How to Boost the Activity of the Monolayer Pt Supported on TiC Catalysts for Oxygen Reduction Reaction: A Density Functional Theory Study


Abstract
1. Introduction
2. The Calculation Details
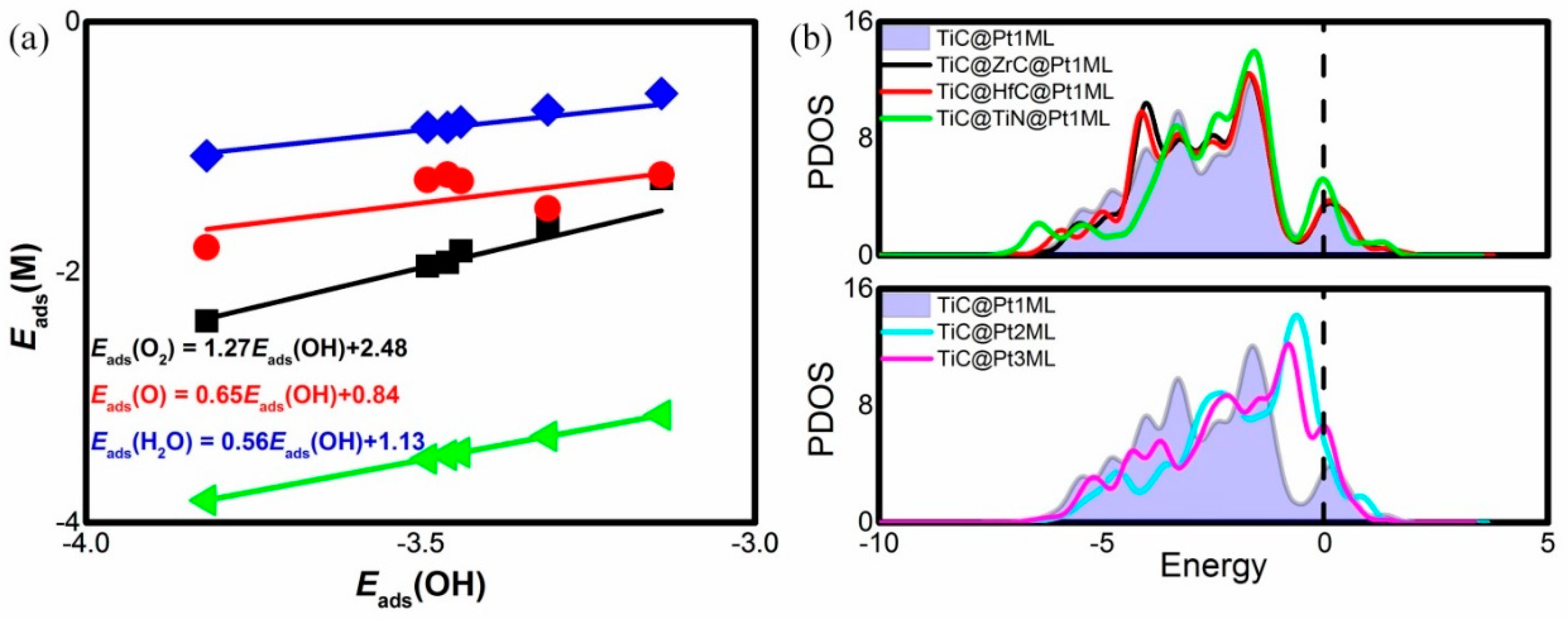
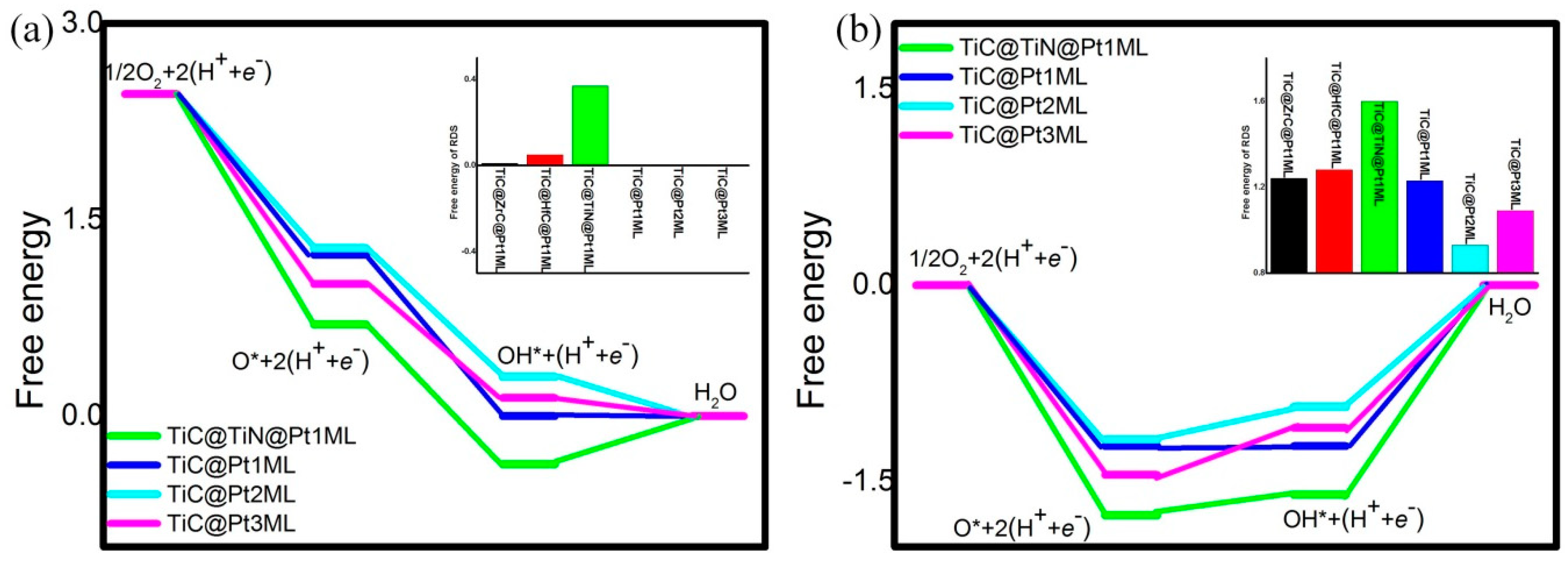
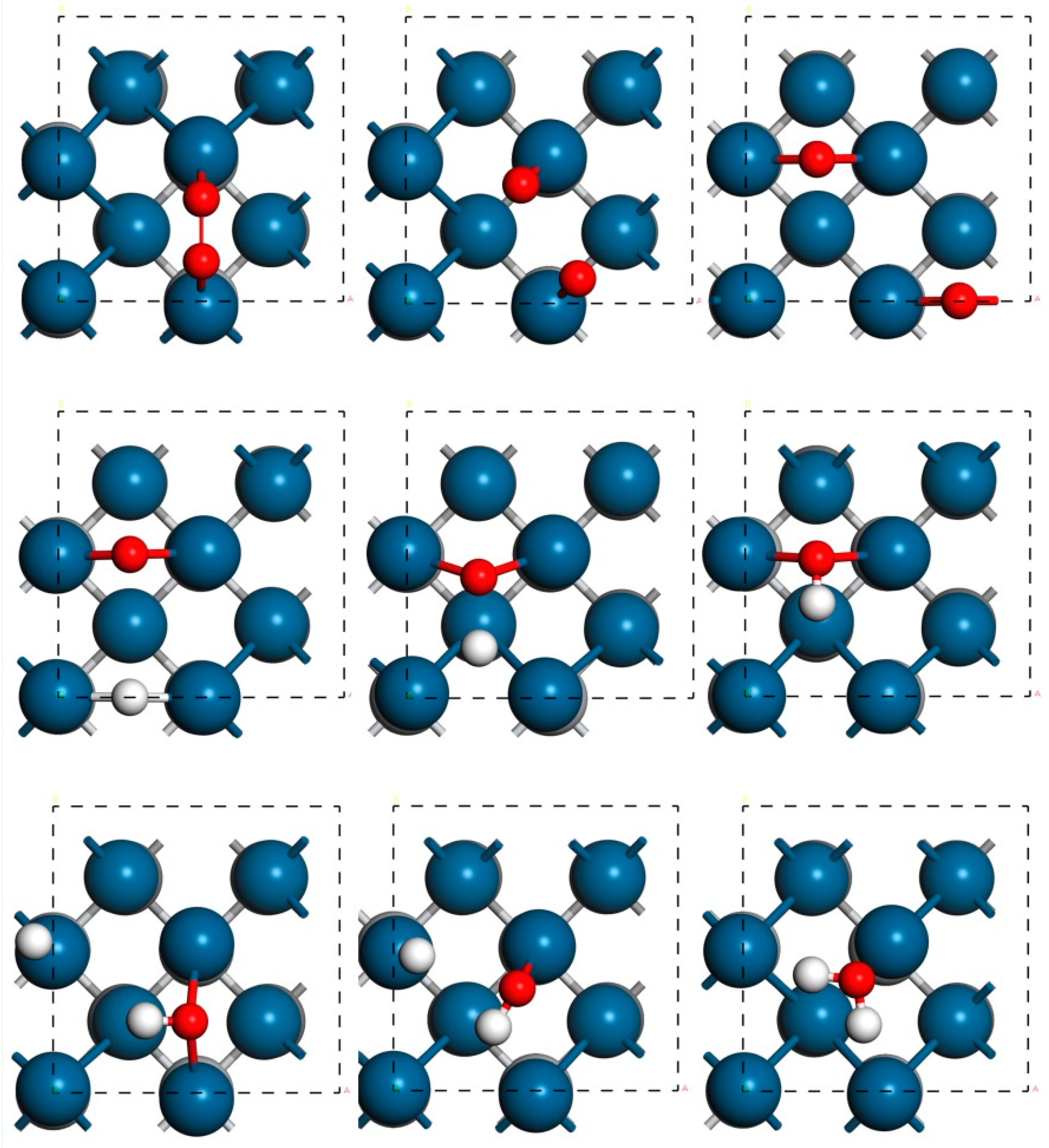
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stacy, J.; Regmi, Y.N.; Leonard, B.; Fan, M. The recent progress and future of oxygen reduction reaction catalysis: A review. Renew. Sustain. Energy Rev. 2017, 69, 401–414. [Google Scholar] [CrossRef]
- Chen, D.; Xu, Y.; Tade, M.O.; Shao, Z. General Regulation of Air Flow Distribution Characteristics within Planar Solid Oxide Fuel Cell Stacks. ACS Energy Lett. 2017, 2, 319–326. [Google Scholar] [CrossRef]
- Wei, T.; Zhang, Z.; Zhu, Z.; Zhou, X.; Wang, Y.; Wang, Y.; Zhuang, Q. Recycling of waste plastics and scalable preparation of Si/CNF/C composite as anode material for lithium-ion batteries. Ionics 2019. [Google Scholar] [CrossRef]
- Zhang, L.; Cai, Z.; Yao, Z.; Ji, L.; Sun, Z.; Yan, N.; Zhang, B.; Xiao, B.; Du, J.; Zhu, X.-Q.; et al. A striking catalytic effect of facile synthesized ZrMn2 nanoparticles on the de/rehydrogenation properties of MgH2. J. Mater. Chem. A 2019, 7, 5626–5634. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Debe, M.K. Electrocatalyst approaches and challenges for automotive fuel cells. Nat. Cell Boil. 2012, 486, 43–51. [Google Scholar] [CrossRef]
- Xiao, B.B.; Jiang, X.B.; Jiang, Q. Density functional theory study of oxygen reduction reaction on Pt/Pd3Al(111) alloy electrocatalyst. Phys. Chem. Chem. Phys. 2016, 18, 14234–14243. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.B.; Jiang, X.B.; Yang, X.L.; Zheng, F. The segregation resistance of the Pt2ML/Os/Pd3Al sandwich catalyst for oxygen reduction reaction: a density functional theory study. Phys. Chem. Chem. Phys. 2016, 18, 30174–30182. [Google Scholar] [CrossRef]
- Xie, S.; Choi, S.-I.; Lu, N.; Roling, L.; Herron, J.A.; Zhang, L.; Park, J.; Wang, J.; Kim, M.J.; Xie, Z.; et al. Atomic Layer-by-Layer Deposition of Pt on Pd Nanocubes for Catalysts with Enhanced Activity and Durability toward Oxygen Reduction. Nano Lett. 2014, 14, 3570–3576. [Google Scholar] [CrossRef]
- Choi, R.; Choi, S.-I.; Choi, C.H.; Nam, K.M.; Woo, S.I.; Park, J.T.; Han, S.W.H. Designed synthesis of well-defined Pd@Pt core-shell nanoparticles with controlled shell thickness as efficient oxygen reduction electrocatalysts. Chem. A Eur. J. 2013, 19, 8190–8198. [Google Scholar] [CrossRef]
- Yang, L.; Vukmirovic, M.B.; Su, D.; Sasaki, K.; Herron, J.A.; Mavrikakis, M.; Liao, S.; Adzic, R.R. Tuning the Catalytic Activity of Ru@Pt Core–Shell Nanoparticles for the Oxygen Reduction Reaction by Varying the Shell Thickness. J. Phys. Chem. C 2013, 117, 1748–1753. [Google Scholar] [CrossRef]
- Todoroki, N.; Watanabe, H.; Kondo, T.; Kaneko, S.; Wadayama, T. Highly Enhanced Oxygen Reduction Reaction Activity and Electrochemical Stability of Pt/Ir(111) Bimetallic Surfaces. Electrochim. Acta 2016, 222, 1616–1621. [Google Scholar] [CrossRef]
- Tian, X.; Tang, H.; Luo, J.; Nan, H.; Shu, T.; Du, L.; Zeng, J.; Liao, S.; Adzic, R.R. High-Performance Core–Shell Catalyst with Nitride Nanoparticles as a Core: Well-Defined Titanium Copper Nitride Coated with an Atomic Pt Layer for the Oxygen Reduction Reaction. ACS Catal. 2017, 7, 3810–3817. [Google Scholar] [CrossRef]
- Hwu, H.H.; Chen, J.G. Surface Chemistry of Transition Metal Carbides. ChemInform 2005, 36, 185–212. [Google Scholar] [CrossRef]
- Ou, Y.; Cui, X.; Zhang, X.; Jiang, Z. Titanium carbide nanoparticles supported Pt catalysts for methanol electrooxidation in acidic media. J. Power Sources 2010, 195, 1365–1369. [Google Scholar] [CrossRef]
- Park, H.-U.; Lee, E.; Kwon, Y.-U. TiC supported Pt-based nanoparticles: Facile sonochemical synthesis and electrocatalytic properties for methanol oxidation reaction. Int. J. Hydrogen Energy 2017, 42, 19885–19893. [Google Scholar] [CrossRef]
- Kimmel, Y.C.; Yang, L.; Kelly, T.G.; Rykov, S.A.; Chen, J.G. Theoretical prediction and experimental verification of low loading of platinum on titanium carbide as low-cost and stable electrocatalysts. J. Catal. 2014, 312, 216–220. [Google Scholar] [CrossRef]
- Chiwata, M.; Kakinuma, K.; Wakisaka, M.; Uchida, M.; Deki, S.; Watanabe, M.; Uchida, H. Oxygen Reduction Reaction Activity and Durability of Pt Catalysts Supported on Titanium Carbide. Catalysts 2015, 5, 966–980. [Google Scholar] [CrossRef]
- Roca-Ayats, M.; García, G.; Galante, J.; Peña, M.A.; Martínez-Huerta, M.; Huerta, M.V.M. Electrocatalytic stability of Ti based-supported Pt3Ir nanoparticles for unitized regenerative fuel cells. Int. J. Hydrogen Energy 2014, 39, 5477–5484. [Google Scholar] [CrossRef]
- Escudero-Escribano, M.; Verdaguer-Casadevall, A.; Malacrida, P.; Grønbjerg, U.; Knudsen, B.P.; Jepsen, A.K.; Rossmeisl, J.; Stephens, I.E.L.; Chorkendorff, I. Pt5Gd as a Highly Active and Stable Catalyst for Oxygen Electroreduction. J. Am. Chem. Soc. 2012, 134, 16476–16479. [Google Scholar] [CrossRef]
- Stamenkovic, V.R.; Mun, B.S.; Arenz, M.; Mayrhofer, K.J.J.; Lucas, C.A.; Wang, G.; Ross, P.N.; Marković, N.M. Trends in electrocatalysis on extended and nanoscale Pt-bimetallic alloy surfaces. Nat. Mater. 2007, 6, 241–247. [Google Scholar] [CrossRef]
- Kuttiyiel, K.A.; Sasaki, K.; Choi, Y.; Su, D.; Liu, P.; Adzic, R.R. Nitride stabilized PtNi core-shell nanocatalyst for high oxygen reduction activity. Nano Lett. 2012, 12, 6266–6271. [Google Scholar] [CrossRef] [PubMed]
- Escaño, M.C.S. First-principles calculations of the dissolution and coalescence properties of Pt nanoparticle ORR catalysts: The effect of nanoparticle shape. Nano Res. 2015, 8, 1689–1697. [Google Scholar] [CrossRef]
- Regmi, Y.N.; Waetzig, G.R.; Duffee, K.D.; Schmuecker, S.M.; Thode, J.M.; Leonard, B.M. Carbides of group IVA, VA and VIA transition metals as alternative HER and ORR catalysts and support materials. J. Mater. Chem. A 2015, 3, 10085–10091. [Google Scholar] [CrossRef]
- Seifitokaldani, A.; Savadogo, O.; Perrier, M. Density Functional Theory (DFT) Computation of the Oxygen Reduction Reaction (ORR) on Titanium Nitride (TiN) Surface. Electrochim. Acta 2014, 141, 25–32. [Google Scholar] [CrossRef]
- Kim, H.; Cho, M.K.; Kwon, J.A.; Jeong, Y.H.; Lee, K.J.; Kim, N.Y.; Kim, M.J.; Yoo, S.J.; Jang, J.H.; Kim, H.-J.; et al. Highly efficient and durable TiN nanofiber electrocatalyst supports. Nanoscale 2015, 7, 18429–18434. [Google Scholar] [CrossRef]
- Wang, C.; Chi, M.; Li, D.; Strmcnik, D.; Van Der Vliet, D.; Wang, G.; Komanicky, V.; Chang, K.-C.; Paulikas, A.P.; Tripkovic, D.; et al. Design and Synthesis of Bimetallic Electrocatalyst with Multilayered Pt-Skin Surfaces. J. Am. Chem. Soc. 2011, 133, 14396–14403. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Xiao, H.; Goddard, W.A. Reaction Mechanisms for the Electrochemical Reduction of CO2 to CO and Formate on the Cu(100) Surface at 298 K from Quantum Mechanics Free Energy Calculations with Explicit Water. J. Am. Chem. Soc. 2016, 138, 13802–13805. [Google Scholar] [CrossRef]
- Strasser, P.; Koh, S.; Anniyev, T.; Greeley, J.; More, K.; Yu, C.; Liu, Z.; Kaya, S.; Nordlund, D.; Ogasawara, H.; et al. Lattice-strain control of the activity in dealloyed core–shell fuel cell catalysts. Nat. Chem. 2010, 2, 454–460. [Google Scholar] [CrossRef]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Delley, B. Hardness conserving semilocal pseudopotentials. Phys. Rev. B 2002, 66, 155125. [Google Scholar] [CrossRef]
- Xiao, B.B.; Liu, H.Y.; Jiang, X.B.; Yu, Z.D.; Jiang, Q. A bifunctional two dimensional TM3(HHTP)2 monolayer and its variations for oxygen electrode reactions. RSC Adv. 2017, 7, 54332–54340. [Google Scholar] [CrossRef]
- Xiao, B.; Zhu, H.; Liu, H.; Jiang, X.; Jiang, Q. The Activity Improvement of the TM3(hexaiminotriphenylene)2 Monolayer for Oxygen Reduction Electrocatalysis: A Density Functional Theory Study. Front. Chem. 2018, 6, 351. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Li, S.; Zhang, Y.; Chu, X.; Yang, Z. Density functional study on the mechanism for the highly active palladium monolayer supported on titanium carbide for the oxygen reduction reaction. J. Chem. Phys. 2016, 144, 204703. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, Z. TiC and TiN supported platinum monolayer as high-performance catalysts for CO oxidation: A DFT study. J. Chem. Phys. 2018, 149, 054705. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Wang, G. Comparison of Reaction Energetics for Oxygen Reduction Reactions on Pt(100), Pt(111), Pt/Ni(100), and Pt/Ni(111) Surfaces: A First-Principles Study. J. Phys. Chem. C 2013, 117, 6284–6292. [Google Scholar] [CrossRef]
- Wang, S.; Chu, X.; Zhang, X.; Zhang, Y.; Mao, J.; Yang, Z. A First-Principles Study of O2 Dissociation on Platinum Modified Titanium Carbide: A Possible Efficient Catalyst for the Oxygen Reduction Reaction. J. Phys. Chem. C 2017, 121, 21333–21342. [Google Scholar] [CrossRef]
- Sha, Y.; Yu, T.H.; Merinov, B.V.; Shirvanian, P.; Goddard, W.A. Mechanism for Oxygen Reduction Reaction on Pt3Ni Alloy Fuel Cell Cathode. J. Phys. Chem. C 2012, 116, 21334–21342. [Google Scholar] [CrossRef]
- Baran, J.D.; Grönbeck, H.; Hellman, A. Analysis of Porphyrines as Catalysts for Electrochemical Reduction of O2 and Oxidation of H2O. J. Am. Chem. Soc. 2014, 136, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Hammer, B.; Nørskov, J. Theoretical surface science and catalysis—Calculations and concepts. Adv. Catal. 2000, 45, 71–129. [Google Scholar]
- Xin, H.; Linic, S. Communications: Exceptions to the d-band model of chemisorption on metal surfaces: The dominant role of repulsion between adsorbate states and metal d-states. J. Chem. Phys. 2010, 132, 221101. [Google Scholar] [CrossRef]
- Liu, W.; Lian, J.S.; Jiang, Q. Theoretical Study of C2H2 Adsorbed on Low-Index Cu Surfaces. J. Phys. Chem. C 2007, 111, 18189–18194. [Google Scholar] [CrossRef]
- Gao, G.; Bottle, S.E.; Du, A. Understanding the activity and selectivity of single atom catalysts for hydrogen and oxygen evolution via ab initial study. Catal. Sci. Technol. 2018, 8, 996–1001. [Google Scholar] [CrossRef]
- Viswanathan, V.; Heine, A.H.; Rossmeisl, J.; Nørskov, J.K. Universality in Oxygen Reduction Electrocatalysis on Metal Surfaces. ACS Catal. 2012, 2, 1654–1660. [Google Scholar] [CrossRef]
- Shao, M.; Shoemaker, K.; Peles, A.; Kaneko, K.; Protsailo, L. Pt Monolayer on Porous Pd−Cu Alloys as Oxygen Reduction Electrocatalysts†. J. Am. Chem. Soc. 2010, 132, 9253–9255. [Google Scholar] [CrossRef] [PubMed]
- Sha, Y.; Yu, T.H.; Merinov, B.V.; Shirvanian, P.; Goddard, W.A. Oxygen Hydration Mechanism for the Oxygen Reduction Reaction at Pt and Pd Fuel Cell Catalysts. J. Phys. Chem. Lett. 2011, 2, 572–576. [Google Scholar] [CrossRef]
- Yang, B.; Burch, R.; Hardacre, C.; Headdock, G.; Hu, P. Understanding the Optimal Adsorption Energies for Catalyst Screening in Heterogeneous Catalysis. ACS Catal. 2013, 4, 182–186. [Google Scholar] [CrossRef]
- Shang, C.; Liu, Z.-P. Origin and Activity of Gold Nanoparticles as Aerobic Oxidation Catalysts in Aqueous Solution. J. Am. Chem. Soc. 2011, 133, 9938–9947. [Google Scholar] [CrossRef]
- Greeley, J.; Stephens, I.E.L.; Bondarenko, A.S.; Johansson, T.P.; Hansen, H.A.; Jaramillo, T.F.; Rossmeisl, J.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T. Alloys of platinum and early transition metals as oxygen reduction electrocatalysts. Nat. Chem. 2009, 1, 552–556. [Google Scholar] [CrossRef]
- Stephens, I.E.L.; Bondarenko, A.S.; Grønbjerg, U.; Rossmeisl, J.; Chorkendorff, I. Understanding the electrocatalysis of oxygen reduction on platinum and its alloys. Energy Environ. Sci. 2012, 5, 6744. [Google Scholar] [CrossRef]
- Lee, D.H.; Lee, W.J.; Lee, W.J.; Kim, S.O.; Kim, Y.-H. Theory, Synthesis, and Oxygen Reduction Catalysis of Fe-Porphyrin-Like Carbon Nanotube. Phys. Lett. 2011, 106, 175502. [Google Scholar] [CrossRef]
- Favaro, M.; Ferrighi, L.; Fazio, G.; Colazzo, L.; Di Valentin, C.; Durante, C.; Sedona, F.; Gennaro, A.; Agnoli, S.; Granozzi, G. Single and Multiple Doping in Graphene Quantum Dots: Unraveling the Origin of Selectivity in the Oxygen Reduction Reaction. ACS Catal. 2015, 5, 129–144. [Google Scholar] [CrossRef]
- Jia, Y.; Zhang, L.; Du, A.; Gao, G.; Chen, J.; Yan, X.; Brown, C.L.; Yao, X. Defect Graphene as a Trifunctional Catalyst for Electrochemical Reactions. Adv. Mater. 2016, 28, 9532–9538. [Google Scholar] [CrossRef]
- Man, I.C.; Su, H.-Y.; Hansen, H.A.; Martinez, J.I.; Inoglu, N.G.; Kitchin, J.; Jaramillo, T.F.; Nørskov, J.K.; Rossmeisl, J.; Calle-Vallejo, F.; et al. Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces. ChemCatChem 2011, 3, 1159–1165. [Google Scholar] [CrossRef]
- Zhang, P.; Hou, X.; Liu, L.; Mi, J.-L.; Dong, M. Two Dimensional π-Conjugated Metal Bis(dithiolene) Complex Nanosheets as Selective Catalysts for Oxygen Reduction Reaction. J. Phys. Chem. C 2015, 119, 28028–28037. [Google Scholar] [CrossRef]
- Lang, X.Y.; Han, G.F.; Xiao, B.B.; Gu, L.; Yang, Z.Z.; Wen, Z.; Zhu, Y.F.; Zhao, M.; Li, J.C.; Jiang, Q. Mesostructured Intermetallic Compounds of Platinum and Non-Transition Metals for Enhanced Electrocatalysis of Oxygen Reduction Reaction. Adv. Funct. Mater. 2015, 25, 230–237. [Google Scholar] [CrossRef]
- Hunt, S.T.; Milina, M.; Alba-Rubio, A.C.; Hendon, C.H.; Dumesic, J.A.; Román-Leshkov, Y. Self-assembly of noble metal monolayers on transition metal carbide nanoparticle catalysts. Science 2016, 352, 974–978. [Google Scholar] [CrossRef]
- Tsai, H.-C.; Lee, Y.-J.; Merinov, B.V.; Wu, P.-W.; Yu, T.H.; Goddard, W.A.; Hsieh, Y.-C.; Wu, Y.-H.; Chen, S.-Y.; Adzic, R.R. DFT Study of Oxygen Reduction Reaction on Os/Pt Core–Shell Catalysts Validated by Electrochemical Experiment. ACS Catal. 2015, 5, 1568–1580. [Google Scholar] [CrossRef]
- Wang, J.X.; Inada, H.; Wu, L.; Zhu, Y.; Choi, Y.M.; Liu, P.; Zhou, W.-P.; Adzic, R.R. Oxygen Reduction on Well-Defined Core−Shell Nanocatalysts: Particle Size, Facet, and Pt Shell Thickness Effects. J. Am. Chem. Soc. 2009, 131, 17298–17302. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst System | Eads(O2) | Eads(O) | Eads(OH) | Eads(H2O) |
|---|---|---|---|---|
| TiC@Pt1ML | −1.83 | −1.27 | −3.44 | −0.8 |
| TiC@ZrC@Pt1ML | −1.92 | −1.22 | −3.46 | −0.84 |
| TiC@HfC@Pt1ML | −1.95 | −1.26 | −3.49 | −0.84 |
| TiC@TiN@Pt1ML | −2.39 | −1.80 | −3.82 | −1.07 |
| TiC@Pt2ML | −1.25 | −1.22 | −3.14 | −0.57 |
| TiC@Pt3ML | −1.63 | −1.49 | −3.31 | −0.70 |
| Catalyst System | d Band Center | Mulliken Charge | Dbef | Daft | d |
|---|---|---|---|---|---|
| TiC@Pt1ML | −2.85 | −0.225 | 3.04 | 3.01 | 2.80 |
| TiC@ZrC@Pt1ML | −2.78 | −0.275 | 3.06 | 3.00 | 2.81 |
| TiC@HfC@Pt1ML | −2.80 | −0.275 | 3.06 | 3.00 | 2.80 |
| TiC@TiN@Pt1ML | −2.40 | −0.108 | 3.07 | 3.00 | 2.75 |
| TiC@Pt2ML | −1.83 | −0.107 | 3.06 | 3.09 | 3.32 |
| TiC@Pt3ML | −2.05 | −0.029 | 2.93 | 3.07 | 3.28 |
| Catalyst System | U = 0 V | U = 1.23 V | ||||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R1 | R2 | R3 | |
| TiC@Pt1ML | −1.23 | −1.23 | 0 | −1.23 | 0 | 1.23 |
| TiC@ZrC@Pt1ML | −1.18 | −1.29 | 0.01 | −1.18 | −0.06 | 1.24 |
| TiC@HfC@Pt1ML | −1.22 | −1.29 | 0.05 | −1.22 | −0.06 | 1.28 |
| TiC@TiN@Pt1ML | −1.76 | −1.07 | 0.37 | −1.76 | 0.16 | 1.60 |
| TiC@Pt2ML | −1.18 | −0.98 | −0.30 | −1.18 | 0.25 | 0.93 |
| TiC@Pt3ML | −1.45 | −0.87 | −0.14 | −1.45 | 0.36 | 1.09 |
| Catalyst System | O2→2O | O + H→OH | OH + H→H2O | |||
|---|---|---|---|---|---|---|
| Ea | Er | Ea | Er | Ea | Er | |
| TiC@Pt1ML | 1.05 | −0.30 | 0.46 | −1.08 | 1.04 | −0.22 |
| TiC@TiN@Pt1ML | 0.76 | −0.70 | 0.55 | −0.90 | 1.41 | 0.68 |
| TiC@Pt2ML | 1.28 | −0.97 | 0.69 | −0.46 | 0.91 | 0.04 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, H.; Liu, H.; Yang, L.; Xiao, B. How to Boost the Activity of the Monolayer Pt Supported on TiC Catalysts for Oxygen Reduction Reaction: A Density Functional Theory Study. Materials 2019, 12, 1560. https://doi.org/10.3390/ma12091560
Zhu H, Liu H, Yang L, Xiao B. How to Boost the Activity of the Monolayer Pt Supported on TiC Catalysts for Oxygen Reduction Reaction: A Density Functional Theory Study. Materials. 2019; 12(9):1560. https://doi.org/10.3390/ma12091560
Chicago/Turabian StyleZhu, Hui, Houyi Liu, Lei Yang, and Beibei Xiao. 2019. "How to Boost the Activity of the Monolayer Pt Supported on TiC Catalysts for Oxygen Reduction Reaction: A Density Functional Theory Study" Materials 12, no. 9: 1560. https://doi.org/10.3390/ma12091560
APA StyleZhu, H., Liu, H., Yang, L., & Xiao, B. (2019). How to Boost the Activity of the Monolayer Pt Supported on TiC Catalysts for Oxygen Reduction Reaction: A Density Functional Theory Study. Materials, 12(9), 1560. https://doi.org/10.3390/ma12091560