observed that the interaction energy of H
2O (−60.9 kJ mol
−1) adsorption on HKUST−1 is higher than that of H
2S (−52.25 kJ mol
−1), hinting that H
2O predominated. Gutiérrez-Sevillano et al. [
81] developed H
2S models to analyze whether a model that provides a precise dipole moment could predict an adsorption behavior (i.e., strong interaction of H
2S with HKUST−1), in agreement with the experimental observation. The authors applied generic force fields for the material under consideration and found that the results appear in line with other relevant adsorption studies [
52]. Moreover, DFT simulations showed that H
2O, on the metal centers of HKUST−1, is energetically favored over the adsorption of H
2S. Ab initio molecular dynamics of the molecules adsorbed on the model cluster show that the distance between H
2S molecules and the metal center of HKUST−1 is longer by approximately 2.6 Å in comparison to H
2O. Overall, the observations obtained from the simulation studies are in direct contradiction with the experimental results, which suggest the dominance of H
2S physical adsorption on HKUST−1, thereby failing to explain that H
2S converts Cu in HKUST−1 to black CuS in the presence of H
2O. It is worth noticing that although the theoretical studies carried out so far consider the host-guest interactions, this is not the case for the thermodynamics governing the reaction, which are needed to explain the CuS formation. In concluding, experimental studies denote that H
2S molecules show stronger affinity for Cu atoms in the center of HKUST−1, which make them capable of displacing H
2O molecules.
Several works can be found in the literature have investigated the interaction between H
2S molecules and HKUST−1 experimentally. For example, Ethiraj et al. [
89] corroborated the structure collapse of HKUST−1 due to the formation of CuS. They reported that at higher equilibrium pressures (20–60 mbar), HKUST−1 exhibits a structural dilapidation possibly to owing to the conversion of Cu into CuS, while at lower equilibrium pressures (under 5 mbar) a stepwise structural distortion occurs. Alvarez et al. [
83] suggested a similar adsorption mechanism for H
2O molecules, concluding that the presence of H
2O also compromises the stability of the HKUST−1.
Li et al. [
84] studied the removal of H
2S using HKUST−1 by activating the sample prior to H
2S adsorption tests. The authors reported the positive effect of preheating treatment on HKUST−1due to the release of air and solvent molecules. In particular, the breakthrough capacity improved by 38% at 180 °C (optimum activation temperature) in comparison to the material that did not undergo activation and dropped by 10% at activation temperature of 200 °C. In addition, breakthrough tests demonstrated that the breakthrough capacity of HKUST−1 accreted with increasing temperature (from 30 to 80 °C). The aforementioned annotations highlighted the antagonistic adsorption between these two polar molecules (H
2S/H
2O) and that an increase in either adsorption temperature or activation temperature can lead to reducing the amount of adsorbed H
2O on the sorbent, thus improving the H
2S uptake. In the opposite direction, Peterson et al. [
85], who also experimentally investigated the same system under dry (0% water) and wet conditions (80% H
2O), reported that H
2S adsorption capacity was not affected in both cases. Therefore, the contradicting results imply that there is room for better understanding the role of humidity in desulfurization using HKUST−1.
Zhang et al. [
86] prepared a series of amine modified HKUST−1 via impregnation to remove H
2S at ambient temperature. It was found that HKUST−1 could maintain its structure following modification with tertiary amine triethanolamine (TEA). However, this was not the case for amine mono-ethanolamine (MEA) and secondary amine diethanolamine (DEA) modification which led to the dilapidation of the HKUST−1’s structure, probably owing to the strong interactions occurred. Breakthrough results demonstrated that HKUST−1 functionalized by TEA exhibited higher H
2S uptake (2.74 mmol g
−1) in comparison to that of the parent sample (1.67 mmol g
−1). Moreover, modification with DEA and MEA resulted in the decrease of H
2S uptake to 0.58 mmol g
−1 and 0.83 mmol g
−1, respectively. Simulations showed that the binding energies of H
2S adsorption on TEA/HKUST−1 were larger in comparison to those on pristine material, which presumably had a positive effect on H
2S adsorption. The improved binding energy of H
2S on the TEA/HKUST−1 can most likely be ascribed to the hydroxyl of TEA.
6. H2S Capture via Universitetet I Oslo MOF (UiO-66)
UiO-66 (Universitetet I Oslo) is a MOF comprised of [Zr
6O
4(OH)
4] clusters (octahedra) that are 12-fold connected with adjacent octahedra through BDC struts (linkers), resulting in a highly face centered cubic structure (
Figure 12) [
93] and one may find a few works in the literature that studied the removal of H
2S using UiO-66 MOFs.
Li et al. [
94] performed molecular simulations to investigate the adsorption performance of the pristine UiO-66(Zr) and its functionalized derivatives in capturing sulfur from binary gas mixtures. UiO-66-(COOH)
2 and UiO-66-COOH exhibited the highest H
2S adsorption capacity in comparison to that of other tested sorbents presumably due to their higher adsorption isosteric heats. The isosteric heat of adsorption at infinite dilution and radial distribution functions imply that the hydrophilic groups and polar H
2S molecules strongly interact with one another, promoting H
2S uptake.
Bhatt et al. [
95] revealed a series of isoreticular rare earth MOFs, namely RE-fcu-MOFs, and argued that these were highly stable and eclectic towards H
2S molecules, with a face centered cubic (fcu) underlying topology (
Figure 13). Having distinct pore-aperture sizes ranging from 4.7–6.4 Å and different functionalities these materials were studied for the removal of H
2S from CO
2 and CH
4 containing gases using multiple cyclic adsorption breakthrough tests. In particular, the breakthrough tests were conducted on three isoreticular fcu-MOFs with different in length organic linkers (between 5 and 8.5 Å), using a simulated gas mixture with compounds typically found in natural gas streams CO
2/H
2S/CH
4 (5%/5%/90%) under various adsorption-desorption conditions. Among the three selected Re-fcu-MOFs, the ones that were assembled by using the naphthalene moiety seemed to exhibit improved H
2S uptake highlighting the crucial role of the pore system and functionality. Moreover, the fcu-MOF demonstrated decent H
2S uptake with an increased H
2S/CO
2 selectivity, outperforming conventional materials (i.e., zeolites and activated carbons) in many aspects. It was also stated that the sufficient H
2S/CO
2 and H
2S/CH
4 selectivity of these materials can afford great opportunities to produce H
2S, limiting the impurities of other compounds, such as CO
2, to acceptable levels.
Recently, Daraee et al. [
96] prepared TOUO-x nano composites (TiO
2/UiO-66, x: 1, 3 and 5 wt.% of TiO
2) and investigated their sulfur uptake performance. The results were then evaluated and compared with pristine UiO-66 at two different temperatures (30 and 50 °C) under space velocity of 30,000 h
−1 and H
2S concentration of 4000 ppm in the gas stream. Breakthrough results showed that rising temperature resulted in lower H
2S uptake for the nanocomposites under consideration, hinting that the physical adsorption mechanism predominates. TOUO−1 outperformed the other tested materials (0.21 g S g
−1) probably due to the higher SSA (1171 m
2 g
−1) in comparison to TOUO-3 (986 m
2 g
−1) and TOUO-5 (652 m
2 g
−1). Moreover, it is claimed that the insertion of metal active sites of titanium on UiO-66 contributed to the enhancement of the properties of the synthesized nano-composite adsorbents. Energy-dispersive X-ray spectroscopy (EDX) gave evidence for the presence of sulfur suggesting that there is a strong interaction between the sulfur molecules and the sorbent, probably due to chemisorption. However, the authors reported that following two cycles of H
2S adsorption tests, the capacity of TOUO−1 was almost fully regenerated (its capacity decreased by approximately 3–5%), which somehow contradicts the fundamental knowledge that chemisorption results in irreversible structure transformation.
Trying to investigate the antagonistic adsorption between CO
2 and H
2S, Huang et al. [
97] synthesized core-shell-structure H
2S imprinted polymers (PMo
12@UiO-66@H
2S-MIPs) based on the surface of UiO-66 modified by phosphomolybdic acid hydrate. Initially, it was reported that using H
2O as substitution template for H
2S can overcome issues associated with the toxic and instable nature of H
2S. It was shown that PMo
12@UiO-66@H
2S-MIPs exhibited increased H
2S uptake (24.05 mg g
−1) in comparison to that of the carrier PMo
12@UiO-66, indicating that the desulfurization performance of the latter was further enhanced by the H
2S imprinted polymers. Moreover, PMo
12@UiO-66@H
2S-MIPs showed high H
2S uptake at room temperature even in the presence of humidity and had decent separation towards H
2S/CO
2. A noteworthy finding was that air purge regenerated PMo
12@UiO-66@H
2S-MIPs at 180 °C and O
3 treatment and ambient temperature demonstrated stable desulfurization ability after six cycles. The authors observed the transformation of H
2S into sulfur following adsorption, as well as that the PMo
12 was the redox agent in the desulfurization process.
8. Additional Works
Peng et al. [
102] performed GCMC simulations, wherein the dispersive interactions between atoms are delineated by directly applying the universal force field (UFF) from Rappe et al. [
103], for zeolites, porous carbons and MOFs to remove H
2S and CO
2 from ternary mixtures (i.e., CH
4, CO
2, H
2S). It was reported that by considering only the desulfurization capacity, zeolites are superior to porous carbons and MOFs, whilst for concurrent desulfurization, MOFs outperformed the other two types of sorbents. MOFs also experienced easier regeneration in comparison to zeolites, since the adsorbed molecules can be easily and quickly released by increasing temperature. Moreover, by increasing the amount of H
2S in the bulk phase, the selectivities of MOFs decrease with a slower rate, in comparison to those of zeolites, which shows that the MOFs are better candidates for the desulfurization of gases with impurities in high concentrations. Along these lines, socMOF and indium-based rho-zMOF, which is representative of zeolite-like MOFs (zMOFs) with a topology of rho-zeolite, have been found to exhibit high selectivity (100 and 170, respectively) and capacity (1.8 and 2.6 mmol g
−1).
In an attempt to gain better insights into the desulfurization performance of MOFs, Chen et al. [
104] probed the contribution of each fragment of these hybrid sorbents (metal center structures, metal ions, organic linker) to the adsorption of sulfur compounds (H
2S, dimethyl sulfide (CH
3SCH
3), ethyl mercaptan (CH
3CH
2SH)) using DFT. The authors reported that the capture of sulfur compounds on organic linkers, NH
2-BDC, BDC (1,4-benzenedicarboxylate), and NDC (dimethyl 2,6-naphthalenedicarboxylate) is realized through physical adsorption. NH
2-BDC exhibited the strongest binding strength owing to the functional amino-group, which enhances the polarity in the linker and thereby improves its interaction with sulfur. Moreover, metal centers with structure M and M-M in HKUST−1 and MOF-74 showed strong binding strength. The chemical interaction between the metal centers of HKUST−1 and MOF-74 and the adsorbed sulfur compounds was evidenced by the fact that the sorbents exhibited their weakest binding energy in −51.8 kJ mol
−1. Meanwhile, it was revealed that MOFs containing Fe demonstrated stronger binding with sulfur compounds in comparison to those containing Cu and Zn. However, the strong binding of H
2S to Fe also implies a risk of iron sulfide formation. Overall, the adsorption strength of MOFs with sulfur species followed the order of: MOFs with coordinatively unsaturated sites > MOFs with NH
2-BDC linker > MOFs with saturated metal center > MOFs with the organic linkers without substituent group (
Figure 14).
Liu et al. [
105] probed, both experimentally and theoretically, the adsorption mechanism and stability of eleven different MOFs when challenged with H
2S separation. The H
2S breakthrough tests and the experimental N
2 isotherms were conducted at 25 and −196 °C, respectively. Most of these MOF-based materials exhibited on-off increased capacity and selectivity to H
2S/CO
2. In particular, Mg-MOF-74, MIL−101(Cr), UiO-66, ZIF-8, and Ce-BTC experienced complete reversible physical sorption, while irreversible H
2S chemisorption was observed for Cu-BTC, Zn-MOF-74 and MOF-5 (albeit, leading to high H
2S uptake and H
2S/CO
2 selectivity). Nevertheless, as is usually the case, the presence of end products (e.g., ZnS, CuS) may afflict the structure of the sorbents and limit their further exploitation. Regarding HKUST−1, Cu-BDC(ted)
0.5, Zn-MOF-74, MIL−100(Fe) gel, and MOF-5, and despite the fact that they experienced the same irreversible chemical adsorption, their structures remained stable. It was also observed that in the case of MIL−100(Fe) the redox reaction between Fe and H
2S (i.e., H
2S reduces Fe(III) to Fe(II)) can result in the formation of octasulfur (S8). The authors tried to shed light into the possible H
2S adsorption mechanisms, using DFT, molecular dynamics and dynamic separation experiments, for Mg-MOF-74, MIL−101(Cr), UiO-66, ZIF-8, and Ce-BTC. It was reported that the chemical composition of the MOF holds a key role in the H
2S adsorption. For instance, in MIL−101(Cr) and Mg-MOF-74 with open metal sites, there are electrostatic interactions between the unsaturated coordination sites and the H
2S molecules (Μ
δ+·SH
2δ−), resulting in the physisorption of H
2S. On the other hand, in MOFs with no evident access to the open metal sites, H
2S capture was realized through CH·π hydrogen bond, where H
2S is the donor and the organic linker (aromatic rings) are the acceptors. In respect to the Ce-BTC sorbent, which affords access to open metal sites, high band gap (1.16 eV) in the highest occupied molecular orbital (HOMO)-lowest unoccupied molecular orbital (LUMO) energy levels of the p orbital from H
2S and f orbital from Ce, leads to the physisorption of the target molecule (H
2S). Eventually, Cu-BDC(ted), which contains a metal oxide cluster paddlewheel-type [Cu
2(O
2C-)], experienced an H
2S/amino group interaction that can stabilize the structure and produce amine-hydrosulfide adduct.
Eddaoui et al. [
106] introduced fluorinated MOFs in polymer membrane with a view to remove acid gases (H
2S and CO
2) from natural gas. Namely, they employed NbOFFIVE−1-Ni (KAUST-7) and AlFFIVE−1-Ni (KAUST-8) with a general formula of [M
1(M
2F
x)(pyrazine)
2]
n where M
1 is Ni, and M
2F
x are the pillars [NbOF
5]
2− and [AlF
5]
2−, respectively. The results of adsorption isotherms for H
2S, CO
2, and CH
4 at 35 °C showed increased CO
2/CH
4 selectivity and improved capacity in capturing H
2S molecules for both fluorinated materials, as well as adequate stability following H
2S exposure. Thereby, selective transport of these gases via a membrane process was studied. The authors used 6FDA-DAM polyimide polymer matrices for the synthesis of mixed-matrix membrane (MMM), and they showed that incorporating these fluorinated MOFs in these polymer matrices resulted in enhanced diffusion for the acid gases (CO
2/H
2S) in comparison to that of CH
4. In this regard, KAUST-7 and KAUST-8 exhibited improved molecular diffusion, through the membranes presumably due to the well-defined channels that provided.
In a very promising work, Belmabkhout et al. [
107] investigated different fluorinated MOF-based materials for the removal of H
2S and CO
2 with a view to upgrade biogas, natural gas, and refinery-off-gas. It was reported that the fluorinated MOF, AlFFIVE−1-Ni allowed the synchronous and equally selective removal of these acid gases (i.e., H
2S and CO
2) from streams with high CH
4 concentrations in a single adsorption step. On the other hand, SIFSIX-2-Ni-I exhibited preferential binding only towards H
2S molecules. A significant finding was that AlFFIVE−1-Ni demonstrated 100% regenerating efficiency at mild conditions (105 °C). Overall, simultaneous removal of both H
2S and CO
2 can be attained by means of the integrated favorable sites for H
2S and CO
2 uptake in a confined pore system.
Belmabkhout et al. [
108] also studied In
3+-, Fe
3+-, Ga
3+-, and the newly isolated Al
3+-based isostructural square octahedral (soq)-MOFs with a view to produce high-quality hydrocarbons (CH
4, C
3H
8 and n-C
4H
10, and olefins), from H
2S-containing gas streams (
Figure 15). It was reported that only Al
3+- and Ga
3+-based soc-samples could exhibit structural stability during the interaction with H
2S molecules. Pure-component adsorption isotherms were measured for H
2S, CO
2, and CH
4 in Ga-soc-MOF at ambient temperature and, differently from CO
2 and CH
4, H
2S exhibited steep adsorption. The authors also conducted breakthrough experiments for a ternary rich in CH
4 mixture (i.e., 5% of H
2S, 5% of CO
2, 90% of CH
4) for six adsorption-desorption cycles under various conditions in a cyclic fashion, which is relevant to temperature and vacuum swing regeneration. Complete recyclability of the MOF was accomplished at 160 °C under He flow. H
2S breakthrough time of 40 min, which was considerably longer in comparison to CH
4 (0 min) and CO
2 (5 min) breakthrough times, illustrates very high H
2S/CH
4 and H
2S/CO
2 selectivities. It was also claimed that Al-soc-MOF exhibited reasonable degree of structural stability for temperatures up to 300 °C and relative humidity (RH) as high as 95%. Nevertheless, the H
2O stability of the Ga-soc-MOF was not mentioned.
Lee et al. [
109] investigated, both experimentally and theoretically, the chemisorption of H
2S using tree different MOFs (MOF−199, MOF-5, and UiO-66-NH
2), two covalent-organic polymers (COPs: CBAP−1 (EDA) and CBAP−1 (DETA)), and two commercial sorbents (Carbopack-X and activated carbon (AC)) in an attempt to evaluate the adsorptive performance of these samples (inlet stream partial H
2S pressure: 10 ppm in 1 bar of N
2, temperature = 25 °C). Despite the fact that the adsorption capacity at 100% breakthrough volume (BTV) has been mentioned as the most common fashion to assess the performance of a sorbent, the authors employed the 10% BTV for that purpose, which has been claimed as a more practical performance metric [
110,
111]. Most adsorbents exhibit a steep rise in breakthrough levels following the initiatory adsorption event (≤10% breakthrough). That said, the actual potential of an adsorbent may be evaluated more accurately with the earlier attainment of breakthrough level (e.g., 10%). The authors noted that MOF−199 exhibited a noticeable advantage in terms of the adsorptive performances determined at BTV10 over the other tested samples. In particular, the materials followed the order MOF−199 > MOF-5 > AC > UiO-66-NH
2 > CBAP−1 (EDA) > CBAP−1 (DETA) > Carbopack-X. Considering the isotherm modeling, it was shown that MOF−199 can effectively adsorb H
2S molecules by means of formation of irreversible chemical bonds with Cu-Cu site bridge. A significant divergence in the partition coefficients between the experimental and theoretical data was also noted, probably because the crystal lattice of MOF−199 is more stable under real-world conditions. Finally, the authors hypothesized that Mg/Cu-based dual metal site MOFs could be a way to surpass the present limitations (i.e., collapsed framework due to chemisorption) in H
2S adsorption. At this point it should be stressed that, to the best of our knowledge, evaluating capacity at 10% BTV is not necessarily panacea. Capacity at full breakthrough volume gives the equilibrium capacity and has a thermodynamic meaning, while capacity at 10% BTV attempts to simulate the impact of the mass transfer zone in a real separation process. Nevertheless, considering the MOFs are unused or fully regenerated in a powdered form, then the mass transfer zone in the lab breakthrough experiments is in any case not representative.
Li et al. [
112] conducted a computational study on composite materials based on Cu-TDPAT MOF and ionic liquids. The MOF was comprised of metal oxide clusters in a paddlewheel-type form [Cu
2(O
2C-)] linked to a 2.4.6-tris(3,5-dicarboxylphenylamino)−1,3,5-triazine (TDPAT) linker. Structurally, it has a three-dimensional network with tree different pores, as well as shortened tetrahedral and octahedral cages with sizes of 9.1, 12.0, 17.2 Å, respectively. In respect to ionic liquids, the authors altered the anions (i.e., bis[(trifluoromethyl)-sulfonyl]imide [TF
2N]ˉ, tetrafluoroborate [BF
4]ˉ, chloride [Cl]ˉ, with a view to selectively adsorb H
2S in a binary mixture of H
2S/CH
4, but they maintained the same cation [BMIM]
+ (1-n-butyl-3-methylimidazolium). Results showed that the anions, exhibited an affinity towards copper atoms while cations were accommodated nearby N atom from triazine. Regarding the [BF
4]ˉ, it was the only anion that located close to the N atom from amino group. Radial distribution functions (RDF) demonstrated distances between anions and copper atoms in the order of [Cl]ˉ < [BF
4]ˉ < [PF
6]ˉ < [TF
2N]ˉ, being in consonance with the trend of ionic radius. Consequently, it can be concluded that the size of the anions holds a key role in the accommodation of ionic liquids into the pores of MOF. Moreover, [Cl]ˉ anion was only found on the tetrahedral cavity, probably since it is sterically blocked, whilst the other anions occupied all pores. In addition, the isosteric heats of adsorption were measured at infinite dilution (Q°
st) for CH
4 and H
2S for both Cu-TDPAT and Cu-TDPAT-composites. Furthermore, computational observations from simulated adsorption selectivities displayed that the increased selectivity of H
2S over CH
4 was ascribed to the fact that H
2S, contrary to CH
4, is polar. In conclusion, this type of materials based on ionic liquids could be an alternative for H
2S capture as they have the potential to protect the metal cluster with open metal sites, preventing the structural disintegration by nucleophilic onset of H
2S molecules. However, the regenerability of these materials under the appropriate conditions was not investigated in this work.
Sánchez-González et al. [
113] mentioned the use of Mg-CUK−1 MOF (CUK = Cambridge University-KRICT), which is built from a 2,4-pyridinecarboxylate (2,4-pdc) linker and Mg(II) octahedral centers, linked to infinite chains of [Mg
3(μ
3-OH)]
5+ clusters by μ
3-OH groups placed inside the pore walls, for the removal of acidic gases, such as H
2S, in the presence of relative humidity. In particular, to elucidate the impact of humidity, the authors attempted to characterize the adsorption/desorption properties of H
2O. According to the H
2O adsorption isotherms, the highest H
2O uptake was almost 19.1 mmol g
−1 and was not affected after several adsorption/desorption cycles. Afterwards, they tried to delineate the H
2S sequestration performance on activated Mg-CUK−1 MOF following several adsorption/desorption cycles. Results showed that the H
2S capture increased with increasing the H
2S concentration. Namely, at 6% volume the H
2S capture reached at 1.41 mmol g
−1, while at 15% volume was equal to 1.41 mmol g
−1. Furthermore, with a view to corroborate that this solid can maintain its crystallinity after H
2S exposure, the authors conducted five adsorption-desorption cycles combined with PXRD analyses. It was found that the Mg-based material maintained its capacity in capturing H
2S between the cycles, suggesting that the structure was not afflicted during the process. Finally, GCMC results demonstrated that H
2S molecules were mainly bonded via hydrogen bonding (μ
3-OH···SH
2). In this case, the hydroxyl group, which is located at the wall of the MOF, is the proton donor. At higher H
2S loadings the aforementioned interaction becomes weaker (−23.3 kJ mol
−1), which explains the easiness of the Mg-CUK−1’s regeneration between adsorption/desorption cycles.
Zárate et al. [
114] investigated H
2S capture in an Al(III)-based MOF, named MIL-53(Al)-TDC (TDC = 2,5-thiophenedicarboxylate) by performing both experimental and theoretical tests. Structurally, this material contains [AlO
4-trans-(μ-OH)
2] octahedra in which Al(III) center is coordinated by six oxygen atoms from four different TDC linkers and two hydroxyl (μ-OH) groups. The capture performance of MIL-53(Al)-TDC was firstly evaluated by breakthrough tests conducted at 30 °C, 1 bar and 5% of H
2S volume, the results of which suggested increased H
2S capture of 18.13 mmol g
−1. Similar results (18.1 mmol g
−1) were obtained by kinetic gravimetric H
2S tests. Following H
2S exposure, the MOF seems to retain its framework, which is something that PXRD analysis corroborates. In addition, thermogravimetric analysis (TGA) showed the complete removal of H
2S molecules at the relatively low temperature of 65 °C. Afterwards, five adsorption/desorption cycles were carried out (5% of H
2S volume), exhibiting a basically constant H
2S regeneration capacity (~18.5 mmol g
−1) for MIL-53(Al)-TDC. This reversibility indicated weak interactions between H
2S molecules and the pores of MOFs. To corroborate these results the authors conducted DRIFT tests and GCMC simulations. The computational study demonstrated that H
2S molecules interact inside the pores via hydrogen bonding (μ-OH·SH
2). Furthermore, H
2S molecules attract one another (adsorption band at ~3491 cm
−1) through dipole–dipole interactions, which constitute the main reason of increased H
2S capture. As expected, H
2S molecules also interact with the organic linker (thiophene). Finally, the low H
2S adsorption enthalpy (−23.2 kJ mol
−1), justifies the weak nature of these interactions.
Generally, the literature shows that MOFs with metal complexes μx-OH (μ3-OH for Mg-CUK−1, μ2-OH for MIL-47(V), MIL−125(Ti), and MIL-53(Cr)) exhibit respectable H2S reversibility at ambient temperature. In respect to MIL-53(Al)-TDC (μ-OH), although it demonstrates considerably high H2S uptake, it bears a major shortcoming, which is the high-temperature reactivation process (activated at 200 °C for 4 h under a flow of dry N2 gas) compared to the room-temperature regeneration conditions.
The last work presented herein was carried out by Mohideen et al. [
115], who reported the synthesis and construction of kag-MOF−1 [Zn(HCN
4)
2·(H
2O)]·1,4(H
2O), which is crystallized in a hexagonal system, comprised of two crystallographically independent Zn cations, in an octahedral environment. The authors showed that kag-MOF−1 due to its high localized charge density, which is attributed to the presence of relatively inexpensive tetrazolate organic building units, and one-dimensional channels with small in size pore-apertures, has great potential in removing acid gases such as H
2S.
Synopsizing,
Table 1 presents information regarding the textural properties (specific surface area, pore size, and pore volume), experimental conditions used and H
2S adsorption capacity (mmol g
−1) of selected MOFs mentioned herein. The adsorption mechanisms and the factors governing selectivity are key when dealing with H
2S capture on MOFs. H
2S can interact with the open metals forming sulfides (e.g., CuS, ZnS), which results in the collapse of their structure, as in the case of HKUST−1 (MOF−199) and ZIF-8. Moreover, these sorbents, display low H
2S adsorption performance in comparison to the MIL family.
In this regard, large pore MIL−100(Cr), MIL−101(Cr) and MIL−101(Cr)-4F demonstrate high SSA, rigid structure and considerably high H2S; a drawback is the poor reversibility of these materials. On the other hand, despite the fact that small pore MIL-47(V) and MIL-53(Al, Cr) show reduced capturing performance in comparison to that of the large pore MILs, they are more flexible, and their structure can be partially recovered. Furthermore, MIL-53(Al)-TDC show considerable high H2S uptake at low temperature (30 °C) and pressure (0.1 MPa) as well as noteworthy reversibility and decent moisture stability. However, high reactivation conditions are required.
When evaluated against MILs, UiO-66 also illustrate a relatively low H2S adsorption capacity, though, but it seems to effectively resist to metal sulfide formation. The incorporation of -NH2 can further improve its H2S capturing performance, but it negatively affects its reversibility. Another MOF with high H2S uptake is Ni-MOF-74 which has also the advantage of a stable structure when exposed to H2S molecules. Notwithstanding, in the presence of water, the H2S adsorption capacity decreases. A major shortcoming is also the high reactivation conditions. Conversely, Mg-CUK−1 can be reactivated under low conditions, but its capacity in capturing H2S is limited (3.03 mmol g−1).
Finally, to reinforce the comparative hypostasis of this work,
Figure 16 shows the H
2S capture capacities of common adsorbents (zeolites, metal oxides and activated carbons) and some of the most promising MOFs mentioned in the literature. It is evident that MOFs can be considered as one of the most interesting and promising candidates for the removal of toxic and corrosive H
2S molecules as they outperform the classic porous sorbents, owing to their enhanced chemical properties and functionality of the linkers and cavity dimensions.
10. Connection between Functions and Structural Features of MOFs
In this final section, we focus on the H2S capture properties of the MOFs and the connection between the removal abilities and the structures. We provide a summary of the unique structural features of MOFs while we emphasize the relationship between the structure and physical properties including pore profile, SSA, and thermal and mechanical stability.
As mentioned above, the main reason MOFs exhibit multiple functions is their diverse crystalline structures with tunable pore sizes. MOFs are constructed from metal nodes that are coordinated by rigid and aromatic organic linkers which are mostly carboxylic acid or nitrogen-containing linkers. Modifiable surfaces and tunable pores for specific applications (e.g., gas separation) can be achieved by judiciously selecting the appropriate metal clusters and organic linkers. The defined MOF structures offer the opportunity of investigating the relationship between the structure and physical properties, which is crucial for the design of MOFs with desirable functionality.
The functional sites of MOFs can be chemically produced by the pore surfaces. For instance, amine functional groups that can recognize small molecules should be incorporated into the organic ligands. However, it is critical to carefully select the metals and organic linkers as they bear different properties and may be incompatible compromising the performance of the material. Furthermore, the pore size of MOFs ranges between microporous to microporous, which facilitates the coexistence of diverse molecules such as metal complexes, polymers, metal atoms etc. These guest species can considerably enhance the functionality of MOFs (multiple functions). In a theoretical work, Gutiérrez-Sevillano et al. [
81] studied the adsorption of H
2S on MOF-5 and reported lower heat of adsorption for MOF-5 (approximately −15 kJ mol
−1) in comparison to that of HKUST−1 (approximately −30 kJ mol
−1) due to wider pores of the MOF-5.
Even though different MOF structures have been reported so far, they often suffer from weak stability, namely acid base stability, H
2O stability, and thermal stability, hindering their use in industrial applications. For example, Alvarez et al. [
83] concluded that the presence of H
2O, which usually coexists with H
2S in gas streams, compromises the HKUST−1’s stability since the metal-coordinated linkers are replaced by H
2O or OHˉ. Generally, since many MOFs bear hydrophilic properties, they strongly interact with H
2O, which means that even low amounts of H
2O can break the coordination bonds, resulting in structural degradation. Consequently, a method to enhance the stability of the material is to strengthen the coordination bonds between metal nodes and organic linker. In this regard, H
2O-stable MOFs, such as MIL−101 family, ZIFs, and zirconium-based carboxylates among others, have been extensively probed for desulfurization in the presence of moisture. Along these lines, new schemes by constructing hydrophobic surfaces or interfaces to improve H
2O-stability have also been developed recently [
122,
123,
124,
125]. A noteworthy work was conducted by Bae et al. who treated MOF−199 with O
2 plasma and maintained the porosity of the MOFs following hours of H
2O exposure, thus improving the surface polarity of the MOF [
126].
An even more challenging task is to construct MOFs with decent acid/base stability. Essentially, hard/soft acid/base (HSAB) theory states that soft Lewis acids react faster and form stronger bonds with soft Lewis bases, whereas hard Lewis acids react faster and form stronger bonds with hard Lewis bases [
75,
127]. However, considerable progress has been made in developing MOFs with acid/base stability. A widespread method is to combine high oxidation state metal ions with carboxylate linkers (i.e., hard acid and hard base) to generate strong bonds with resistance to chemical attacks. Another common method to eliminate H
2S via an acid/base reaction is the use of different amines for modifying MOFs and to eventually improve H
2S uptake, owing to their available nitrogen sites and high affinity for acid gases. The amine loading aims to simultaneously increase the SSA and affinity towards H
2S molecules without hampering diffusion and accessibility of H
2S molecules in the internal pores and surface of the MOFs. That said, controlled grafting at molecular precision is critical to prevent the blockage of the pores, and therefore forestall the access of the functional moieties inside the pore volume, while the amine groups are attached to the open metal sites [
128]. However, this method is not efficient in competitive CO
2/H
2S adsorption since both gases are electron acceptors (Lewis acids) and they have similar effect with amine.
The matter of thermal stability of MOFs is also of paramount importance as high temperatures are also required for different applications (e.g., activation of the sorbent prior to H
2S adsorption). This parameter is typically determined by the node-ligand bond strength and the number of ligands connected to each metal node. High valence metal ions such as Al
3+, Cr
3+, Zr
4+ usually generate materials that can resist to high temperatures. Typically, most MOFs are stable up to 350–400 °C [
28].
Another important factor for practical applications is mechanical stability of MOFs under vacuum or pressure. Even though high SSA and porosity of MOFs can be beneficial for an adsorption process, they also make MOFs less stable from a mechanical perspective. Permanent and highly ordered porosity and high SSAs provide the basis for the multiple functionalities of MOFs. While high SSA is typically beneficial to gas adsorption processes, the pore volume of MOFs is least as important since it affords the physical upper limit of the amount of the stored gas. Microporous MOFs with pore diameter lower than 2 nm have demonstrated increased adsorption capacities for various gases including H
2S. In addition, high micropore volumes and large SSAs are desirable for many application, as narrow pores inhibit the insertion of large unwanted impurities. The mesoporous MOFs MIL−100(Cr) and MIL−101(Cr) have also been investigated for desulfurization processes with interesting results [
49,
50]. Different methods have arisen that are efficient at constructing and tuning the pores of MOFs. Trying to elongate the length of linkers is a common and efficient strategy to achieve this task. In principle, the linear extension of organic linkers can improve pore size of MOFs in each network. However, sometimes, the openness of the structure can lead to interpenetration, thus preventing the space from being accessible [
32].






















{kind=link}
{kind=link}
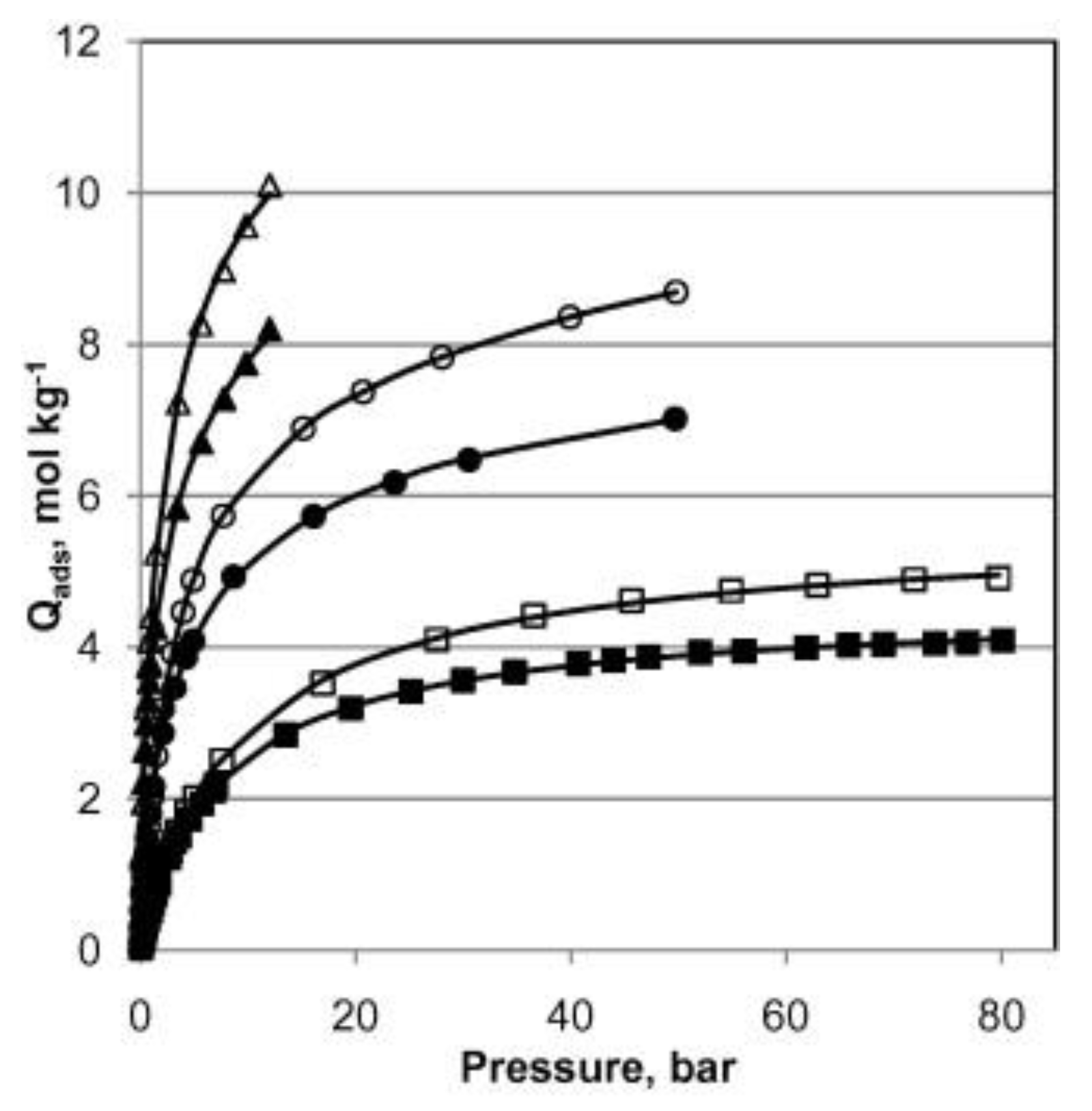{kind=link}
{kind=link}
{kind=link}
{kind=link}
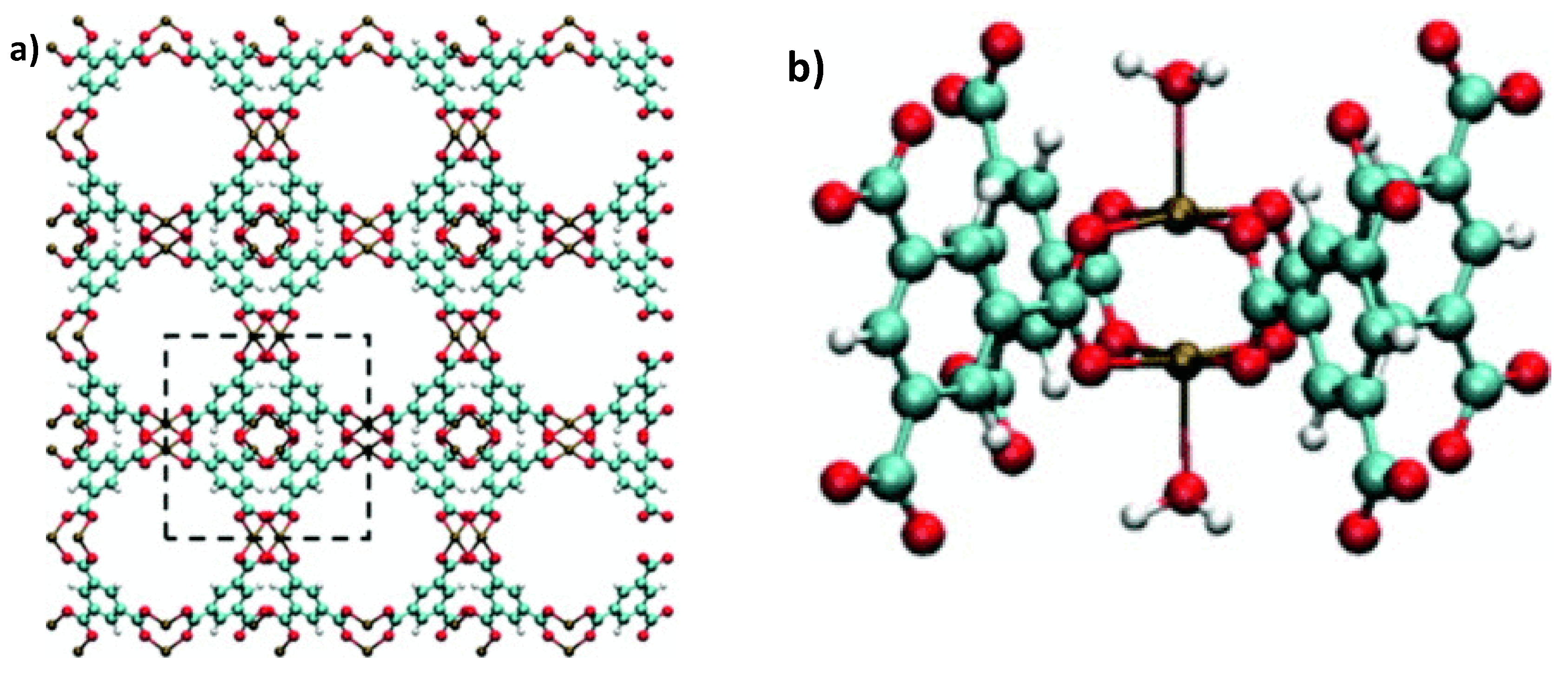{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
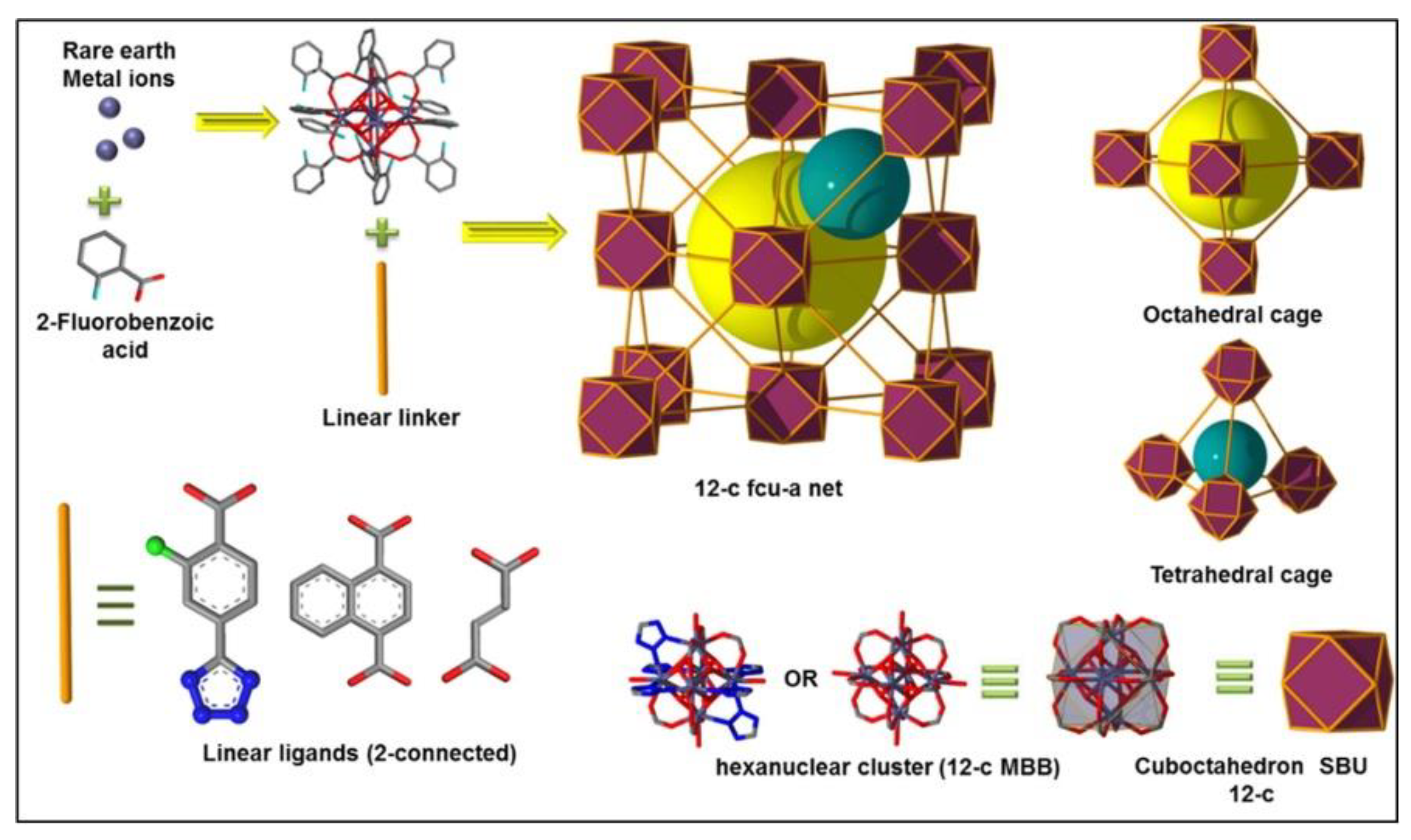{kind=link}
{kind=link}
{kind=link}
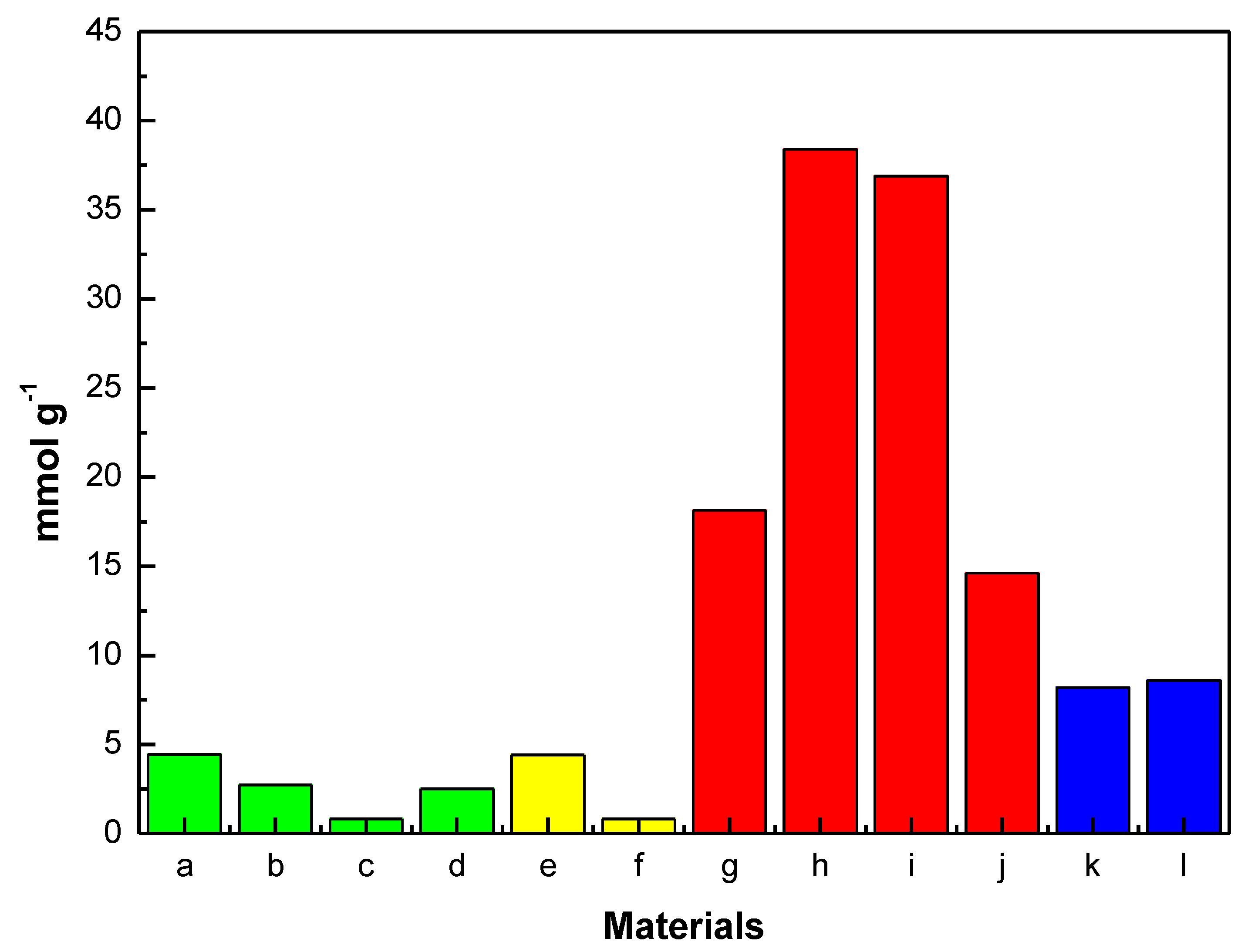{kind=link}