Effect of In Situ Grown SiC Nanowires on the Pressureless Sintering of Heterophase Ceramics TaSi2-TaC-SiC



Abstract
1. Introduction
- The decrease of grain size of metastable β-ZrO2 below 300 nm stabilized the phase and inhibited its spontaneous martensitic transformation and low-temperature degradation;
- The stability of β-ZrO2 showed a linear dependence on the concentration of stabilizing agents;
- The highest stability of β-ZrO2 was achieved by mixing trivalent and pentavalent stabilizers;
- Inhomogeneous distribution of stabilizer particles increased the susceptibility of β-ZrO2 to martensitic transformation;
- Slow cooling and isothermal dwelling during the heating-cooling cycles promoted the nucleation and growth of the thermodynamically stable α-ZrO2 phase;
- Dopant cations with radii smaller than Zr4+ reduced the lattice parameters, destabilized the fluorite structure, and promoted the formation of vacancies on non-metallic sublattice. As a result, the diffusion coefficients increased, lowering the ceramic solidification temperature and shortening the time of sintering, which allowed for the production of ceramics with a relative density close to one and enhanced mechanical properties.
2. Materials and Methods
2.1. Calculations
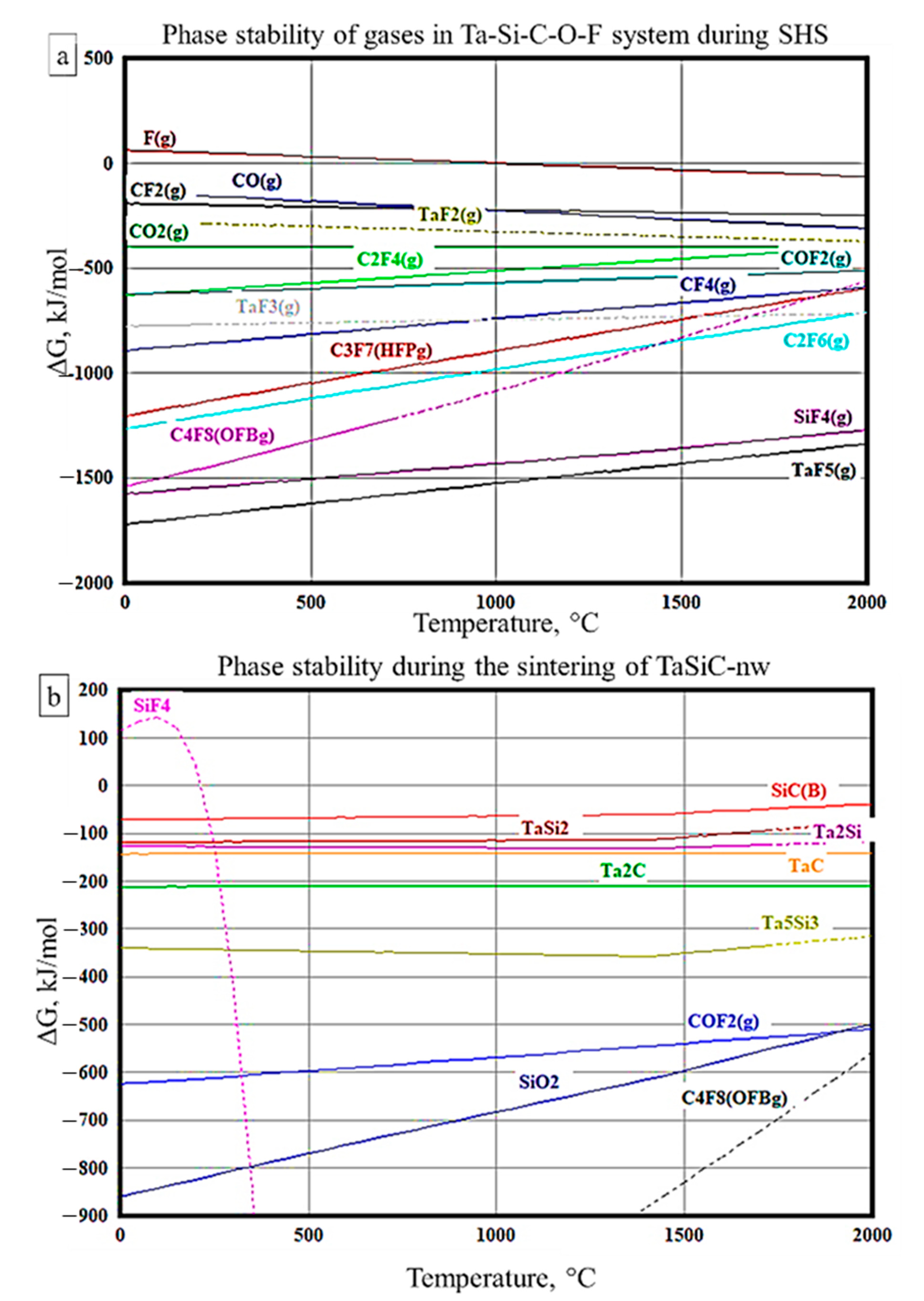
2.1.1. Calculations of Phase Stability (Ellingham Diagrams)
2.1.2. Construction of Grand Potential Phase Diagrams
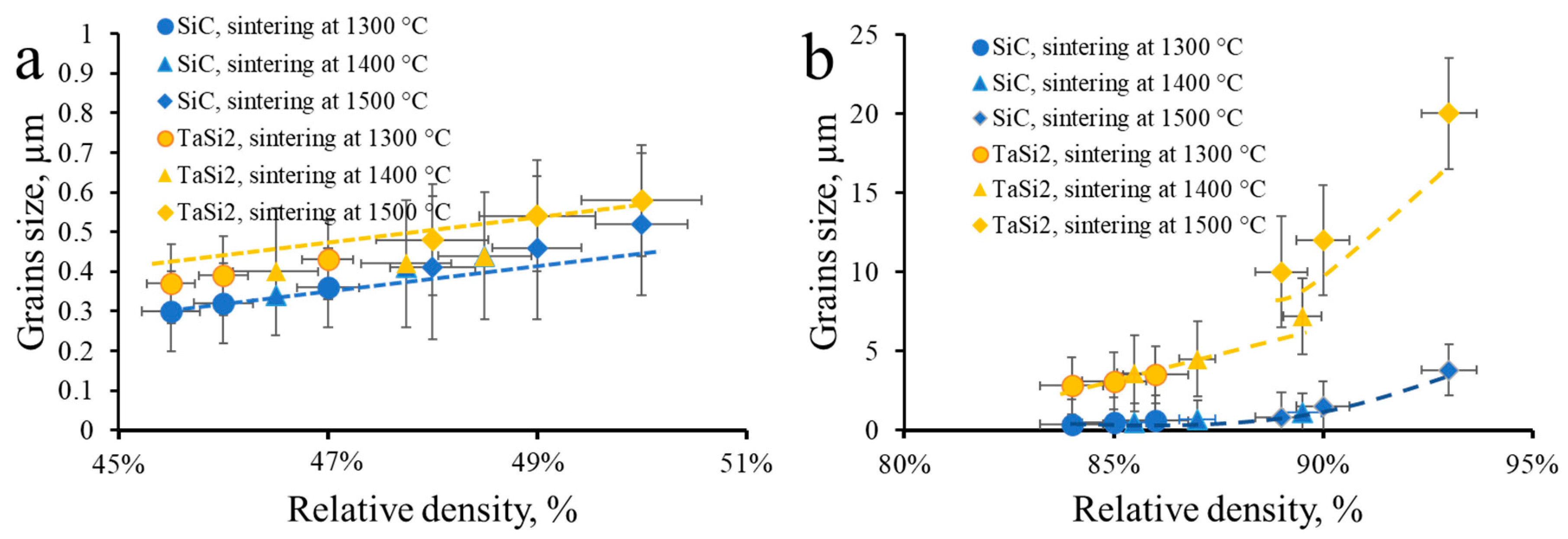
2.1.3. Analysis of the Sintering Path via Grain Size Versus Relative Density Relationships
2.2. Experimental
2.2.1. Preparation of Composite Powders
2.2.2. Pressing of Composite Ceramic Powders
2.2.3. Sintering Experiments
2.2.4. Microstructural Investigations and Mechanical Testing of Sintered Specimens
3. Results and Discussion
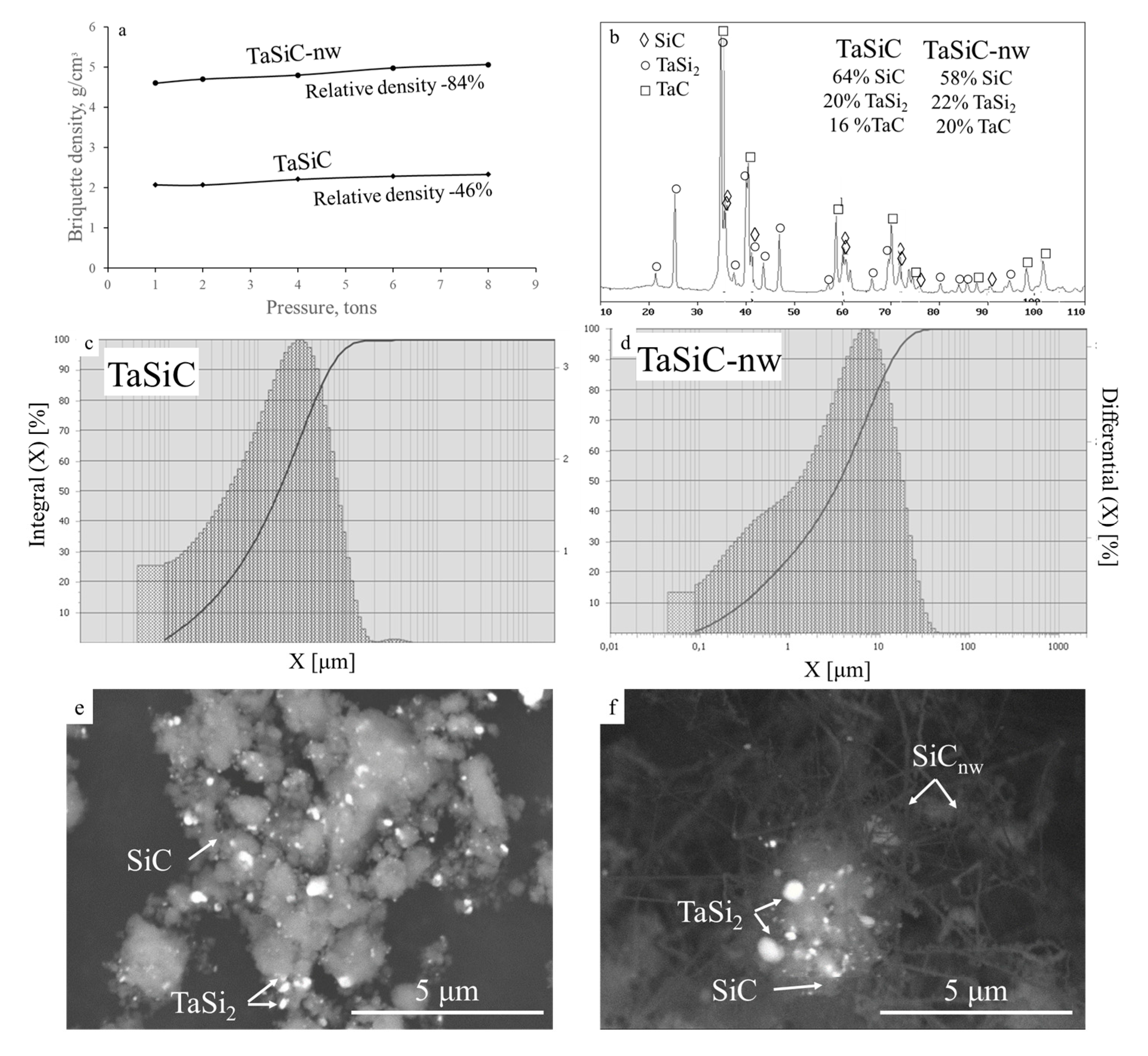
3.1. Characterization of the Ceramic Powders
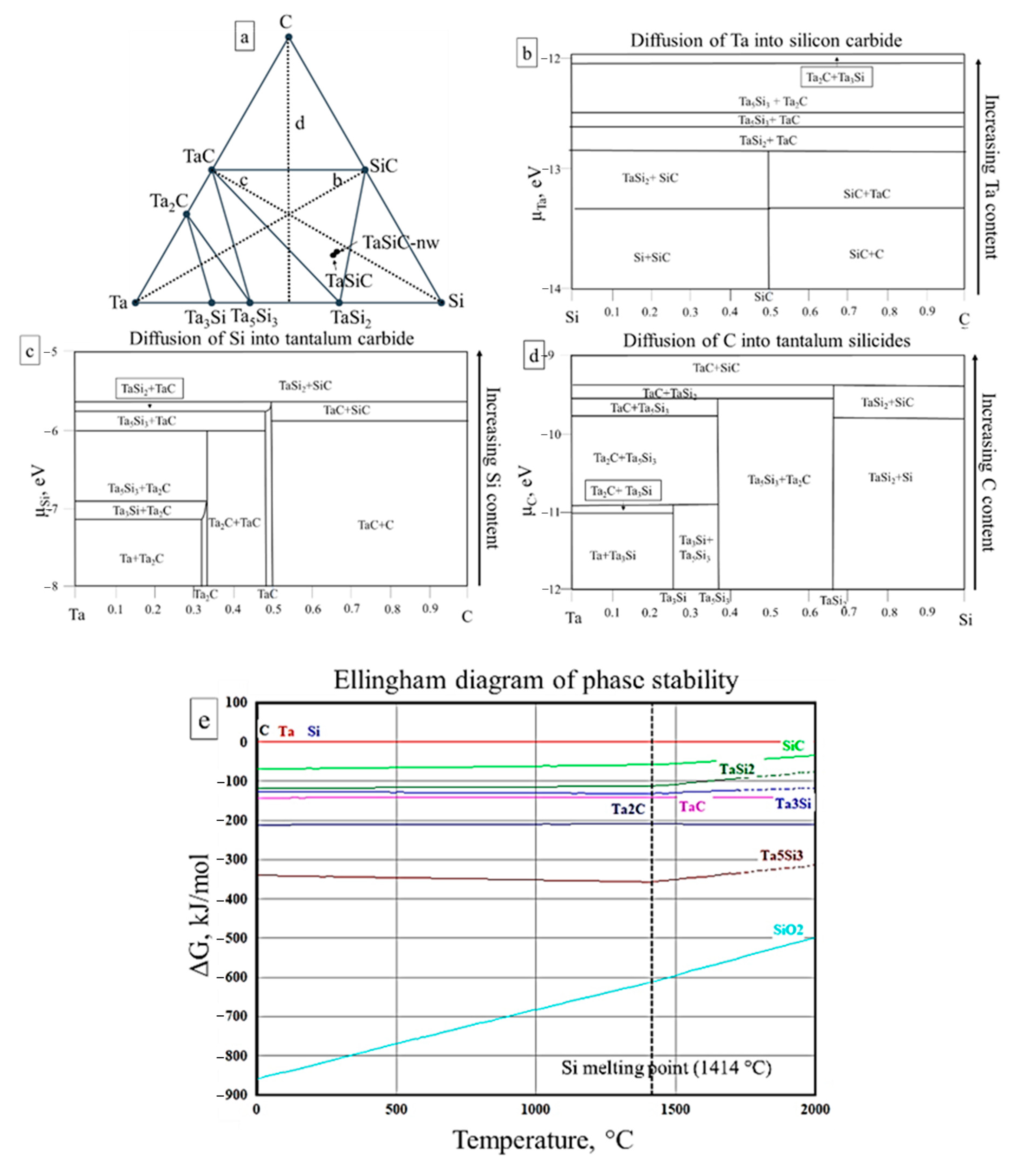
3.2. Sintering of TaSiC Composition: Thermodynamic Analysis
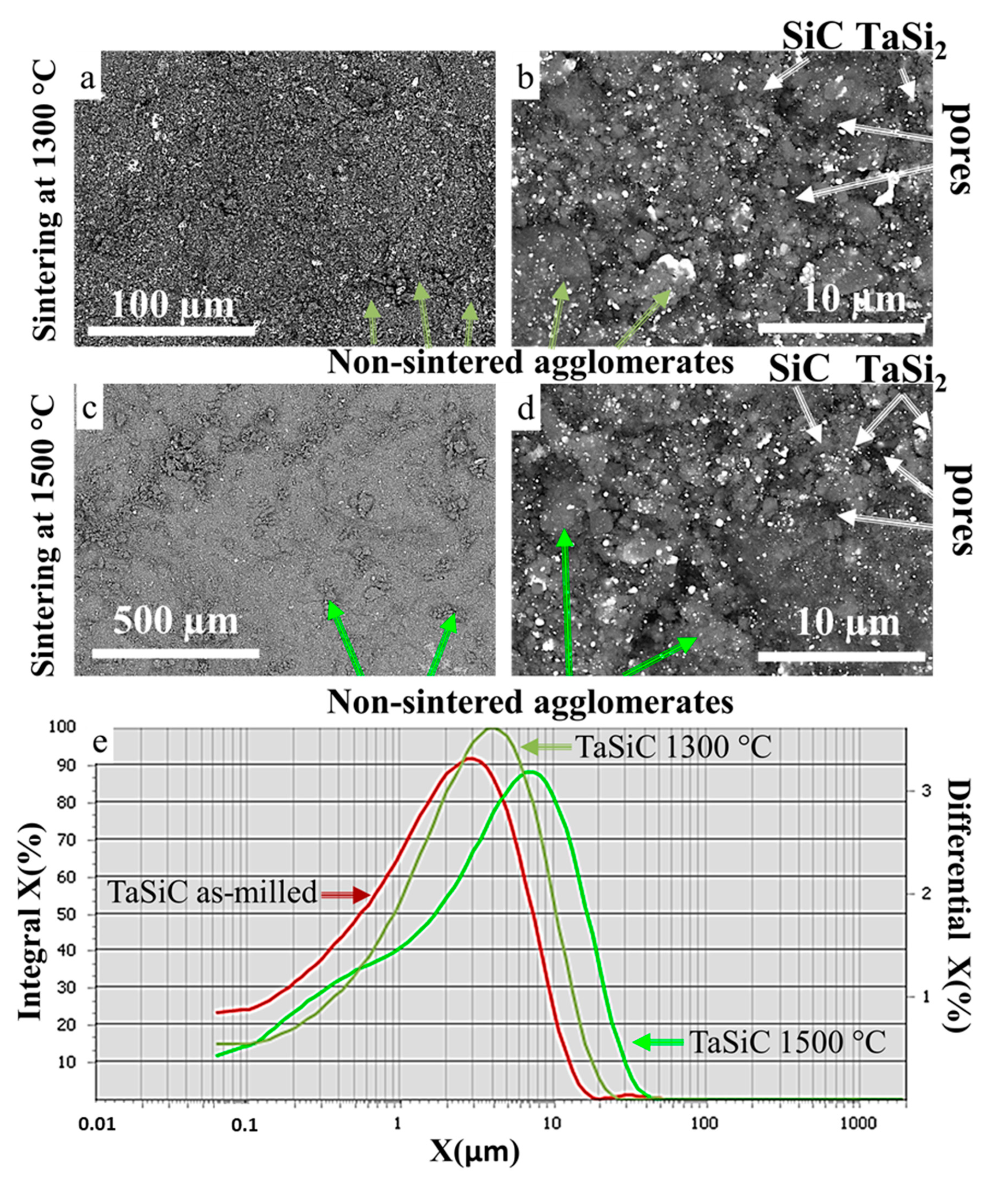
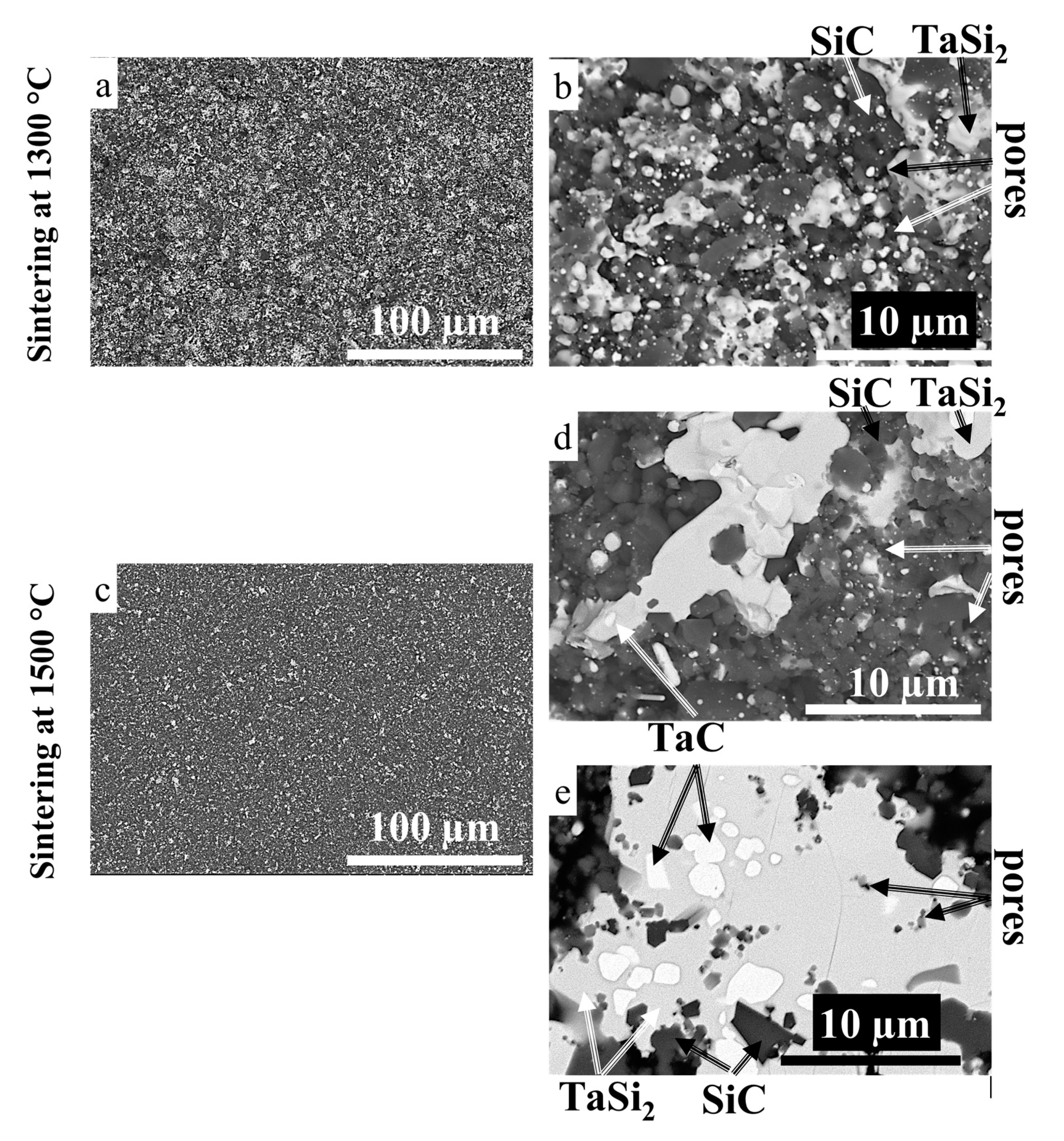
3.3. Sintering of TaSiC Composition: Microstructural Investigation and Mechanical Testing
3.4. Sintering of TaSiC-nw Composition: Thermodynamic Analysis
3.5. Sintering of TaSiC-nw Composition: Microstructural Investigation and Mechanical Testing
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Padture, N.P. In Situ-Toughened Silicon Carbide. J. Am. Ceram. Soc. 1994, 77, 519–523. [Google Scholar] [CrossRef]
- Zhou, Y.; Tanaka, H.; Otani, S.; Bando, Y. Low-Temperature Pressureless Sintering of alpha-SiC with Al4C3-B4C-C Additions. J. Am. Ceram. Soc. 2004, 82, 1959–1964. [Google Scholar] [CrossRef]
- Zhan, G.-D.; Mitomo, M.; Tanaka, H.; Kim, Y.-W. Effect of Annealing Conditions on Microstructural Development and Phase Transformation in Silicon Carbide. J. Am. Ceram. Soc. 2004, 83, 1369–1374. [Google Scholar] [CrossRef]
- Morales, A.M.; Lieber, C.M. A Laser Ablation Method for the Synthesis of Crystalline Semiconductor Nanowires. Science 1998, 279, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Seeger, T.; Kohler-Redlich, P.; Rühle, M. Synthesis of Nanometer-Sized SiC Whiskers in the Arc-Discharge. Adv. Mater. 2000, 12, 279–282. [Google Scholar] [CrossRef]
- Otoishi, S.; Tange, Y. Effect of a Catalyst on the Formation of SiC Whiskers from Polycarbosilane. Nickel Ferrite as a Catalyst. Bull. Chem. Soc. Jpn. 1999, 72, 1607–1613. [Google Scholar] [CrossRef]
- Addamiano, A. Preparation and properties of 2H SiC crystals. J. Cryst. Growth 1982, 58, 617–622. [Google Scholar] [CrossRef]
- Krishnarao, R.V.; Godkhindi, M.M.; Mukunda, P.G.I.; Chakraborty, M. Direct Pyrolysis of Raw Rice Husks for Maximization of Silicon Carbide Whisker Formation. J. Am. Ceram. Soc. 1991, 74, 2869–2875. [Google Scholar] [CrossRef]
- Yang, W.; Araki, H.; Kohyama, A.; Hu, Q.; Suzuki, H.; Noda, T. Growing SiC Nanowires on Tyranno-SA SiC Fibers. J. Am. Ceram. Soc. 2004, 87, 733–735. [Google Scholar] [CrossRef]
- Verspui, G.; Knippenberg, W.; Bootsma, G. Lanthanum-stimulated high-temperature whisker growth of α-SiC. J. Cryst. Growth 1972, 12, 97–105. [Google Scholar] [CrossRef]
- Salama, I.A.; Quick, N.R.; Kar, A. Laser synthesis of carbon-rich SiC nanoribbons. J. Appl. Phys. 2003, 93, 9275–9281. [Google Scholar] [CrossRef]
- Yang, W.; Araki, H.; Hu, Q.; Ishikawa, N.; Suzuki, H.; Noda, T. In situ growth of SiC nanowires on RS-SiC substrate(s). J. Cryst. Growth 2004, 264, 278–283. [Google Scholar] [CrossRef]
- Yunlong, Z.; Ming, H.; Xiangge, Q.; XiaoGang, S. The influence of additive content on microstructure and mechanical properties on the Csf/SiC composites after annealed treatment. Appl. Surf. Sci. 2013, 279, 71–75. [Google Scholar] [CrossRef]
- Vorotilo, S.; Potanin, A.Y.; Loginov, P.A.; Shvindina, N.V.; Levashov, E.A. Combustion synthesis of SiC-based ceramics reinforced by discrete carbon fibers with in situ grown SiC nanowires. Ceram. Int. 2020, 46, 7861–7870. [Google Scholar] [CrossRef]
- Hao, S.; Cui, L.; Jiang, D.; Han, X.; Ren, Y.; Jiang, J.; Liu, Y.; Liu, Z.; Mao, S.; Wang, Y.; et al. A Transforming Metal Nanocomposite with Large Elastic Strain, Low Modulus, and High Strength. Science 2013, 339, 1191–1194. [Google Scholar] [CrossRef]
- Fang, T.-H.; Chang, W.-J.; Chiu, J.-W. Study on coalescent properties of ZnO nanoclusters using molecular dynamics simulation and experiment. Microelectron. J. 2006, 37, 722–727. [Google Scholar] [CrossRef]
- Zhang, H.; Banfield, J.F. Thermodynamic analysis of phase stability of nanocrystalline titania. J. Mater. Chem. 1998, 8, 2073–2076. [Google Scholar] [CrossRef]
- Koparde, V.N.; Cummings, P.T. Molecular Dynamics Simulation of Titanium Dioxide Nanoparticle Sintering. J. Phys. Chem. B 2005, 109, 24280–24287. [Google Scholar] [CrossRef]
- Naicker, P.K.; Cummings, P.T.; Zhang, H.; Banfield, J.F. Characterization of Titanium Dioxide Nanoparticles Using Molecular Dynamics Simulations. J. Phys. Chem. B 2005, 109, 15243–15249. [Google Scholar] [CrossRef]
- Filyukov, D.V.; Brodskaya, E.N.; Piotrovskaya, E.M.; De Leeuw, S.W. Molecular-dynamics simulation of nanoclusters of crystal modifications of titanium dioxide. Russ. J. Gen. Chem. 2007, 77, 10–16. [Google Scholar] [CrossRef]
- Koparde, V.N.; Cummings, P.T. Phase Transformations during Sintering of Titania Nanoparticles. ACS Nano 2008, 2, 1620–1624. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-W.; Shih, C.-F.; Behera, R.K.; Hsu, W.-D. Investigation of initial stages of nano-ceramic particle sintering using atomistic simulations. Surf. Coat. Technol. 2013, 231, 316–322. [Google Scholar] [CrossRef]
- Chen, I.; Wang, X.-H. Sintering dense nanocrystalline ceramics without final-stage grain growth. Nature 2000, 404, 168–171. [Google Scholar] [CrossRef]
- Cameron, C.P.; Raj, R. Grain-Growth Transition During Sintering of Colloidally Prepared Alumina Powder Compacts. J. Am. Ceram. Soc. 1988, 71, 1031–1035. [Google Scholar] [CrossRef]
- Li, F.; Pan, J. Modelling “Nano-Effects” in Sintering. In Sintering: Mechanisms of Convention Nanodensification and Field Assisted Processes; Castro, R., Benthem, K., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 17–34. [Google Scholar]
- Guo, X.; Maier, J. Grain Boundary Blocking Effect in Zirconia: A Schottky Barrier Analysis. J. Electrochem. Soc. 2001, 148, E121–E126. [Google Scholar] [CrossRef]
- Kosmac, T.; Oblak, C.; Jevnikar, P.; Funduk, N.; Marion, L. Strength and reliability of surface treated Y-TZP dental ceramics. J. Biomed. Mater. Res. 2000, 53, 304–313. [Google Scholar] [CrossRef]
- Fang, Z.Z.; Wang, H. Densification and grain growth during sintering of nanosized particles. Int. Mater. Rev. 2008, 53, 326–352. [Google Scholar] [CrossRef]
- Wakai, F.; Yoshida, M.; Kashyap, B.P. Influence of Particle Arrangement on Coarsening during Sintering of Three Spherical Particles. J. Ceram. Soc. Jpn. 2006, 114, 974–978. [Google Scholar] [CrossRef][Green Version]
- Wang, H.; Fang, Z.Z.; Hwang, K.S. Kinetics of Initial Coarsening During Sintering of Nanosized Powders. Met. Mater. Trans. A 2011, 42, 3534–3542. [Google Scholar] [CrossRef]
- Fang, J.; Thompson, A.M.; Harmer, M.P.; Chan, H.M. Effect of Yttrium and Lanthanum on the Final Stage Sintering Behavior of Ultrahigh Purity Alumina. J. Am. Ceram. Soc. 2005, 80, 2005–2012. [Google Scholar] [CrossRef]
- Chen, P.-L.; Chen, I.W. Sintering of Fine Oxide Powders: II, Sintering Mechanisms. J. Am. Ceram. Soc. 1997, 80, 637–645. [Google Scholar] [CrossRef]
- Lee, H.; Speyer, R.F. Pressureless Sintering of Boron Carbide. J. Am. Ceram. Soc. 2003, 86, 1468–1473. [Google Scholar] [CrossRef]
- Lange, F.F. Powder Processing Science and Technology for Increased Reliability. J. Am. Ceram. Soc. 1989, 72, 3–15. [Google Scholar] [CrossRef]
- Li, J.; Sun, X. Synthesis and sintering behavior of a nanocrystalline α-alumina powder. Acta Mater. 2000, 48, 3103–3112. [Google Scholar] [CrossRef]
- Zhu, D.; Miller, R.A. Sintering and creep behavior of plasma-sprayed zirconia- and hafnia-based thermal barrier coatings. Surf. Coat. Technol. 1998, 108, 114–120. [Google Scholar] [CrossRef]
- Cocks, A.C.F. Overview no. 117 The structure of constitutive laws for the sintering of fine grained materials. Acta Met. Mater. 1994, 42, 2191–2210. [Google Scholar] [CrossRef]
- Durán, P.; Tartaj, J.; Fernández, J.F.; Villegas, M.; Moure, C. Crystallisation and sintering behaviour of nanocrystalline Y-TZP powders obtained by seeding-assisted chemical coprecipitation. Ceram. Int. 1999, 25, 125–135. [Google Scholar] [CrossRef]
- Theunissen, G.A.S.M.; Winnubst, A.J.A.; Burggraaf, A.J. Sintering kinetics and microstructure development of nanoscale Y-TZP ceramics. J. Eur. Ceram. Soc. 1993, 11, 315–324. [Google Scholar] [CrossRef]
- Bertsch, A.; Jiguet, S.; Renaud, P. Microfabrication of ceramic components by microstereolithography. J. Micromech. Microeng. 2003, 14, 197–203. [Google Scholar] [CrossRef]
- Nygren, M.; Shen, Z. On the preparation of bio-, nano- and structural ceramics and composites by spark plasma sintering. Solid State Sci. 2003, 5, 125–131. [Google Scholar] [CrossRef]
- An, L.; Chan, H.M.; Soni, K.K. Control of calcium hexaluminate grain morphology in in-situ toughened ceramic composites. J. Mater. Sci. 1996, 31, 3223–3229. [Google Scholar] [CrossRef]
- Heule, M.; Gauckler, L.J. Gas sensors fabricated from ceramic suspensions by micromolding in capillaries. Adv. Mater. 2001, 13, 1790–1793. [Google Scholar] [CrossRef]
- Yang, H.; Deschatelets, P.; Brittain, S.T.; Whitesides, G.M. Fabrication of high performance ceramic microstructures from a polymeric precursor using soft lithography. Adv. Mater. 2001, 13, 54–58. [Google Scholar] [CrossRef]
- Nakonieczny, D.S.; Ziębowicz, A.; Paszenda, Z.K.; Krawczyk, C. Trends and perspectives in modification of zirconium oxide for a dental prosthetic applications—A review. Biocybern. Biomed. Eng. 2017, 37, 229–245. [Google Scholar] [CrossRef]
- Ju, H.-F.; Ning, K.; Lu, K. Sintering behaviors of micron-sized ceramic rod features. Acta Mater. 2018, 144, 534–542. [Google Scholar] [CrossRef]
- Jain, A.; Hautier, G.; Ong, S.P.; Moore, C.J.; Fischer, C.C.; Persson, K.A.; Ceder, G. Formation enthalpies by mixing GGA and GGA +U calculations. Phys. Rev. B 2011, 84, 045115. [Google Scholar] [CrossRef]
- Ong, S.P.; Wang, L.; Kang, B.; Ceder, G. The Li-Fe-P-O2 phase diagram from first principles calculations. Chem. Mater. 2008, 20, 1798–1807. [Google Scholar] [CrossRef]
- Ong, S.P.; Jain, A.; Hautier, G.; Kang, B.; Ceder, G. Thermal stabilities of delithiated olivine MPO4 (M=Fe, Mn) cathodes investigated using first principles calculations. Electrochem. Commun. 2010, 12, 427–430. [Google Scholar] [CrossRef]
- Richards, W.D.; Miara, L.J.; Wang, Y.; Kim, J.C.; Ceder, G. Interface Stability in Solid-State Batteries. Chem. Mater. 2015, 28, 266–273. [Google Scholar] [CrossRef]
- Folch, R.; Plapp, M. Quantitative phase-field modeling of two-phase growth. Phys. Rev. E 2005, 72, 011602. [Google Scholar] [CrossRef]
- Lu, Z.; Ciucci, F. Metal Borohydrides as Electrolytes for Solid-State Li, Na, Mg, and Ca Batteries: A First-Principles Study. Chem. Mater. 2017, 29, 9308–9319. [Google Scholar] [CrossRef]
- Bernard-Granger, G.; Guizard, C. New relationships between relative density and grain size during solid-state sintering of ceramic powders. Acta Mater. 2008, 56, 6273–6282. [Google Scholar] [CrossRef]
- Vorotilo, S.; Levashov, E.A.; Kurbatkina, V.V.; Kovalev, D.Y.; Kochetov, N.A. Self-propagating high-temperature synthesis of nanocomposite ceramics TaSi2-SiC with hierarchical structure and superior properties. J. Eur. Ceram. Soc. 2018, 38, 433–443. [Google Scholar] [CrossRef]
- Bondarev, A.V.; Vorotilo, S.; Shchetinin, I.; Levashov, E.; Shtansky, D. Fabrication of Ta-Si-C targets and their utilization for deposition of low friction wear resistant nanocomposite Si-Ta-C-(N) coatings intended for wide temperature range tribological applications. Surf. Coat. Technol. 2019, 359, 342–353. [Google Scholar] [CrossRef]
- Vorotilo, S.; Polosova, E.D.; Levashov, E.A. Peculiarities of the Synthesis of High-Temperature TaSi2–SiC Ceramics Reinforced in situ by Discrete Silicon Carbide Nanofibers. Russ. J. Non Ferr. Met. 2019, 60, 169–172. [Google Scholar] [CrossRef]
- Lin, Z.; Wang, L.; Zhang, J.; Guo, X.-Y.; Yang, W.; Mao, H.-K.; Zhao, Y. Nanoscale twinning-induced elastic strengthening in silicon carbide nanowires. Scr. Mater. 2010, 63, 981–984. [Google Scholar] [CrossRef]
- Miyamoto, T.; Kaneko, R.; Miyake, S. Tribological characteristics of amorphous carbon films investigated by point contact microscopy. J. Vac. Sci. Technol. B Microelectron. Nanometer Struct. 1991, 9, 1336. [Google Scholar] [CrossRef]
- Gutmanas, E.Y.; Gotman, I. Reactive synthesis of ceramic matrix composites under pressure. Ceram. Int. 2000, 26, 699–707. [Google Scholar] [CrossRef]
- Laurila, T.; Molarius, J.; Kivilahti, J.K. Interfacial reactions in the Si/TaC/Cu system. Microelectron. Eng. 2004, 71, 301–309. [Google Scholar] [CrossRef]
- Feng, J.C.; Naka, M.; Schuster, J.C. Phase formation and diffusion path of SiC/Ta/SiC joint. J. Mater. Sci. Lett. 1997, 16, 1116–1117. [Google Scholar] [CrossRef]
- Chen, J.S.; Kolawa, E.; Nicolet, M.-A.; Ruiz, R.P.; Baud, L.; Jaussaud, C.; Madar, R. Reaction of Ta thin film with single crystalline (001) β-SiC. J. Appl. Phys. 1994, 76, 2169–2175. [Google Scholar] [CrossRef]
- Cao, Y.; Pérez-García, S.A.; Nyborg, L. Interface Reactions and Electrical Properties of Ta/4H-SiC Contacts. Mater. Sci. Forum 2007, 556, 713–716. [Google Scholar] [CrossRef]
- Cao, Y.; Nyborg, L. Contact Formation on Silicon Carbide by Use of Nickel and Tantalum in a Materials Science Point of View. In Properties and Applications of Silicon Carbide; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Mogilevsky, P.; Gutmanas, E.Y. Solid State Reactions and Surface Diffusion in SiC-Ta and SiC-Nb Powder Systems. Mater. Sci. Forum 1994, 155, 553–556. [Google Scholar] [CrossRef]
- Imahori, J.; Oku, T.; Murakami, M. Diffusion barrier properties of TaC between Si and Cu. Thin Solid Films 1997, 301, 142–148. [Google Scholar] [CrossRef]
- Laurila, T.; Vuorinen, V.; Kivilahti, J.K. Analyses of interfacial reactions at different levels of interconnection. Mater. Sci. Semicond. Process. 2004, 7, 307–317. [Google Scholar] [CrossRef]
- Laurila, T.; Zeng, K.; Kivilahti, J.K.; Molarius, J.; Riekkinen, T.; Suni, I. Tantalum carbide and nitride diffusion barriers for Cu metallization. Microelectron Eng. 2002, 60, 71–80. [Google Scholar] [CrossRef]
- Laurila, T.; Zeng, K.; Seppälä, A.; Molarius, J.; Suni, I.; Kivilahti, J.K. Reliability of Tantalum Based Diffusion Barriers between Cu and Si. MRS Proc. 2000, 612, D7.4.1. [Google Scholar] [CrossRef]
- Bhanumurthy, K.; Schmid-Fetzer, R. Interface reactions between silicon carbide and metals (Ni, Cr, Pd, Zr). Composites Part A 2001, 32, 569–574. [Google Scholar] [CrossRef]
- Laurila, T.; Zeng, K.; Molarius, J.; Riekkinen, T.; Suni, I.; Kivilahti, J. Effect of oxygen on the reactions in Si/Ta/Cu and Si/TaC/Cu systems. Microelectron. Eng. 2002, 64, 279–287. [Google Scholar] [CrossRef]
- Laurila, T.; Zeng, K.; Kivilahti, J.K.; Molarius, J.; Suni, I. TaC as a diffusion barrier between Si and Cu. J. Appl. Phys. 2002, 91, 5391–5399. [Google Scholar] [CrossRef]
- Fahrenholtz, W.G.; Wuchina, E.J.; Lee, W.E.; Zhou, Y. Ultra-High Temperature Ceramics: Materials for Extreme Environment Applications; Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Lang, F.; Yamaguchi, H.; Nakagawa, H.; Sato, H. Thermally Stable Bonding of SiC Devices with Ceramic Substrates: Transient Liquid Phase Sintering Using Cu/Sn Powders. J. Electrochem. Soc. 2013, 160, D315–D319. [Google Scholar] [CrossRef]
- Ko, I.-Y.; Bae, S.-K.; Yoon, J.-K.; Shon, I.-J. Rapid synthesis and consolidation of nanostructured TaSi2–SiC–Si3N4 composite from mechanically activated powders by high-frequency induction-heated combustion. J. Alloys Compd. 2010, 504, 548–551. [Google Scholar] [CrossRef]
- Jun, H.S.; Kim, K.N.; Park, K.Y.; Woo, S.I. Thermal degradation of polytetrafluoroethylene in flowing helium atmosphere II. Product distribution and reaction mechanism. Korean J. Chem. Eng. 1995, 12, 183–187. [Google Scholar] [CrossRef]
- Koch, E.-C. Metal-Fluorocarbon-Pyrolants IV: Thermochemical and Combustion Behaviour of Magnesium/Teflon/Viton (MTV). Propellants Explos. Pyrotech. 2002, 27, 340–351. [Google Scholar] [CrossRef]
- Watanabe, N.; Nakajima, T.; Touhara, H. Graphite Fluorides; Elsevier: Amsterdam, The Netherlands, 1988. [Google Scholar]
- García, A.N.; Viciano, N.; Font, R.C. Products obtained in the fuel-rich combustion of PTFE at high temperature. J. Anal. Appl. Pyrolysis 2007, 80, 85–91. [Google Scholar] [CrossRef]
- Nersisyan, G.A.; Nikogosov, V.N.; Kharatyan, S.L.; Merzhanov, A.G. Chemical transformation mechanism and combustion regimes in the system silicon-carbon-fluoroplastic. Combust. Explos. Shock Waves 1992, 27, 720–724. [Google Scholar] [CrossRef]
- Pienaar, A.D.; Wagener, J.B.; Crouse, P.L. Niobium and tantalum separation by gas-phase fluorination. Int. J. Miner. Process. 2012, 114, 7–10. [Google Scholar] [CrossRef]
- Butler, J.N. The Thermal Decomposition of Octafluorocyclobutane. J. Am. Chem. Soc. 1962, 84, 1393–1398. [Google Scholar] [CrossRef]
- Li, X.; Ling, L.; Hua, X.; Fukasawa, M.; Oehrlein, G.S.; Barela, M.; Anderson, H.M. Effects of Ar and O2 additives on SiO2 etching in C4F8-based plasmas. J. Vac. Sci. Technol. A 2003, 21, 284–293. [Google Scholar] [CrossRef]
- Lu, P.; German, R. Multiple grain growth events in liquid phase sintering. J. Mater. Sci. 2001, 36, 3385–3394. [Google Scholar] [CrossRef]
- Bernard-Granger, G.; Guizard, C. Apparent Activation Energy for the Densification of a Commercially Available Granulated Zirconia Powder. J. Am. Ceram. Soc. 2007, 90, 1246–1250. [Google Scholar] [CrossRef]
- Bernard-Granger, G.; Guizard, C.; San-Miguel, L. Sintering Behavior and Optical Properties of Yttria. J. Am. Ceram. Soc. 2007, 90, 2698–2702. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Grain Growth Mechanism | Densification Controlled by Grain Boundary (gb) Diffusion | Densification Controlled by Volume (v) Diffusion |
|---|---|---|
| Grain growth controlled by the grain boundaries (gb) | (5) | (6) |
| Grain growth controlled by diffusion at the pores surface (s) | (7) | (8) |
| Grain growth controlled by the pores with a bulk diffusion (d) pathway | (9) | (10) |
| Grain growth controlled by the pores with a gas-phase (g) diffusion pathway plus an evaporation/condensation limiting step (e/c) | + (11) | (12) |
| Grain growth controlled by the pores with a gas-phase (g) diffusion pathway plus a diffusion limiting step (d) | G= (13) | (14) |
| Grain Growth Mechanism | Densification by Grain Boundary Diffusion | Densification by Volume Diffusion | ||
|---|---|---|---|---|
| TaSi2 | SiC | TaSi2 | SiC | |
| TaSiC composition, sintered at 1300–1500 °C | ||||
| Grain growth controlled by grain boundaries | 0.9926 | 0.9942 | 0.9813 | 0.9854 |
| Grain growth controlled by pore surface diffusion | 0.9341 | 0.9487 | 0.9494 | 0.9513 |
| Grain growth controlled by pore volume diffusion pathway | 0.9248 | 0.9273 | 0.9789 | 0.9183 |
| Grain growth controlled by pore gas-phase diffusion with e/c control | 0.9383 | 0.9339 | 0.9747 | 0.9284 |
| Grain growth controlled by pore gas-phase diffusion pathway with diffusion control | 0.9297 | 0.8679 | 0.9646 | 0.9175 |
| TaSiC-nw composition, sintered at 1300–1400 °C | ||||
| Grain growth controlled by grain boundaries | 0.9879 | 0.9903 | 0.9739 | 0.9630 |
| Grain growth controlled by pore surface diffusion | 0.9450 | 0.9504 | 0.9382 | 0.9429 |
| Grain growth controlled by pore volume diffusion pathway | 0.9203 | 0.9199 | 0.9378 | 0.9272 |
| Grain growth controlled by pore gas-phase diffusion with evaporation/condensation control | 0.9308 | 0.9272 | 0.9420 | 0.9507 |
| Grain growth controlled by pore gas-phase diffusion pathway with diffusion control | 0.9297 | 0.8679 | 0.9646 | 0.9175 |
| TaSiC-nw composition, sintered at 1500 °C | ||||
| Grain growth controlled by grain boundaries | 0.9236 | 0.9308 | 0.9739 | 0.9930 |
| Grain growth controlled by pore surface diffusion | 0.9551 | 0.9431 | 0.9382 | 0.9429 |
| Grain growth controlled by pore volume diffusion pathway | 0.9309 | 0.9123 | 0.9878 | 0.9572 |
| Grain growth controlled by pore gas-phase diffusion with evaporation/condensation control | 0.9528 | 0.9371 | 0.9423 | 0.9307 |
| Grain growth controlled by pore gas-phase diffusion pathway with diffusion control | 0.9197 | 0.8871 | 0.9541 | 0.9275 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vorotilo, S.; Patsera, E.; Shvindina, N.; Rupasov, S.; Levashov, E. Effect of In Situ Grown SiC Nanowires on the Pressureless Sintering of Heterophase Ceramics TaSi2-TaC-SiC. Materials 2020, 13, 3394. https://doi.org/10.3390/ma13153394
Vorotilo S, Patsera E, Shvindina N, Rupasov S, Levashov E. Effect of In Situ Grown SiC Nanowires on the Pressureless Sintering of Heterophase Ceramics TaSi2-TaC-SiC. Materials. 2020; 13(15):3394. https://doi.org/10.3390/ma13153394
Chicago/Turabian StyleVorotilo, Stepan, Evgeniy Patsera, Natalya Shvindina, Sergei Rupasov, and Evgeniy Levashov. 2020. "Effect of In Situ Grown SiC Nanowires on the Pressureless Sintering of Heterophase Ceramics TaSi2-TaC-SiC" Materials 13, no. 15: 3394. https://doi.org/10.3390/ma13153394
APA StyleVorotilo, S., Patsera, E., Shvindina, N., Rupasov, S., & Levashov, E. (2020). Effect of In Situ Grown SiC Nanowires on the Pressureless Sintering of Heterophase Ceramics TaSi2-TaC-SiC. Materials, 13(15), 3394. https://doi.org/10.3390/ma13153394