Reactivity of Different Crystalline Surfaces of C3S During Early Hydration by the Atomistic Approach



Abstract
1. Introduction
2. Methods and Modeling Approach
Model Construction
3. Results and Discussion
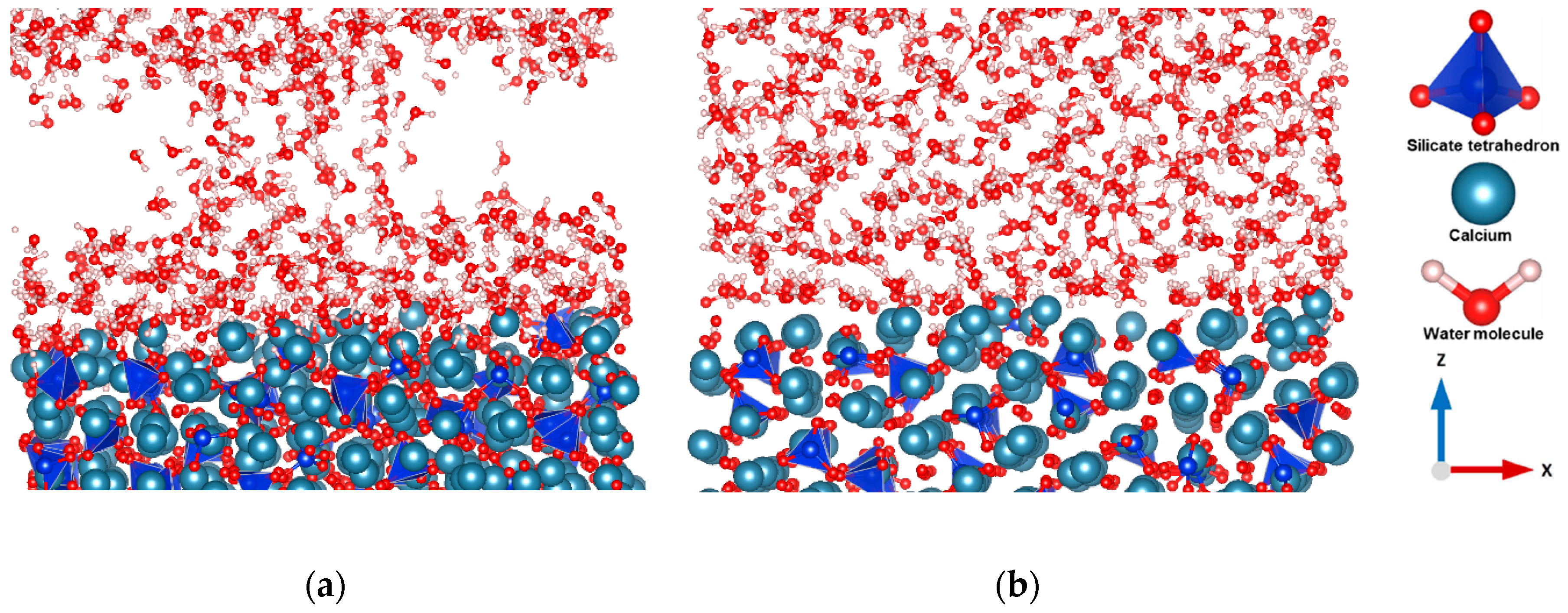
3.1. Hydration of C3S
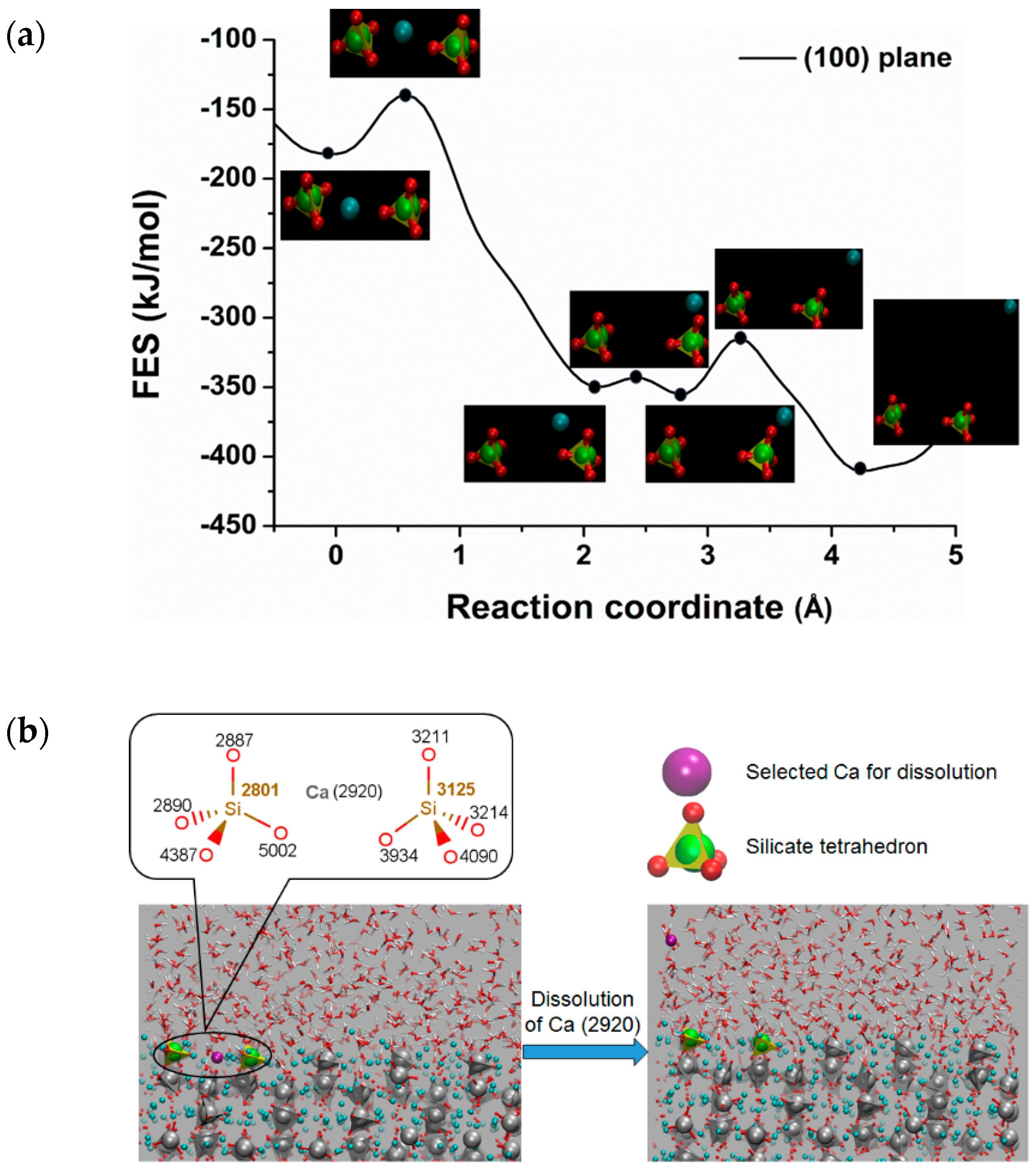
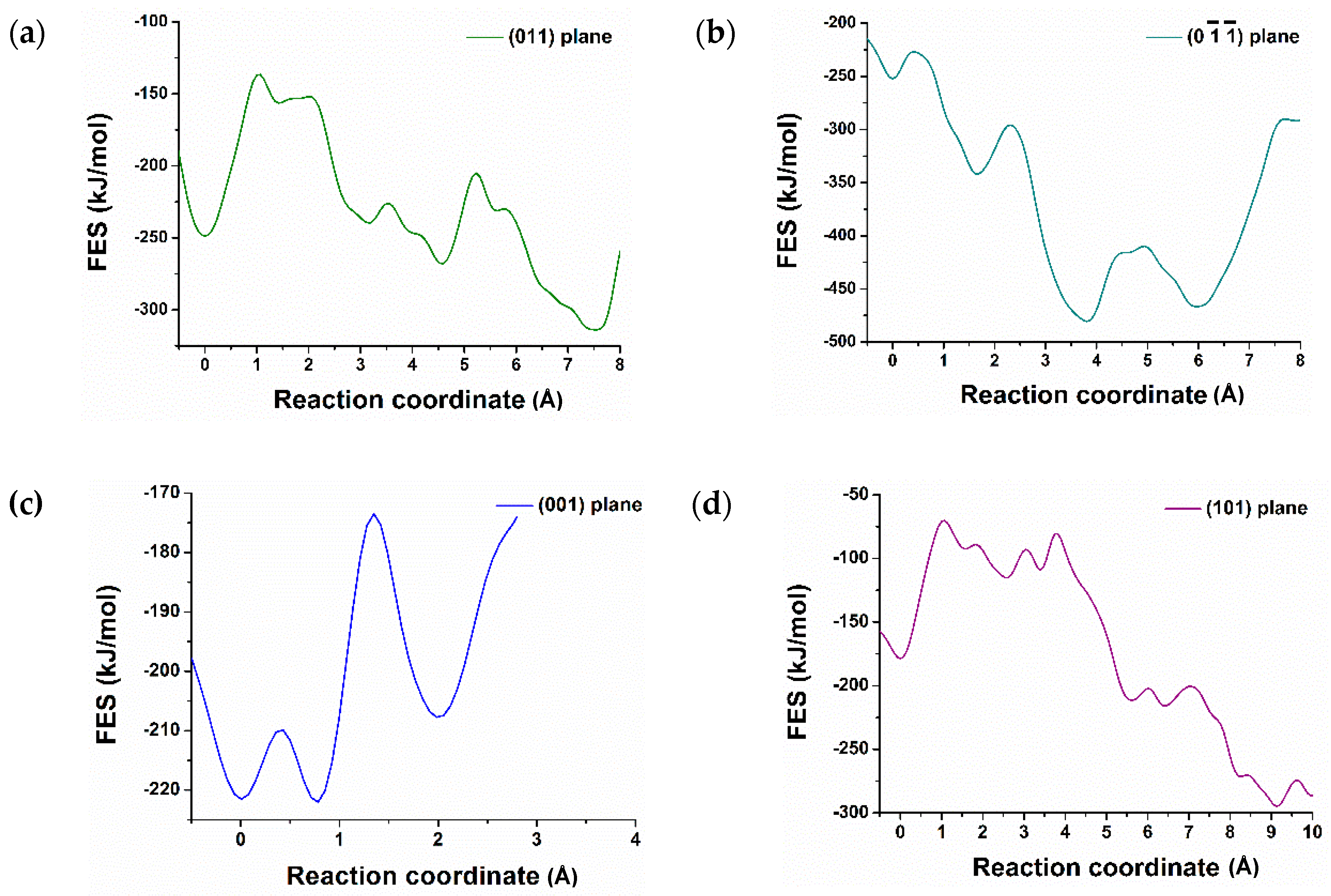
3.2. Dissolution of Calcium from C3S
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Plane of C3S | Orthogonal Cell Dimension (Å3 / 10-30 m3) | No of Atoms in the Simulation Cell |
|---|---|---|
| (100) | 56.49, 34.39, 37.06 | 5799 |
| (101) | 32.96, 34.39, 59.06 | 6003 |
| (001) | 28.37, 50.45, 49.91 | 5514 |
| (010) | 32.28, 37.78, 47.66 | 5115 |
| () | 32.77, 38.35, 48.37 | 5115 |
| (110) | 38.35, 43.35, 46.45 | 6538 |
| () | 38.35, 43.35, 46.45 | 6536 |
| (011) | 32.77, 47.71, 47.81 | 5250 |
| () | 32.77, 47.71, 47.81 | 5248 |
| MD | Molecular Dynamics |
|---|---|
| C3S | Tricalcium silicate (Ca3SiO5, alite) |
| C-S-H | Calcium Silicate Hydrates |
| ReaxFF | Reactive Force Field |
| LAMMPS | Large-scale Atomic/Molecular Massively Parallel Simulator |
| metaD | Metadynamics |
| TS | Transition State |
| CVs | Collective Variables |
| FES | Free Energy Surfaces |
| VNL | Virtual Nano Lab |
| hftn | Hessian-free truncated Newton algorithm |
| nvt | Nose-Hoover thermostat |
| npt | Nose-Hoover pressure barostat |
References
- Hewlett, P.; Hewlett, P.C.; Lea, F.M. Lea’s Chemistry of Cement and Concrete, 4th ed.; Hewlett, P.C., Ed.; Butterworth-Heinemann: Oxford, UK, 2003; ISBN 9780080535418. [Google Scholar]
- Taylor, H.F.W. Cement Chemistry, 2nd ed.; Thomas Telford: London, UK, 1997; ISBN 0727725920. [Google Scholar]
- Worrell, E.; Price, L.; Martin, N.; Hendriks, C.; Meida, L.O. Carbon dioxide emissions from the global cement industry. Annu. Rev. Energy. Environ. 2001, 26, 303–329. [Google Scholar] [CrossRef]
- Prabhu, A.; Gimel, J.-C.; Ayuela, A.; Arrese-Igor, S.; Gaitero, J.J.; Dolado, J.S. A multi-scale approach for percolation transition and its application to cement setting. Sci. Rep. 2018, 8, 15830. [Google Scholar] [CrossRef] [PubMed]
- Jennings, H.M.; Johnson, S.K. Simulation of Microstructure Development During the Hydration of a Cement Compound. J. Am. Ceram. Soc. 1986, 69, 790–795. [Google Scholar] [CrossRef]
- Koenders, E.A.B. Simulation of Volume Changes in Hardening Cement-Based Materials; Technische Universiteit Delft: Delft, The Netherlands, 1997; ISBN 90-407-1499-1. [Google Scholar]
- Van Breugel, K. Numerical simulation of hydration and microstructural development in hardening cement-based materials (I) theory. Cem. Concr. Res. 1995, 25, 319–331. [Google Scholar] [CrossRef]
- Chenoweth, K.; van Duin, A.C.T.; Goddard, W.A. ReaxFF reactive force field for molecular dynamics simulations of hydrocarbon oxidation. J. Phys. Chem. A 2008, 112, 1040–1053. [Google Scholar] [CrossRef]
- Fogarty, J.C.; Aktulga, H.M.; Grama, A.Y.; van Duin, A.C.T.; Pandit, S.A. A reactive molecular dynamics simulation of the silica-water interface. J. Chem. Phys. 2010, 132, 174704. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.; Deng, W.-Q.; van Duin, A.C.T.; Goddard, W.A. ReaxFF(MgH) reactive force field for magnesium hydride systems. J. Phys. Chem. A 2005, 109, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Chenoweth, K.; Cheung, S.; van Duin, A.C.T.; Goddard, W.A.; Kober, E.M. Simulations on the thermal decomposition of a poly(dimethylsiloxane) polymer using the ReaxFF reactive force field. J. Am. Chem. Soc. 2005, 127, 7192–7202. [Google Scholar] [CrossRef] [PubMed]
- Barducci, A.; Bonomi, M.; Parrinello, M. Metadynamics. WIREs Comput. Mol. Sci. 2011, 1, 826–843. [Google Scholar] [CrossRef]
- Manzano, H.; Durgun, E.; López-Arbeloa, I.; Grossman, J.C. Insight on Tricalcium Silicate Hydration and Dissolution Mechanism from Molecular Simulations. ACS Appl. Mater. Interfaces 2015, 7, 14726–14733. [Google Scholar] [CrossRef]
- Van Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: A Reactive Force Field for Hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef]
- Manzano, H.; Pellenq, R.J.M.; Ulm, F.-J.; Buehler, M.J.; van Duin, A.C.T. Hydration of calcium oxide surface predicted by reactive force field molecular dynamics. Langmuir 2012, 28, 4187–4197. [Google Scholar] [CrossRef]
- Laio, A.; Gervasio, F.L. Metadynamics: A method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 2008, 71, 126601. [Google Scholar] [CrossRef]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef]
- Schneider, J.; Hamaekers, J.; Chill, S.T.; Smidstrup, S.; Bulin, J.; Thesen, R.; Blom, A.; Stokbro, K. ATK-ForceField: A new generation molecular dynamics software package. Model. Simul. Mater. Sci. Eng. 2017, 25, 85007. [Google Scholar] [CrossRef]
- Stradi, D.; Jelver, L.; Smidstrup, S.; Stokbro, K. Method for determining optimal supercell representation of interfaces. J. Phys. Condens. Matter 2017, 29, 185901. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Tuckerman, M.E.; Alejandre, J.; López-Rendón, R.; Jochim, A.L.; Martyna, G.J. A Liouville-operator derived measure-preserving integrator for molecular dynamics simulations in the isothermal–isobaric ensemble. J. Phys. A Math. Gen. 2006, 39, 5629–5651. [Google Scholar] [CrossRef]
- Salah Uddin, K.M.; Middendorf, B. Atomistic modeling of early Hydration of C3S. In Computational Modelling of Concrete Structures: Proceedings of the Conference on Computational Modelling of Concrete and Concrete Structures (EURO-C 2018), Bad Hofgastein, Austria, 26 February–1 March, 2018; Meschke, G., Pichler, B., Rots, J.G., Eds.; CRC Press/Balkema: Leiden, The Netherlands, 2018; ISBN 9781315182964. [Google Scholar]
- Salah Uddin, K.M.; Middendorf, B. Insight into the Reactivity of Different Crystalline Phases of C3S during Early Hydration by the Atomistic Approach. In Proceedings of the 20th International Conference on Building Materials, Bauhaus-Universität, Weimar, Germany, 12–14 September 2018. [Google Scholar]




| Crystal Plane of C3S | The Atomic ID of the Selected Ca for Dissolution | Free Energy of Activation kJ/mol | Free Energy Change kJ/mol | Thermodynamic Properties |
|---|---|---|---|---|
| () | 2994 | 25.60 | −214.20 | Exergonic |
| (100) | 2920 | 46.00 | −225.90 | Exergonic |
| (011) | 3013 | 112.80 | −65.00 | Exergonic |
| (101) | 3048 | 108.40 | −116.10 | Exergonic |
| (001) | 1957 | 50.00 | +14.00 | Endergonic |
| (010) | 2471 | 169.70 | +36.00 | Endergonic |
| () | 2352 | 267.10 | +202.10 | Endergonic |
| (110) | 3043 | 319.10 | +291.50 | Endergonic |
| () | 2998 | 584.70 | +502.00 | Endergonic |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salah Uddin, K.M.; Middendorf, B. Reactivity of Different Crystalline Surfaces of C3S During Early Hydration by the Atomistic Approach. Materials 2019, 12, 1514. https://doi.org/10.3390/ma12091514
Salah Uddin KM, Middendorf B. Reactivity of Different Crystalline Surfaces of C3S During Early Hydration by the Atomistic Approach. Materials. 2019; 12(9):1514. https://doi.org/10.3390/ma12091514
Chicago/Turabian StyleSalah Uddin, K. M., and Bernhard Middendorf. 2019. "Reactivity of Different Crystalline Surfaces of C3S During Early Hydration by the Atomistic Approach" Materials 12, no. 9: 1514. https://doi.org/10.3390/ma12091514
APA StyleSalah Uddin, K. M., & Middendorf, B. (2019). Reactivity of Different Crystalline Surfaces of C3S During Early Hydration by the Atomistic Approach. Materials, 12(9), 1514. https://doi.org/10.3390/ma12091514