Phase Transition in Iron Thin Films Containing Coherent Twin Boundaries: A Molecular Dynamics Approach

Abstract
1. Introduction
2. Simulation Methodology
3. Results
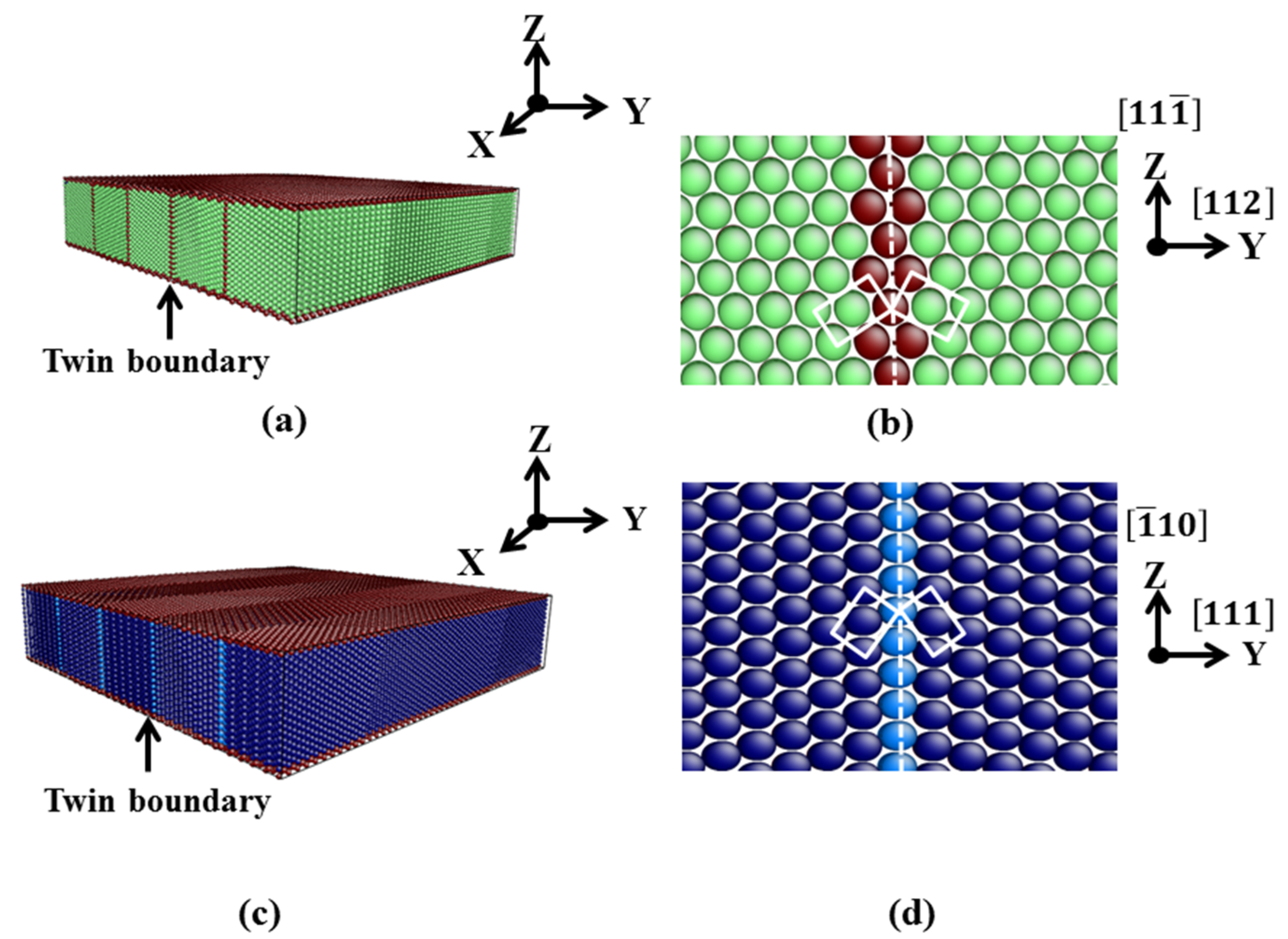
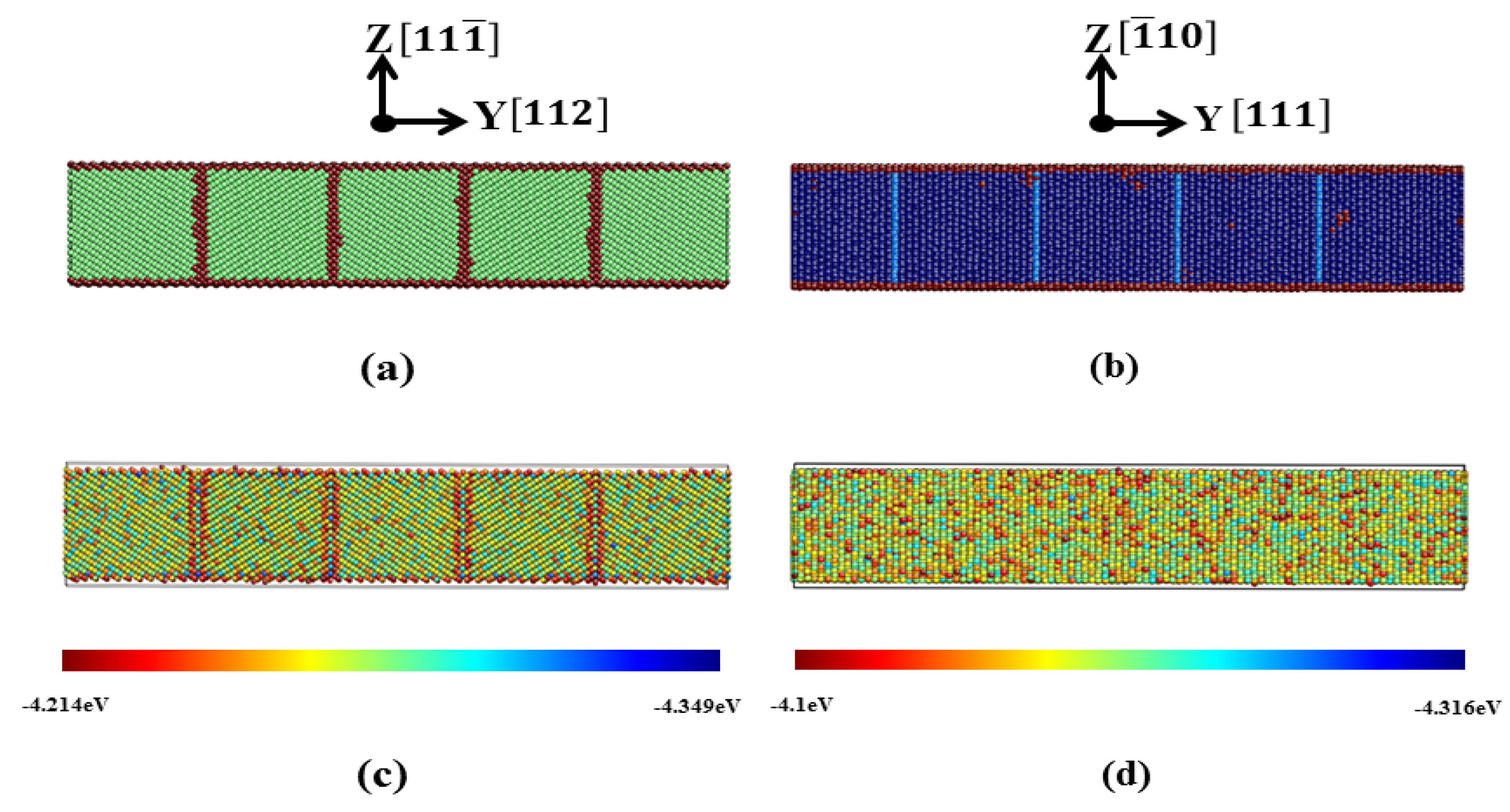
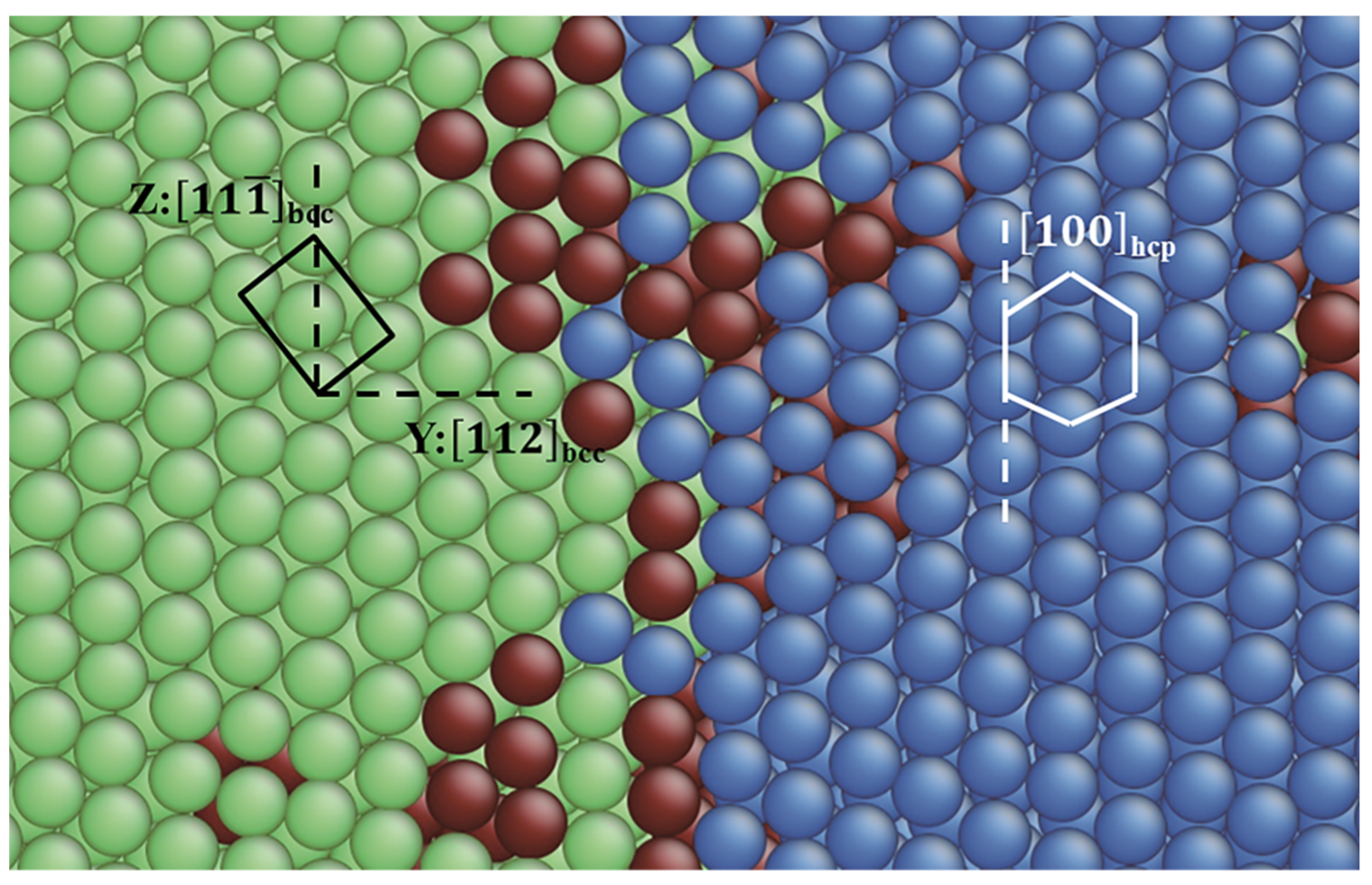
3.1. TB in the bcc and fcc Structure
3.2. Austenitic Transition
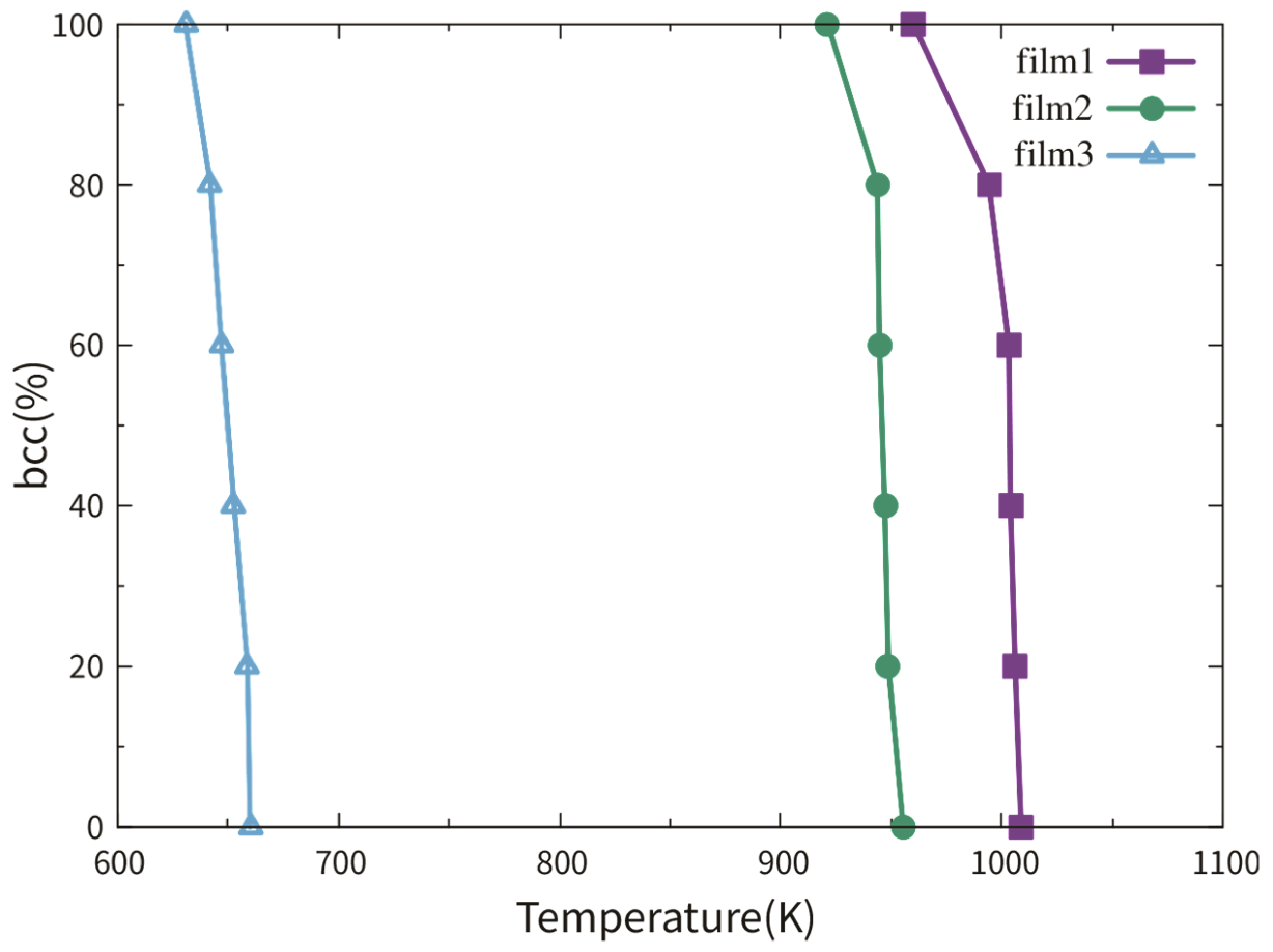
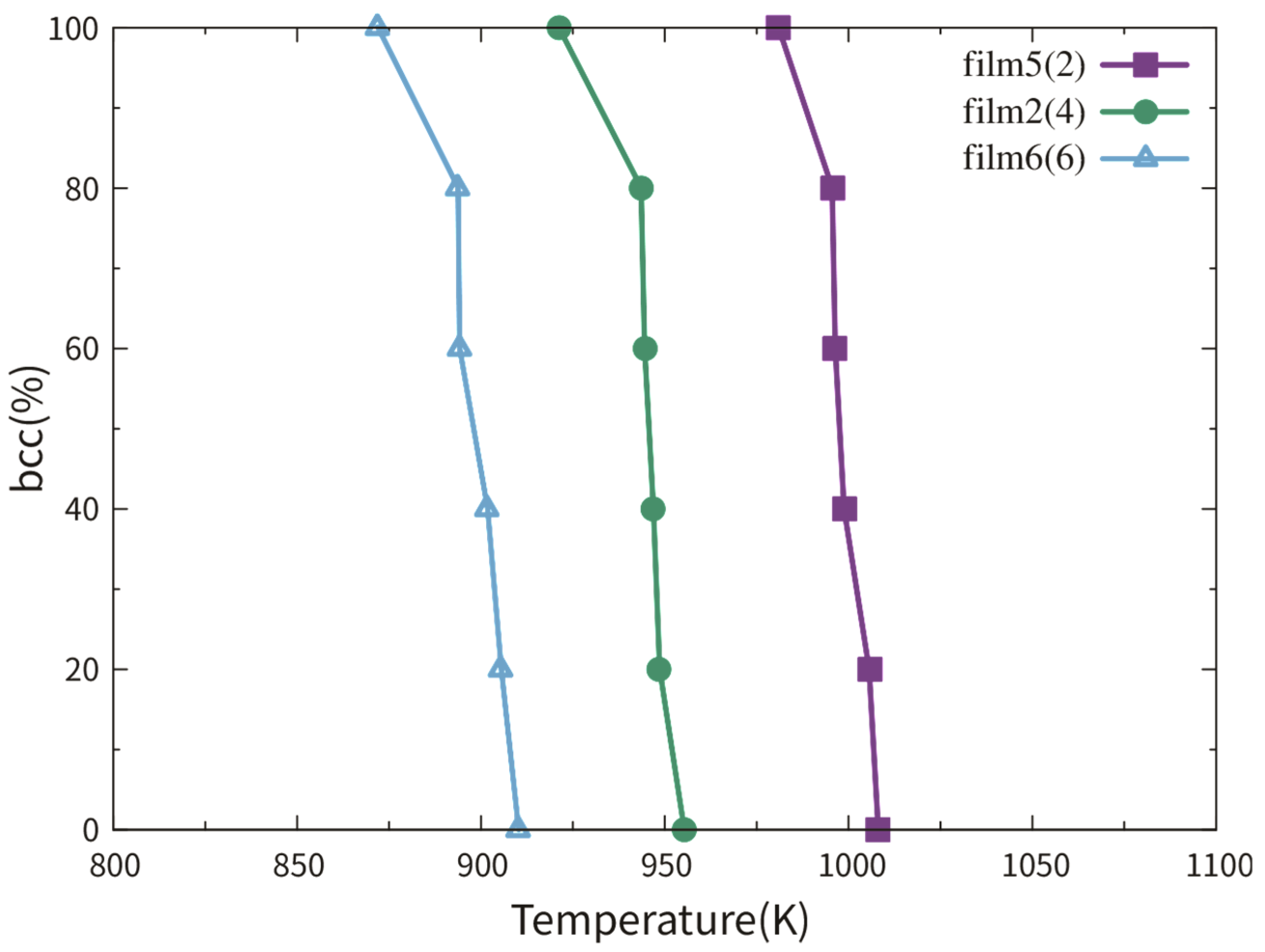
3.2.1. Dependence on Film Thickness
3.2.2. Dependence on TB Fraction
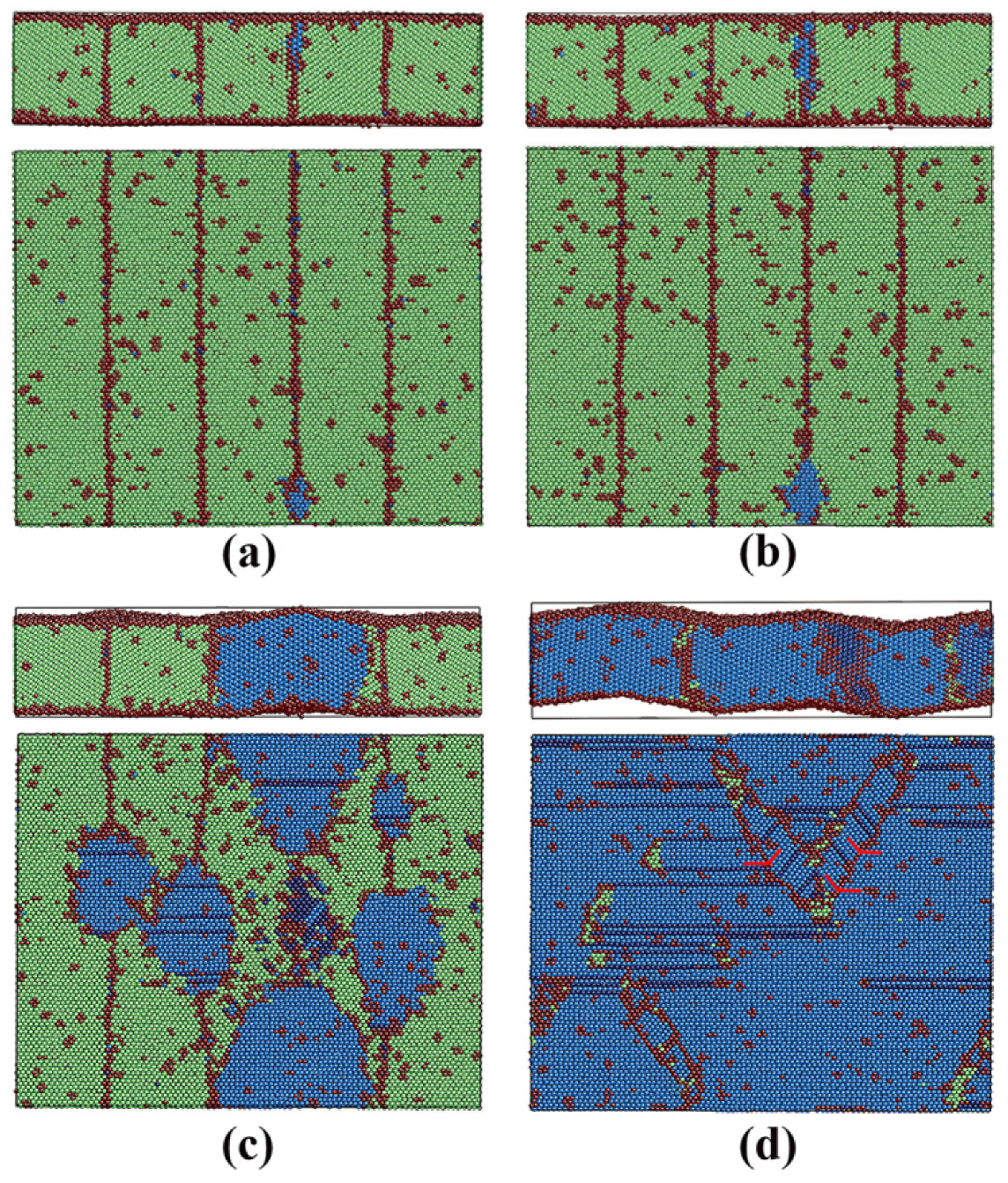
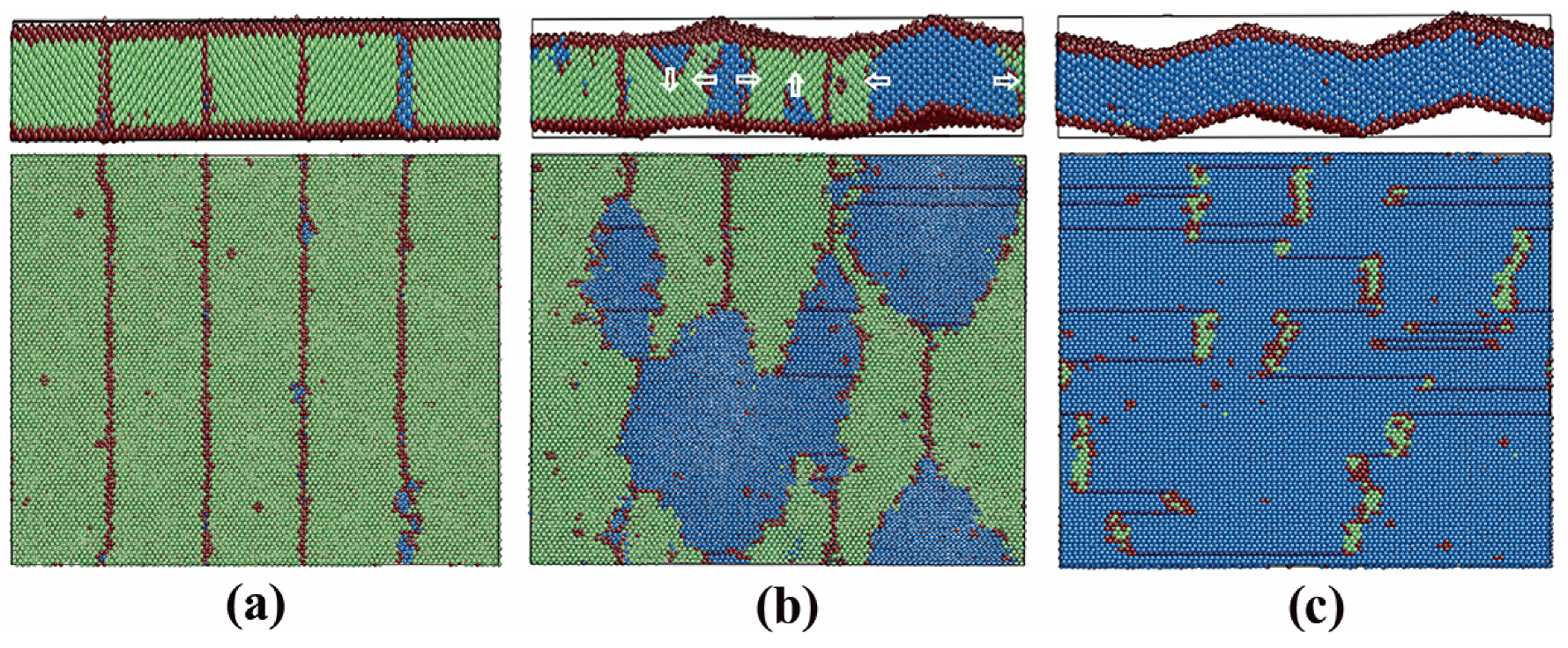
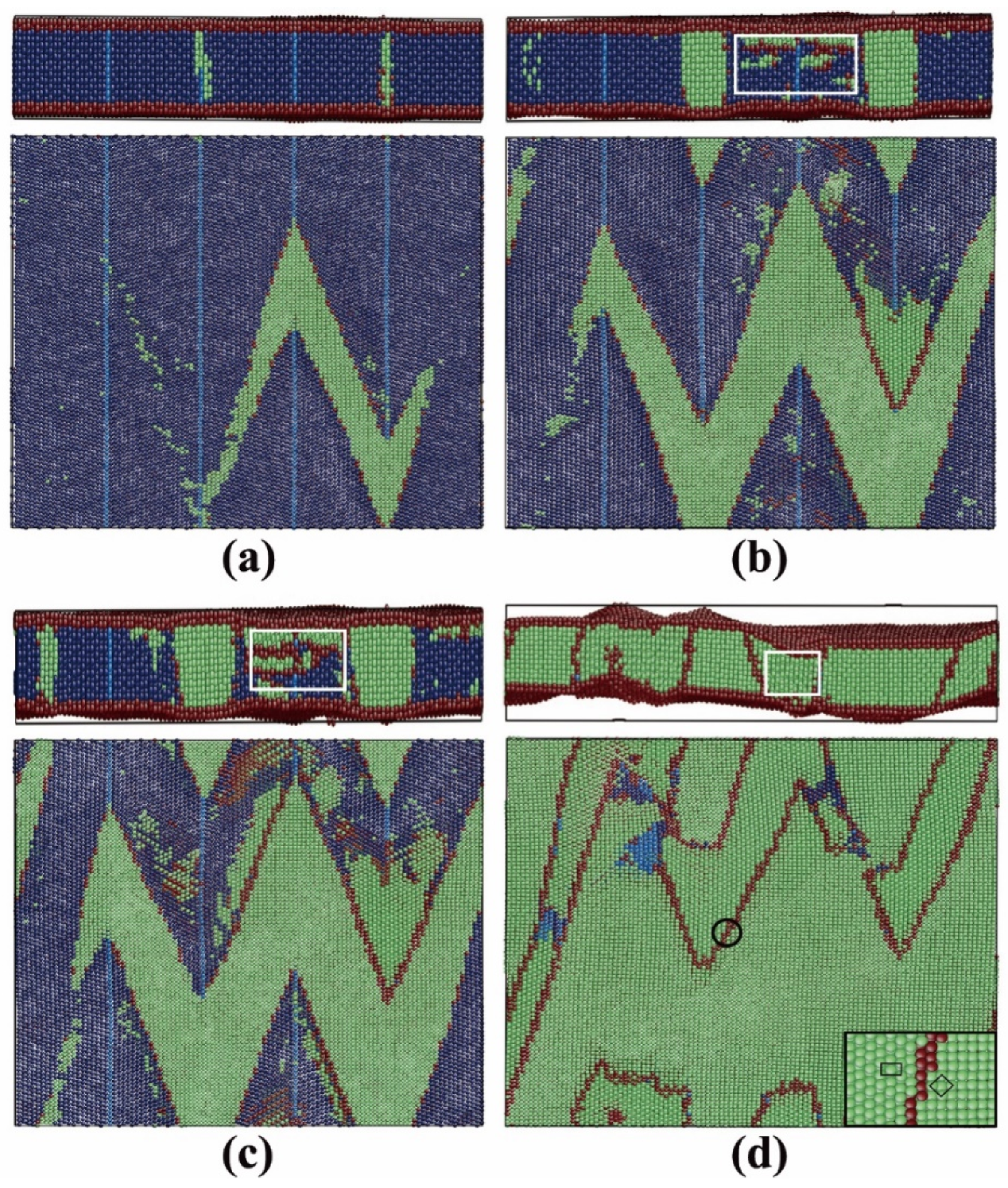
3.2.3. Pathway of the Austenitic Phase Transition
3.3. Martensitic Phase Transition
3.3.1. Dependence on Film Thickness
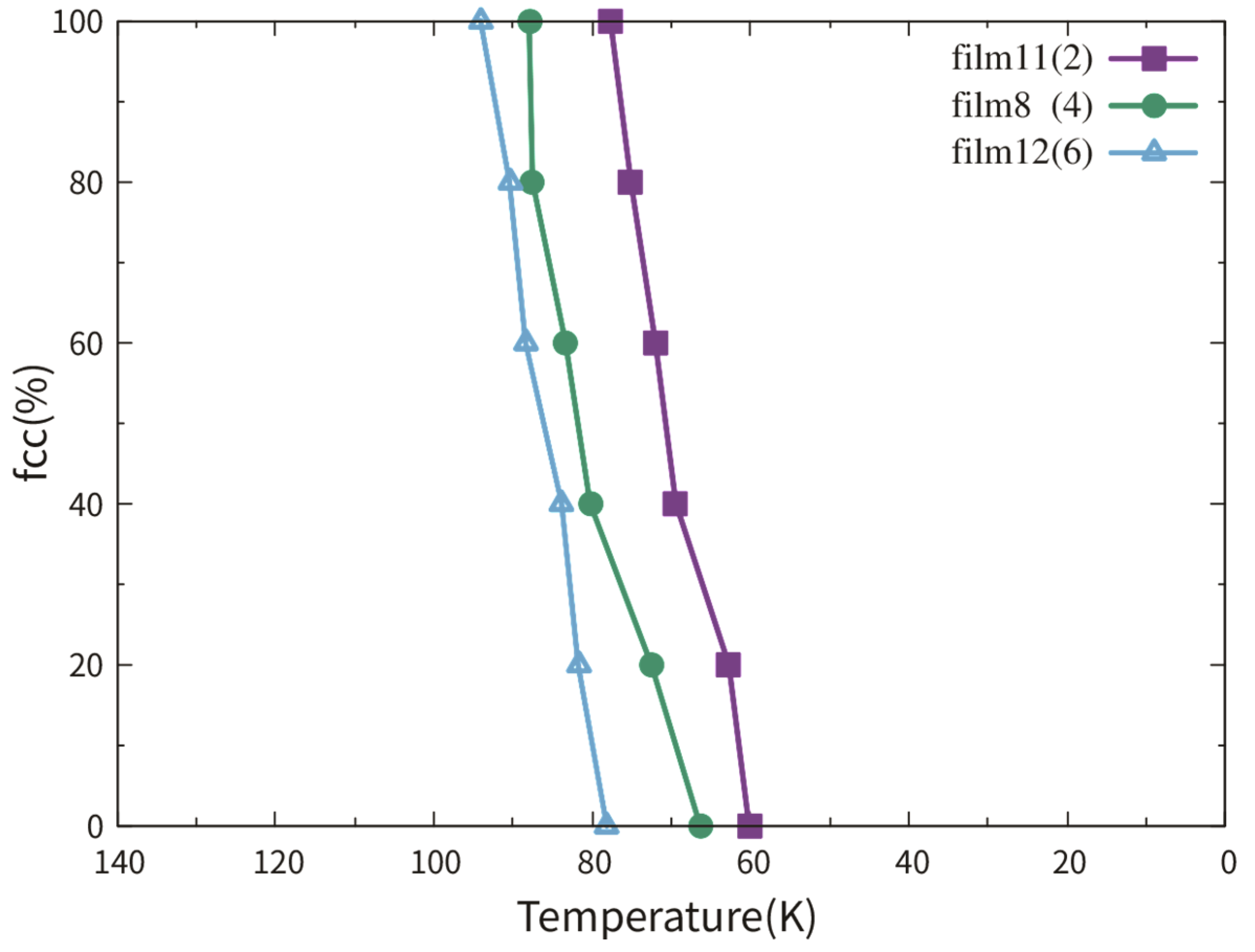
3.3.2. Dependence on the TB Fraction
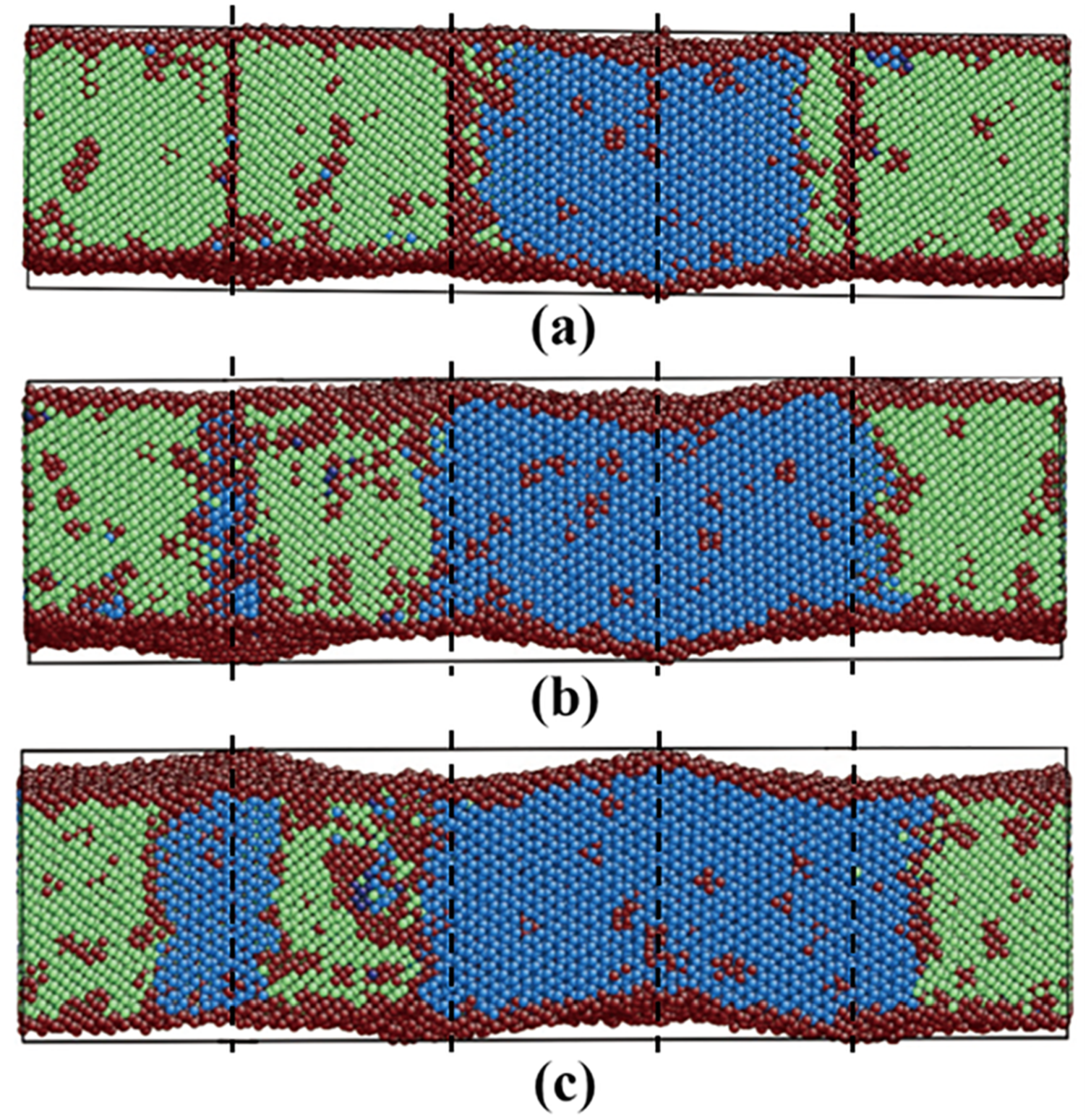
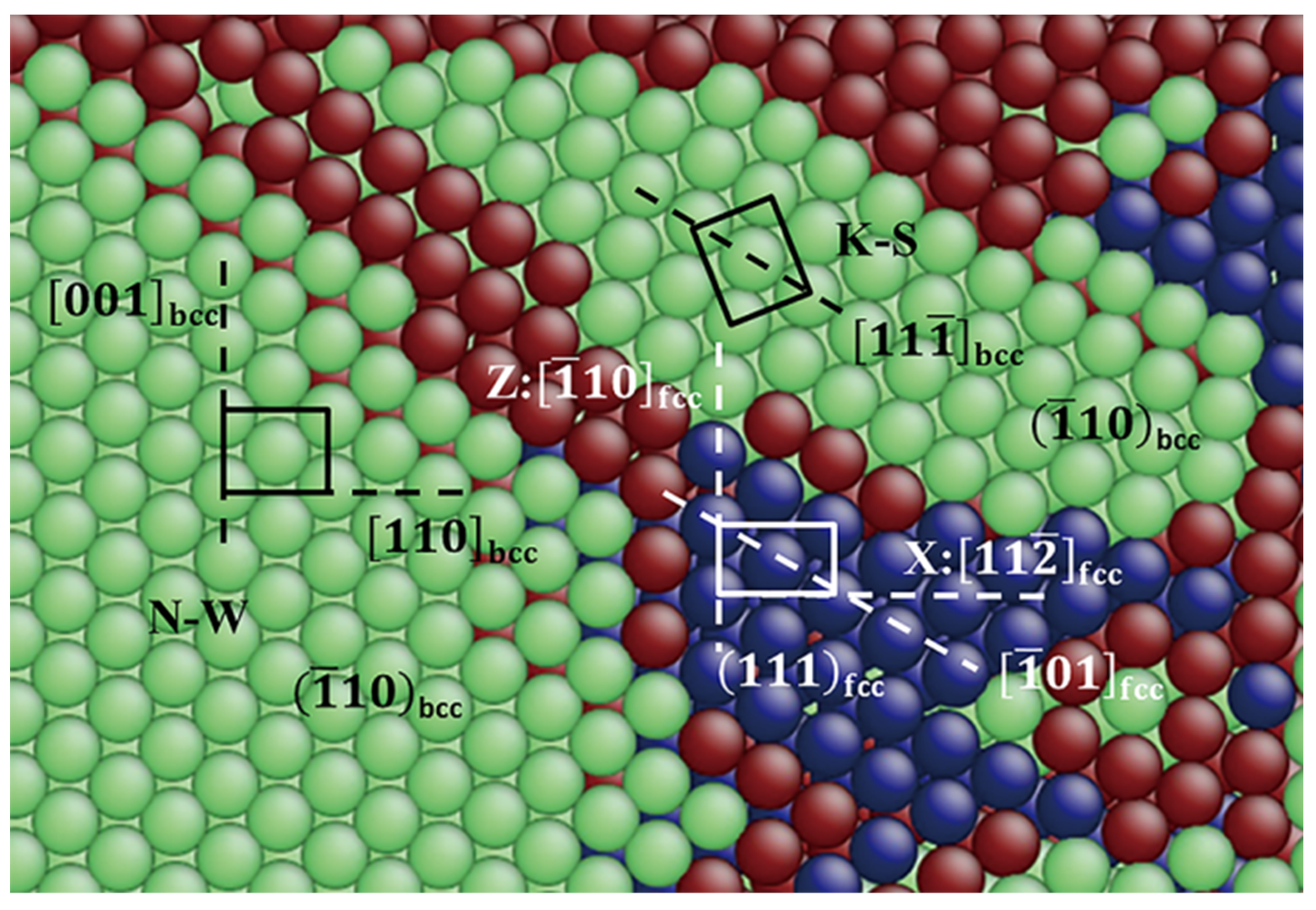
3.3.3. Pathway of the Martensitic Phase Transition
3.4. Transition Temperature Hysteresis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Type | T (K) | SFE (mJ/m2) | Finite-Size System | Phase Transition | Calculation Speed |
|---|---|---|---|---|---|---|
| Meyer–Entel | EAM | 550 ± 50 [27] | −54 [14] | Yes | Yes | fast |
| Müller | Bond-order | 1030 [44] | - | No [44] | No | low |
| Lee | MEAM | 1050;1075 [39] | 37.5 [14] | - | - | - |
References
- Pepperhoff, W.; Acet, M. Constitution and Magnetism of Iron and its Alloys; Springer: Berlin, Germany, 2001. [Google Scholar]
- Pereloma, E.; Edmonds, D.V. Phase transformations in steels. In Diffusionless Transformations, High Strength Steels, Modelling and Advanced Analytical Techniques; Woodhead Publishing Limited: Cambridge, UK, 2012; Volume 2. [Google Scholar]
- Yang, X.S.; Sun, S.; Wu, X.L.; Ma, E.; Zhang, T.Y. Dissecting the mechanism of martensitic transformation via atomic-scale observations. Sci. Rep. 2014, 4, 6141. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, X.; Sun, X.; Wang, Y.; Zuo, L. Twinning and martensite in a 304 austenitic stainless steel. Mater. Sci. Eng. A 2012, 552, 514–522. [Google Scholar] [CrossRef]
- Memmel, N.; Detzel, T. Growth, structure and stability of ultrathin iron films on Cu (001). Surf. Sci. 1994, 307, 490–495. [Google Scholar] [CrossRef]
- Cuenya, B.R.; Doi, M.; Löbus, S.; Courths, R.; Keune, W. Observation of the fcc-to-bcc Bain transformation in epitaxial Fe ultrathin films on Cu 3 Au (001). Surf. Sci. 2001, 493, 338–360. [Google Scholar] [CrossRef]
- Sandoval, L.; Urbassek, H.M. Solid–solid phase transitions in Fe nanowires induced by axial strain. Nanotechnology 2009, 20, 325704. [Google Scholar] [CrossRef]
- Sandoval, L.; Urbassek, H.M. Finite-size effects in Fe-nanowire solid−solid phase transitions: A molecular dynamics approach. Nano Lett. 2009, 9, 2290–2294. [Google Scholar] [CrossRef]
- Wang, B.; Sak-Saracino, E.; Sandoval, L.; Urbassek, H.M. Martensitic and austenitic phase transformations in Fe–C nanowires. Model. Simul. Mater. Sci. Eng. 2014, 22, 45003. [Google Scholar] [CrossRef]
- Wang, B.; Urbassek, H.M. Computer simulation of strain-induced phase transformations in thin Fe films. Model. Simul. Mater. Sci. Eng. 2013, 21, 085007. [Google Scholar] [CrossRef]
- Sak-Saracino, E.; Urbassek, H.M. Effect of uni- and biaxial strain on phase transformations in Fe thin films. Int. J. Comput. Mater. Sci. Eng. 2016, 5, 1650001. [Google Scholar] [CrossRef]
- Meiser, J.; Urbassek, H.M. Influence of the crystal surface on the austenitic and martensitic phase transition in pure iron. Crystals 2018, 8, 469. [Google Scholar] [CrossRef]
- Meiser, J.; Urbassek, H.M. Martensitic transformation of pure iron at a grain boundary: Atomistic evidence for a two-step Kurdjumov-Sachs–Pitsch pathway. AIP Adv. 2016, 6, 085017 . [Google Scholar] [CrossRef]
- Karewar, S.; Sietsma, J.; Santofimia, M. Effect of pre-existing defects in the parent fcc phase on atomistic mechanisms during the martensitic transformation in pure Fe: A molecular dynamics study. Acta Mater. 2018, 142, 71–81. [Google Scholar] [CrossRef]
- Cahn, J.W. Nucleation on dislocations. Acta Met. 1957, 5, 169–172. [Google Scholar] [CrossRef]
- Sharma, H.; Sietsma, J.; Offerman, S.E. Preferential nucleation during polymorphic transformations. Sci. Rep. 2016, 6, 30860. [Google Scholar] [CrossRef] [PubMed]
- Meiser, J.; Urbassek, H.M. Dislocations help initiate the α–γ phase transformation in iron—An atomistic study. Metals 2019, 9, 90. [Google Scholar] [CrossRef]
- Yonezawa, T.; Suzuki, K.; Ooki, S.; Hashimoto, A. The effect of chemical composition and heat treatment conditions on stacking fault energy for Fe-Cr-Ni austenitic stainless steel. Met. Mater. Trans. A 2013, 44, 5884–5896. [Google Scholar] [CrossRef]
- Shibuta, Y.; Takamoto, S.; Suzuki, T. A Molecular dynamics study of the energy and structure of the symmetric tilt boundary of iron. ISIJ Int. 2008, 48, 1582–1591. [Google Scholar] [CrossRef]
- Papon, A.M.; Simon, J.P.; Guyot, P.; Desjonquères, M.C. Calculation of {112} twin and stacking fault energies in b.c.c. transition metals. Philos. Mag. B 1979, 40, 159–172. [Google Scholar] [CrossRef]
- Grässel, O.; Krüger, L.; Frommeyer, G.; Meyer, L.W. High strength Fe-Mn-(Al, Si) TRIP/TWIP steels development—properties—application. Int. J. Plast. 2000, 16, 1391–1409. [Google Scholar] [CrossRef]
- Barbier, D.; Gey, N.; Allain, S.; Bozzolo, N.; Humbert, M. Analysis of the tensile behavior of a TWIP steel based on the texture and microstructure evolutions. Mater. Sci. Eng. A 2009, 500, 196–206. [Google Scholar] [CrossRef]
- Wang, K.; Chen, J.; Zhang, X.; Zhu, W. Interactions between coherent twin boundaries and phase transition of iron under dynamic loading and unloading. J. Appl. Phys. 2017, 122, 105107.1–105107.11. [Google Scholar] [CrossRef]
- Tamura, S. Notes on pseudo-twinning in ferrite and on the solubility of carbon in alpha-iron at the A1 point. J. Iron St. Inst. 1927, 115, 747–753. [Google Scholar]
- Zhuang, Y.Z.; Wu, C.H. Annealing twin in α-iron. Acta Metall. Sin. 1958, 3, 55–61. [Google Scholar]
- Fang, X.; Zhang, K.; Guo, H.; Wang, W.; Zhou, B. Twin-induced grain boundary engineering in 304 stainless steel. Mater. Sci. Eng. A 2008, 487, 7–13. [Google Scholar] [CrossRef]
- Engin, C.; Sandoval, L.; Urbassek, H.M. Characterization of Fe potentials with respect to the stability of the bcc and fcc phase. Model. Simul. Mater. Sci. Eng. 2008, 16, 35005. [Google Scholar] [CrossRef]
- Meyer, R.; Entel, P. Martensite-austenite transition and phonon dispersion curves of Fe1-xNix studied by molecular-dynamics simulations. Phys. Rev. B 1998, 57, 5140–5147. [Google Scholar] [CrossRef]
- Sak-Saracino, E.; Urbassek, H.M. Temperature-induced phase transformation of Fe1-xNix alloys: Molecular-dynamics approach. Eur. Phys. J. B 2015, 88, 169. [Google Scholar] [CrossRef]
- Wang, B.; Sak-Saracino, E.; Gunkelmann, N.; Urbassek, H.M. Molecular-dynamics study of the α γ phase transition in Fe–C. Comput. Mater. Sci. 2014, 82, 399–404. [Google Scholar] [CrossRef]
- Song, H.; Hoyt, J. A molecular dynamics study of heterogeneous nucleation at grain boundaries during solid-state phase transformations. Comput. Mater. Sci. 2016, 117, 151–163. [Google Scholar] [CrossRef]
- Rayne, J.A.; Chandrasekhar, B.S. Elastic constants of iron from 4.2 to 300 K. Phys. Rev. 1961, 122, 1714–1716. [Google Scholar] [CrossRef]
- Wang, B.; Urbassek, H.M. Phase transitions in an Fe system containing a bcc/fcc phase boundary: An atomistic study. Phys. Rev. B 2013, 87, 104108. [Google Scholar] [CrossRef]
- Yang, Z.; Johnson, R.A. An EAM simulation of the α-γ iron interface. Model. Simul. Mater. Sci. Eng. 1993, 1, 707. [Google Scholar] [CrossRef]
- Murr, L.E. Interfacial Phenomena in Metals and Alloys; Addison-Wesley: Boston, MA, USA, 1975. [Google Scholar]
- Spencer, M.J.S.; Hung, A.; Snook, I.K.; Yarovsky, I. Further studies of iron adhesion: (111) surfaces. Surf. Sci. 2002, 515, L464–L468. [Google Scholar] [CrossRef]
- Tyson, W.; Miller, W. Surface free energies of solid metals: Estimation from liquid surface tension measurements. Surf. Sci. 1977, 62, 267–276. [Google Scholar] [CrossRef]
- Rigsbee, J.; Aaronson, H. The interfacial structure of the broad faces of ferrite plates. Acta Met. 1979, 27, 365–376. [Google Scholar] [CrossRef]
- Lee, T.; Baskes, M.I.; Valone, S.M.; Doll, J.D. Atomistic modeling of thermodynamic equilibrium and polymorphism of iron. J. Phys. Condens. Matter 2012, 24, 225404. [Google Scholar] [CrossRef]
- Dick, A.; Hickel, T.; Neugebauer, J. The effect of disorder on the concentration-dependence of stacking fault energies in Fe1-xMnx-a first principles study. Steel Res. Int. 2009, 80, 603–608. [Google Scholar]
- Bleskov, I.; Hickel, T.; Neugebauer, J.; Ruban, A. Impact of local magnetism on stacking fault energies: A first-principles investigation for fcc iron. Phys. Rev. B 2016, 93, 214115. [Google Scholar] [CrossRef]
- Sandoval, L.; Urbassek, H.M.; Entel, P. The Bain versus Nishiyama–Wassermann path in the martensitic transformation of Fe. New J. Phys. 2009, 11, 103027. [Google Scholar] [CrossRef]
- Urbassek, H.M.; Sandoval, L. Molecular dynamics modeling of martensitic transformations in steels. In Phase Transformations in Steels; Diffusionless Transformations, High Strength Steels, Modelling and Advanced Analytical Techniques; Pereloma, E., Edmonds, D.V., Eds.; Woodhead Publishing Limited: Cambridge, UK, 2012; Volume 2, pp. 433–463. [Google Scholar]
- Müller, M.; Erhart, P.; Albe, K. Analytic bond-order potential for bcc and fcc iron—Comparison with established embedded-atom method potentials. J. Phys. Condens. Matter 2007, 19, 326220. [Google Scholar] [CrossRef]
- Dosset, J.L.; Boyer, H.E. Practical Heat Treating: Second Edition; ASM International: MaterialsPark, OH, USA, 2006; pp. 22–24. [Google Scholar]
- Hirel, P. Atomsk: A tool for manipulating and converting atomic data files. Comput. Phys. Commun. 2015, 197, 212–219. [Google Scholar] [CrossRef]
- Faken, D.; Jonsson, H. Systematic analysis of local atomic structure combined with 3D computer graphics. Comput. Mater. Sci. 1994, 2, 279–286. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the open visualization tool. Model. Simul. Mater. Sci. Eng. 2009, 18, 15012. [Google Scholar] [CrossRef]
- Stukowski, A.; Albe, K. Extracting dislocations and non-dislocation crystal defects from atomistic simulation data. Model. Simul. Mater. Sci. Eng. 2010, 18, 85001. [Google Scholar] [CrossRef]
- Li, J. AtomEye: An efficient atomistic configuration viewer. Model. Simul. Mater. Sci. Eng. 2003, 11, 173–177. [Google Scholar] [CrossRef]
- Available online: http://lammps.sandia.gov/ (accessed on 15 August 2020).
- Sainath, G.; Choudhary, B.K. Deformation behaviour of body centered cubic iron nanopillars containing coherent twin boundaries. Philos. Mag. 2016, 96, 3502–3523. [Google Scholar] [CrossRef][Green Version]
- Finnis, M.W.; Sinclair, J.E. A simple empirical N-body potential for transition metals. Philos. Mag. A 1984, 50, 45–55. [Google Scholar] [CrossRef]
- Nakashima, H.; Takeuchi, M. Grain boundary energy and structure of α-Fe symmetric tilt boundary. Tetsu Hagane 2000, 86, 357–362. [Google Scholar] [CrossRef]
- Wang, B.; Urbassek, H.M. Atomistic dynamics of the bccfcc phase transition in iron: Competition of homo- and heterogeneous phase growth. Comput. Mater. Sci. 2014, 81, 170–177. [Google Scholar] [CrossRef]
- Engin, C.; Urbassek, H.M. Molecular-dynamics investigation of the fcc bcc phase transformation in Fe. Comput. Mater. Sci. 2008, 41, 297–304. [Google Scholar]
- Burgers, W. On the process of transition of the cubic-body-centered modification into the hexagonal-close-packed modification of zirconium. Physica 1934, 1, 561–586. [Google Scholar] [CrossRef]
- Kurdjumov, G.V.; Sachs, G. Über den mechanismus der stahlhärtung. Z. Phys. 1930, 64, 325–343. [Google Scholar] [CrossRef]
- Fukino, T.; Tsurekawa, S. In-situ SEM/EBSD observation of α/γ phase transformation in Fe-Ni alloy. Mater. Trans. 2008, 49, 2770–2775. [Google Scholar] [CrossRef]
- Nishiyama, Z. Mechanism of transformation from face-centred to body-centred cubic lattice. Sci. Rep. Tohoku Imp. Univ. 1934, 23, 637. [Google Scholar]
- Chen, A.Y.; Ruan, H.H.; Wang, J.; Chen, H.L.; Wang, Q.; Li, Q.; Lu, J. The influence of strain rate on the microstructure transition of 304 stainless steel. Acta Mater. 2011, 59, 3697–3709. [Google Scholar] [CrossRef]
- Olson, G.; Cohen, M. A mechanism for the strain-induced nucleation of martensitic transformations. J. Less Common Met. 1972, 28, 107–118. [Google Scholar] [CrossRef]
- Olson, G.B.; Cohen, M. A general mechanism of martensitic nucleation: Part II. FCC → BCC and other martensitic transformations. Metall. Mater. Trans. A 1976, 7, 1905–1914. [Google Scholar]
- Olson, G.B.; Cohen, M. A Perspective on martensitic nucleation. Annu. Rev. Mater. Res. 1981, 11, 1–32. [Google Scholar] [CrossRef]
- Olson, G.B.; Cohen, M. Dislocation theory of martensitic transformations. Dislocat. Solids. 1986, 7, 295–403. [Google Scholar]
- Pitsch, W. The martensite transformation in thin foils of iron foils of iron-nitrogen alloys. Philos. Mag. 1959, 4, 577–584. [Google Scholar] [CrossRef]
- Johnston, H.L.; Arnold, C.S.; Venus, D. Thickness-dependent fcc to bcc structural change in iron films: Use of a 2-ML Ni/W(110) substrate. Phys. Rev. B 1997, 55, 13221–13229. [Google Scholar] [CrossRef]












| Film | x | y | z | Δx (Å) | Δy (Å) | Δz (Å) | T | N |
|---|---|---|---|---|---|---|---|---|
| 1 | [0] | [112] | [] | 202.9 | 210.9 | 89.4 | 4 | 324000 |
| 2 | [0] | [112] | [] | 202.9 | 210.9 | 44.7 | 4 | 162000 |
| 3 | [0] | [112] | [] | 202.9 | 210.9 | 22.4 | 4 | 81000 |
| 4 | [0] | [112] | [] | 202.9 | 210.9 | 44.7 | 0 | 162000 |
| 5 | [0] | [112] | [] | 202.9 | 210.9 | 44.7 | 2 | 162000 |
| 6 | [0] | [112] | [] | 202.9 | 210.9 | 44.7 | 6 | 162000 |
| Film | x | y | z | Δx (Å) | Δy (Å) | Δz (Å) | T | N |
|---|---|---|---|---|---|---|---|---|
| 7 | [11] | [111] | [] | 216.9 | 210.9 | 88.7 | 4 | 323136 |
| 8 | [11] | [111] | [] | 216.9 | 210.9 | 44.4 | 4 | 161568 |
| 9 | [11] | [111] | [] | 216.9 | 210.9 | 20.9 | 4 | 80640 |
| 10 | [11] | [111] | [] | 216.9 | 210.9 | 44.4 | 0 | 161568 |
| 11 | [11] | [111] | [] | 216.9 | 210.9 | 44.4 | 2 | 161568 |
| 12 | [11] | [111] | [] | 216.9 | 210.9 | 44.4 | 6 | 161568 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.; Jiang, Y.; Xu, C. Phase Transition in Iron Thin Films Containing Coherent Twin Boundaries: A Molecular Dynamics Approach. Materials 2020, 13, 3631. https://doi.org/10.3390/ma13163631
Wang B, Jiang Y, Xu C. Phase Transition in Iron Thin Films Containing Coherent Twin Boundaries: A Molecular Dynamics Approach. Materials. 2020; 13(16):3631. https://doi.org/10.3390/ma13163631
Chicago/Turabian StyleWang, Binjun, Yunqiang Jiang, and Chun Xu. 2020. "Phase Transition in Iron Thin Films Containing Coherent Twin Boundaries: A Molecular Dynamics Approach" Materials 13, no. 16: 3631. https://doi.org/10.3390/ma13163631
APA StyleWang, B., Jiang, Y., & Xu, C. (2020). Phase Transition in Iron Thin Films Containing Coherent Twin Boundaries: A Molecular Dynamics Approach. Materials, 13(16), 3631. https://doi.org/10.3390/ma13163631