Facile Synthesis of Co3O4@CoO@Co Gradient Core@Shell Nanoparticles and Their Applications for Oxygen Evolution and Reduction in Alkaline Electrolytes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Synthesis of Co3O4/CoO/Co and Co3O4@Pt Nanoparticles
2.2. Materials Characterization
2.3. Electrochemical Analysis
3. Results
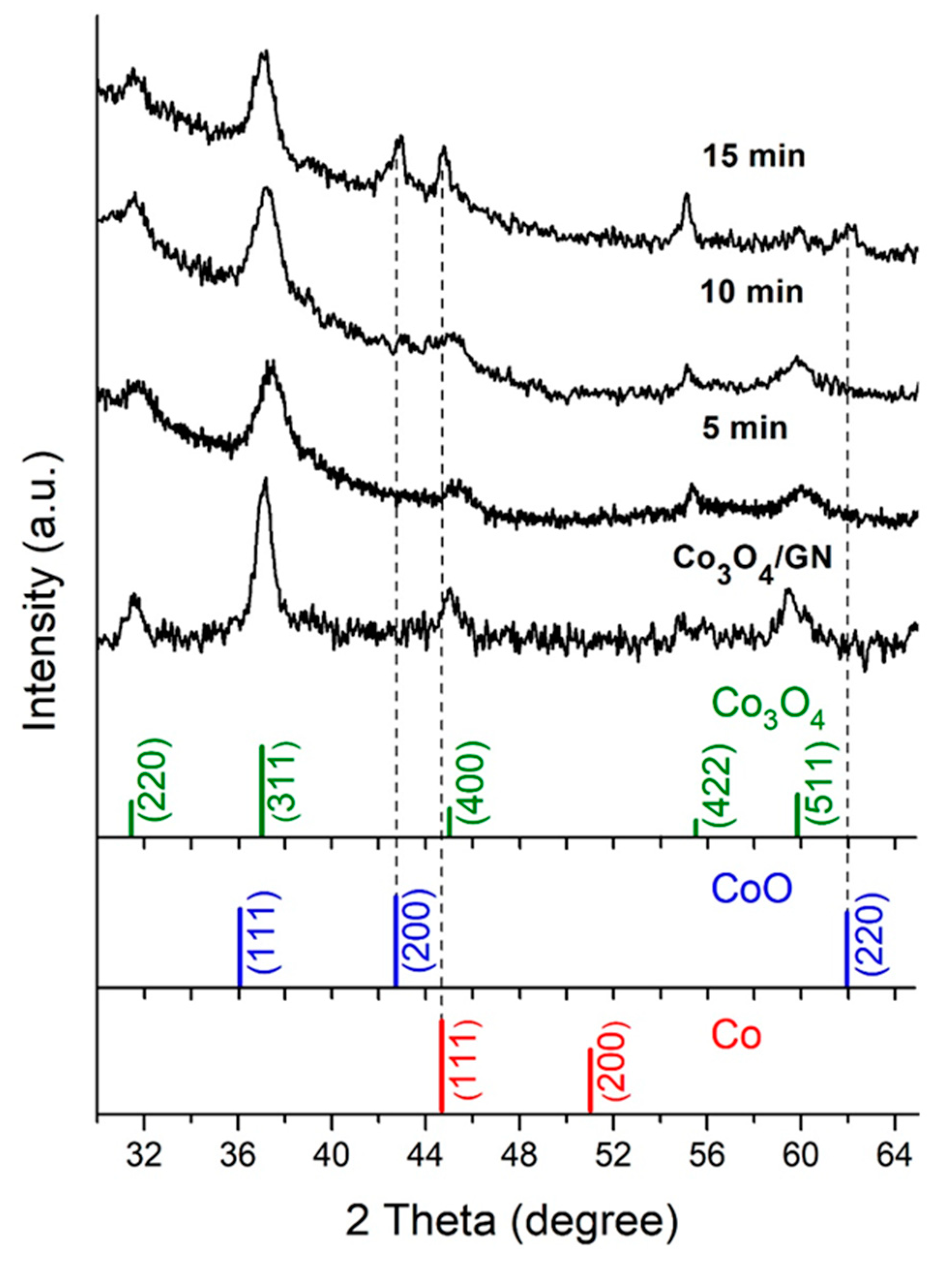
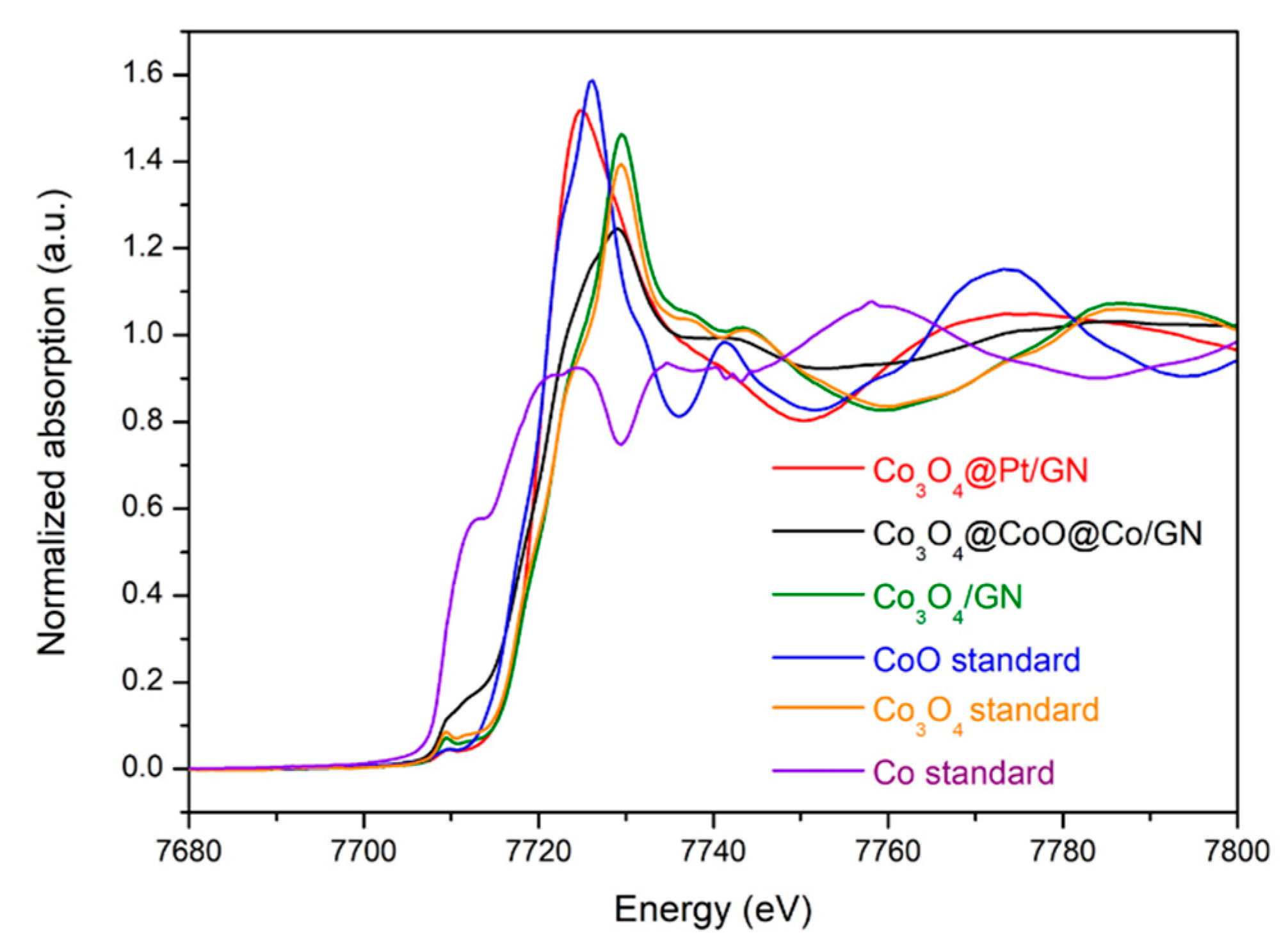
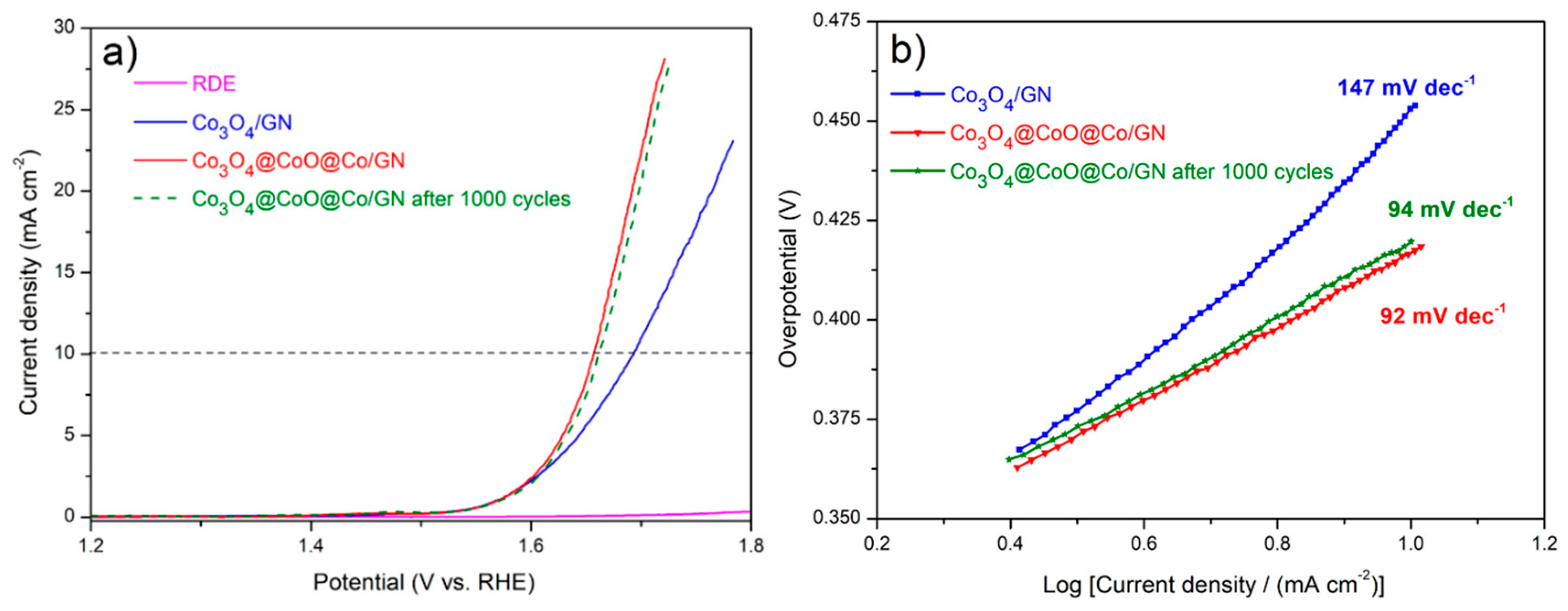
3.1. Characterizations of Co3O4@CoO@Co for OER Activities
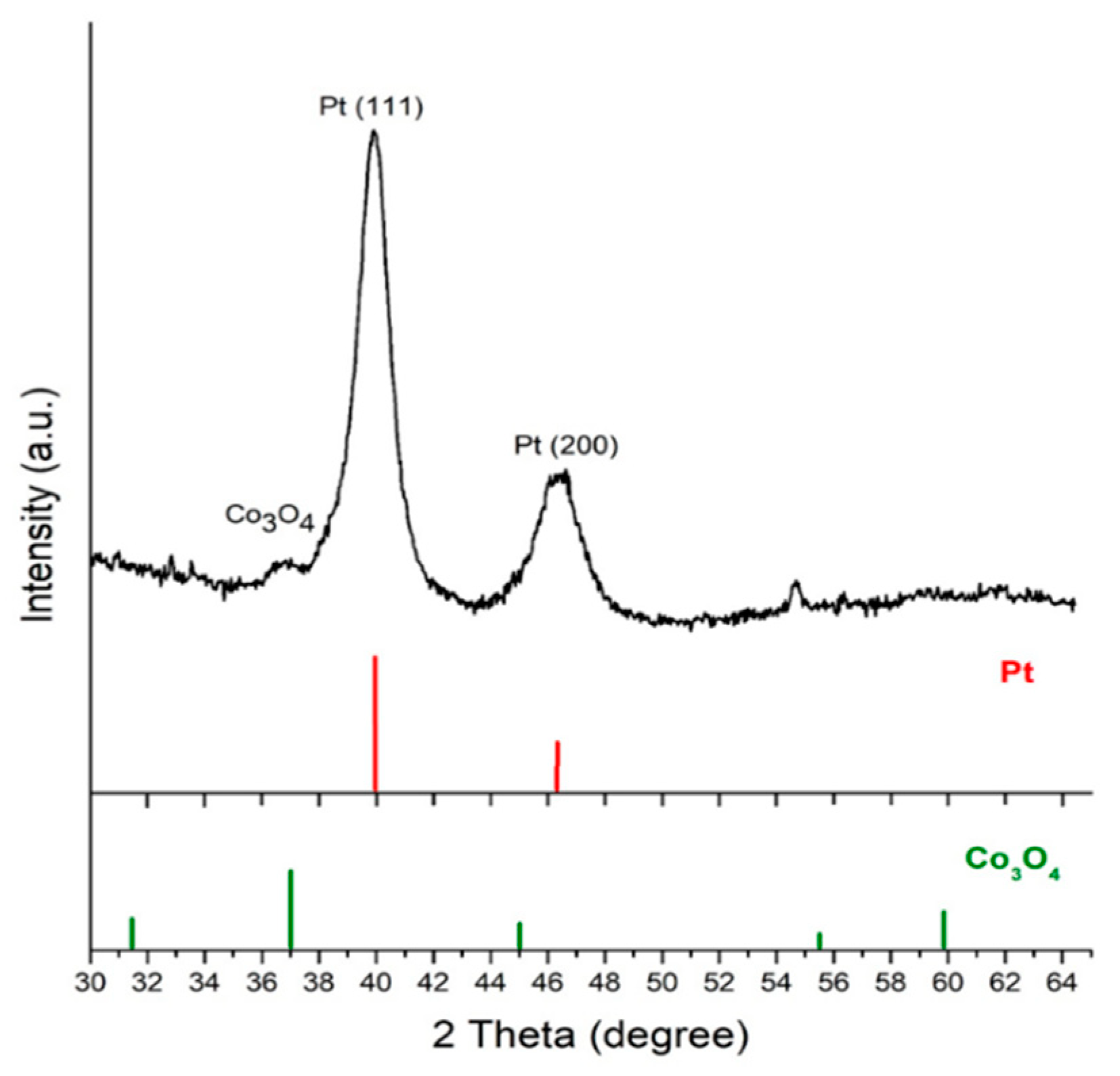
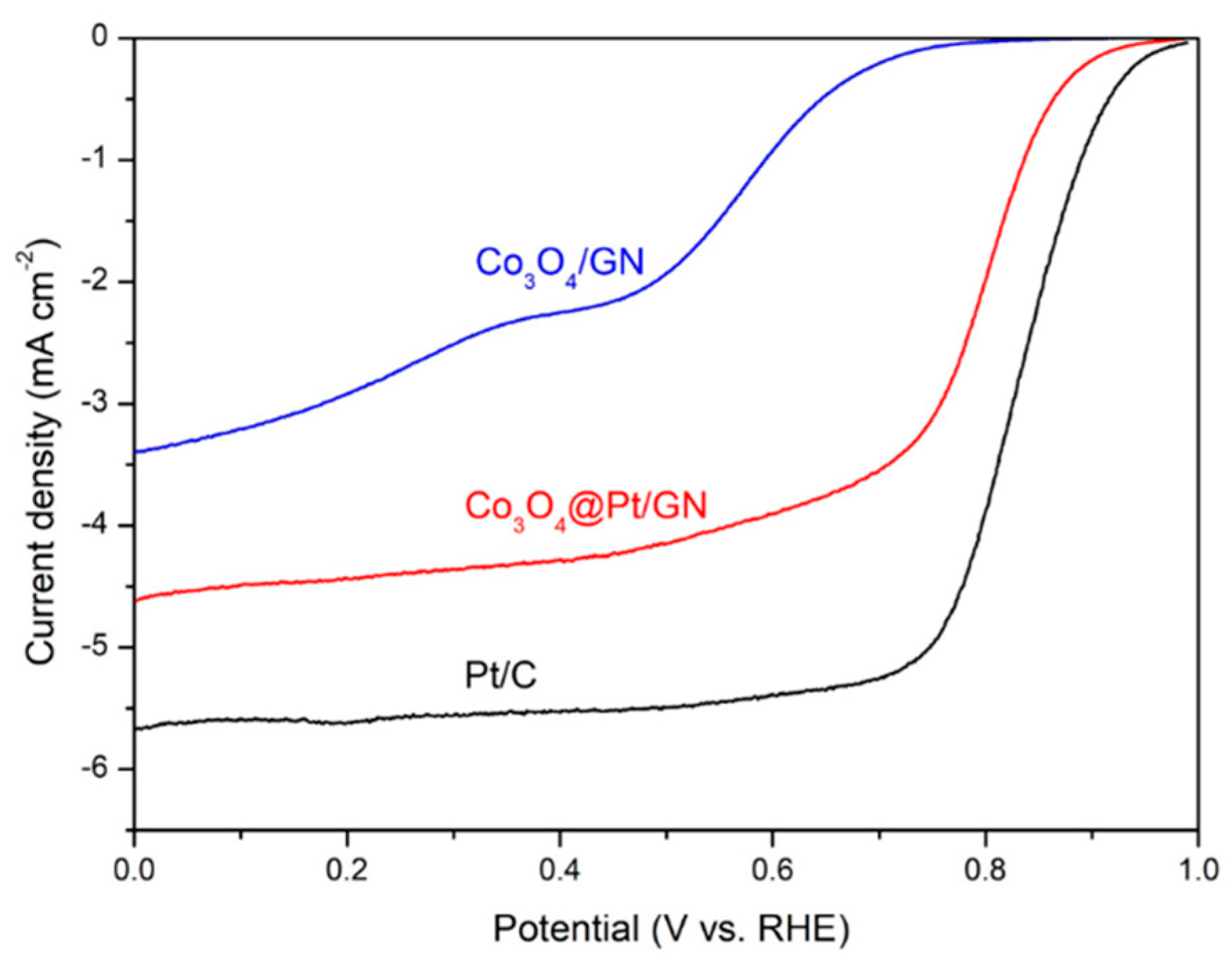
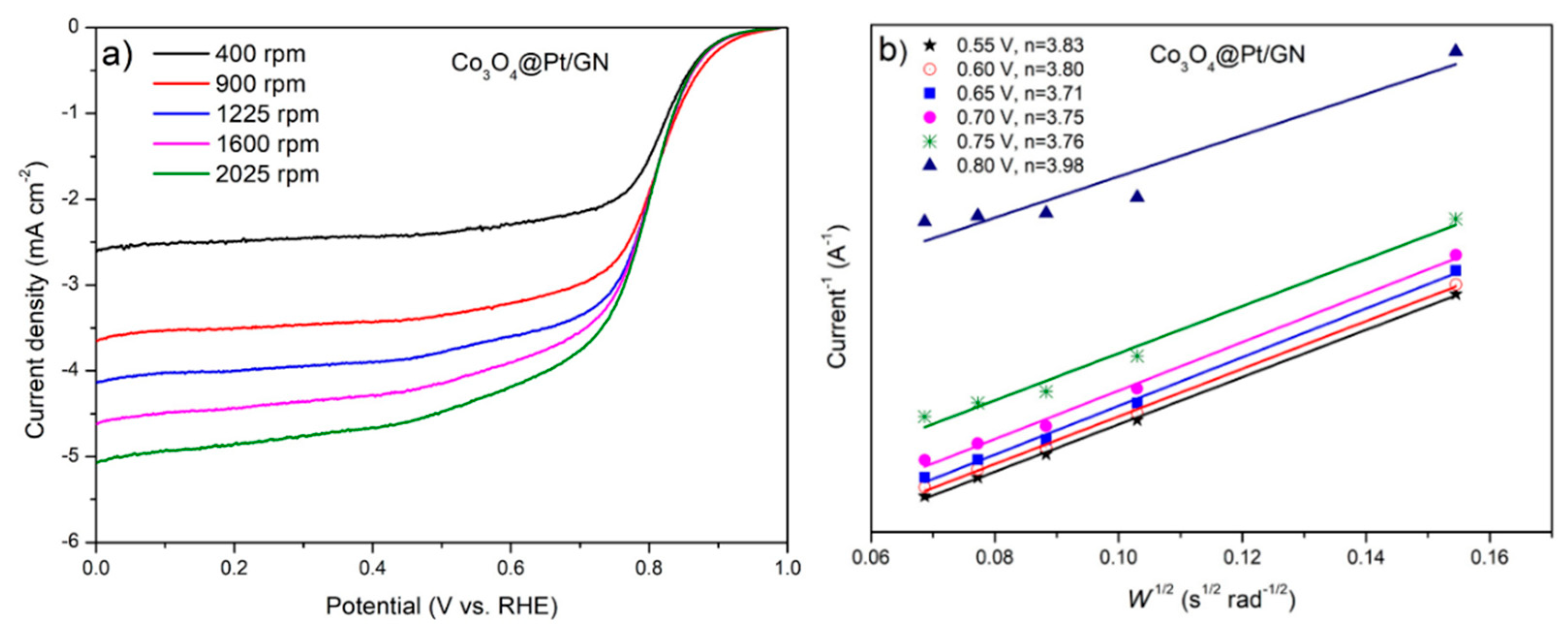
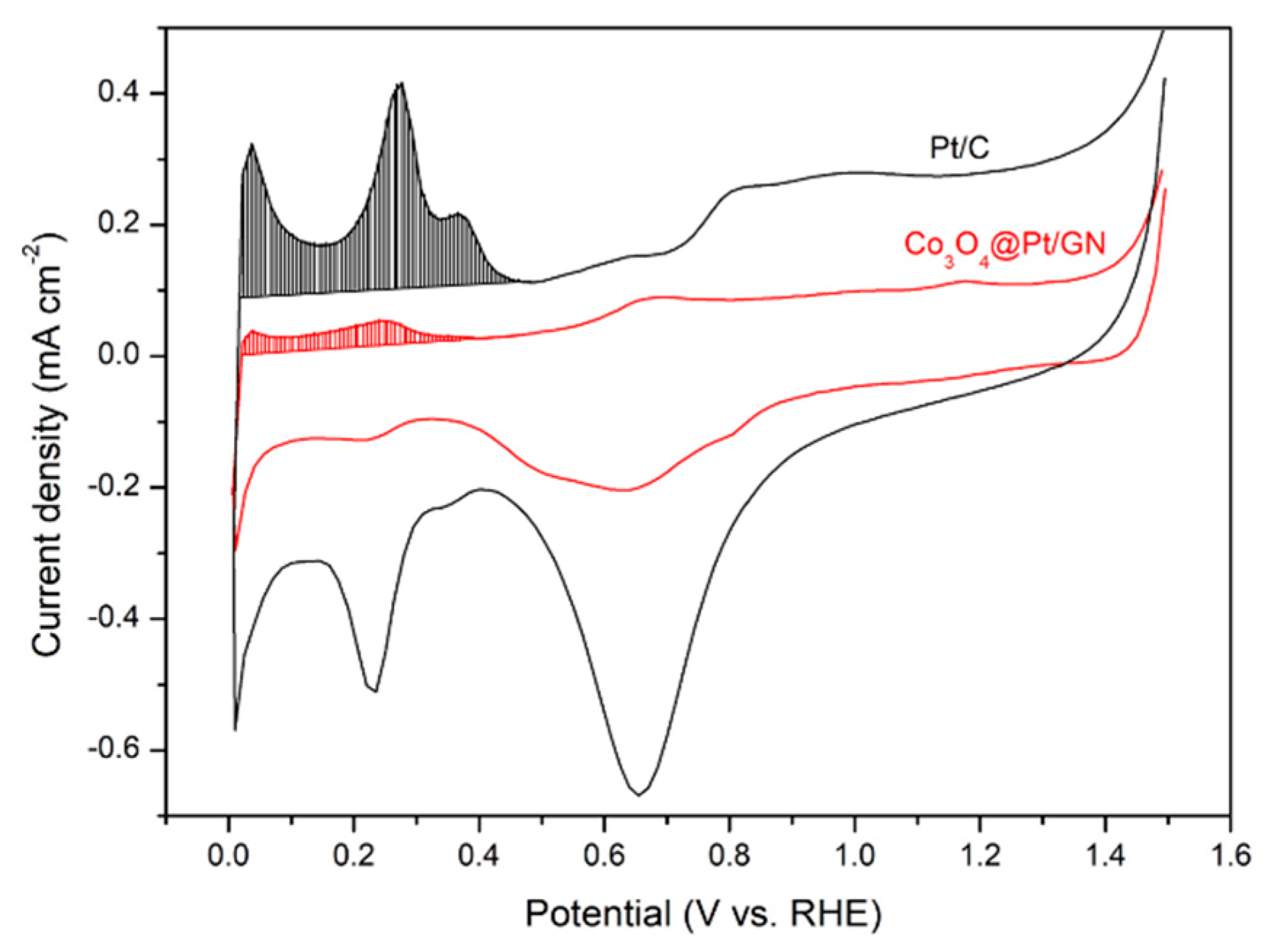
3.2. Characterization of Co3O4@Pt/GN for ORR Activities
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gorlin, Y.; Jaramillo, T.F. A bifunctional nonprecious metal catalyst for oxygen reduction and water oxidation. J. Am. Chem. Soc. 2010, 132, 13612–13614. [Google Scholar] [CrossRef] [PubMed]
- Sunarso, J.; Glushenkov, A.; Torriero, A.; Howlett, P.; Chen, Y.; MacFarlane, D.; Forsyth, M. Bi-functional water/oxygen electrocatalyst based on PdO-RuO2 composites. J. Electrochem. Soc. 2013, 160, H74–H79. [Google Scholar] [CrossRef]
- Kasian, O.; Geiger, S.; Stock, P.; Polymeros, G.; Breitbach, B.; Savan, A.; Ludwig, A.; Cherevko, S.; Mayrhofer, K.J.J. On the origin of the improved ruthenium stability in RuO2-IrO2 mixed oxides. J. Electrochem. Soc. 2016, 163, F3099–F3104. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Xiao, F.; Wang, S. Facile synthesis of heterostructured nickel/nickel oxide wrapped carbon fiber: Flexible bifunctional gas evolving electrode for highly efficient overall water splitting. ACS Sustainable Chem. Eng. 2017, 5, 529–536. [Google Scholar] [CrossRef]
- Suliman, M.A.; Suliman, M.H.; Adam, A.; Basheer, C.; Yamani, Z.H.; Qamar, M. Interfacial coupling of amorphous cobalt boride with g-C3N4 nanosheets for superior oxygen evolution reaction. Mater. Lett. 2020, 268, 127593. [Google Scholar] [CrossRef]
- Ali, Z.; Mehmood, M.; Ahmed, J.; Majeed, A.; Thebo, K.H. CVD grown defect rich-MWCNTs with anchored CoFe alloy nanoparticles for OER activity. Mater. Lett. 2020, 259, 126831. [Google Scholar] [CrossRef]
- Pan, S.; Yu, J.; Zhang, Y.; Li, B. Pulsed laser deposited Cr-doped CoFe2O4 thin film as highly efficient oxygen evolution reaction electrode. Mater. Lett. 2020, 262, 127027. [Google Scholar] [CrossRef]
- Chang, Y.M.; Wu, P.W.; Wu, C.Y.; Hsieh, Y.C. Synthesis of La0.6Ca0.4Co0.8Ir0.2O3 perovskite for bi-functional catalysis in an alkaline electrolyte. J. Power Sources 2009, 189, 1003–1007. [Google Scholar] [CrossRef]
- Liang, Y.; Li, Y.; Wang, H.; Zhou, J.; Wang, J.; Regier, T.; Dai, H. Co3O4 nanocrystals on graphene as a synergistic catalyst for oxygen reduction reaction. Nat. Mater. 2011, 10, 780–786. [Google Scholar] [CrossRef]
- Okoli, C.; Kuttiyiel, K.A.; Sasaki, K.; Su, D.; Kuila, D.; Mahajan, D.; Adzic, R.R. Highly dispersed carbon supported PdNiMo core with Pt monolayer shell electrocatalysts for oxygen reduction reaction. J. Electrochem. Soc. 2018, 165, J3295–J3300. [Google Scholar] [CrossRef]
- Wang, X.; Huang, W.; Liao, S.; Li, B. High oxygen reduction activity of TM13@Pt134 and TM12N@Pt134 (TM = Ti, V, Mn, Fe, Co, Ni, and Cu) core-shell electrocatalysts studied by first-principles theory. Mater. Chem. Phys. 2018, 212, 378–384. [Google Scholar] [CrossRef]
- Mukerjee, S. Role of structural and electronic properties of Pt and Pt alloys on electrocatalysis of oxygen reduction. J. Electrochem. Soc. 1995, 142, 1409. [Google Scholar] [CrossRef]
- García-Contreras, M.A.; Fernández-Valverde, S.M.; Basurto-Sánchez, R. Investigation of oxygen reduction in alkaline media on electrocatalysts prepared by the mechanical alloying of Pt, Co, and Ni. J. Appl. Electrochem. 2015, 45, 1101–1112. [Google Scholar] [CrossRef]
- Zhdanov, V.; Kasemo, B. Kinetics of electrochemical O2 reduction on Pt. Electrochem. Commun. 2006, 8, 1132–1136. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Tian, X.; Wang, J.; Xu, P.; Xiao, F.; Wang, S. Facile synthesis of N-doped porous carbon encapsulated bimetallic PdCo as a highly active and durable electrocatalyst for oxygen reduction and ethanol oxidation. J. Mater. Chem. A 2017, 5, 10876. [Google Scholar] [CrossRef]
- Alegre, C.; Sebastián, D.; Gálvez, M.E.; Baquedano, E.; Moliner, R.; Aricò, A.S.; Baglio, V.; Lázaro, M.J. N-doped carbon xerogels as Pt support for the electro-reduction of oxygen. Materials 2017, 10, 1092. [Google Scholar] [CrossRef]
- Lee, Y.J.; Hsieh, Y.C.; Tsai, H.C.; Lu, I.T.; Wu, Y.H.; Yu, T.H.; Lee, J.F.; Merinov, B.V.; Goddard, W.A., III; Wu, P.W. Dealloyed Pt2Os nanoparticles for enhanced oxygen reduction reaction in acidic electrolytes. Appl. Catal. B Environ. 2014, 150–151, 636–646. [Google Scholar] [CrossRef]
- Wang, J.X.; Ma, C.; Choi, Y.; Su, D.; Zhu, Y.; Liu, P.; Si, R.; Vukmirovic, M.B.; Zhang, Y.; Adzic, R.R. Kirkendall effect and lattice contraction in nanocatalysts: A new strategy to enhance sustainable activity. J. Electrochem. Soc. 2011, 133, 13551–13557. [Google Scholar] [CrossRef]
- Wang, H.; Yin, S.; Eid, K.; Li, Y.; Xu, Y.; Li, X.; Xue, H.; Wang, L. Fabrication of mesoporous cage-bell Pt nanoarchitectonics as efficient catalyst for oxygen reduction reaction. ACS Sustain. Chem. Eng. 2018, 6, 11768–11774. [Google Scholar] [CrossRef]
- Kim, O.-H.; Cho, Y.-H.; Jeon, T.-Y.; Kim, J.W.; Cho, Y.-H.; Sung, Y.-E. Realization of both high-performance and enhanced durability of fuel cells: Pt-exoskeleton structure electrocatalysts. ACS Appl. Mater. Interfaces 2015, 7, 14053–14063. [Google Scholar] [CrossRef]
- Mardle, P.; Du, S. Annealing behaviour of Pt and PtNi nanowires for proton exchange membrane fuel cells. Materials 2018, 11, 1473. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Igarashi, H.; Watanabe, M. Enhancement of the electrocatalytic O2 reduction on Pt-Fe alloys. J. Electroanal. Chem. 1999, 460, 258–262. [Google Scholar] [CrossRef]
- Koh, S.; Yu, C.; Mani, P.; Srivastava, R.; Strasser, P. Activity of ordered and disordered Pt-Co alloy phases for the electroreduction of oxygen in catalysts with multiple coexisting phases. J. Power Sources 2007, 172, 50–56. [Google Scholar] [CrossRef]
- Hwang, S.J.; Yoo, S.J.; Jang, S.; Lim, T.-H.; Hong, S.A.; Kim, S.-K. Ternary Pt-Fe-Co alloy electrocatalysts prepared by electrodeposition: Elucidating the roles of Fe and Co in the oxygen reduction reaction. J. Phys. Chem. C 2011, 115, 2483–2488. [Google Scholar] [CrossRef]
- Zhang, C.; Sandorf, W.; Peng, Z. Octahedral Pt2CuNi uniform alloy nanoparticle catalyst with high activity and promising stability for oxygen reduction reaction. ACS Catal. 2015, 5, 2296–2300. [Google Scholar] [CrossRef]
- Wang, C.; Li, D.; Chi, M.; Pearson, J.; Rankin, R.B.; Greeley, J.; Duan, Z.; Wang, G.; Vliet, D.V.D.; More, K.L.; et al. Rational development of ternary alloy electrocatalysts. J. Phys. Chem. Lett. 2012, 3, 1668–1673. [Google Scholar] [CrossRef]
- Wanjala, B.N.; Fang, B.; Shan, S.; Petkov, V.; Zhu, P.; Loukrakpam, R.; Chen, Y.; Luo, J.; Yin, J.; Yang, L.; et al. Design of ternary nanoalloy catalysts: Effect of nanoscale alloying and structural perfection on electrocatalytic enhancement. Chem. Mater. 2012, 24, 4283–4293. [Google Scholar] [CrossRef]
- Cui, C.; Gan, L.; Heggen, M.; Rudi, S.; Strasser, P. Compositional segregation in shaped Pt alloy nanoparticles and their structural behaviour during electrocatalysis. Nat. Mater. 2013, 12, 765–771. [Google Scholar] [CrossRef]
- Wang, D.; Xin, H.L.; Hovden, R.; Wang, H.; Yu, Y.; Muller, D.A.; Di Salvo, F.J.; Abruña, H.D. Structurally ordered intermetallic platinum–cobalt core–shell nanoparticles with enhanced activity and stability as oxygen reduction electrocatalysts. Nat. Mater. 2013, 12, 81–87. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, W.; Wang, J.; Minett, A.; Lo, V.; Liu, H.; Chen, J. Mesoporous hollow PtCu nanoparticles for electrocatalytic oxygen reduction reaction. J. Mater. Chem. A 2013, 1, 2391–2394. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Theoretical surface science and catalysis—calculations and concepts. Adv. Catal. 2000, 45, 71–129. [Google Scholar]
- Santos, E.; Quaino, P.; Schmickler, W. On the electrocatalysis of nanostructures: Monolayers of a foreign atom (Pd) on different substrates M(111). Electrochim. Acta 2010, 55, 4346–4352. [Google Scholar] [CrossRef]
- Sun, Y.; Hsieh, Y.-C.; Chang, L.-C.; Wu, P.-W.; Lee, J.-F. Synthesis of Pd9Ru@Pt nanoparticles for oxygen reduction reaction in acidic electrolytes. J. Power Sources 2015, 277, 116–123. [Google Scholar] [CrossRef]
- Kuo, C.-C.; Chou, S.-C.; Chang, Y.-C.; Hsieh, Y.-C.; Wu, P.-W.; Wu, W.-W. Core-shell Pd9Ru@Pt on functionalized graphene for methanol electrooxidation. J. Electrochem. Soc. 2018, 165, H365–H373. [Google Scholar] [CrossRef]
- Brankovic, S.R.; Wang, J.X.; Adžić, R.R. Metal monolayer deposition by replacement of metal adlayers on electrode surfaces. Surf. Sci. 2001, 474, L173–L179. [Google Scholar] [CrossRef]
- Zhang, J.; Mo, Y.; Vukmirovic, M.B.; Klie, R.; Sasaki, K.; Adzic, R.R. Platinum monolayer electrocatalysts for O2 reduction: Pt monolayer on Pd(111) and on carbon-supported Pd nanoparticles. J. Phys. Chem. B 2004, 108, 10955–10964. [Google Scholar] [CrossRef]
- Takasu, Y.; Oohori, K.; Yoshinaga, N.; Sugimoto, W. An examination of the oxygen reduction reaction on RuO2-based oxide coatings formed on titanium substrates. Catal. Today 2009, 146, 248–252. [Google Scholar] [CrossRef]
- Chen, D.; Chen, C.; Baiyee, Z.M.; Shao, Z.; Ciucci, F. Nonstoichiometric oxides as low-cost and highly-efficient oxygen reduction/evolution catalysts for low-temperature electrochemical devices. Chem. Rev. 2015, 115, 9869–9921. [Google Scholar] [CrossRef]
- Chang, Y.M.; Chang, Y.F.; Wu, P.W.; Wu, C.Y.; Lin, P. Synthesis and characterization of La0.6Ca0.4Co0.8Ru0.2O3 for oxygen reduction reaction in an alkaline electrolyte. J. Electrochem. Soc. 2010, 157, B900–B905. [Google Scholar] [CrossRef]
- Mathumba, P.; Fernandes, D.M.; Matos, R.; Iwuoha, E.I.; Freire, C. Metal oxide (Co3O4 and Mn3O4) impregnation into S, N-doped graphene for oxygen reduction reaction (ORR). Materials 2020, 13, 1562. [Google Scholar] [CrossRef]
- Dhavale, V.; Kurungot, S. Tuning the performance of low-Pt polymer electrolyte membrane fuel cell electrodes derived from Fe2O3@Pt/C core—shell catalyst prepared by an in situ anchoring strategy. J. Phys. Chem. C 2012, 116, 7318–7326. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Choi, B.; Kim, Y.-B. Development of highly active bifunctional electrocatalyst using Co3O4 on carbon nanotubes for oxygen reduction and oxygen evolution. Sci. Rep. 2018, 8, 2543. [Google Scholar] [CrossRef] [PubMed]
- Hassan, D.; El-safty, S.; Khalil, K.A.; Dewidar, M.; Abu El-magd, G. Carbon supported engineering NiCo2O4 hybrid nanofibers with enhanced electrocatalytic activity for oxygen reduction reaction. Materials 2016, 9, 759. [Google Scholar] [CrossRef]
- Cao, X.; Jin, C.; Lu, F.; Yang, Z.; Shen, M.; Yang, R. Electrochemical properties of MnCo2O4 spinel bifunctional catalyst for oxygen reduction and evolution reaction. J. Electrochem. Soc. 2014, 161, H296–H300. [Google Scholar] [CrossRef]
- Xu, D.; Mu, C.; Xiang, J. Carbon-encapsulated Co3O4@CoO@Co nanocomposites for multifunctional applications in enhanced long-life lithium storage, supercapacitor and oxygen evolution reaction. Electrochim. Acta 2016, 220, 322–330. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, T.; Jiang, K.; Da, P.; Peng, Z.; Tang, J.; Kong, B.; Cai, W.B.; Yang, Z.; Zheng, G. Reduced mesoporous Co3O4 Nanowires as efficient water oxidation electrocatalysts and supercapacitor electrodes. Adv. Energy Mater. 2014, 4, 1400696. [Google Scholar] [CrossRef]
- Hsieh, Y.-C.; Chang, L.-C.; Wu, P.-W.; Chang, Y.-M.; Lee, J.-F. Displacement reaction of Pt on carbon-supported Ru nanoparticles in hexachloroplatinic acids. Appl. Catal. B 2011, 103, 116–127. [Google Scholar] [CrossRef]
- Hang, L.; Sun, Y.-Q.; Men, D.; Shengwen, L.; Zhao, Q.; Cai, W.; Li, Y. Hierarchical micro/nanostructured C doped Co/Co3O4 hollow spheres derived from PS@Co(OH)2 for oxygen evolution reaction. J. Mater. Chem. A 2017, 5, 11163–11170. [Google Scholar] [CrossRef]
- Shi, F.; Geng, Z.; Huang, K.; Liang, Q.; Zhang, Y.; Sun, Y.; Cao, J.; Feng, S. Cobalt nanoparticles/black phosphorus nanosheets: An efficient catalyst for electrochemical oxygen evolution. Adv. Sci. 2018, 5, 1800575. [Google Scholar] [CrossRef]
- Paulus, U.A.; Schmidt, T.J.; Gasteiger, H.A.; Behm, R.J. Oxygen reduction on a high-surface area Pt/Vulcan carbon catalyst: A thin-film rotating ring-disk electrode study. J. Electroanal. Chem. 2001, 495, 134–145. [Google Scholar] [CrossRef]
- Kumar, M.; Raju, M.; Asokan, A.; Selvaraj, S.; Kalita, G.; Narayanan, T.; Sahu, A.K.; Pattanayak, D. Nitrogen doped graphene as metal free electrocatalyst for efficient oxygen reduction reaction in alkaline media and its application in anion exchange membrane fuel cells. J. Electrochem. Soc. 2016, 163, F848–F855. [Google Scholar] [CrossRef]
- Qaseem, A.; Chen, F.; Wu, X.; Johnston, R.L. Pt-free silver nanoalloy electrocatalysts for oxygen reduction reaction in alkaline media. Catal. Sci. Technol. 2016, 6, 3317–3340. [Google Scholar] [CrossRef]
- Li, Z.P.; Yu, X.Y.; Paik, U. Facile preparation of porous Co3O4 nanosheets for high-performance lithium ion batteries and oxygen evolution reaction. J. Power Sources 2016, 310, 41–46. [Google Scholar] [CrossRef]
- Xing, X.; Liu, R.L.; Liu, S.Q.; Xiao, S.; Xu, Y.; Wang, C.; Wu, D.Y. Surfactant-assisted hydrothermal synthesis of cobalt oxide/nitrogen-doped graphene framework for enhanced anodic performance in lithium ion batteries. Electrochim. Acta 2016, 194, 310–316. [Google Scholar] [CrossRef]
- Xie, G.; Chen, B.; Jiang, Z.; Niu, X.; Cheng, S.; Zhen, Z.; Jiang, Y.; Rong, H.; Jiang, Z.J. High catalytic activity of Co3O4 nanoparticles encapsulated in a graphene supported carbon matrix for oxygen reduction reaction. RSC Adv. 2016, 6, 50349–50357. [Google Scholar] [CrossRef]
- Zhang, T.; He, C.; Sun, F.; Ding, Y.; Wang, M.; Peng, L.; Wang, J.; Lin, Y. Co3O4 nanoparticles anchored on nitrogen-doped reduced graphene oxide as a multifunctional catalyst for H2O2 reduction, oxygen reduction and evolution reaction. Sci. Rep. 2017, 7, 43638. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chou, S.-C.; Tso, K.-C.; Hsieh, Y.-C.; Sun, B.-Y.; Lee, J.-F.; Wu, P.-W. Facile Synthesis of Co3O4@CoO@Co Gradient Core@Shell Nanoparticles and Their Applications for Oxygen Evolution and Reduction in Alkaline Electrolytes. Materials 2020, 13, 2703. https://doi.org/10.3390/ma13122703
Chou S-C, Tso K-C, Hsieh Y-C, Sun B-Y, Lee J-F, Wu P-W. Facile Synthesis of Co3O4@CoO@Co Gradient Core@Shell Nanoparticles and Their Applications for Oxygen Evolution and Reduction in Alkaline Electrolytes. Materials. 2020; 13(12):2703. https://doi.org/10.3390/ma13122703
Chicago/Turabian StyleChou, Shih-Cheng, Kuang-Chih Tso, Yi-Chieh Hsieh, Bo-Yao Sun, Jyh-Fu Lee, and Pu-Wei Wu. 2020. "Facile Synthesis of Co3O4@CoO@Co Gradient Core@Shell Nanoparticles and Their Applications for Oxygen Evolution and Reduction in Alkaline Electrolytes" Materials 13, no. 12: 2703. https://doi.org/10.3390/ma13122703
APA StyleChou, S.-C., Tso, K.-C., Hsieh, Y.-C., Sun, B.-Y., Lee, J.-F., & Wu, P.-W. (2020). Facile Synthesis of Co3O4@CoO@Co Gradient Core@Shell Nanoparticles and Their Applications for Oxygen Evolution and Reduction in Alkaline Electrolytes. Materials, 13(12), 2703. https://doi.org/10.3390/ma13122703